

Abstract
Various boron-containing drugs have been approved for clinical use over the past two decades, and more are currently in clinical trials. The increasing interest in boron-containing compounds is due to their unique binding properties to biological targets; for example, boron substitution can be used to modulate biological activity, pharmacokinetic properties, and drug resistance. In this perspective, we aim to comprehensively review the current status of boron compounds in drug discovery, focusing especially on progress from 2015 to December 2020. We classify these compounds into groups showing anticancer, antibacterial, antiviral, antiparasitic and other activities, and discuss the biological targets associated with each activity, as well as potential future developments.
Keywords: Boron-containing compounds, Covalent inhibitors, Prodrug, Linker components
Abbreviations: ACTs, artemisinin combination therapies; ADCs, Acinetobacter-derived cephalosporinases; AML, acute myeloid leukemia; AMT, aminopterin; BLs, β-lactamases; BNCT, boron neutron capture therapy; BNNPs, boron nitride nanoparticles; BNNTs, boron nitride nanotubes; CEs, carboxylesterases; CIA, collagen-induced arthritis; ClpP, casein protease P; COVID-19, coronavirus disease 2019; cUTI, complex urinary tract infection; dCTPase, dCTPase pyrophosphatase; GSH, glutathione; HADC1, class I histone deacetylase; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; LeuRS, leucyl-tRNA synthetase; MBLs, metal β-lactamases; Mcl-1, myeloid cell leukemia 1; MDR-TB, multidrug-resistant tuberculosis; MERS, Middle East respiratory syndrome; MIDA, N-methyliminodiacetic acid; MM, multiple myeloma; Mtb, Mycobacterium tuberculosis; MTX, methotrexate; NA, neuraminidase; NS5B, non-nucleoside polymerase; OBORT, oxaborole tRNA capture; QM, quinone methide; OPs, organophosphate; PBA, phenylboronic acid; PDB, Protein Data Bank; PPI, protein–protein interaction; RA, rheumatoid arthritis; ROS, reactive oxygen species; SaClpP, Staphylococcus aureus caseinolytic protease P; SARS-CoV-2, syndrome coronavirus 2; SBLs, serine β-lactamases; SERD, selective estrogen receptor downregulator; SHA, salicyl hydroxamic acid; TB, tuberculosis; TTR, transthyretin; U4CR, Ugi 4-component reaction
Graphical abstract
Boron derivatives are playing an increasingly important role on drug discovery campaigns. Here, we reviewed the use and pharmacological properties of boronic acids as therapeutic agents for various diseases.

1. Introduction
The application of boron in medicine dates back to the early 19th century, when boric acid (B[OH]3) was used as a mild antiseptic. However, boron derivatives were long neglected thereafter as a result of largely unfounded claims that they are unstable and toxic. In recent years, there has been an upsurge of interest in the therapeutic potential of boron-based drugs1, 2, 3, 4, 5 since the approval of bortezomib (1), a boronic acid-containing drug6, by the U.S. Food and Drug Administration (FDA) in 2003. Since then, tavaborole (2) has been approved for the treatment of onychomycosis7, ixazomib (3) for the treatment of multiple myeloma (MM)8, crisaborole (4) for the treatment of atopic dermatitis9 and vaborbactam (5) for the treatment of bacterial infections10 (Fig. 1). Other boron-containing drug candidates are also in various stages of clinical trials11, 12, 13, 14, 15, 16, 17, 18, 19(Table 1).
Figure 1.

