

Abstract
The application of induced pluripotent stem cells (iPSCs) in advanced therapies is increasing at pace, but concerns remain over their clinical safety profile. We report the first-ever application of doggybone DNA (dbDNA) vectors to generate human iPSCs. dbDNA vectors are closed-capped linear double-stranded DNA gene expression cassettes that contain no bacterial DNA and are amplified by a chemically defined, current good manufacturing practice (cGMP)-compliant methodology. We achieved comparable iPSC reprogramming efficiencies using transiently expressing dbDNA vectors with the same iPSC reprogramming coding sequences as the state-of-the-art OriP/EBNA1 episomal vectors but, crucially, in the absence of p53 shRNA repression. Moreover, persistent expression of EBNA1 from bacterially derived episomes resulted in stimulation of the interferon response, elevated DNA damage, and increased spontaneous differentiation. These cellular activities were diminished or absent in dbDNA-iPSCs, resulting in lines with a greater stability and safety potential for cell therapy.
Keywords: induced pluripotent stem cells, reprograming, non-viral, doggybone DNA vectors, DNA damage response, interferon response
Graphical abstract
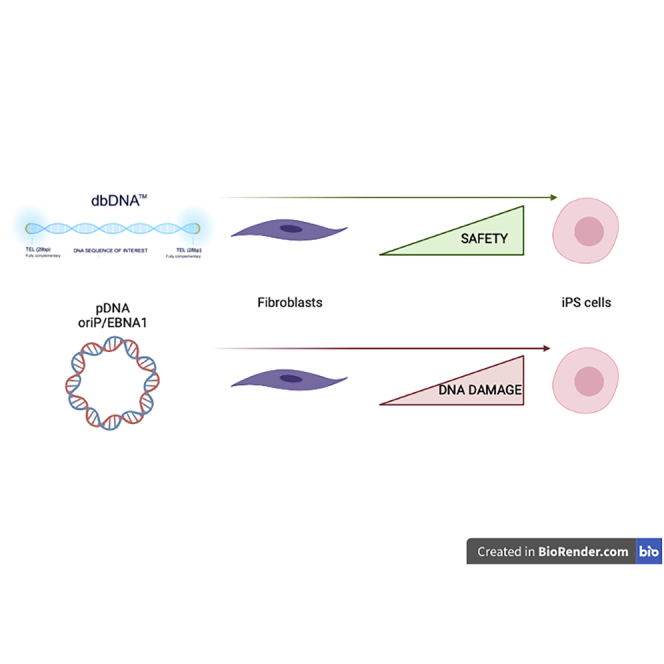
Thornton et al. have applied novel doggybone cDNA technology to generate transiently maintained gene delivery vectors containing no bacterial motifs to generate induced pluripotent stem cells (iPSCs). When compared to episomal OriP/EBNA1, our vectors generated safer, more stable colonies at an equivalent efficiency.
Introduction
The first descriptions of induced pluripotent stem cell (iPSC) reprograming of human somatic cells involved ectopic expression of the gatekeeper transcription factor OCT4 in combination with other key pluripotency-associated factors, SOX2, Nanog, KLF4, and LIN28, combining to induce and maintain the pluripotent state.1,2 It is also clear that pro-mitotic factors such as C-MYC/L-MYC and/or repression of cell cycle checkpoints, such as p53, are conduits to creating a compliant cellular state for iPSC reprograming.3,4 The process of iPSC reprograming is mechanistically classified into initiation, maturation, and stabilization phases.5 The initiation stage is stochastic, with progression requiring mesenchymal to epithelial transition (MET) and epigenetic reprogramming, including X chromosome reactivation.6 At the stabilization phase, bona fide iPSC colonies transition to a state of transgene independence. Overall, iPSC reprogramming remains inefficient as a process, reaching maximal reprogramming efficiencies up to 4%, although the reasons for this are not fully understood.7 However, it is clear that the number, dosage, relative stoichiometry, and persistence of transgenes in a somatic cell play a critical role. In early experiments retroviruses were used for transgene delivery, although the genomic integration of reprogramming factor payload has significant safety implications.1,8 The generation of iPSCs for research purposes is largely dominated by the use of Sendai virus vectors for gene delivery and direct application of mRNA or protein.9, 10, 11 However, episomal OriP/EBNA1 non-viral vectors are predominantly used in the clinical cell therapy domain.12 Overall, efficient transgene delivery, persistent but transient transgene expression over the reprogramming period, and reliable, footprint-free removal of transgenic material remain the critical factors for effective iPSC reprogramming.
Clinical trials to date have used a cocktail of 3 OriP/EBNA1 episomal plasmids expressing SOX2/KLF4, LIN28/L-MYC, and OCT4/shp53, respectively, to produce iPSC banks.13 We have employed doggybone DNA (dbDNA) vectors to deliver the same 3 expression cassettes except for the absence of both OriP/EBNA1 and short hairpin RNA designed against p53 (shp53) sequences to elicit iPSC reprogramming of human dermal fibroblasts at an efficiency and time frame equivalent to the state-of-the-art. Moreover, dbDNA vectors contain no bacterial sequences and batches contain no traceable endotoxin, reducing immune challenge to transfected cells.
