

Abstract
Genetic diseases that affect the cardiovascular system are relatively common and include cardiac channelopathies, cardiomyopathies, aortopathies, hypercholesterolemias, and structural diseases of the heart and great vessels. The rapidly expanding availability of clinical genetic testing leverages decades of research into the genetic origins of these diseases, helping inform diagnosis, clinical management, and prognosis. Although a number of guidelines and statements detail best practices for cardiovascular genetic testing, there is a paucity of pediatric-focused statements addressing the unique challenges in testing in this vulnerable population. In this scientific statement, we seek to coalesce the existing literature around the use of genetic testing for cardiovascular disease in infants, children, and adolescents.
Keywords: AHA Scientific Statements; cardiomyopathies; channelopathies; connective tissue diseases; counseling, genetic; predictive genetic testing
Genetic testing plays an important role in the diagnosis and management of heritable cardiovascular diseases, including cardiac channelopathies, cardiomyopathies, aortopathies, hypercholesterolemias, and structural diseases of the heart and great vessels.
INTRODUCTION AND SURVEY OF THE LITERATURE
With broadening availability of clinical genetic testing, pediatric patients are increasingly undergoing genetic testing for cardiovascular diseases. This raises complex and important issues of consent, family concerns and values, family risk communication, and timing and scope of testing, among others. In the absence of robust clinical evidence to inform practice, this statement draws on existing clinical practice guidelines, scientific statements, expert consensus statements, and key studies in the field to synthesize recommendations for these issues. These include prior statements from the American Heart Association, Heart Rhythm Society, American College of Medical Genetics and Genomics, and European Society of Cardiology. Literature that was in English and was human subject based or clinically focused was included. As a result of the rapid evolution of genetic medicine, literature was limited primarily to those published in or after 2010. Statements or position articles on the ethical, legal, and psychosocial implications of pediatric genetic testing were included from 1995 or later with emphasis on more recent adaptations. Given the wide array of genetic abnormalities that can be associated with cardiovascular disease, the scope of this statement focuses on cardiovascular diseases that are traditionally considered non-syndromic and have predominantly cardiac involvement.
BASIC PRINCIPLES OF CARDIOVASCULAR GENETIC TESTING
Clinical cardiovascular genetic testing includes methods to analyze genetic alterations at the gene/chromosome or DNA sequence level.1 There are 2 categories of genetic testing most common to cardiology, offering differing resolution: cytogenetic and molecular genetic testing. The former involves analysis at the chromosome level, for example, chromosome microarray; the latter involves Sanger sequencing or next-generation sequencing technologies to identify abnormalities at the nucleotide level. The clinical indication drives testing, with cytogenetics used largely to evaluate suspected syndromic congenital heart defects (CHDs) in which extracardiac abnormalities are identified. Conversely, molecular genetic testing is used more widely for monogenic cardiac diseases such as cardiomyopathies and channelopathies. Next-generation sequencing technologies are becoming more common, primarily gene panels and exome/genome sequencing, as first-tier tests for many diseases.
The goal of clinical genetic testing is to identify genetic variation known to cause disease, although the presence of a genetic variant does not always manifest in disease (variable penetrance) and the same variant can manifest in different ways clinically (variable expressivity). Variants are classified on the basis of health consequence across a spectrum of anticipated pathogenicity from benign and likely benign to likely pathogenic and pathogenic.2 Variants that are unable to be classified in this manner are designated variants of uncertain significance, which should be reviewed with caution in practice.3,4 The predicted effect of a variant on health is probabilistic according to the disease, genetic origins, gene-environment interactions, and penetrance/expressivity. The clinical context under which genetic testing is obtained is critical to the interpretation of findings, which makes the clinical evaluation and accurate phenotypic classification key to determining the scope of genetic testing.4 Typically, genetic testing should be limited to genes associated with the clinical phenotype identified, whereas broader genetic testing such as exome/genome sequencing may be considered in cases that do not meet diagnostic criteria for a single, well-defined syndrome but a genetic origin is suspected.
DIAGNOSTIC AND RISK-PREDICTIVE GENETIC TESTING
In general, genetic testing is performed for diagnostic or risk-predictive purposes (Figure 1). In diagnostic genetic testing, a positive genetic test confirms an existing clinical suspicion for disease in a patient. In risk-predictive genetic testing, if a pathogenic/likely pathogenic (P/LP) variant found in a proband is identified in an asymptomatic family member, that identifies the carrier as being at risk of developing the disease. Risk-predictive genetic testing is the basis for family cascade testing.5 Cascade testing involves testing additional at-risk relatives of an affected family member in enlarging circles of relatedness. That is, the identification of genetically at-risk individuals triggers genetic testing of their first-degree relatives, leading to a cascading expansion of testing throughout the family. Those confirmed to carry the family’s P/LP variant (genetic risk) are then referred for longitudinal screening to assess for phenotypic evolution and family counseling. Those who test negative can typically be reassured that they have not inherited the familial genetic risk. Finally, given the evolving nature of variant pathogenicity classification, in particular variants of uncertain significance, continued follow-up is important to re-evaluate variant pathogenicity over time.
Figure 1. Algorithm for diagnostic and risk-predictive genetic testing.
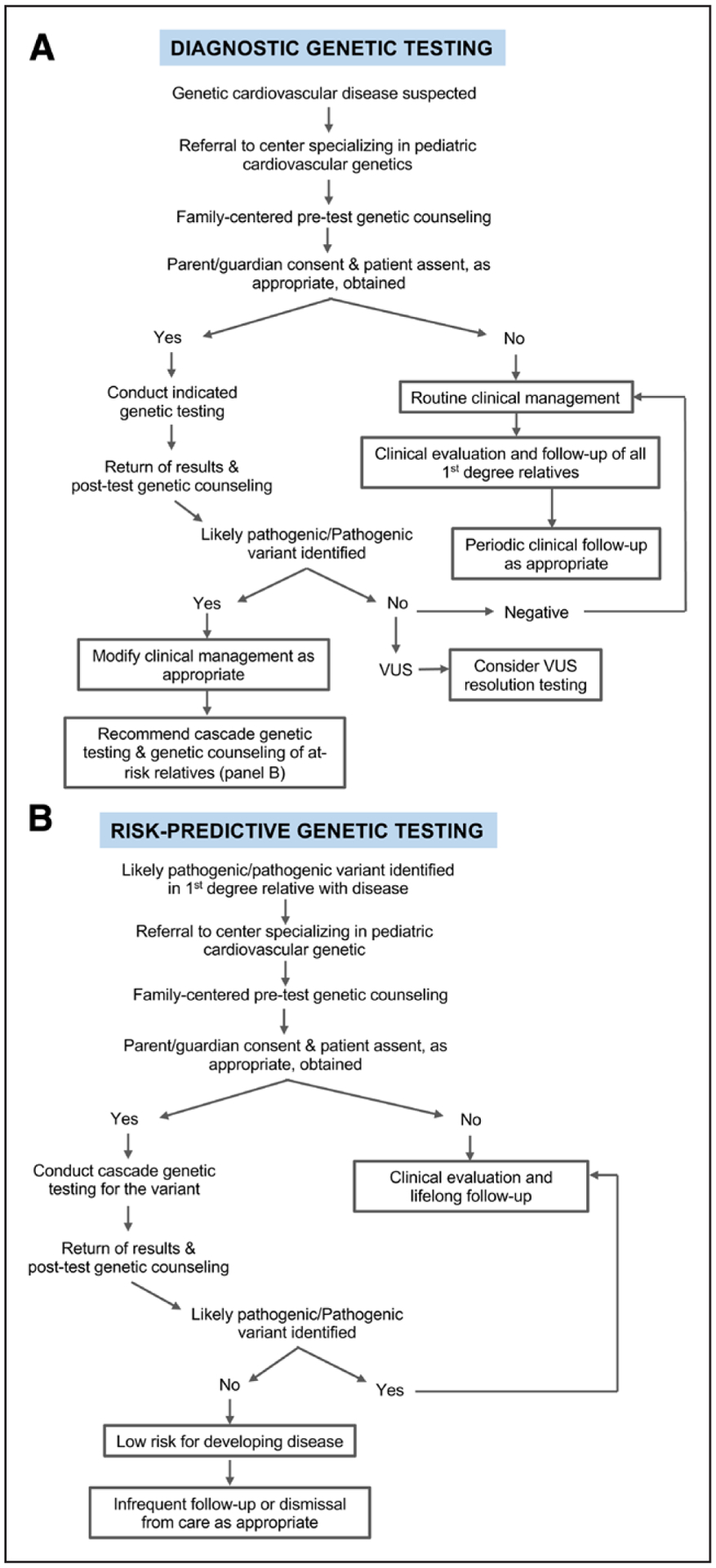
A, Algorithm for conducting diagnostic genetic testing in pediatric patients with heritable cardiovascular disease. B, Algorithm for conducting risk-predictive genetic testing in a family following identification of a likely pathogenic variant in a proband. VUS indicates variants of uncertain significance.