Timeline and structures of FDA-approved boron-containing drugs.
Table 1.
Boron-containing drugs in clinical trials.
| Name | Structure | Conditions or disease | Phase |
|---|---|---|---|
| Talabostat (PT100, 6) |  |
Advanced malignant solid neoplasm Recurrent malignant solid neoplasm |
II |
| Dutogliptin (PHX-1149, 7) | 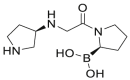 |
Acute myocardial infarction Acute myocardial ischemia STEMI-ST elevation myocardial infarction |
II |
| GSK656, 8 | 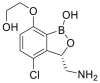 |
Tuberculosis | II |
| Acoziborole (SCYX-7158; AN5568, 9) | 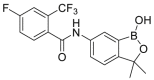 |
Trypanosomiasis, African Gambiense trypanosomiasis Sleeping sickness |
II III |
| AN2898, 10 | 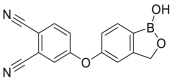 |
Dermatitis, atopic | II |
| Taniborbactam, 11 | 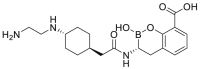 |
Urinary tract infections Acute pyelonephritis |
III |
| QPX7728, 12 |  |
Bacterial infections | I |
| GSK2878175, 13 |  |
Hepatitis C, chronic | I |
A key reason for the increasing research and development interest in boron-containing drugs is probably that boron compounds can easily interconvert between neutral trigonal planar sp2 and tetrahedral sp3 hybridization states, and thus can adopt a diverse range of binding modes during target engagement20. Many proteins have been co-crystallized with boron-containing compounds, and structural data are available in the Protein Data Bank (PDB). Boronic acids can form trigonal covalent21, tetragonal covalent22 or bidentate covalent adducts23 with nucleophiles such as serine, lysine, tyrosine, threonine and cysteine residues in target proteins, and are thus useful as high-affinity ligands with low molecular mass. Moreover, adduct formation is reversible, and so the random covalent modification of unintended targets by covalent warheads can be minimized24. Furthermore, the two hydroxyl groups of boronic acids provide four lone pairs and two hydrogen bond donors, thus offering six opportunities to form direct or water-mediated hydrogen bond contacts with key amino acid residues. These hydrogen bonds can have a high degree of covalency; i.e., the hydrogen bonds are short and the interaction energy between boronic acid and the target protein is large. Multiple hydrogen bonds are effective to enhance the affinity of the ligand (or inhibitor) for the target and to facilitate binding even in the presence of drug resistance-related mutations25,26. In addition, the boron moiety can interact with metal ions in enzymes (Fig. 2)27. These attributes are valuable when designing candidate drugs to work in the context of selection pressures for drug resistance.
Figure 2.

Diverse structures of boron compounds and their interaction modes with biological targets.
The boronic acid pharmacophore is an established glycan-binding functional group that forms reversible complexes with diol groups commonly found in sugar molecules and glycoproteins28, 29, 30, 31. Exploiting this behavior, multimeric boronic acids have been designed to target pathogens’ extracellular glycans, both to overcome inherent cell envelope barriers and to prevent the development of drug resistance32. Boronic acid-containing prodrugs of hydroxylated drug molecules have also been developed to reduce first-pass metabolism and thus to increase bioavailability33, 34, 35. In addition, boronic acids, together with catechols and cis-alkyl diol partners, have been explored as linker components for ligands and proteins to address challenging drug targets36, 37, 38. The reaction between reactive oxygen species (ROS) and aromatic boronic acids has been exploited for the construction of ROS-triggered boronic acid prodrugs that are selectively activated under pathological conditions, thereby improving targeting and reducing toxicity39, 40, 41. Boron clusters that contain plural boron atoms are also useful in drug design42. For example, carborane with a high content of 10B, which absorbs thermal neutrons, is stable under biological conditions and can penetrate the cell membrane, making it an excellent candidate for boron neutron capture therapy (BNCT)43,44.
Thus, boron compounds appear to have great potential for development of next-generation target-specific drugs. In this perspective, we aim to comprehensively review the current status of boron compounds in drug discovery, focusing especially on progress from 2015 to December 2020. We will consider compounds showing anticancer, antibacterial, antiviral, antiparasitic and other activities in separate sections, and discuss the biological targets associated with each activity, as well as potential future developments.
2. Boron-containing anticancer agents
Cancer is the second leading cause of death and is the cause of one-sixth of all deaths worldwide. There were 9.6 million deaths from cancer in 201845. At present, chemotherapy is still the main treatment method, but anticancer drugs typically cause toxicity, have side effects, and eventually encounter drug resistance46,47. Thus, there is a continuing demand for improved anticancer drugs. One design approach is to utilize specific abnormalities of the tumor microenvironment, such as overexpressed enzymes or high levels of reactive oxygen species (ROS), to target therapeutic agents. In this context, boronic acids serve as excellent covalent ligands, and have been used in ROS-triggered prodrugs. Boronic acid-based prodrugs have also been used to improve the pharmacokinetic properties of anti-estrogens based on their glycan-binding capability. BNCT is also used to treat cancer, based on the ability of boron to capture thermal neutrons. Here we will consider boron-based anticancer agents in terms of their targets.
2.1. Agents targeting key enzymes
Bortezomib, a break-through MM treatment, is a reversible boronic acid inhibitor, which preferentially targets the proteasomal β5 active site. The drug blocks the catalytic β5 site by binding covalently to the hydroxyl group of the N-terminal threonine residue. While bortezomib is a first-line therapy for multiple myeloma, most patients develop drug resistance, leading to disease recurrence48. Overexpression of class I histone deacetylase (HDAC1) in MM cells confers resistance to bortezomib, but importantly, HDAC1 inhibitors could reverse bortezomib resistance49. Zhou et al.50 synthesized a series of peptide boronate derivatives as HDAC/proteasome dual inhibitors by substituting zinc-binding groups of HDAC inhibitors onto solvent-exposed sites of bortezomib (Fig. 3). The most promising inhibitor 14, in which the zinc-binding moiety originates from the HDAC inhibitor entinostat (15) and the covalent-binding group originates from bortezomib, exhibited more potent proteasome inhibition than bortezomib (IC50 values of 1.1 vs. 19.4 nmol/L) and showed good selectivity for HDAC1 (IC50 = 255 nmol/L). Compound 14 showed antiproliferative activity against MM cell lines RPMI-8226, U266 and KM3 with IC50 values of 6.66, 4.31 and 10.1 nmol/L, respectively. Notably, it showed potent antiproliferative activity towards the bortezomib-resistant MM cell line KM3/BTZ with an IC50 value of 8.98 nmol/L, a level far exceeding that achieved by bortezomib (226 nmol/L) or even the combination of entinostat and bortezomib (1:1, 98.0 nmol/L). Dual proteasome/HDAC inhibitors are promising candidates for the treatment of proteasome inhibitor-resistant MM, and further developments in this area can be expected.
Figure 3.

