

Summary
In this issue of Neuron, Shi et al. show a protective role for the low-density lipoprotein receptor (LDLR) in tau pathology. Brain overexpression of LDLR lowers apolipoprotein E (apoE), suppresses microglia activation, preserves myelin, and ameliorates neurodegeneration, pointing the way toward potential new therapies.
Emerging evidence underscores the contribution of neuroimmune and neurovascular mechanisms in the pathogenesis of neurodegenerative diseases. Microglia activation, blood brain-barrier (BBB) disruption, exacerbation of amyloid-β (Aβ) and tau-mediated neurodegeneration, as well as compromised white matter integrity, are associated with the apolipoprotein E (APOE) gene variant APOE4, which increases the risk of Alzheimer’s disease (AD) up to 15-fold (Mahley and Huang, 2012). The approval of a drug to deplete Aβ in the brain has ushered in a new era of possibility for the development of a toolbox of therapeutics targeting immune and vascular mechanisms to maximize therapeutic benefit in AD and related dementias. Shi et al. (2021) now report that overexpression of the low-density lipoprotein receptor (LDLR), which binds apoE, suppresses microglia activation and neurodegeneration, and preserves myelin integrity in tauopathy (Figure 1).
Figure 1:
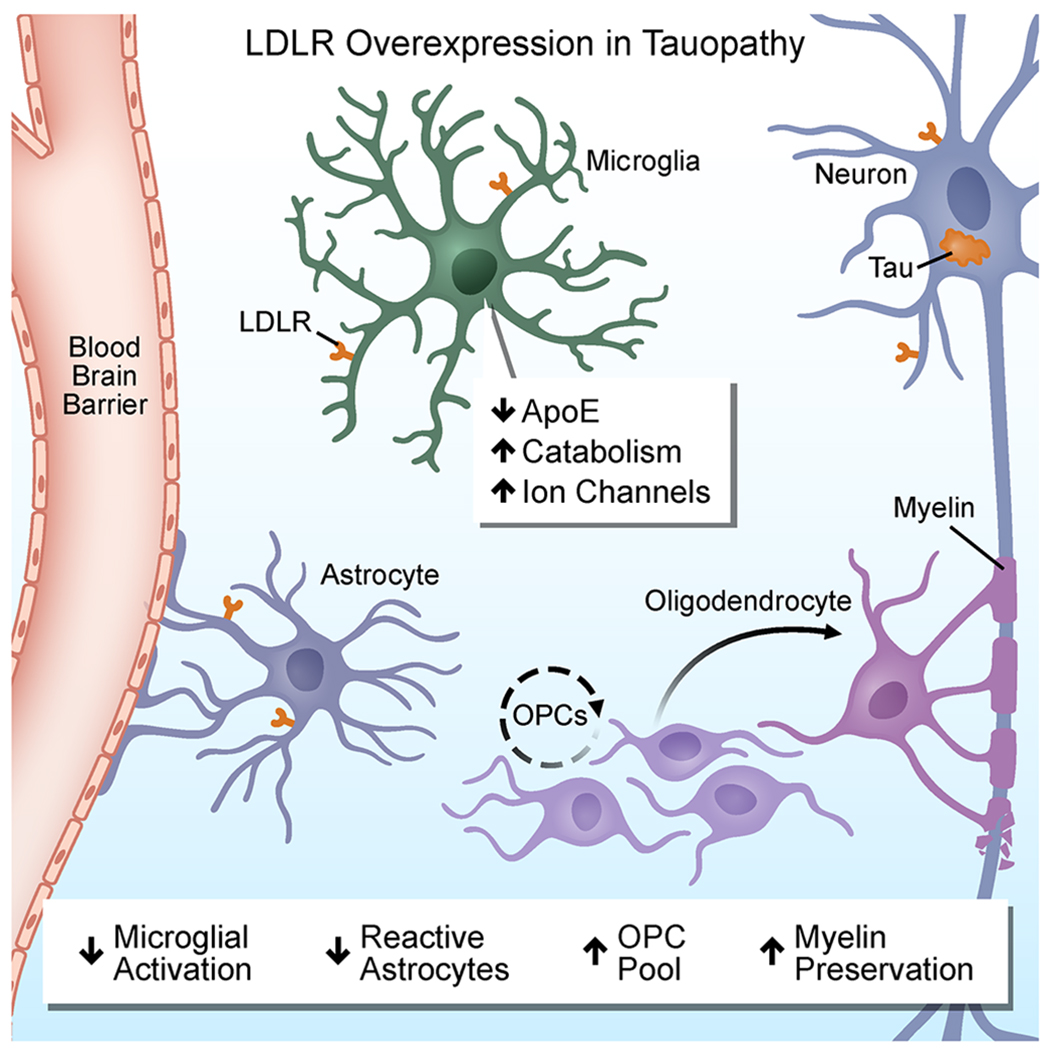
Protective effects of LDLR overexpression in tauopathy.