CHILDREN AS A VULNERABLE POPULATION
Genetic testing in children warrants special consideration, and focus must be kept on the best interest of the child. Given that nearly all cardiovascular diseases have incomplete penetrance and some degree of variable expressivity, positive risk-predictive genetic testing cannot determine whether a child will truly develop disease or predict how the disease will manifest.6,7 This is particularly critical in children, for whom the balance of benefits and harms of genetic testing always needs to be considered. Multiple professional societies have concluded that, unless there is a clinical intervention appropriate in childhood for the disease in question, parents should consider delaying risk-predictive testing for adult-onset conditions until their child reaches adulthood or is old enough to provide assent and to participate in decision-making.6–8 In contrast to consent, which typically can be provided only by individuals at the legal age of consent (≥18 years of age), assent is the expression of approval and willingness to participate in testing by a child who is too young to give informed consent but old enough to understand the rationale for and implications of genetic testing. The age at which a child can reliably provide assent depends on the child and his/her ability to understand both the rationale and implications of genetic testing. These principles of delaying genetic testing until assent or consent can be obtained cannot be universally applied indiscriminately to heritable cardiovascular diseases because presentation can occur during early childhood. Thus, prompt diagnosis in childhood allows clinical management, including medications and other clinical interventions, which may improve outcomes. In certain circumstances, it may be reasonable to consider testing minors for adult-onset conditions, specifically to provide anticipatory lifestyle guidance at an earlier age or to alleviate psychological distress attributable to uncertainty. Children should be involved in the assent or consent process to the extent possible, and adolescents should be offered the opportunity for private discussion.8
Referrals to specialized, multidisciplinary clinical cardiovascular genetics programs should be considered when possible after the diagnosis of a suspected cardiovascular genetic disease and before the initiation of genetic testing. The increase in telehealth capabilities of many centers has opened the door for virtual visits, which can supplement traditional in-person evaluation. These programs provide integrated evaluation and management with comprehensive assessments, including personal and family histories, physical examination, cardiac testing interpretation, pretest and posttest genetic counseling, psychological assessment, and cascade testing for at-risk relatives. Typically, these programs include close collaboration between adult and pediatric cardiologists, genetic counselors, geneticists, behavioral health specialists, and others to ensure optimal care provision for families with heritable cardiovascular disorders.5 This multidisciplinary team–based approach is important to integrate complex clinical evaluations with evolving genetic testing platforms for informative pretest and posttest genetic counseling. Furthermore, because syndromic disease with extracardiac manifestations may present as purely cardiac disease early in life, a multidisciplinary team is best suited to identify and manage these cases. Currently, access to these centers, and to cardiovascular genetic expertise more broadly, is not as great as the widespread availability of genetic testing services accessible to providers. This highlights the need for increased provider education on cardiovascular genetics and the need for additional genetic counselors and physician providers trained in heritable cardiovascular disease to bridge this gap in knowledge and care.
Pretesting counseling should be provided to all patients in an age-appropriate manner and to parents/guardians before genetic testing. Counseling should cover the reasons for genetic testing and the diagnostic limitations and should anticipate the outcomes of testing and their potential impact on care of all outcomes. Care should be taken to evaluate for possible differences in the perspective of the child, when age appropriate, and their parent/guardian. Similarly, posttest genetic counseling should be provided to all patients to discuss findings, the clinical implications of the results, and whether risk-predictive genetic testing is warranted and to address any questions. Pretest and posttest counseling should be done by providers with experience and expertise in this area.
In more expansive genetic testing, such as exome and genome sequencing, there is a higher chance of discovering secondary findings that may be clinically relevant yet are unrelated to the initial indication for testing. Such variants found in genes identified by American College of Medical Genetics and Genomics guidelines as being clinically actionable are considered secondary variants and are included in the genetic test results returned to the provider.9 Because secondary findings have potential medical implications for children and their relatives, this should be clearly discussed during the informed consent process, and parents and guardians should decide whether to receive these results.
CARDIAC CHANNELOPATHIES
The cardiac channelopathies (long QT syndrome, Brugada syndrome, short QT syndrome, and catecholaminergic polymorphic ventricular tachycardia [CPVT]) are a group of genetic diseases that increase the risk for arrhythmias and sudden death.10 A number of genes have been associated with cardiac channelopathies, with varying degrees of supporting evidence (Table 1). These have been recently reappraised, with reclassification of some disease-associated genes now considered to have disputed or limited evidence.12,13,16 In some cases, unambiguous long QT syndrome or Brugada syndrome may result from several/many common variants (oligogenic/polygenic); thus, inheritance is not always strictly mendelian.17,18
Table 1.