Results
dbDNAs are closed-capped linear, double-stranded DNA gene delivery vectors that have already shown efficacy in scalable lentivirus and adeno-associated virus (AAV) production,14 CAR-T cell generation,15 and DNA vaccine technologies.16 In the latter study, Allen and colleagues showed that, unlike bacterially amplified plasmid, dbDNA per se did not stimulate a TLR9-mediated innate immune response in transfected cells. We speculated that dbDNA vectors would maintain transgene expression in human dermal fibroblasts (hDFs) for sufficient time, after a single transfection, to facilitate iPSC reprograming. Taking a reductionist approach, dbDNA vectors were designed and manufactured with transgenes equivalent to the current state-of-the-art iPSC reprogramming OriP/EBNA1 episomal plasmids,4 with the omission of shp53 and OriP/EBNA1 sequences to increase the clinical safety specification of dbDNA vectors (Figures 1Ai and 1Aii). In our hands, preliminary colony formation during iPSC reprograming occurs between 16 and 23 days irrespective of the gene delivery vector used or conditions of reprograming (feeder maintained or feeder-free). To assess the longevity of transgene expression we first nucleofected adult hDFs with equivalent quantities of GFP-expressing dbDNA or proTLx-K, the parental plasmid template for dbDNA production prior to bacterial DNA removal. A regression of transgene activity over time by flow cytometry was then determined (Figures 1Bi and 1Bii; Figure S1). There were consistently more GFP+ cells, with higher median fluorescence intensity, in dbDNA transfected cells compared to plasmid from day 1 through to day 21, suggesting that dbDNA vectors could result in expression of transgenes for a sufficient period to effect somatic cell reprograming.
Figure 1.

dbDNA vectors generate iPSCs at an efficiency equivalent to OriP-EBNA1 vectors
(Ai) Schematic depicting the process of dbDNA vector production. (Aii) Schematic representation of iPSC-reprogramming dbDNA vector constructs used in this study. (B) Evaluation of GFP gene expression in nhDFs after nucleofection of equivalent vector quantities of proTLX-eGFP or dbDNA-eGFP vectors (day [D]1–21) by fluorescence-activated cell sorting (FACS) analysis of GFP expression (Bi) and median fluorescence intensity (Bii). Analyses were performed from D1 to D21. n = 3 experiments ± SEM. (C) Comparison of iPSC reprogramming with equivalent dbDNA and OriP-EBNA1 vectors. At D21 emerging colonies appeared morphologically similar (Ci), stained positive for alkaline phosphatase (AP) (Cii), and formed similar numbers of colonies (Ciii). Scale bar represents 100 μm. n = 4 independent biological repeats ± SEM, Student’s t test.
This encouraged us to apply dbDNA vectors lacking p53-repressing shRNA (shp53) and OriP/EBNA1 sequences but otherwise mirroring the expression cassettes of the episomal plasmid OriP/EBNA1 system currently used in clinical trial protocols.4 The dbDNA vectors were initially independently transfected into HEK293T cells to confirm transgene expression (Figures S1Bi and S1Biii). Normal adult hDFs (nhDFs) were transfected with proTLx-K plasmid, dbDNA, or OriP/EBNA1 vectors, and the emergence of alkaline phosphatase (AP)-positive colonies was assessed after 21 days (Figures 1Ci–1Ciii). As expected, there were no colonies formed after plasmid transfection, but dbDNA and OriP/EBNA1 produced AP+ colonies at an efficiency of 0.1%–0.2% with no obvious difference in the time taken for colony formation. Both dbDNA-iPSCs and EBNA1-iPSCs adhered to established quality control (QC) standards for pluripotency including expression of endogenous pluripotency transcripts, protein, cell surface antigen, and the ability to achieve tri-lineage differentiation (Figures 2Ai, 2Aii, and 2B; Figure S2). dbDNA-iPSCs and EBNA1-iPSCs were subjected to a well-established neural differentiation protocol17 and produced broadly equivalent numbers of nestin+ neural progenitor cells and βIII-tubulin+ mature neurons during the expected time frames (Figure 2C). We were unable to detect the presence of dbDNA vector after passage (P10) in genomic preps from iPSC lines, although OriP/EBNA1 episomes remained detectable (Figure 2D). To prove the robustness of dbDNA-iPSC methodology, we generated iPSC lines from a range of hDFs obtained from normal adolescent donors and those affected by the childhood neurological disease variants CLN3, CLN6, and CLN7 of Batten disease. Moreover, we were able to generate iPSC lines from fresh and long-term cryopreserved hDFs (>10 years) as well as from hDFs subjected to extended passage (>P30), including some that were refractory to EBNA1-iPSC reprograming (Table 1). Throughout our extensive comparative evaluations, it became clear dbDNA-iPSCs developed colonies with a reduced propensity for spontaneous differentiation in comparison to OriP-EBNA1-produced colonies. Cell cycle analyses using propidium iodide (PI) staining and flow cytometry showed increased numbers of dbDNA-iPSCs in G0/G1 than EBNA1-iPSCs (Figure 3A; Figure S4), indicative of a slower cell cycle progression. This is likely a consequence of increased p53 activity in dbDNA-iPSCs due to a lack of shp53 target repression, so we evaluated protein levels of the p53 target and cell cycle checkpoint inhibitor p21. As expected, we found increased p21 protein in dbDNA-iPSCs compared to EBNA1-iPSCs by western blot (Figures 3Bi and 3Bii; Figure S5). Analyses of large numbers of colonies under rigidly comparable conditions also showed that dbDNA-iPSC colonies contained significantly fewer SSEA1+ cells and reduced APnull colony area (Figures 3C and 3D; Figures S6i and S6ii). These data strongly imply that dbDNA-iPSC colonies are less likely to spontaneously differentiate in maintenance culture without losing the capacity to respond appropriately to exogenous differentiation cues. We therefore propose that dbDNA-iPSCs would be more compliant with robust pluripotent stem cell amplification protocols necessary to generate cell banks for cell therapies.
Figure 2.

dbDNA-iPSCs are pluripotent and capable of multi-lineage differentiation
(A) Pluripotency comparison of dbDNA-iPSCs, EBNA1-iPSCs, and hESCs (Shef3) by RT-PCR specific to endogenous pluripotency markers (Ai). (Aii) Immunocytochemistry for transgene-specific (OCT4, SOX2) and endogenous (NANOG, TRA-1-81, TRA-1-60) pluripotency markers. Scale bar represents 100 μm. (B) Embryoid body formation and replating (16 days) followed by immunocytochemistry analysis of markers of the three germ lineages, β-iii-tubulin (neurectoderm), SOX17 (endoderm), and α-SMA (mesoderm). Scale bar represents 100 μm. (C) Targeted differentiation of dbDNA-iPSCs and EBNA1-iPSCs to nestin+ neural progenitor cells and βIII-tubulin+ mature neurons. Scale bar represents 100 μm. (D) PCR for detection of vector presence in identical-passage iPSCs produced by dbDNA and oriP-EBNA1 vectors.