LDLRs are a group of structurally related multifunctional endocytic cell surface receptors highly expressed within the CNS that have known roles in AD pathobiology (Andersen and Willnow, 2006). Two members of this receptor family, LDLR-related protein 1 (LRP1) and LDLR bind apoE and influence its levels in the brain. LRP1 binds apoE-Aβ complexes, tissue-type plasminogen activator, myelin, and tau to regulate Aβ clearance, promote BBB disruption, myelin phagocytosis, and tau endocytosis and spread, respectively (Chang et al., 2021; Herz, 2003). In neurons and astrocytes, LDLR controls cholesterol transport and metabolism of apoE (Andersen and Willnow, 2006). ApoE is elevated in the cerebrospinal fluid (CSF) of LDLR deficient mice (Andersen and Willnow, 2006), suggesting that functional receptor activity controls apoE levels. Since LDLR lowers apoE levels in the CNS, the Holtzman lab previously generated an elegant animal model for overexpression of LDLR in the brain under the control of the prion promoter (reviewed in Chen et al., 2021b). They showed that LDLR overexpression in the brain reduces apoE and significantly attenuates Aβ-mediated pathology in the APP/PS1 AD mouse model. Similar to Aβ-mediated pathology, apoE4 also exacerbates tau-mediated neurodegeneration (reviewed in Chen et al., 2021b). However, whether overexpression of LDLR could be protective against tauopathy was unknown.
To address this question, Shi et al. (2021) crossed LDLR overexpressing mice with the P301S mouse model of tauopathy. LDLR overexpression reduced apoE levels in the CSF of P301S/LDLR mice by 90%, which was accompanied by reduced brain atrophy in P301S/LDLR mice. LDLR was detected in multiple cell types in the CNS of P301S/LDLR mice including microglia, astrocytes, and neurons. Microglia expression of LDLR was demonstrated in both cell autonomous systems and in the brain of P301S/LDLR mice and was associated with apoE reduction in microglia. Synaptic loss in hippocampal CA3 region was also reduced as assessed by immunohistochemistry for PSD-95 and electron microscopy. LDLR overexpression shifted p-tau towards an early disease phenotype and reduced phosphorylated tau (p-tau) in the CSF of P301S/LDLR mice. These findings identified a beneficial role for brain overexpression of LDLR in reducing apoE and features of neurodegeneration in an animal model of tauopathy.