Genes Associated With Pediatric-Onset Cardiovascular Disease
| Genes associated with disease* | ||||
|---|---|---|---|---|
| Definitive evidence† | Moderate evidence | Limited evidence | Diagnostic testing yield among pediatric cases, % | |
| Channelopathies | ||||
| Brugada syndrome | SCN5A | ABCC9, ANK2, CACNA1C, CACNA2D1, CACNB2, FGF12, GPD1L, HCN4, KCND3, KCNE3, KCNE5, KCNH2, KCNJ8, RANGRF, PKP2, SCN10A, SCN1B. SCN2B, SCN3B, SEMA3A, SLMAP, TRPM4 | ≈30 | |
| CPVT | RYR2, CASQ2 | TRDN, CALM1, CALM2, CALM3 | TECRL | ≈60 |
| Long QT syndrome | KCNQ1, KCNH2, SCN5A, CALM1, CALM2, CALM3, TRDN | CACNA1C | AKAP9, ANK2, CAV3, KCNE1, KCNE2, KCNJ2, KCNJ5, SCN4B, SNTA1 | ≈75 |
| Short QT syndrome | KCNQ1, KCNH2, KCNJ2 | Unknown | ||
| Cardiomyopathies | ||||
| Arrhythmogenic right ventricular cardiomyopathy | PKP2, DSC2, DSG2, DSP, JUP, DES, TMEM43 | ≈80‡ | ||
| Dilated cardiomyopathy | TTN, LMNA, MYH7, TNNT2, BAG3, RBM20, TNNC1, TNNI3, TPM1, SCN5A, PLN, TMEM43, FLNC | ≈30 | ||
| Hypertrophic cardiomyopathy | MYBPC3, MYH7, TNNI3, TNNT2, TPM1, ACTC1, MYL2, MYL3, PRKAG2, LAMP2, GLA | ≈70 | ||
| Left ventricular noncompaction cardiomyopathy | MYH7, MYBPC3, TTN, LBD3LBD3 | ≈10 | ||
| Restrictive cardiomyopathy | MYH7, TNNI3 | ≈60 | ||
| Familial hypercholesterolemia | ||||
| Familial hypercholesterolemia | LDLR, APOB, PCSK9 | ≈95 | ||
| Aortopathies | Definitive/strong evidence§ | Moderate evidence | Limited evidence/unknown | Diagnostic testing yield among adult cases, %|| |
| Familial TAA and dissection | ACTA2, MYH11, MYLK, LOX, PRKG1 | FOXE3, MFAP5 | HCN4, MAT2A | ≈20 |
| Loeys-Dietz syndrome | TGFBR1, TGFBR2, SMAD3, TGFB2 | SMAD2 | TGFB3 | ≈80 |
| Marfan syndrome | FBN1 | ≈80 | ||
| Vascular Ehlers-Danlos syndrome | COL3A1 | Unknown | ||
| Other: progressive | BGN | Unknown | ||
| Other: nonprogressivefl¶ | EFEMP2 | CBS, COL4A5, ELN, FBN2, FLNA, NOTCH1, PKD1, PKD2, SLC2A10, SMAD4, SKI | Unknown | |
CPVT indicates catecholaminergic polymorphic ventricular tachycardia; and TAA, thoracic aortic aneurysm.
Based on Heart Rhythm Society/European Heart Rhythm Association guidelines.11
Meeting revised task force criteria as definite arrhythmogenic right ventricular cardiomyopathy cases.14
Based on ClinGen (MONDO:0019625) assessment of strength of association with disease.15
As a result of limited pediatric data, yield in adults meeting clinical criteria (Marfan syndrome and Loeys-Dietz syndrome) or with autosomal dominant family history of familial TAA and dissection.
Potentially diagnostic genes in patients who are typically ascertained as a result of other noncardiac clinical features and do not routinely have progressive aortic disease.
Diagnostic genetic testing for pediatric probands should occur when the diagnosis is considered to be likely and should entail a disease-specific gene panel. Cascade risk-predictive genetic testing of relatives to identify at-risk individuals with a P/LP variant should be variant specific and accompany appropriate clinical evaluation. Long QT syndrome and short QT syndrome may be diagnosed with an ECG, which can be reliably interpreted at any age and should precede diagnostic genetic testing. Risk-predictive genetic testing of children for a P/LP variant identified in a family member can identify children at risk of developing disease later in life. CPVT is diagnosed with exercise testing, which is not feasible in infants or young children. In very young relatives of CPVT probands, diagnostic genetic testing and initiation of β-blockers for genotype-positive children before a clinical diagnosis is made may be required. As with long QT syndrome and short QT syndrome, risk-predictive genetic testing of children for a P/LP variant identified in a family member can identify those at risk of developing disease. Evaluation for Brugada syndrome in children is complex in that there is age-related penetrance and the majority of adult probands do not host a P/LP variant. Thus, all first-degree relatives of an affected individual should undergo clinical evaluation. Fever or pharmacological drug challenge with a sodium channel blocker may unmask a diagnostic Brugada syndrome ECG in individuals without a diagnostic ECG at baseline. Although pediatric-specific guidelines are not available, drug challenge is usually reserved for children with a suspicious but not diagnostic ECG at rest or for adolescents who are genotype positive.19 Should a P/LP variant be found in a family member with Brugada syndrome, risk-predictive genetic testing of children for the identified variant can identify those at risk of developing disease. Research-based genetic testing with exome or genome sequencing can be considered in cases that remain genotype-negative. Because of complexities of conducting genomic research in children, this should be done in the setting of a multidisciplinary team with expertise in both clinical cardiovascular genetics and pediatric genetic/genomic research.
Children resuscitated from a cardiac arrest with no clear cause should undergo repeated clinical evaluations at a center with pediatric electrophysiology and cardiovascular genetics expertise. Genetic testing with a comprehensive arrhythmia panel can be considered in children without a clear clinical phenotype if done in this setting. In the event of a pediatric sudden unexplained death, channelopathy genetic testing can be considered after the exclusion of other causes of sudden pediatric death. In some cases, genetic testing, including cardiomyopathy or sudden unexplained death in epilepsy genes, may be considered given the history and postmortem evaluation. Clinical evaluation of the biological parents and siblings of the decedent may also be undertaken.
Age at pediatric genetic testing should be tailored to the likelihood of disease onset during childhood, the potential for mortality during childhood, the availability of therapies that can mitigate risk of mortality, and the family’s values and preferences. Because this approach is highly individualized to the patient and family, appropriateness of pediatric diagnostic genetic testing should be evaluated on a case-by-case basis. Obtaining diagnostic genetic testing when the child is old enough to provide assent is reasonable; however, testing earlier in life can be considered to facilitate risk-predictive genetic testing in the family if a P/LP variant is identified. This is particularly true in channelopathies, which carry a high positive yield on diagnostic genetic testing (ie, long QT syndrome and CPVT). Similarly, risk-predictive genetic testing should be considered earlier than the age of assent, particularly when disease is likely to manifest early in life and therapies or preventive measures can be implemented for children identified as being genetically at risk. For example, in a family with genotype-positive CPVT, genetic testing of children may occur early in life because of the possibility of life-threatening arrhythmias in childhood and the availability of effective β-blocker therapy.
CARDIOMYOPATHIES
Heritable cardiomyopathies represent primary myocardial diseases that are clinically and genetically heterogeneous. A relatively common cardiovascular disease in adults with a prevalence of at least 1 in 250 to 500 individuals, cardiomyopathy can present at any age, including among infants and children. Clinical features can include life-threatening arrhythmias, heart failure, and sudden cardiac death, with significant phenotypic diversity. The genetic basis of the inherited cardiomyopathies (Table 1) can be established in the majority of pediatric patients with yield of genetic testing dependent on various factors, including family history, age at disease onset, and phenotypic severity. In most inherited cardiomyopathies, the pattern of inheritance is autosomal dominant, but autosomal recessive inheritance and X-linked inheritance are also observed.