Table 1.
Human dermal fibroblasts reprogrammed to iPSC lines in direct comparative experiments using dbDNA and EBNA1 vectors
| Dermal fibroblast source | NCL developmental classification | Passage fibroblast number | Starting material | Time of cryopreservation |
|---|---|---|---|---|
| Normal | neonate | P32 | cryopreserved | <1 year |
| Normal | juvenile | P16 | cryopreserved | <1 year |
| Normal | juvenile | P9 | cryopreserved | <1 year |
| CLN3-A1 | juvenile | P3 | fresh | N/A |
| ∗CLN3-A2 | juvenile | P3 | fresh | N/A |
| CLN3-B | juvenile | P7 | fresh | N/A |
| CLN3-C | juvenile | P38 | cryopreserved | 12 years |
| CLN6-A | juvenile | P8 | cryopreserved | 15 years |
| CLN7-A | juvenile | P12 | cryopreserved | 15 years |
| CLN7-B | juvenile | P7 | cryopreserved | 15 years |
hDFs refractory to EBNA1-mediated iPSC reprogramming but successful with dbDNA. NCL, Neuronal ceroid lipofuscinosis.
Figure 3.

dbDNA-iPSCs have cell cycle dynamics different from EBNA1-iPSCs and are less prone to spontaneous differentiation
(A) Comparison of dbDNA-iPSCs and EBNA1-iPSCs by cell cycle by flow cytometry after propidium iodide incorporation (n = 3 independent biological repeats ± SEM). (B) Comparison of dbDNA-iPSCs and EBNA1-iPSC by western blot for p21 with β-actin control (Bi) and densitometric quantification of 3 independent iPSC experiments (Bii) (n = 3 ± SEM). ∗∗p < 0.01, Student’s paired t test. (C) Comparison of dbDNA-iPSC and EBNA1-iPSC colony spontaneous differentiation by live-cell flow cytometry for the presence of SSEA1 antigen, indicative of exit from pluripotency. (D) Comparison of dbDNA-iPSCs and EBNA1-iPSCs by AP staining. ImageJ analysis is represented as AP+ area relative to AP− colony area (n = 3 independent biological repeats ± SEM). ∗∗∗∗p < 0.0001, Student’s paired t test.
To gain molecular insights into our observational data we performed a gene expression microarray on three dbDNA-iPSC and three EBNA1-iPSC lines (P25–30), using a human embryonic stem cell (hESC) line, SHEF3, as a pluripotency gold standard and parental hDFs as a negative control. Correlogram and Volcano plot (Figures 4Ai and 4Aii; Figure S7) and principal component analysis (PCA) (Figures 4Bi and 4Bii) indicated that dbDNA-iPSC transcriptome was more similar to hESCs than EBNA1-iPSCs. Hierarchical clustering of transcripts associated with either pluripotency (Figure 4Ci) or early differentiation (Figure 4Cii) indicated that dbDNA-iPSCs were again more similar to hESCs than EBNA1-iPSCs. Of note, OCT4 transcript (POU5F1) was low in dbDNA-iPSCs, although this was not reflected in our RT-PCR and immunocytochemistry data (Figures 2Ai and 2Aii). This may have been due to the detection of OCT4 transgene expression as a result of EBNA1 episome retention, although this was not experimentally verified. It was clear that EBNA1-iPSC colonies are enriched for all the analyzed transcripts, indicative of early mesendodermal and neurectodermal differentiation compared to dbDNA-iPSCs and hESCs cultured under standard pluripotency maintenance conditions. Furthermore, PANTHER analysis of transcripts overrepresented in EBNA1-iPSCs compared to dbDNA-iPSCs returned “cellular differentiation” as the top term (Figure 4Di). This could be as either a consequence of transition to a pre-differentiation state, although one might expect this to favor a defined developmental lineage, or a result of sporadic spontaneous differentiation. We validated our transcriptomic data using qRT-PCR on RNA harvested from an independent reprograming experiment, showing a significant upregulation of mesendoderm-associated transcripts for goosecoid, brachyury (T), and SOX17 (Figure 4Dii; Figures S8i and S8ii). We conclude that EBNA1-iPSCs may have a primed phenotype, prone to differentiation, whereas dbDNA-iPSCs have a more robust pluripotent phenotype.
Figure 4.

dbDNA-iPSCs are more similar to hESCs than EBNA1-iPSCs, which have a propensity for differentiation
(A) Transcriptomic comparison of dbDNA-iPSCs and EBNA1-iPSCs. (Ai) Correlogram for all cell types across all probed genes. (Aii) Volcano plot representing differential gene expression between dbDNA- and oriP-EBNA1-iPSCs. (B) Principal component analysis (PCA) determining relative expression differences between iPSCs and hESCs. (Bi) PC1 and PC2 comparison incorporates the highest proportion of microarray variation. (Bii) PC1 and PC3 to incorporate the remaining variation between all cell types in the microarray analysis. Microarray probes that had an FDR > 0.05 and a fold-change ≤ 1.5 were determined for both vectors. (C) Hierarchical clustering heatmap of pluripotency-related transcripts (Ci) and transcripts associated with early stage tri-lineage differentiation (Cii). (Di) Cellular differentiation is the highest relating biological process when relating EBNA1-iPSCs to dbDNA-iPSCs, applying PANTHER analysis. (Dii) Transcriptomic data were validated by qRT-PCR for selected (Goosecoid, Brachyury, SOX17) transcripts associated with early differentiation of pluripotent stem cells (n = 3 independent biological repeats ± SEM). ∗p < 0.05, ∗∗p < 0.001, ∗∗∗∗p < 0.0001, Student’s paired t test.