How does LDLR overexpression protect in tauopathy? Genetic and transcriptomic studies have pinned APOE at the nexus of microglia activation in neurodegenerative diseases. ApoE regulates microglial transcriptional signatures, which drive neurodegeneration in tauopathy (reviewed in Chen et al., 2021b). Using single-nucleus RNA sequencing (snRNA-seq) Shi et al. (2021) identified the microglial transcriptomic profiles in P301S mice overexpressing LDLR, and found that LDLR overexpression dramatically decreased a subset of activated microglia with transcriptional programs linked to neurodegeneration including the disease-associated microglial (DAM) genes Apoe, Spp1, Itgax and Trem2. Concomitantly, LDLR overexpression in P301S mice induced a robust transcriptional reprogramming of a new microglia subset termed Cluster 3. Microglia upregulated genes in Cluster 3 encoded ion channels and neurotransmitter receptors, such as the voltage-gated K+ and Na+ channels Kcnc1, Kcnc3, Scn2a, NMDARs, GABAARs and mGluRs, while expression of genes involved in antigen processing and microglia activation, such as Apoe, H2-K1, and C1qc were reduced. Given the inherent low coverage of the microglial transcriptome in snRNA-seq data, further validation using single-cell RNA-sequencing analyses in larger cohorts of mice and comparisons with human microglia gene signatures will be required to determine the significance of these cell clusters in neurodegeneration. Together, these findings indicate that LDLR may attenuate tau pathology in part through decreasing DAM genes, as well as regulating microglia-neuron interactions by altering the milieu of ion channels and neurotransmitters.
Are the effects of LDLR overexpression mediated only by lowering apoE? Shi et al. (2021) investigated the interplay between LDLR and apoE by comparing the effects of LDLR overexpression to those of apoE genetic depletion in P301S mice. Both P301S/LDLR and P301S:Apoe−/− mice had similar reduction in p-tau in the CSF, suggesting that the effects of LDLR on p-tau are apoE-dependent. Accordingly, p-tau in the CSF correlated with apoE levels in P301S/LDLR mice. In addition, using snRNA-seq, the authors established that both mouse strains showed a greater degree of gene expression changes in microglia than in neurons or astrocytes. However, subcluster analysis revealed both distinct and shared microglia populations in P301S/LDLR and P301S:Apoe−/− mice. The P301S:Apoe−/− mice did not show expansion of the microglia Cluster 3, suggesting that enhanced neurotransmitter and ion channel expression was an apoE-independent mechanism downstream of LDLR overexpression. In contrast, apoE deficiency was primarily associated with upregulation of lysosomal catabolic pathways and downregulation of anabolic pathways specific for mTOR and glycolysis. Overexpression of LDLR also promoted microglia catabolism with an intermediate decrease of mTOR activation compared to apoE depletion. Disease-associated genes were also reduced in microglia in both P301S/LDLR and P301S:Apoe−/− mice. These findings suggest that while microglial immunometabolism and downregulation of the DAM signature are likely apoE-dependent functions of LDLR, microglia polarization towards neurotransmitter release and ion channel production occurs through an apoE-independent mechanism. Future studies are needed to determine the relative contribution of apoE-dependent and apoE-independent functions of LDLR that mediate its neuroprotective effects.
How does LDLR overexpression affect oligodendrocyte lineage cells and myelination? Myelin perturbations and impaired oligodendrocyte functions are associated with cognitive decline and AD pathogenesis (Mathys et al., 2019). Shi et al. (2021) make the exciting discovery that apoE and LDLR regulate myelin integrity associated with expansion of the oligodendrocyte progenitor cell (OPC) population in aged mice and in the context of tauopathy. In tauopathy, demyelination was detected in the hippocampus and corpus callosum in the P301S mice. Both P301S/LDLR and P301S:apoe−/− mice had increased density of NG2/CSPG4 positive OPCs in the hippocampus and in the corpus callosum associated with increased myelination. Notably, even in the absence of tauopathy, OPC density and myelin basic protein expression increased in LDLR overexpressing and Apoe−/− mice, potentially due to intrinsic regulation of OPC differentiation and myelination by apoE/LDLR. Electron microscopy revealed decreased demyelinated axons in P301S/Apoe−/− and P301S/LDLR mice as compared to age-matched P301S mice. Future experiments are needed to test whether LDLR overexpression protects from demyelination by preventing oligodendrocyte cell death or promoting oligodendrocyte differentiation to myelinating cells. It is conceivable that in response to myelin loss, adult OPCs expand to generate a reparative progenitor pool capable of rapidly differentiating into mature myelinating oligodendrocytes. Furthermore, uncoupling the effects of apoE on innate immune activation and myelination could provide novel insights on the contribution of APOE to white matter pathology in dementias. Given the potential of enhanced myelination as a therapeutic strategy to alleviate cognitive deficits (Chen et al., 2021a), promyelinating drugs could also be explored in models of tauopathy.