Hypertrophic cardiomyopathy is characterized by isolated ventricular hypertrophy in the absence of identifiable causes. Hypertrophic cardiomyopathy is classically caused by variants in genes encoding sarcomere proteins critical for contractile function.20 Familial dilated cardiomyopathy is characterized by cardiac enlargement and reduced left ventricular systolic function. Dilated cardiomyopathy can present at any age, including at birth and during childhood and adolescence. Arrhythmogenic right ventricular cardiomyopathy (increasingly referred to more broadly as arrhythmic cardiomyopathy) is characterized by progressive fibrofatty replacement of the myocardium, usually affecting the right ventricle but also involving the left ventricle. Arrhythmogenic right ventricular cardiomyopathy can present at any age but typically is seen beginning at 10 years of age. Left ventricular noncompaction cardiomyopathy is characterized by prominent left ventricular trabeculations with deep intertrabecular recesses and thinning of the compact epicardium. Left ventricular noncompaction can be isolated or associated with a dilated or hypertrophic phenotype. Finally, restrictive cardiomyopathy, defined by impaired relaxation of the myocardium, carries a poor prognosis in children, with heart transplantation being the only viable long-term treatment option. The genetic basis of cardiomyopathies is well established for hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and dilated cardiomyopathy, whereas the genetic basis of left ventricular noncompaction and restrictive cardiomyopathy is still emerging.21,22 Clinical diagnosis of cardiomyopathy should be established before diagnostic genetic testing. In general, diagnostic genetic testing for isolated cardiomyopathy should be limited to a panel of genes associated with the specific type of cardiomyopathy, although there is significant phenotypic and genetic overlap between forms of cardiomyopathy, which may make broad-based “pan” panel testing reasonable. Risk-predictive genetic testing of children for a P/LP variant identified in a family with cardiomyopathy can identify those at risk of developing disease.
A number of metabolic and neuromuscular diseases with an associated risk of cardiomyopathy are important to consider in children.23 These include glycogen storage diseases (eg, Pompe and Danon diseases), lysosomal storage diseases (eg, Hurler syndrome and Anderson-Fabry disease), and RASopathies (eg, Noonan and Costello syndromes). Several heritable neuromuscular diseases such as Duchenne and Becker muscular dystrophies are associated with dilated cardiomyopathy and primary mitochondrial disorders caused by genetic variants in either mitochondrial or nuclear genes (eg, Kearns-Sayre syndrome), as well as genetic disease that affects mitochondrial function (eg, Barth syndrome and Friedreich ataxia). The vast majority of these diseases and syndromes are attributable to single-gene mutations that cause disease through a variety of modes of inheritance. Clinical suspicion of these diseases and syndromes should trigger evaluation by a multidisciplinary team with expertise in the management of pediatric cardiomyopathies, medical/metabolic geneticists, and genetic counselors. Ideally, this should be done before diagnostic genetic testing. Although panel-based diagnostic genetic testing can be performed, exome or genome sequencing–based testing may be considered in these individuals because variable expressivity of disease may make targeted panel testing challenging. Trio testing, which involves genetic testing of both biological parents in addition to the child, can be helpful for exome/genome test interpretation. Research-based genetic testing with exome or genome sequencing can be considered. Because of the complexities of conducting genomic research in children, it should be done in the setting of a multidisciplinary team with expertise in both clinical cardiovascular genetics and pediatric genetic/genomic research.
Age at pediatric cardiomyopathy genetic testing should be tailored to the likelihood of disease onset during childhood, the potential for morbidity and mortality during childhood, the availability of therapies, and the family’s values and preferences. Because this approach is highly individualized to the patient and family, appropriateness of pediatric diagnostic genetic testing should be evaluated on a case-by-case basis. Obtaining diagnostic genetic testing when the child is old enough to provide assent is reasonable; however, testing earlier in life can be considered to facilitate risk-predictive genetic testing in the family should a P/LP variant be identified. This is particularly true in cardiomyopathies that carry a high positive yield on diagnostic genetic testing (ie, hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy). Similarly, risk-predictive genetic testing should be considered early in childhood, particularly when disease is likely to manifest before the child is able to provide assent, and therapies or other preventive measures can be implemented for at-risk children.
HERITABLE AORTOPATHIES
Heritable aortopathies encompass a number of different diseases involving the aorta that have a diverse genetic basis (Table 1). Thoracic aortic aneurysm (TAA) is a potentially life-threatening condition occurring as an isolated finding or as part of a genetic syndrome. Early recognition of TAA or risk for TAA enables timely surveillance and medical management and decreases morbidity and mortality. Although focused on the adult patient, American Heart Association/American College of Cardiology and European Society of Cardiology consensus guidelines have syndrome-specific imaging recommendations and guidelines for surgical intervention.24,25 In the pediatric population, TAA is genetic, with up to 35% of individuals having an identifiable P/LP variant typically inherited in an autosomal dominant fashion.15,26 The transforming growth factor-β pathway, extracellular matrix, and smooth muscle cell components of the aorta are important causes of heritable aortopathy. Connective tissue disorders associated with TAA include Marfan syndrome, Loeys-Dietz syndrome, and vascular Ehlers-Danlos syndrome. Genes important in aortic smooth muscle homeostasis are important causes of nonsyndromic TAA. Progressive aortic dilation is a highly penetrant feature of connective tissue disorders despite variable expressivity of other systemic features. In contrast, the penetrance of aortic dilation in isolated familial TAA is more variable and shows sex bias, affecting men more frequently.
Clinical evaluation by medical geneticists or other specialists in the diagnosis of connective tissue disorders is important for the evaluation of connective tissue disorders and the identification of other genetic syndromes associated with TAA.26,27 Although young children with TAA are more likely to have a genetic syndromic cause of disease, there is significant overlap in age at presentation. Furthermore, there is overlap in clinical manifestations of disease in children, which complicates clinical assessment. Thus, diagnostic panels including genes associated with both syndromic and nonsyndromic TAA are typically preferred. FBN1-specific testing for Marfan syndrome may be considered in individuals who fulfill diagnostic criteria with subsequent panel testing if found to be FBN1-variant negative.15 Deletion/duplication analysis is a mechanism of disease in a small subset (eg, ≈5% in Marfan syndrome). Genetic testing is important even in individuals fulfilling clinical criteria for connective tissue disorders because gene-specific management guidelines exist. Negative genetic testing in TAA provides useful risk stratification information.
Diagnostic genetic testing should be initiated at the time of diagnosis of TAA or in individuals with suspicious connective tissue disorder features. Pretest and posttest counseling and interpretation of genetic testing results in the context of the patient’s phenotype are essential. American College of Cardiology/American Heart Association and European Society of Cardiology guidelines also recommend cardiac screening in first-degree relatives of patients with TAA. If a P/LP variant is identified in the proband, risk-predictive genetic testing should be offered to relatives.24,25,27
FAMILIAL HYPERCHOLESTEROLEMIA
Familial hypercholesterolemias (FHs) are the most common inherited cardiovascular disease, affecting ≈1 in 250 individuals, yet are vastly underdiagnosed. FH is characterized by severely elevated low-density lipoprotein cholesterol (LDL-C) levels and, if left untreated, confers high risk for premature atherosclerotic cardiovascular disease. If FH is treated at young ages, the risk for atherosclerotic cardiovascular disease can be drastically reduced.28 FH is most commonly caused by the presence of a P/LP variant in the LDLR gene, with disease-causing variants in other genes being less common (Table 1). FH is inherited in an autosomal dominant fashion and encompasses a spectrum of clinical phenotypes based on the number of P/LP variants.29 Heterozygous FH is typically caused by the presence of 1 P/LP variant in 1 of the 3 primary FH genes. Homozygous FH is caused by biallelic P/LP variants, usually in LDLR; however, rare autosomal recessive forms exist. LDL-C levels can overlap between individuals with 1 or 2 pathological variants associated with FH. For individuals <20 years of age, an LDL-C >130 mg/dL indicates a high likelihood of heterozygous FH.30 LDL-C levels vary for individuals with true homozygous FH, compound heterozygous FH, and double heterozygous FH, but generally, LDL-C levels ranging from 350 to 500 mg/dL indicate a high likelihood that an individual has 2 pathological variants associated with FH. Polygenic hypercholesterolemias have also been described in up to 30% of patients clinically suspected to have monogenic FH.31
It is imperative to identify individuals with FH in childhood so that lipid-lowering therapies and lifestyle interventions can be established. Left untreated, children with FH are at high risk for atherosclerotic cardiovascular disease in early to middle adulthood attributable to the cumulative burden of elevated LDL-C levels. Children with FH who initiate a statin have a lower risk of atherosclerotic cardiovascular disease compared with their affected parents.32 The American Academy of Pediatrics recommends universal lipid screening for all children at 9 to 11 years of age and as early as 2 years of age in the setting of newly diagnosed FH33 within the family. Markedly elevated LDL-C levels, particularly in nonobese children, should raise suspicion for FH. Specific guidelines for clinical FH genetic testing in children are based on LDL-C levels and family history.29 Children with a strong clinical suspicion for FH should be offered genetic testing for diagnosis. Furthermore, if a P/LP variant is found in an individual with FH, risk-predictive genetic testing should be performed in the family, including children.