Design of HDAC/proteasome dual inhibitor 14.
dCTPase pyrophosphatase 1 (dCTPase) is overexpressed in multiple carcinomas and is involved in promoting tumor cell growth and in maintenance of stemness51. To exploit the therapeutic potential of dCTPase inhibition, Llona-Minguez and co-workers52 screened a proprietary compound collection of 5500 compounds via HTS-adapted Malachite Green assay and identified a hit compound 16 that inhibits dCTPase with an IC50 value of 0.057 μmol/L. Subsequent optimization led to the identification of boronic acid derivative 17 that showed enhanced cellular efficacy (EC50 = 0.046 μmol/L) and synergized with cytidine analogues in killing HL60 leukemia cells. But unfortunately, compound 17 showed only moderate aqueous solubility (52 μmol/L) and low plasma stability (11% remaining after 4 h). To improve the ADME properties of compound 17, they masked the boronic acid functionality with N-methyliminodiacetic acid (MIDA) ester to provide derivative 18, which displayed greater aqueous solubility (>100 μmol/L), improved plasma stability (86% left after 4 h) and enhanced cellular permeability, while exhibiting comparable dCTPase inhibitory activity (IC50 = 0.047 μmol/L, Fig. 4). Intriguingly, compound 18 also presented a favorable characteristic of CYP inhibition, with only modest inhibition of CYP2C.
Figure 4.

Design of dCTPase inhibitor 18.
Working with AAA + chaperones such as ClpX, casein protease P (ClpP) maintains cellular homeostasis by degrading defective proteins. Human ClpP is highly expressed in acute myeloid leukemia (AML) cells. Therefore, hClpP inhibitors or activators could be promising candidates for therapy of AML53,54. α-Aminoboronic acids were considered to have the potential to covalently inhibit hClpP activity, as they are reported to be selective inhibitors of Mycobacterium tuberculosis (Mtb) ClpP55. On the basis of the Ugi 4-component reaction (U4CR), Tan et al.56 built a α-aminoboronic acid virtual library using BIOVIA Pipeline Pilot and filtered the resulting U4CR library against the active site of hClpP to find covalent inhibitors. They identified three α-aminoboronic acids represented by compound 19, which inhibited hClpP in the low micromolar range. Importantly, in contrast to the potent hClpP inhibitor AV167 (IC50 = 1.54 μmol/L) previously reported by Hackl et al.57, these compounds also inhibited hClpXP (Fig. 5). Therefore, α-aminoboronic acid should be a promising template for the development of inhibitors of hClpXP.
Figure 5.