We next sought to interrogate the variance between the EBNA1-iPSC and dbDNA-iPSC molecular phenotypes. A total of 1,449 transcripts were significantly upregulated and 1,409 downregulated in EBNA1-iPSCs compared to dbDNA-iPSCs in our microarray dataset. Hierarchical clustering of the 1,409 overrepresented transcripts in dbDNA-iPSCs (p ≤ 0.05, q ≤ 0.05, and fold-change ≥ 1.5) showed no significant similarity of either EBNA1-iPSCs or dbDNA-iPSCs with hESCs, the key enriched Reactome term being “cell cycle” (Figures 5Ai and Aii). This is consistent with our previous data on alterations to cell cycle in EBNA1-iPSCs likely due to p53 repression. The reverse analysis of the 1,449 significantly upregulated transcripts in EBNA1-iPSCs compared to dbDNA-iPSCs (p ≤ 0.05, q ≤ 0.05, and fold-change ≥ 1.5) showed that dbDNA-iPSCs are more similar to hESCs than EBNA1-iPSCs, implying that many of these transcripts upregulated in EBNA1-iPSCs are not compatible with ground-state pluripotency (Figure 5Bi). Combined analyses of this transcript group, applying Reactome, gene set enrichment analysis (GSEA), MSigDB, and gene ontology (GO) tools, showed an enrichment of terms associated with pluripotent stem cell differentiation such as “endodermal-mesodermal cell signaling,” “positive regulation of axonogenesis,” and “embryonic skeletal system development” (Tables S1 and S2). This further corroborates our previous data showing that EBNA1-iPSCs present indications of spontaneous tri-lineage differentiation. The largest represented term in Reactome with the most significant p value was “immune system,” for which the major component was “interferon signaling” (Figures 5Bii, 5Ci, and 5Cii; Figure S9i). Interferons (IFNs) are the common potentiators of the cellular innate immune response to foreign DNA, in macrophages and other cells, through IRF, NF-kappaB, and STAT1 transcription factors (reviewed by Atianand and Fitzgerald18). We identified IFN activators/potentiators in our transcriptomic database and found these to be upregulated with high penetrance in EBNA1-iPSCs compared to dbDNA-iPSCs and hESCs (Figure 5Di). Transcriptomic data were independently confirmed by qRT-PCR targeting selected transcripts of type I and II IFN signal transducers. Interestingly, there was no change in IRF3 expression, the predominant anti-microbial dsDNA transcription factor, or its co-factors. However, there was upregulation of components of the IRF7 DNA sensor complex including MyD88, IRAK1, IRAK4, and IRF1, the signal transducer of the secondary type I IFN response (Figure 5Dii; Figure S9ii). Type I IFNs potentiate through IFNGR-JAK/STAT1 signaling, but EBNA1 is also known to amplify IFN-mediated STAT1 activation.19 We also noted elevated STAT1 transcript 48 h and 72 h after transfection of HEK293T cells (Figure 5Diii). Collectively, these data provide evidence that, unlike dbDNA vectors, pEBNA1 episomes induce a foreign dsDNA IFN response in transfected cells that is likely further amplified by EBNA1 expression.
Figure 5.

EBNA1-iPSCs have elevated interferon signaling and differentiation cues compared to dbDNA-iPSCs and hESCs
(A) Comparison of transcripts upregulated in dbDNA-iPSCs compared to EBNA1-iPSCs as a hierarchical heatmap analysis (Ai) or graphical representation of biological processes most significantly aligned with the overrepresented probes (Aii) in Reactome for EBNA1-iPSCs. (B) Comparison of transcripts upregulated in EBNA1-iPSCs compared to dbDNA-iPSCs as a hierarchical heatmap analysis (Bi) or graphical representation of biological processes most significantly aligned with the overrepresented probes (Bii) in Reactome for EBNA1-iPSCs. (C) Identification of interferon signaling pathway by Reactome analysis (Ci) focusing on interferon signaling (Cii). (D) Comparison of transcripts upregulated in EBNA1-iPSCs compared to dbDNA-iPSCs as a hierarchical heatmap analysis (Di) as the most significantly upregulated signaling pathway in EBNA1-iPSCs compared to dbDNA-iPSCs. (Dii) qRT-PCR analysis of transcripts involved in interferon signaling in dbDNA-iPSCs and EBNA1-iPSCs (n = 3 independent biological repeats ± SEM). p < 0.05. (Diii) A STAT1 transcript time course comparison on nhDFs (n = 3 independent biological repeats ± SEM).
As well as amplifying the type I IFN response, there is evidence that prolonged expression of EBNA1 can result in host cell genomic DNA damage, likely through a reactive oxygen species (ROS)-mediated mechanism.20 To assess vector-induced genomic DNA damage, we transfected hDFs with molar equivalents of pEBNA1-GFP and dbDNA-GFP and then induced ROS by treating with low-dose H2O2 (50 μM; Figures S10, S11, and S12). Three days post-transfection we carried out comet and γH2AX DNA damage comparisons on GFP+ cells. Remarkably, we saw significant increases in all indicators of DNA damage (comet head and length, tail DNA, and tail moment,; Figures 6A and 6Bi–6Biv) and DNA damage response (nuclear γH2AX puncta area; Figures 6Ci and 6Cii) in pEBNA1-GFP-transfected cells compared to dbDNA-GFP. We conclude that the increase in ROS-mediated DNA damage is caused by DNA fragmentation as a result of an EBNA1-agonized mechanism. This has fundamental safety implications in the use of EBNA1 vectors to generate iPSCs for clinical application and strongly suggests that dbDNA vectors would be a safer and more efficacious transgene delivery system.
Figure 6.