Although multiple clinical diagnostic schemas exist for FH, diagnostic genetic testing has been described as the gold standard for diagnosis and has the potential to improve FH diagnosis rates and to provide prognostic data and accurate risk assessment. Patients with an FH clinical phenotype may have negative genetic testing for the 3 main genes and may have an FH phenotype resulting from other potential causes, including polygenic variation. Clinical testing for polygenic causes of hypercholesterolemias is not yet widely clinically available and will require studies of clinical utility.31
CONGENITAL HEART DISEASE
Collectively, CHDs are the most common form of birth defect, affecting nearly 1% of all live births. Syndromic forms of CHD have a well-established genetic cause (eg, aneuploidies, copy number variants and single-gene mendelian disorders) but make up only ≈20% to 30% of CHD cases. The genetic architecture of nonsyndromic (isolated) CHD is more complex, also including copy number variants and single-gene disorders in addition to more complex inheritance. Details of the genetic landscape are emerging with the widespread application of next-generation sequencing modalities in large CHD cohorts. Early studies of familial CHD implicated genes encoding cardiac transcription factors, signaling/adhesion molecules, and structural proteins in the pathogenesis of nonsyndromic CHD. These studies highlight that CHD is a largely genetically heterogeneous disorder, encompassing individual genes characterized by low population attributable risk (ie, the proportion of disease explained by a particular gene). Unique categories of genes are highly enriched in CHD cases with a damaging genetic variant such as chromatin-modifying genes. The likelihood of encountering a damaging de novo variant in any gene is substantially increased if the child has an extracardiac anomaly and neurodevelopmental defect.34,35 Although chromatin-modifying genes are highly enriched in dominant forms of CHD, cilia-related genes are significantly enriched for damaging rare recessive genotypes, particularly compound heterozygous genotypes. Last, the combination of inherent variable expressivity and the difficulties in identifying syndromic features in newborns/infants further supports the need for more standardized clinical and genetic testing assessments.36
Whom to test, when, and with what modality remain contentious issues, especially in light of emerging rapid next-generation sequencing technologies. Options for genetic testing in known syndromic forms of CHD are well established,37,38 but significant practice variation exists in their application, especially in infants with apparently isolated nonsyndromic CHD. In general, the clinical phenotype drives the choice of genetic testing modalities. Cytogenetic genetic testing (eg, microarrays) or gene panel testing should be used for diagnostic genetic testing in infants and children in whom syndromic CHD is suspected on the basis of the differential diagnosis. Diagnostic gene panel testing or exome and genome sequencing may have an emerging role in patients with isolated or apparently isolated nonsyndromic CHD. Trio testing, which involves genetic testing of both biological parents in addition to the child, can be helpful for exome/genome test interpretation and may be considered. Given that the distinction between syndromic and isolated CHD is often subtle, a high level of suspicion and careful, prospective evaluation are warranted in cases of presumed isolated CHD for extracardiac anomalies or neurodevelopmental features. Determination of best practices requires further investigation; however, emerging evidence of modestly increased diagnostic yields in nonsyndromic tetralogy of Fallot (with associated absent pulmonary valve, aortic arch or pulmonary artery anomalies, and aortopulmonary collaterals) and heterotaxy syndromes suggests that lesionspecific recommendations may emerge, although the overall yield is <10% of cases.37 Recent evidence suggests that ≈10% of syndromic and nonsyndromic CHD cases may be attributed to de novo variants in the coding genome, with a similar proportion potentially attributable to de novo variants in noncoding areas of the genome.36,39 Last, chromosomal microarrays have diagnostic value for patients with CHD, particularly complex forms of CHD, and gene panels have an emerging diagnostic role. Moving forward, early studies showing diagnostic value in rapid trio-based genome sequencing among critically ill newborns40 suggest that this methodology may ultimately supplant current genetic testing modalities.
SPECIAL CASES
Prenatal Genetic Testing
The decision to pursue prenatal genetic testing is complex and nuanced. A number of statements have highlighted the need to prioritize the personal goals and preferences of the mother, in close partnership with a multidisciplinary care team, in the decision to pursue prenatal genetic testing.41,42 Practice guidelines outline prenatal screening and testing options that should be offered for aneuploidy.43 We restrict our comments here to families in whom there is a known heritable cause of cardiac disease or fetuses with a detected cardiac abnormality. Risk-predictive prenatal genetic testing can be undertaken when a parent has a cardiovascular genetic trait and an identified causal variant.44,45 In such instances, prenatal genetic testing can be used either to avoid a pregnancy with a fetus inheriting that variant (preimplantation genetic diagnosis) or during the pregnancy to test the developing fetus (prenatal diagnosis).
Decisions about pursuing diagnostic prenatal genetic testing for cardiovascular conditions require prenatal genetic counseling expertise and should consider the specific cardiac disorder and the limitations of prenatal ultrasound and fetal echocardiography. Because cardiovascular genetic traits such as many cardiomyopathies may exhibit incomplete penetrance and widely variable expressivity, counseling about outcomes is challenging and should be approached in an individualized manner with a multidisciplinary team approach. The detection of a major CHD in a fetus is a specialized case in that practice guidelines from professional societies for genetics or obstetrics and gynecology outline recommendations for offering prenatal genetic testing in cases of fetal birth defects.41–43 Chromosomal microarray is offered unless the pattern of anomalies is highly suggestive of aneuploidy or would require molecular panel testing for diagnosis. Studies examining the results of prenatal exome sequencing are ongoing46; the application of genome sequencing and the expansion of noninvasive prenatal genetic testing via cell free DNA ensure that prenatal genetic testing will continue to be a rapidly changing field with requirement for multidisciplinary coordination of care and expertise.