The structure of inhibitor 19.
Myeloid cell leukemia 1 (Mcl-1) is a prosurvival protein that is overexpressed by tumor cells and is associated with resistance to apoptosis, being involved in multiple protein–protein interactions (PPIs). Akçay et al.58 used a docking method to design the first reversible covalent inhibitors targeting Lys234 of Mcl-1 by placing a boronic acid carbonyl warhead in noncovalent Mcl-1 inhibitors and selecting those that positioned the warhead carbonyl within ∼3 Å of Lys234 (Fig. 6). Biochemical and cellular examination revealed an increased activity with incorporation of the warhead; for instance, more than 24-fold enhancement of the EC50 value towards MOLP-8 cell line was detected between noncovalent inhibitor 20 (>11 μmol/L) and the corresponding warhead-containing 21 (0.46 μmol/L). Further alkylation of the indole nitrogen of this scaffold afforded 22 with significantly enhanced efficacy (EC50 = 0.075 μmol/L). These reversible covalent inhibitors induced caspase activation in a manner dependent on Mcl-1 without showing general cytotoxicity, and Lys234 was confirmed to be the residue involved in the covalent modification by means of point-mutation studies combined with mass spectrometry.
Figure 6.

Structure-based design identified covalent Mcl-1 inhibitors targeting Lys234: Docked structure of inhibitor 21 (PDB ID:3WIX) and structures of compounds 20–22.
2.2. Agents targeting reactive oxygen species
Reactive oxygen species (ROS) are a family of redox-active small molecules, such as hydrogen peroxide (H2O2) and oxygen radicals, which are widely present in biological systems. Oxidative stress due to abnormal production of H2O2 can lead to the development and progression of serious diseases such as cancer, inflammation, obesity and diabetes59. Boronic acids and their esters are cleaved by H2O2 and other ROS to produce the corresponding alcohol and boric acid, which is considered non-toxic to humans (Fig. 7)60. In drug development, boronic/boronate groups are often used to replace the hydroxyl groups of active agents in order to achieve selective release of drugs in the tumor microenvironment. The phenylboronic/boronate functionalities are connected to the active drug structure through a direct C–N/C–O bond or a self-immolative spacer (such as a carbamate linkage). Oxidation of such a spacer triggers a self-immolative cascade, leading to the release of the active drug (Fig. 7)61.
Figure 7.

Oxidation of boronic prodrugs to release the active drug.
Chen et al.62 developed several new aromatic nitrogen mustards with boronic esters or boronic acids as ROS-based activators to improve the selectivity for tumor cells. These prodrugs are not deleterious to normal cells owing to the electron-withdrawing effect of the boronate group, but H2O2 in cancer cells cleaves the boronic ester or boronic acid to a hydroxyl group, which increases electron release to the nitrogen of the mustard moiety, affording a highly electrophilic aziridinium ring that induces DNA cross-linking (Fig. 8). These prodrugs caused 40%–80% cell death of primary leukemic lymphocytes but less than 25% apoptosis of normal lymphocytes. The most potent compound (23) displayed an IC50 value of around 5 μmol/L in CLL cells. In further work63, various aromatic substituents were incorporated into compound 23 to improve the biophysical properties, and compound 24 potently induced apoptosis in CLL lymphocytes with IC50 values between 3 and ∼778 nmol/L. In addition, 24 was more cytotoxic toward breast-cancer MDA-MB-468 cells (IC50 = 3.43 μmol/L) than clinically used chlorambucil (IC50 = 48.7 μmol/L) or melphalan (IC50 = 34.44 μmol/L). More importantly, compound 24 is highly selective for cancer cells with no apparent toxicity to normal lymphocytes at the highest concentration tested (10 μmol/L). It efficiently inhibited tumor growth in an MDA-MB-468-derived xenograft mouse model without apparent side effects.
Figure 8.