EBNA1-iPSCs have elevated DNA damage compared to dbDNA-iPSCs
nhDFs nucleofected with dbDNA and OriP-EBNA1 vectors were assessed for evidence of DNA damage by comet assay. (A) Representative micrographs of nuclear comets (n > 100 images, scale bar represents 100 μm). (B) Graphical representation of data generated with CaspLab of nhDFs transfected with dbDNA and OriP-EBNA1 including head DNA (Bi), tail DNA (Bii), comet length (Biii), and tail moment (Biv) (n > 100 images, error bars represent ±SEM of 3 independent experiments). ∗∗∗∗p < 0.0001, ∗∗p < 0.01, ∗p < 0.05, Student’s paired t test. (C) nhDFs nucleofected with dbDNA-GFP and OriP-EBNA1-GFP vectors were assessed for evidence of DNA damage response by nuclear γH2AX puncta staining, (Ci) GFP+ cells were identified and then γH2AX puncta counted for transfected cells. (Cii) Average puncta per cells counted (>200 nuclei per treatment; scale bar represents 100 μm) graphically represented and compared to untreated nhDFs and those treated with 100 μM H2O2 as a positive control. (>200 nuclei per treatment, error bars represent ±SEM). ∗∗p < 0.01.
Clinical translation of dbDNA iPSC reprograming would require current good manufacturing practice (cGMP) compliance under feeder-free conditions using xenofree, chemically defined reagents. We devised such a protocol and showed proof of principle by carrying out an independent iPSC reprograming experiment under such conditions. hDFs were reprogramed to iPSCs with dbDNA and OriP/EBNA1 vectors in a research laboratory environment but with cGMP-compliant xenofree reagents in the absence of feeder cells. iPSC colonies emerged under similar time frames, and efficiencies and were stabilized by P12 (Figure 7A). Feeder-free dbDNA-iPSCs expressed pluripotency proteins SOX2 and OCT4 and presented equivalent levels of SSEA3 cell surface antigen (Figures 7B and 7C; Figures S13 and S14), significantly higher endogenous transcript levels of LIN28, and significantly lower levels of transcripts associated with differentiation and type I IFN response (Figure 7D). These data support our existing analyses that feeder-free dbDNA-iPSCs produced with cGMP-compliant reagents are more stable and compliant with scale-up than EBNA1-iPSCs.
Figure 7.

dbDNA-iPSCs generated under feeder-free, xenofree, cGMP-compliant conditions
(A) dbDNA vectors generated colonies under feeder-free conditions at a time and relative efficiency comparable to OriP-EBNA1. (B) Immunohistochemistry for SOX2 and OCT4 on feeder-free dbDNA-iPSCs at P12. (C) Flow cytometry for SSEA3 pluripotency antigen shows equivalent median fluorescence intensity for dbDNA-iPSCs and EBNA1-iPSCs (P12, error bars represent ± SEM of 3 independent experiments). p = 0.623546, Student’s paired t test. (D) qRT-PCR analyses of gene expression in dbDNA-iPSCs compared to EBNA1-iPSCs (error bars represent ±SEM of 3 independent experiments). ∗∗∗p < 0.0005, ∗∗p < 0.01, ∗p < 0.05, Student’s paired t test.
Discussion
The greatest aspiration for iPSC technologies is their application in regenerative medicine therapies, but methodological evolution since 2007 has tended to compromise safety over efficacy. The groundbreaking work of Takahashi and colleagues has culminated in a first-in-man phase I clinical trial using iPSC-derived retinal pigmented epithelia to treat macular degeneration.13 The standardized clinical protocols in this clinical trial use iPSC banks generated with 3 episomal OriP/EBNA1 plasmids co-expressing SOX2-2A-KLF4, LIN28-2A-L-MYC, and OCT4 and short-hairpin knockdown of p53 (shp53).13 Both L-MYC and EBNA1 have significant oncogenic potential, and loss of the p53 mitotic checkpoint is one of the most common genetic events in oncogenesis. Consequently, iPSC banks require rigorous time-consuming and costly safety evaluations, resulting in preclusive delays for autologous cell therapies.
The ideal protocol for iPSC reprograming would be to introduce a single dose of transient protein-coding sequences (DNA or mRNA) that perseveres sufficiently to affect the induction, stabilization, and transgene-independent maintenance of pluripotency and is then rapidly lost with no genetic footprint. Previous transient gene expression approaches using plasmid, minicircle,21,22 or mRNA10 required multiple, time-consuming and costly transfections to effect iPSC reprograming of somatic cells. The current clinically translatable state-of-the-art is the OriP/EBNA1-based episomal plasmid system. Episomes attach to, and segregate with, chromosomes during mitosis, thereby extending expression time in highly proliferative cells. However, these vectors have been shown to persist in iPSC lines for >10 passages,23 making autologous therapies logistically challenging by extending the pipeline. Moreover, bacteria-derived CpG motifs on plasmid DNA induce an IFN-mediated innate immune response (transduced by NF-kappaB, IRF3, 7, and 9) in transfected cells, and the implications of expressing the Epstein-Barr virus protein EBNA1 are not fully understood.
We have utilized dbDNA vectors, closed-capped linear double-stranded DNA gene expression cassettes that contain no bacterial DNA, to generate iPSCs. We mirrored the 3-vector system under the assumption that this previously successful vector combination confers an appropriate stoichiometry of transgene expression. The exception was that we chose to remove the shp53 short hairpin RNA sequence to increase the safety profile of these vectors. There is clear evidence that p53 repression increases iPSC reprograming efficiency,3 but this comes at the expense of the cell’s genomic integrity. Babos and co-workers described the production of hypertranscribing hyperproliferating iPSCs (HHCs) secondary to the activation of topoisomerases, which significantly improved reprograming efficiencies in fibroblasts.24 The p53 protein has been demonstrated to positively regulate topoisomerase expression, providing positive implications for the maintenance of wild-type p53 expression during reprogramming relating to iPSC quality.25 Using dbDNA vectors, we were able to generate iPSCs at efficiency comparable to OriP/EBNA1 vectors across a multitude of fresh and cryopreserved donor hDFs. Our extensive dataset shows, in direct comparisons, that dbDNA-iPSCs are more robust in continued culture and less likely to spontaneously differentiate than EBNA1-iPSCs.