At-Home Genetic Testing
At-home genetic testing takes 2 forms: direct-to-consumer genetic testing and consumer-directed (or consumer-initiated) clinical testing, which may or may not be done in a Clinical Laboratory Improvement Amendments–approved laboratory.47 These are in contrast to genetic testing in which the tissue sample is collected at home for subsequent testing in a Clinical Laboratory Improvement Amendments–approved laboratory (eg, a commercial diagnostic laboratory obtaining a DNA sample for testing with at-home saliva collection kit). Direct-to-consumer genetic testing typically reveals information about ancestry but also risks for certain medical conditions such as coronary artery disease. Direct-to-consumer testing of children for disorders with adult onset, including cardiovascular ones, remains ethically controversial.47,48 Consumer-directed clinical testing includes tests that evaluate for genetic traits that present in childhood and may be done in a Clinical Laboratory Improvement Amendments–approved laboratory. One currently marketed gene panel covers a number of pediatric traits with cardiovascular involvement such as fatty acid oxidation defects, Pompe disease, and Marfan syndrome.47 Genetic counseling is included with the testing. Recent evidence has suggested that direct-to-consumer genetic testing has a high prevalence of false-positive results, which highlights the importance of validating findings in a College of American Pathologists/Clinical Laboratory Improvement Amendments–approved laboratory in the context of comprehensive genetic counseling.49
Secondary Genetic Findings
The American College of Medical Genetics and Genomics put forth criteria, now widely adopted, for which genes should be disclosed when found to contain so-called secondary variants,50 defined as variants with medical relevance unrelated to the reason that genetic testing was ordered. Currently, variants in 59 genes are reported as secondary findings, including genes for cardiomyopathies, channelopathies, aortopathies, and FH. Typically, individuals undergoing exome or genome testing (or their parents/guardians) must opt in or out of the return of secondary findings. Although superficially similar to positive findings arising for primary indications, secondary findings are distinct. This derives in part from bayesian statistical considerations of prior probabilities.51 Recent experiences with large-scale sequencing of cohorts of healthy individuals have revealed issues with variant curation, especially challenging for long QT syndrome genes,52 and disease penetrance being lower than anticipated according to studies of affected individuals and families.53,54 Discovery of secondary findings for cardiovascular traits should trigger a clinical evaluation relevant for the specific disorder.55 Although a completely negative evaluation for an adult may effectively end active surveillance, the age-dependent penetrance for many of the cardiovascular traits may necessitate ongoing surveillance in children. In addition, broad genetic testing such as exome and genome sequencing obtained in children with cardiovascular findings may yield actionable secondary genetic findings in genes not associated with cardiovascular disease. This highlights the importance of approaching broad genetic testing in concert with a multidisciplinary team capable of both providing comprehensive pretest genetic counseling, which includes the possibility of secondary findings, and interpreting and acting on a secondary finding.
WHAT THE PATIENT AND FAMILY SHOULD KNOW
When pursuing genetic testing in children, health care providers who order the genetic testing should engage families to address and discuss several topics, including information about the test itself, types of results, and utility of the test (Figure 2). Health care providers with expertise in genetic testing should facilitate these pretest discussions.4,5 Before considering genetic testing, providers should work with families so that they understand the reason for genetic testing and its diagnostic limitations. For example, testing might not provide definitive or clinically useful information as a result of inconclusive results and incomplete knowledge of genetic causes for many cardiac diseases. Providers should work with families to help them understand the types of possible results and the anticipated plan for using them for individual and family care.4 Timing of genetic testing should be discussed to maximize benefits and to limit potential harms or impacts on child autonomy. Last, clinical providers should discuss alternatives when risk cannot be confirmed with genetic testing.
Figure 2. Diagram illustrating key aspects/topics to be considered when providing pretesting and posttesting genetic counseling for pediatric patients with heritable cardiovascular disease.

Counseling should cover the mechanics of the testing (A); the clinical impact of the results (B); and the ethical, legal, and socioeconomic concerns of the testing (C).
Families may be hesitant about genetic testing in children because of perceived costs, limited insurance coverage, and fears about genetic discrimination or adverse psychosocial effects. These concerns should be addressed before testing. In the United States, information on the Genetic Information Nondiscrimination Act should be provided when relevant. Indeed, the Genetic Information Nondiscrimination Act and the Health Insurance Portability and Accountability Act provide protection against forms of genetic discrimination and third-party disclosure. Similar laws and resources are in existence in other countries and the World Health Organization. Cost and insurance coverage of genetic testing are important considerations that should be addressed with the family. Adverse psychosocial effects of genetic risks in children are possible, although there have not been studies confirming increased negative effects in children. Ideally, family-centered counseling should include the anticipated psychosocial effects of genetic testing results on children and their families and ways to adapt to risk healthfully. Continued follow-up is important to re-evaluate variant pathogenicity, which can change over time.
CONCLUSIONS
Cardiovascular genetic testing in children has an important diagnostic role in determining the risk of developing heritable cardiovascular disease (Table 2). Although genetic testing can be considered in children who are suspected or at risk of having cardiovascular disease, special emphasis should be placed on appropriate multidisciplinary counseling and timing of testing, which is individualized to the patient, the family, and the disease in question. Continued follow-up is important to re-evaluate variant pathogenicity and to address the changing needs of the patient and family as genomic and precision medicine evolves.
Table 2.
Key Points and Recommendations for Pediatric Cardiovascular Genetic Testing
| Genetic testing should always involve pretesting and posttesting counseling that is comprehensive and ideally occurs in or in partnership with a specialized multidisciplinary setting. |
| Diagnostic genetic testing should be considered only in children with a high likelihood of disease. |
| Risk-predictive genetic testing should be performed in children after identification of a P/LP variant in a family member with disease. |
| The timing of genetic testing in children should take into account disease-specific considerations of disease penetrance, the likelihood of pediatric disease presentation, the availability of effective therapies or lifestyle modifications, and the possibility of psychological distress in the family attributable to uncertainty. |
| Continued follow-up of genetic test results is important to re-evaluate or confirm variant pathogenicity over time. |
P/LP indicates pathogenic/likely pathogenic.
Acknowledgments
This statement was approved by the American Heart Association Science Advisory and Coordinating Committee on May 21, 2021, and the American Heart Association Executive Committee on June 21, 2021. A copy of the document is available at https://professional.heart.org/statements by using either “Search for Guidelines & Statements” or the “Browse by Topic” area. To purchase additional reprints, call 215-356-2721 or email Meredith.Edelman@wolterskluwer.com.
Footnotes
The American Heart Association makes every effort to avoid any actual or potential conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the writing panel. Specifically, all members of the writing group are required to complete and submit a Disclosure Questionnaire showing all such relationships that might be perceived as real or potential conflicts of interest.