Targeting ROS-producing cancer cells; the structures of prodrugs 23 and 24.
Compared with normal cells, cancer cells show increased generation of ROS and are also more susceptible to further ROS insults64. Cancer cells, through upregulating antioxidant systems such as glutathione (GSH), adapt to oxidative stress and thus counteract ROS insults65. Quinone methide (QM, 25), an antioxidant inhibitor with anticancer activity, rapidly alkylates GSH, thus inhibiting the cellular antioxidant systems66. Cinnamaldehyde (26) induces ROS generation in cancer cells and inhibits the growth of human cancer cells with minimal cytotoxicity to healthy cells, but poor bioavailability limits its clinical application67. Based on these studies, Noh et al.68 reported a novel hybrid anticancer prodrug 27, which, under oxidative conditions, can release QM and cinnamaldehyde dependent on the activation of H2O2 and acidic pH, respectively, in cancer cells and thus exerts synergistic therapeutic actions (Fig. 9). Firstly, the generation of both QM and cinnamaldehyde in prostate cancer DU145 cells was confirmed. Then they demonstrated that 27 induced a significant reduction (>50%) of the GSH level in DU145 cells, while it had no effect on the GSH level in non-malignant NIH3T3 cells. The hybrid drug 27 exhibits cytotoxicity towards DU145 cells and SW620 cells with IC50 values of 48 and 76 μmol/L, respectively, being significantly more cytotoxic than cinnamaldehyde and a quinone methide generator. Importantly, in xenograft mouse models using SW620 and DU145 cells, 27 displayed more potent anticancer action than the combination of cinnamaldehyde and a quinone methide generator. Its efficacy was comparable to that of the anticancer drug camptotethin at the dose of 2 mg/kg.
Figure 9.

A dual-stimulus-responsive hybrid anticancer drug 27.
Apart from prodrug development, selective reactions have also been applied for fluorescence detection of H2O2, gene expression and point-of-care assay. For example, Chang's group69 used boronic esters for the development of H2O2-activated fluorescent probes to explore the physiological roles of H2O2 in living systems. Govan et al.70 developed a genetically encoded gene activation system through the use of a boronate group-substituted estrone molecule. Upon oxidation of the prodrug by H2O2 after binding to the estrogen receptor, active estrone is released, thereby activating gene expression.
2.3. Agents targeting estrogen receptor
Boronic prodrugs of anti-estrogen compounds significantly reduce the first-pass metabolism of hydroxylated drug molecules, leading to increased bioavailability. It is suggested that boronic acid blocks rapid clearance by forming reversible complexes with 1,2 and 1,3 diol groups of sugar molecules and glycoproteins, thereby facilitating enrichment in plasma as well as making the boronic acid moiety inaccessible to glucuronidation. Moreover, such complexes may serve as a drug reservoir. Endoxifen (28) is a selective estrogen receptor modulator (SERM) that is in clinical trials, but it undergoes rapid first-pass metabolism through O-glucuronidation, leading to poor bioavailability. Zhang et al.34 designed a boronic acid prodrug of endoxifen, ZB483 (29), which produced a 40-fold increase in peak endoxifen concentration and an 80-fold increase in the area under the curve (AUC) value (Fig. 10).
Figure 10.

Metabolic conversion of ZB483 to endoxifen.
Fulvestrant (31), a selective estrogen receptor downregulator (SERD), is indicated for metastatic breast cancer therapy, but poor oral bioavailability has hampered its clinical application. However, substitution of the 3-hydroxyl group of fulvestrant with a boronic acid moiety allowed the compound to evade first-pass metabolism33. After a single oral dose of 8.3 mg/kg in mice, fulvestrant-3 boronic acid (ZB716, 32) afforded far superior half-life (t1/2 = 23.5 h), peak concentration (Cmax = 169.8 ng/mL) and area under the curve (AUC = 2547.1 ng h/mL) in comparison to fulvestrant when given by s.c. injection to mice. At the same time, ZB716 is as potent as fulvestrant in binding ERα (IC50 = 4.1 nmol/L) and downregulating ERα (Fig. 11). Improvement of poor bioavailability can allow physicians to prescribe a lower dose, potentially reducing side effects, increasing patient compliance, and lowering the risk of cancer recurrence. The coordination of boronic acid with diols from sugar molecules has also been exploited to enhance insulin delivery in the treatment of diabetes71.
Figure 11.