Moreover, we present comprehensive data that the combined absence of bacterial DNA sequences and EBNA1 coding sequence decreases cellular inflammation and DNA damage. IFN signaling has recently become a hot topic in influencing stem cell potency and more specifically in destabilizing the pluripotent state and tri-lineage differentiation capacity.26, 27, 28 Our data imply that type I IFN could contribute to priming or inducing spontaneous differentiation in EBNA1-iPSCs that is absent from dbDNA-iPSCs. Although we do not delineate this to the presence of bacterial DNA motifs or the presence of EBNA1 expression, it is a somewhat moot point, as both are absent from our dbDNA vectors. Finally, we are confident that continued EBNA1 expression results in a defective DNA damage response to ROS. In this context, dbDNA vectors have a significant safety and efficacy advantage over the current clinical state-of-the-art OriP/EBNA1 episomes. We propose that a single transfection of three dbDNA vectors expressing OCT4, SOX2/L-MYC, and LIN28/KLF4 could be the safest and most efficacious method of generating iPSC lines for clinical application.
Materials and methods
Doggybone DNA production
Coding sequences for the iPSC reprograming factors were sub-cloned into the proTLx-K template plasmid inside the protelomerase telRL sequences. Verification of correct clones was achieved by complete Sanger sequencing. The template plasmids were denatured with 0.1 M NaOH followed by quenching in reaction buffer (Tris-HCl based) containing oligonucleotide primers and deoxyribonucleotide triphosphates (dNTPs). Phi29 DNA polymerase and pyrophosphatase were then added, and the reaction was incubated at 30°C for ∼30 h. The resulting concatemeric DNA was processed by addition of processing enzymes. protelomerase TelN, restriction enzyme suitable for plasmid backbone cleavage (template dependent), and exonucleases (all enzymes used were sourced from QIAGEN [Beverly. MA, USA] or New England Biolabs [Hitchin, UK]). The dbDNA was purified from the reaction components at appropriate stages of the process through the addition of 500 mM NaCl-100 mM MgCl2 and precipitation using polyethylene glycol (PEG) 8000 (AppliChem, Bredbury, UK). This process was repeated multiple times during the process, resuspending pellets in appropriate buffers with final resuspension in water prior to ethanol precipitation to remove residual salts. Samples were formulated into required storage buffer and then passed though a 0.22-μm filter.
iPSC production
A total of 8 μg of episomal plasmid (pCXLE-hSK, pCXLE-hUL, pCXLE-hOCT3/4-shp53-F, and pCXWB-EBNA1) and 8 μg of dbDNA reprograming vectors (dbDNA-hSK, dbDNA-hUL, dbDNA-OCT4) were prepared. The net weight equivalents of the vectors resulted in a ratio of 1.49:1 (dbDNA:oriP-EBNA1) in terms of vector copies. The vectors were resuspended in Nucleofector solution (Lonza) along with 4.5 × 105 hDFs. The solution was then nucleofected with the Amaxa Nucleofector 2b system (Lonza) before being re-seeded in complete DMEM (DMEM supplemented with 10% FBS, 1% Pen/Strep [penicillin-streptomycin], 1% non-essential amino acids [NEAAs], 4% 200 mM l-glutamine; all Sigma-Aldrich). On day 7 post-nucleofection, the reprograming cells were re-seeded onto an inactive murine embryonic fibroblast (iMEF) feeder layer (Cambridge Bioscience) in complete DMEM. Twenty-four hours after re-plating, the medium was altered to hESC medium supplemented with FGF2 (10 ng/mL) (R&D Systems) until primary colony formation.
Feeder-free reprogramming was performed where the reprograming procedure was carried out as normal but the hESC medium was replaced with Essential 8 (Gibco) and the iMEF feeder layer for rhLaminin521 (Thermo Fisher Scientific).
Reprogramming efficiency
Twenty-one days post-nucleofection, the number of primary colonies was determined via the addition of an AP stain (Sigma). A tablet was dissolved in 10 mL of Dulbecco’s PBS (DPBS) (Sigma) before being added to wells containing primary colonies. The cells were left for 10–20 min in the dark before being analyzed. The efficiency can be determined by the number of primary colonies in proportion to the number of fibroblasts seeded per well.
Pluripotency immunocytochemistry
For fluorescence immunocytochemistry, cells were fixed in 4% paraformaldehyde (PFA) (Sigma). Samples were blocked in 2% BSA in PBS + 0.05% (v/v) Tween 20 (all from Sigma). Antibodies were diluted in blocking buffer (SOX2 1:200 [AF2018], OCT4 1:100 [Ab18976], TRA-1-60 1:200 [Ab16288], TRA-1-81 1:200 [Ab16289], βIII-tubulin 1:200 [MAB1195], α-Smooth muscle actin 1:100 [Ab5694], SOX17 1:60 [AF1924], γ-H2AX 1:1,000 [Ab11174]) and incubated overnight at 4°C. Secondary antibodies (1:500, all Alexa Fluor) were added for 60 min at room temperature. The cells were then washed before visualization.
Quantitative real-time PCR
Total RNA was extracted from cells with the RNeasy kit (QIAGEN) as directed by the manufacturer’s instructions. Random hexamer primers and Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega) were used to reverse transcribe RNA as per the manufacturer’s instructions. For the pluripotency quantitative real-time PCR, the cDNA generated was incorporated into a standard PCR reaction using Q5 DNA polymerase (New England Biolabs) along with endogenous pluripotency primers (10 μM). The samples were then electrophoresed on an agarose gel to determine transcript presence. In quantitative real-time-PCR, triplicate samples were analyzed by the Bio-Rad CFX Connect Real-Time PCR detection system using Fast SYBR Green Master Mix (Thermo Fisher Scientific). Transcript levels were normalized to PABPC4.