Disclosures
| Writing group member | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board | Other |
|---|---|---|---|---|---|---|---|---|
| Andrew P. Landstrom | Duke University School of Medicine, Duke University Medical Center | NIH (PI for and NIH K08 award and co-I for an NIH R01 award)*; Doris Duke Charitable Foundation (PI for the DDCF CSDA Award)* | None | None | None | None | None | None |
| Jeffrey J. Kim | Texas Children’s Hospital–Baylor College of Medicine | CPRIT (site PI for a grant related to mechanisms of chemotherapy-related cardiotoxicity)† | None | None | None | None | None | None |
| Bruce D. Gelb | Icahn School of Medicine at Mount Sinai | Onconova (sponsored research agreement)† | None | None | None | None | None | None |
| Benjamin M. Helm | Indiana University School of Medicine | None | None | None | None | None | None | None |
| Prince J. Kannankeril | Vanderbilt University Medical School | None | None | None | None | None | None | None |
| Christopher Semsarian | University of Sydney (Australia) | None | None | None | None | None | None | None |
| Amy C. Sturm | Geisinger Genomic Medicine Institute | None | None | None | None | None | None | None |
| Martin Tristani-Firouzi | University of Utah | NIH (multiple NIH awards as PI)*; AHA (Children’s SFRN Center director)* | None | None | None | None | None | None |
| Stephanie M. Ware | Indiana University School of Medicine | None | None | None | None | None | None | None |
| Reviewer | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board | Other |
|---|---|---|---|---|---|---|---|---|
| E. Kevin Hall | Yale University | None | None | None | None | None | None | None |
| Salim F. Idriss | Duke Medical Center | None | None | None | None | None | None | None |
| Shaine A. Morris | Texas Children’s Hospital | NHLBI (on a no-cost extension through 2021 for an NHL-BIK23 award studying imaging biomarkers in aortopathy)† | The Marfan Foundation (PI for a current grant from the Marfan Foundation evaluating effects of exercise in Marfan syndrome)† | None | None | None | ACER Therapeutics* | Baylor College of Medicine (BCM is a vendor for many types of genetic testing)† |
| Shubhayan Sanatani | University of British Columbia, British Columbia Children’s Hospital (Canada) | Canadian Heart and Stroke (funding to research CPVT)†; Canadian Institute of Health Research (grant on inherited heart rhythm conditions)† | None | None | None | None | Xenon Pharmaceuticals* | None |
Contributor Information
Andrew P. Landstrom, Duke University, School of Medicine, Duke University Medical Center.
Jeffrey J. Kim, Texas Children’s Hospital–Baylor College of Medicine.
Bruce D. Gelb, Icahn School of Medicine at Mount Sinai.
Benjamin M. Helm, Indiana University, School of Medicine.
Prince J. Kannankeril, Vanderbilt University, Medical School.
Christopher Semsarian, University of Sydney (Australia).
Amy C. Sturm, Geisinger Genomic Medicine Institute.
Martin Tristani-Firouzi, University of Utah.
Stephanie M. Ware, Indiana University, School of Medicine.
REFERENCES
- 1.Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC; on behalf of the American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology. Genetic testing for inherited cardiovascular diseases: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2020;13:e000067. doi: 10.1161/HCG.0000000000000067 [DOI] [PubMed] [Google Scholar]
- 2.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caleshu C, Day S, Rehm HL, Baxter S. Use and interpretation of genetic tests in cardiovascular genetics. Heart. 2010;96:1669–1675. doi: 10.1136/hrt.2009.190090 [DOI] [PubMed] [Google Scholar]
- 4.Arscott P, Caleshu C, Kotzer K, Kreykes S, Kruisselbrink T, Orland K, Rigelsky C, Smith E, Spoonamore K, Larsen Haidle J, et al. A case for inclusion of genetic counselors in cardiac care. Cardiol Rev. 2016;24:49–55. doi: 10.1097/CRD.0000000000000081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmad F, McNally EM, Ackerman MJ, Baty LC, Day SM, Kullo IJ, Madueme PC, Maron MS, Martinez MW, Salberg L, et al. Establishment of specialized clinical cardiovascular genetics programs: recognizing the need and meeting standards: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2019;12:e000054. doi: 10.1161/HCG.0000000000000054 [DOI] [PubMed] [Google Scholar]
- 6.Committee on Bioethics, Committee on Genetics, American College OF Medical Genetics, Genomics Social, Ethical, and Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Pediatrics. 2013;131:620–622. doi: 10.1542/peds.2012-3680 [DOI] [PubMed] [Google Scholar]
- 7.Ross LF, Ross LF, Saal HM, David KL, Anderson RR; American Academy of Pediatrics; American College of Medical Genetics and Genomics. Technical report: ethical and policy issues in genetic testing and screening of children. Genet Med. 2013;15:234–245. doi: 10.1038/gim.2012.176 [DOI] [PubMed] [Google Scholar]
- 8.Botkin JR, Belmont JW, Berg JS, Berkman BE, Bombard Y, Holm IA, Levy HP, Ormond KE, Saal HM, Spinner NB, et al. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet. 2015;97:6–21. doi: 10.1016/j.ajhg.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, et al. ; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10:1932–1963. doi: 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 11.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, et al. ; Heart Rhythm Society (HRS); European Heart Rhythm Association (EHRA). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13:1077–1109. doi: 10.1093/europace/eur245 [DOI] [PubMed] [Google Scholar]
- 12.Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, Feilotter H, Amenta S, Mazza D, Bikker H, et al. An international, multi-centered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation. 2020;141:418–428. doi: 10.1161/CIRCULATIONAHA.119.043132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, Jamal SM, Szybowska M, Morel CF, Bowdin S, et al. ; National Institutes of Health Clinical Genome Resource Consortium. Reappraisal of reported genes for sudden arrhythmic death: evidence-based evaluation of gene validity for Brugada syndrome. Circulation. 2018;138:1195–1205. doi: 10.1161/CIRCULATIONAHA.118.035070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Adès LC, Andelfinger GU, Arnaud P, Boileau C, Callewaert BL, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2018;72:605–615. doi: 10.1016/j.jacc.2018.04.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campuzano O, Fernandez-Falgueras A, Lemus X, Sarquella-Brugada G, Cesar S, Coll M, Mates J, Arbelo E, Jordà P, Perez-Serra A, et al. Short QT syndrome: a comprehensive genetic interpretation and clinical translation of rare variants. J Clin Med. 2019;8:E1035. doi: 10.3390/jcm8071035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. doi: 10.1038/ng.2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lahrouchi N, Tadros R, Crotti L, Mizusawa Y, Postema PG, Beekman L, Walsh R, Hasegawa K, Barc J, Ernsting M, et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation. 2020;142:324–338. doi: 10.1161/CIRCULATIONAHA.120.045956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obeyesekere MN, Klein GJ, Modi S, Leong-Sit P, Gula LJ, Yee R, Skanes AC, Krahn AD. How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011;4:958–964. doi: 10.1161/CIRCEP.111.965947 [DOI] [PubMed] [Google Scholar]
- 20.Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–888. doi: 10.1038/gim.2014.205 [DOI] [PubMed] [Google Scholar]
- 21.van Waning JI, Caliskan K, Hoedemaekers YM, van Spaendonck-Zwarts KY, Baas AF, Boekholdt SM, van Melle JP, Teske AJ, Asselbergs FW, Backx APCM, et al. Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. 2018;71:711–722. doi: 10.1016/j.jacc.2017.12.019 [DOI] [PubMed] [Google Scholar]
- 22.Ross SB, Jones K, Blanch B, Puranik R, McGeechan K, Barratt A, Semsarian C. A systematic review and meta-analysis of the prevalence of left ventricular non-compaction in adults. Eur Heart J. 2020;41:1428–1436. doi: 10.1093/eurheartj/ehz317 [DOI] [PubMed] [Google Scholar]