Design of ZB716.
2.4. Agents with unknown targets
Zhang et al.72 developed a series of 7-propanamide derivatives based on an antimalarial benzoxaborole 33, obtaining a compound 34 with submicromolar antiproliferation activity towards ovarian cancer cells. Further structure−activity relationship studies found that replacement of the phenyl group with biphenyl and introduction of a hydrogen-bond acceptor group led to a significant increase of potency. The most potent compound 35 showed an IC50 value of 21 nmol/L against ovarian cancer cells, with good selectivity between cancerous and normal cells (nearly 200-fold selectivity: SKOV3 vs. WI-38 cells). Compound 35 effectively induced cancer cell apoptosis and inhibited colony formation (Fig. 12). More importantly, compound 35 also demonstrated in vivo efficacy and low toxicity in a tumor xenograft mouse model at dosages of 2 and 10 mg/kg. Although the cellular target of these anticancer benzoxaboroles is not clear, these structures provide a new direction for anticancer drug research.
Figure 12.

The structures of inhibitors 33–35.
2.5. Boron neutron capture therapy
BNCT is currently an important mode of tumor therapy. When 10B-rich agents are selectively accumulated in malignant cells, the 10B atoms absorb low-energy (<0.5 eV) neutrons (thermal neutrons) and disintegrate to afford an alpha (4He) particle and a lithium nucleus (7Li). The alpha particles deposit high energy along a short path length (<10 μm; approximately the diameter of a single cell), and consequently kill tumor cells containing boron without damaging surrounding normal cells73. Clinical studies of BNCT have focused on locally recurring malignant glioma and head and neck cancer. The clinical efficacy of two boron agents, 4-dihydroxyborylphenylalanine (BPA, 36) and sodium mercaptoundecahydrododecaborate (BSH, Na2B12H11SH, 37, Fig. 13), has been confirmed in patients with the above tumors74, 75, 76, 77. However, some issues remain. BSH does not show active targeting or uptake, and has undesired interactions with other biomolecules, while BPA displays poor water solubility, low boron content and lack of specificity78. In recent years, the advent of new in-hospital neutron accelerators has generated renewed interest in BNCT79,80. However, efforts to develop improved boron delivery agents, including amino acids, carbohydrates, and nanoparticles, have not yet made a breakthrough. At present, the best way to further improve the clinical efficacy of BNCT may be to optimize the administration and delivery methods of BPA and BSH81.
Figure 13.

Structures of BPA and BSH.
2.6. Boron nitride nanotubes and nanoparticles
Boron nitride nanotubes (BNNTs) and boron nitride nanoparticles (BNNPs) are currently considered promising candidates for drug delivery. For example, folic acid-functionalized BNNTs are specifically taken up by glioblastoma multiforme, and therefore may be useful to deliver drugs to glioblastoma multiforme in the brain82.
3. Boron-containing antibacterial agents
In the United States alone, more than 2.8 million people acquire an antibiotic-resistant infection every year, resulting in an annual death toll of more than 35,00083. Among infectious diseases in general, tuberculosis (TB) caused by Mtb is the main killer. In 2015, the worldwide incidence of multidrug-resistant tuberculosis (MDR-TB) was approximately 580,000, leading to an estimated 250,000 deaths. The latest available data show that the treatment success rate for MDR-TB is only 56%84. In more than 70 years, only two completely new drugs for the treatment of MDR-TB have entered the market. Therefore, there is an urgent need for new antibacterial agents. In the 1970s, boric acid was reported to reversibly inhibit serine β-lactamases produced by Bacillus cereus85. Boronic acids are regarded as transition-state analogues of β-lactamases and are currently considered promising candidates to combat antibiotic resistance. It was shown that boronic acids selectively kill Mtb through targeting Mtb leucyl-tRNA synthetase (LeuRS) based on the oxaborole tRNA capture (OBORT) mechanism or via chelation of its unique cell wall glycans. Boronic acids have also been developed as covalent binding inhibitors of Staphylococcus aureus caseinolytic protease P (SaClpP). These approaches represent a step towards pathogen-specific, next-generation antibiotics.
3.1. Targeting β-lactamases
β-Lactam antibiotics are currently among the most widely used drugs to treat bacterial infections, but the rapid evolution and spread of antibiotic resistance are having a dramatic impact on their efficacy86. The main mechanism leading to resistance is hydrolysis of the amide bond of the lactam ring by serine β-lactamases (SBLs; Ambler A, C and D) or metal β-lactamases (MBLs; Ambler B), as illustrated in Fig. 14A and B. Both β-lactamase-catalyzed hydrolysis processes involve a high-energy tetrahedral (sp3 hybridized) intermediate49,85,87. One approach to this issue is combined administration of β-lactamase inhibitors with β-lactam antibiotics. Unfortunately, currently commercially available BLIs, such as sulbactam and clavulanic acid, are themselves β-lactams, and bacteria can quickly develop resistance to these chemically similar molecules. In contrast, boronic acid transition-state analogs are a different class of β-lactamase inhibitors (Fig. 14C). The capacity of boron to morph between hybridisation states enables boronate-based inhibitors to mimic substrates of β-lactamases (in their sp2 state), allowing them to bind efficiently to β-lactamases, and they can then react with the SBL nucleophilic serine or MBL-Zn(II)-bridged water to form the sp3 state, mimicking a high-energy intermediate. Thus, boronate-based inhibitors are promising candidates for SBL/MBL inhibition.
Figure 14.