Tri-lineage iPSC differentiation
Human iPSCs maintained on MEFs were trypsinized, resuspended in iPSC medium, and seeded on 6-well ultralow-attachment plates (Corning) with 10 μM Y-27632 (Sigma). Medium was changed on day 4 to DMEM + 20% FBS, and on day 7 the embryoid bodies (EBs) were plated on tissue culture-treated plates coated with 0.1% porcine gelatin (Sigma). EB outgrowths were fixed with 4% PFA 8 days later for immunocytochemical staining.
Western blotting
Cells were scrape into RIPA buffer (Sigma) with the addition of protease inhibitor cocktail (Sigma) at a concentration of 2 × 107 cells/mL. Total protein was quantified by the Bio-Rad protein assay. Protein lysates were resolved by SDS-PAGE and electrotransferred onto Hybond-enhanced chemiluminescence (ECL) nitrocellulose membrane (Amersham Biosciences). The membrane was blocked in 5% BSA in PBS + 0.05% Tween 20 and incubated in primary antibody (OCT4 1:400 [Ab18976], SOX2 1:2,000 [AF2018], LIN28 1:1,000 [AF3757], p21 1:2,000 [2947]; Cell Signaling Technology) overnight at 4°C. The blots were then incubated with their appropriate horseradish peroxidase-conjugated secondary antibody (Dako, 1:2,000) in blocking buffer for 60 min at room temperature. The membrane was developed by ECL (Pierce/Amersham) according to manufacturer’s instructions. Densitometric quantification was performed on unsaturated images in ImageJ (NIH).
Transcriptomic microarray
RNA was isolated from three biological repeats of iPSCs, ESCs, and fibroblasts. The whole-genome gene expression array (Human HT-12 v4 Expression BeadChip) kit (Illumina) was processed with QC assessment by the DKFZ German Cancer Research Center, Heidelberg, Germany. Single-stranded cRNA was derived from inputs of 200 ng of total RNA with the Illumina TotalPrep RNA amplification kit (Life Technologies). Standard Illumina hybridization protocol was used as per manufacturer’s instructions. The data were normalized by quantile normalization using the R function “normalize.quantiles” from the Bioconductor package “Preprocess core.” The R Studio “cor” function was used to generate correlation matrices with the Pearson method. The GPLOTS package in R studio generated heatmaps of normalized data. The “Volcanoplot” function was used in R to generate a volcano “ploheatmapsgplot” to generate a PCA. Reactome, Panther, and GSEA were used to acquire transcription factor enrichment terms.
Comet assay
In brief, ∼1 × 106 cells to be analyzed were mixed with low-melting agarose and added to normal-melting agarose-coated slides. The slides were added to a lysis buffer before being left to dry overnight. The following day, slides underwent electrophoresis and were stained with SYBR Gold (Thermo Fisher) and imaged with a fluorescent microscope. The images were then analyzed for any signs of damage with CaspLab (1.2.3beta2).
Flow cytometry
Cell cycle was assayed in both dbDNA- and oriP-EBNA1-produced iPSCs with PI (Sigma). Cells were picked and mechanically dissociated as much as possible. The cells were then centrifuged at 300 × g for 2 min and the supernatant removed. The cells were resuspended in 2 mL of DPBS before 2 mL of ice-cold 70% ethanol was added dropwise while the cells were vortexing. The cells were left for fixation for 30 min at 4°C. After washes with DPBS, the cells were subjected to 100 μL of a 100 μg/mL stock solution of RNase A. Four hundred microliters of a 50 μg/mL stock of PI was added, and the reaction was kept in the dark for 10 min. The PI staining was then analyzed with a Becton Dickinson (BD) FACScalibur flow cytometer with BD CellQuest Pro software.
Spontaneous differentiation quantification using ImageJ
Spontaneous differentiation was quantified as relative areas of a colony that occluded any AP stain. The iPSC lines were stained with AP staining (Sigma) for 10 min before being imaged for analysis. Any images were analyzed with ImageJ, whereby the area of each colony was determined before the relative area of occluded stain/differentiation was quantified in proportion to the overall area. The analysis was carried out on 3 different iPSC lines across both earlier passages (P8) up to colonies that were passage 32.
Statistical analyses
Percentage data were transformed and pairwise comparisons were conducted between dbDNA-iPSC and EBNA1-iPSC samples by a one-way paired t test, with a Bonferroni post hoc correction.
For transcriptomic analysis, QC analysis was undertaken with quantile normalization on R studio and the “normalize.quantiles” function from the Bioconductor package preprocess Core. After QC analysis, probes from dbDNA- and EBNA1-iPSCs were subjected to a two-way Student’s t test analysis with a cutoff p value of ≤0.05. From this, the significantly different probes were subjected to a Benjamini-Hochberg analysis to determine the false discovery rate (FDR), limiting the possibility of a type 1 error, applying a cutoff of ≤0.05. Once our significantly different probes were compiled (p value ≤ 0.05 and FDR ≤ 0.05) fold-change expression was calculated between the dbDNA- and EBNA1-iPSC samples; probes with a fold-change difference of ≥1.5 were selected for GO analysis using multiple online resources.
Acknowledgments
C.D.T. and S.F. were funded by Touchlight Genetics. T.R.M. received funding from Horizon2020 (BATCure Ref: 666918). Batten disease hDFs were provided by S.E.M. from a repository held at University College London, UK as part of the EU Horizon 2020 BATCure (Ref: 666918) consortium. We thank the microarray unit of the DKFZ Genomics and Proteomics Core Facility for providing the Illumina Whole-Genome Expression Beadchips and related services.
Author contributions
Conceptualization: T.R.M., L.J.C., and J.P.T. Implementation: C.D.T., K.K., L.M.F., S.F., A.R.-M., S.E.M., and A.E.B. Data analysis: C.D.T., T.R.M., and R.P.H. Writing, reviewing, and editing: C.D.T., T.R.M., and L.J.C.
Declaration of interests
L.J.C., K.K., and J.P.T. are employees of Touchlight Genetics Ltd. Touchlight contributed to funding this research in the laboratory of T.R.M.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.09.018.