The findings by Shi et al. (2021) shed new light at the interconnected mechanisms between the brain, immune and metabolic pathways that drive neurodegeneration and demyelination in AD and related tauopathies. The multipronged and rigorous experimental design is a tour-de-force of neuropathology, transcriptomics, and biochemistry aiming to understand the role of apoE and LDLR in neurons and glia in tauopathy. Shi et al. (2021) sets the stage for future gain and loss of function studies to discover the causal relationships among innate immune activation mechanisms and myelination driving tau-mediated neurodegeneration. Since in the mouse model used in the study LDLR overexpression under the prion promoter occurs in multiple cells types, generation of microglia-specific LDLR overexpressing mice could demonstrate the cell specificity in vivo and complement the in vitro microglial cell autonomous LDLR expression. Shi et al. (2021) also opens new directions for exploring the therapeutic efficacy of decreasing apoE by LDLR overexpression beyond its immunomodulatory role. APOE4 increases BBB permeability and disrupts neurovascular regulation leading to increased levels in the CSF and brain of the blood coagulation factor fibrinogen, which is a key vascular driver of demyelination, as well as microglia activation and cognitive impairment in AD mice (Merlini et al., 2019; Ishii and Iadecola, 2020). Studies to examine the potential protective role of LDLR overexpression on apoE4-induced BBB permeability may reveal novel approaches to control cerebrovascular dysfunction in cognitive decline. Multiple complementary therapeutic strategies are currently in development or in clinical trials to block pathogenic tau in neurodegeneration (Chang et al., 2021). Targeting LDLR could be an additional strategy to reduce tau-induced apoE-dependent neurodegeneration. A drug discovery pipeline aimed to increase LDLR in the brain might hold promise to rein in toxic neuroinflammation and reduce apoE-dependent neurodegeneration in AD and related tauopathies.
Footnotes
DECLARATION OF INTERESTS
K.A. is the scientific founder, advisor, and a board director of Therini Bio, Inc. Her interests are managed by the Gladstone Institutes according to its conflict of interest policy.
REFERENCES
- Andersen OM, and Willnow TE (2006). Lipoprotein receptors in Alzheimer’s disease. Trends Neurosci. 29, 687–694. [DOI] [PubMed] [Google Scholar]
- Chang CW, Shao E, and Mucke L (2021). Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Liu K, Hu B, Li RR, Xin W, Chen H, Wang F, Chen L, Li RX, Ren SY, et al. (2021a). Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Strickland MR, Soranno A, and Holtzman DM (2021b). Apolipoprotein E: structural insights and links to Alzheimer disease pathogenesis. Neuron 109, 205–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J (2003). LRP: a bright beacon at the blood-brain barrier. J. Clin. Invest 112, 1483–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, and Iadecola C (2020). Risk factor for Alzheimer’s disease breaks the blood-brain barrier. Nature 581, 31–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, and Huang Y (2012). Apolipoprotein E sets the stage: response to injury triggers neuropathology. Neuron 76, 871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini M, Rafalski VA, Rios Coronado PE, Gill MT, Ellisman M, Muthukumar G, Ryu JK, Syme CA, Davalos D, Seeley WW, et al. (2019). Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in Alzheimer’s disease. Neuron 101, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Andhey PS, Ising C, Wang K, Snipes LL, Boyer K, Lawson S, Yamada K, Qin W, Manis M, et al. (2021). Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]