- 23.Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, Hsu DT, Lin KY, Price JF, Wilkinson JD, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American Heart Association. Circulation. 2019;140:e9–e68. doi: 10.1161/CIR.0000000000000682 [DOI] [PubMed] [Google Scholar]
- 24.Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, et al. ; ESC Committee for Practice Guidelines. 2014 ESC guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult: the Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2873–2926. doi: 10.1093/eurheartj/ehu281 [DOI] [PubMed] [Google Scholar]
- 25.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr, Eagle KA, Hermann LK, Isselbacher EM, Kazerooni EA, et al. ; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine [published correction appears in Circulation. 2010;122:e410]. Circulation. 2010;121:e266–e369. doi: 10.1161/CIR.0b013e3181d4739e [DOI] [PubMed] [Google Scholar]
- 26.Rigelsky CM, Moran RT. Genetics of syndromic and nonsyndromic aortopathies. Curr Opin Pediatr. 2019;31:694–701. doi: 10.1097/MOP.0000000000000836 [DOI] [PubMed] [Google Scholar]
- 27.Verhagen JMA, Kempers M, Cozijnsen L, Bouma BJ, Duijnhouwer AL, Post JG, Hilhorst-Hofstee Y, Bekkers SCAM, Kerstjens-Frederikse WS, van Brakel TJ, et al. ; National Working Group on BAV & TAA. Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives. Int J Cardiol. 2018;258:243–248. doi: 10.1016/j.ijcard.2018.01.145 [DOI] [PubMed] [Google Scholar]
- 28.Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, Ose L, Averna M, Boileau C, Borén J, et al. ; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36:2425–2437. doi: 10.1093/eurheartj/ehv157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, Ballantyne CM, Baum SJ, Bourbon M, Carrié A, Cuchel M, et al. ; Convened by the Familial Hypercholesterolemia Foundation. Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018;72:662–680. doi: 10.1016/j.jacc.2018.05.044 [DOI] [PubMed] [Google Scholar]
- 30.Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, Daniels SR, Gidding SS, de Ferranti SD, Ito MK, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:133–140. doi: 10.1016/j.jacl.2011.03.001 [DOI] [PubMed] [Google Scholar]
- 31.Brown EE, Sturm AC, Cuchel M, Braun LT, Duell PB, Underberg JA, Jacobson TA, Hegele RA. Genetic testing in dyslipidemia: a scientific statement from the National Lipid Association. J Clin Lipidol. 2020;14:398–413. doi: 10.1016/j.jacl.2020.04.011 [DOI] [PubMed] [Google Scholar]
- 32.Luirink IK, Wiegman A, Kusters DM, Hof MH, Groothoff JW, de Groot E, Kastelein JJP, Hutten BA. 20-Year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med. 2019;381:1547–1556. doi: 10.1056/NEJMoa1816454 [DOI] [PubMed] [Google Scholar]
- 33.Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents, National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128(suppl 5):S213–S256. doi: 10.1542/peds.2009-2107C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, et al. ; INTERVAL Study; UK10K Consortium; Deciphering Developmental Disorders Study. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060–1065. doi: 10.1038/ng.3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shikany AR, Landis BJ, Parrott A, Miller EM, Coyan A, Walters L, Hinton RB, Goldenberg P, Ware SM. A comprehensive clinical genetics approach to critical congenital heart disease in infancy. J Pediatr. 2020;227:231–238. e14. doi: 10.1016/j.jpeds.2020.07.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. doi: 10.1038/ng.3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A, et al. ; on behalf of the American Heart Association Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; and Council on Genomic and Precision Medicine. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association [published correction appears in Circulation. 2018;138:e713]. Circulation. 2018;138:e653–e711. doi: 10.1161/CIR.0000000000000606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, Zhou J, Qi H, Patel N, DePalma SR, et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat Genet. 2020;52:769–777. doi: 10.1038/s41588-020-0652-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farnaes L, Hildreth A, Sweeney NM, Clark MM, Chowdhury S, Nahas S, Cakici JA, Benson W, Kaplan RH, Kronick R, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3:10. doi: 10.1038/s41525-018-0049-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson KL, Czerwinski JL, Hoskovec JM, Noblin SJ, Sullivan CM, Harbison A, Campion MW, Devary K, Devers P, Singletary CN. NSGC practice guideline: prenatal screening and diagnostic testing options for chromosome aneuploidy. J Genet Couns. 2013;22:4–15. doi: 10.1007/s10897-012-9545-3 [DOI] [PubMed] [Google Scholar]
- 42.Gregg AR, Skotko BG, Benkendorf JL, Monaghan KG, Bajaj K, Best RG, Klugman S, Watson MS. Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2016;18:1056–1065. doi: 10.1038/gim.2016.97 [DOI] [PubMed] [Google Scholar]
- 43.Practice Bulletin No. 162 summary: prenatal diagnostic testing for genetic disorders. Obstet Gynecol. 2016;127:976–978. doi: 10.1097/AOG.0000000000001405 [DOI] [PubMed] [Google Scholar]
- 44.Charron P, Héron D, Gargiulo M, Feingold J, Oury JF, Richard P, Komajda M. Prenatal molecular diagnosis in hypertrophic cardiomyopathy: report of the first case. Prenat Diagn. 2004;24:701–703. doi: 10.1002/pd.969 [DOI] [PubMed] [Google Scholar]
- 45.Tester DJ, McCormack J, Ackerman MJ. Prenatal molecular genetic diagnosis of congenital long QT syndrome by strategic genotyping. Am J Cardiol. 2004;93:788–791. doi: 10.1016/j.amjcard.2003.11.061 [DOI] [PubMed] [Google Scholar]
- 46.Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, Prigmore E, Keelagher R, Best SK, Carey GK, et al. ; Prenatal Assessment of Genomes and Exomes Consortium. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747–757. doi: 10.1016/S0140-6736(18)31940-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weissman SM, Kirkpatrick B, Ramos E. At-home genetic testing in pediatrics. Curr Opin Pediatr. 2019;31:723–731. doi: 10.1097/MOP.0000000000000824 [DOI] [PubMed] [Google Scholar]
- 48.National Research Council. Direct-to-Consumer Genetic Testing: Summary of a Workshop. National Academies Press; 2011. [PubMed] [Google Scholar]
- 49.ACMG Board of Directors. Direct-to-consumer genetic testing: a revised position statement of the American College of Medical Genetics and Genomics. Genet Med. 2016;18:207–208. doi: 10.1038/gim.2015.190 [DOI] [PubMed] [Google Scholar]
- 50.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255. doi: 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- 51.Biesecker LG. Genomic screening and genomic diagnostic testing: two very different kettles of fish. Genome Med. 2019;11:75. doi: 10.1186/s13073-019-0696-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diebold I, Schön U, Scharf F, Benet-Pagès A, Laner A, Holinski-Feder E, Abicht A. Critical assessment of secondary findings in genes linked to primary arrhythmia syndromes. Hum Mutat. 2020;41:1025–1032. doi: 10.1002/humu.23996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haggerty CM, Murray B, Tichnell C, Judge DP, Tandri H, Schwartz M, Sturm AC, Matsumura ME, Murray MF, Calkins H, et al. Managing secondary genomic findings associated with arrhythmogenic right ventricular cardiomyopathy: case studies and proposal for clinical surveillance. Circ Genom Precis Med. 2018;11:e002237. doi: 10.1161/CIRCGEN.118.002237 [DOI] [PubMed] [Google Scholar]
- 54.Carruth ED, Young W, Beer D, James CA, Calkins H, Jing L, Raghunath S, Hartzel DN, Leader JB, Kirchner HL, et al. Prevalence and electronic health record-based phenotype of loss-of-function genetic variants in arrhythmogenic right ventricular cardiomyopathy-associated genes. Circ Genom Precis Med. 2019;12:e002579. doi: 10.1161/CIRCGEN.119.002579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katz AE, Nussbaum RL, Solomon BD, Rehm HL, Williams MS, Biesecker LG. Management of secondary genomic findings. Am J Hum Genet. 2020;107:3–14. doi: 10.1016/j.ajhg.2020.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]