Supplemental information
References
- 1.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 3.Hong H., Takahashi K., Ichisaka T., Aoi T., Kanagawa O., Nakagawa M., Okita K., Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 5.Samavarchi-Tehrani P., Golipour A., David L., Sung H.K., Beyer T.A., Datti A., Woltjen K., Nagy A., Wrana J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 6.Buganim Y., Faddah D.A., Cheng A.W., Itskovich E., Markoulaki S., Ganz K., Klemm S.L., van Oudenaarden A., Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haridhasapavalan K.K., Borgohain M.P., Dey C., Saha B., Narayan G., Kumar S., Thummer R.P. An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene. 2019;686:146–159. doi: 10.1016/j.gene.2018.11.069. [DOI] [PubMed] [Google Scholar]
- 8.Okita K., Ichisaka T., Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 9.Cho H.J., Lee C.S., Kwon Y.W., Paek J.S., Lee S.H., Hur J., Lee E.J., Roh T.Y., Chu I.S., Leem S.H. Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood. 2010;116:386–395. doi: 10.1182/blood-2010-02-269589. [DOI] [PubMed] [Google Scholar]
- 10.Yakubov E., Rechavi G., Rozenblatt S., Givol D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Commun. 2010;394:189–193. doi: 10.1016/j.bbrc.2010.02.150. [DOI] [PubMed] [Google Scholar]
- 11.Fusaki N., Ban H., Nishiyama A., Saeki K., Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu J., Hu K., Smuga-Otto K., Tian S., Stewart R., Slukvin I.I., Thomson J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandai M., Kurimoto Y., Takahashi M. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017;377:792–793. doi: 10.1056/NEJMc1706274. [DOI] [PubMed] [Google Scholar]
- 14.Karda R., Counsell J.R., Karbowniczek K., Caproni L.J., Tite J.P., Waddington S.N. Production of lentiviral vectors using novel, enzymatically produced, linear DNA. Gene Ther. 2019;26:86–92. doi: 10.1038/s41434-018-0056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop D.C., Caproni L., Gowrishankar K., Legiewicz M., Karbowniczek K., Tite J., Gottlieb D.J., Micklethwaite K.P. CAR T Cell Generation by piggyBac Transposition from Linear Doggybone DNA Vectors Requires Transposon DNA-Flanking Regions. Mol. Ther. Methods Clin. Dev. 2020;17:359–368. doi: 10.1016/j.omtm.2019.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen A., Wang C., Caproni L.J., Sugiyarto G., Harden E., Douglas L.R., Duriez P.J., Karbowniczek K., Extance J., Rothwell P.J. Linear doggybone DNA vaccine induces similar immunological responses to conventional plasmid DNA independently of immune recognition by TLR9 in a pre-clinical model. Cancer Immunol. Immunother. 2018;67:627–638. doi: 10.1007/s00262-017-2111-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.FitzPatrick L.M., Hawkins K.E., Delhove J.M.K.M., Fernandez E., Soldati C., Bullen L.F., Nohturfft A., Waddington S.N., Medina D.L., Bolaños J.P., McKay T.R. NF-κB Activity Initiates Human ESC-Derived Neural Progenitor Cell Differentiation by Inducing a Metabolic Maturation Program. Stem Cell Reports. 2018;10:1766–1781. doi: 10.1016/j.stemcr.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atianand M.K., Fitzgerald K.A. Molecular basis of DNA recognition in the immune system. J. Immunol. 2013;190:1911–1918. doi: 10.4049/jimmunol.1203162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood V.H., O’Neil J.D., Wei W., Stewart S.E., Dawson C.W., Young L.S. Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFbeta signaling pathways. Oncogene. 2007;26:4135–4147. doi: 10.1038/sj.onc.1210496. [DOI] [PubMed] [Google Scholar]
- 20.Gruhne B., Sompallae R., Masucci M.G. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene. 2009;28:3997–4008. doi: 10.1038/onc.2009.258. [DOI] [PubMed] [Google Scholar]
- 21.Jia F., Wilson K.D., Sun N., Gupta D.M., Huang M., Li Z., Panetta N.J., Chen Z.Y., Robbins R.C., Kay M.A. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods. 2010;7:197–199. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Si-Tayeb K., Noto F.K., Sepac A., Sedlic F., Bosnjak Z.J., Lough J.W., Duncan S.A. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev. Biol. 2010;10:81. doi: 10.1186/1471-213X-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlaeger T.M., Daheron L., Brickler T.R., Entwisle S., Chan K., Cianci A., DeVine A., Ettenger A., Fitzgerald K., Godfrey M. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015;33:58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babos K.N., Galloway K.E., Kisler K., Zitting M., Li Y., Shi Y., Quintino B., Chow R.H., Zlokovic B.V., Ichida J.K. Mitigating Antagonism between Transcription and Proliferation Allows Near-Deterministic Cellular Reprogramming. Cell Stem Cell. 2019;25:486–500.e9. doi: 10.1016/j.stem.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gobert C., Skladanowski A., Larsen A.K. The interaction between p53 and DNA topoisomerase I is regulated differently in cells with wild-type and mutant p53. Proc. Natl. Acad. Sci. USA. 1999;96:10355–10360. doi: 10.1073/pnas.96.18.10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eggenberger J., Blanco-Melo D., Panis M., Brennand K.J., tenOever B.R. Type I interferon response impairs differentiation potential of pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2019;116:1384–1393. doi: 10.1073/pnas.1812449116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercado N., Schutzius G., Kolter C., Estoppey D., Bergling S., Roma G., Gubser Keller C., Nigsch F., Salathe A., Terranova R. IRF2 is a master regulator of human keratinocyte stem cell fate. Nat. Commun. 2019;10:4676. doi: 10.1038/s41467-019-12559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witteveldt J., Knol L.I., Macias S. MicroRNA-deficient mouse embryonic stem cells acquire a functional interferon response. eLife. 2019;8:e44171. doi: 10.7554/eLife.44171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.