Mechanisms of β-lactam cleavage by (A) SBLs and (B) MBLs, exemplified by the hydrolysis of a carbapenem. (C) Mode of action of boronate inhibitors for inhibition of SBLs and MBLs.
3.1.1. Inhibitors of SBLs
1-Amido-2-(m-carboxyphenyl)ethane boronic acid derivatives, generated by replacing the carbonyl of the β-lactam ring with boron-containing groups, are potent SBL inhibitors88. Considering that replacement of a phenyl ring by a triazole ring would enable the installation of various R groups to explore the topology of the β-lactamase active site, Caselli et al.89 designed and synthesized a series of highly substituted α-amido-β-triazolylethane boronic acids by utilizing click chemistry (Fig. 15). The resulting compounds showed potent activity against class C SBLs (P99 and PDC-3) with low micromolar to low nanomolar inhibition constants, which were as low as 4 and 23 nmol/L towards PDC-3 and P99, respectively, for the most potent compound 38. These compounds significantly reduced bacterial resistance when used in combination with cefotaxime.
Figure 15.

Design strategy for compound 38.
The resistance of Acinetobacter baumannii to all classes of β-lactam antibiotics has greatly increased over the past decade. Class C Acinetobacter-derived cephalosporinases (ADCs) β-lactamases, especially ADC-7, mainly mediate β-lactam resistance in A. baumannii and are not affected by clinically used BLIs90. Caselli et al.91 continued to employ click chemistry and synthesized a new class of boronic acid transition-state inhibitors as ADC β-lactamase inhibitors, in which the amide group was replaced with triazole. These compounds showed Ki values in the range of 90 nmol/L to 38 μmol/L, with compound 39 being one of the best inhibitors of ADC-7 (Fig. 16). Crystallographic data indicated that the triazole establishes the expected hydrogen bonds with Gln120 and Asn152, maintaining the typical interactions of amidomethaneboronic acid inhibitors. In addition, multiple hydrogen bonds were observed between the O1 atom hydroxyl group of boronic acid and Ser315 and Ser64, and another O atom establishes a covalent bond with phosphate ion. These compounds restored ceftazidime or cefepime activity against classes C and A β-lactamase strains. These results demonstrate that α-triazolylmethaneboronic acid is a good scaffold for ADC β-lactamase inhibitors.
Figure 16.

Planar structure of 39 and X-ray crystal structure of ADC-7/inhibitor complex (PDB code: 6TZJ).
Ness et al.92 synthesized inhibitor 40a with greatly increased affinity for TEM-1 β-lactamases by installing a hydroxyl group on the aromatic ring. Hecker et al.93 envisaged that cyclic boronate ester compounds might show a favorable conformation for enzyme complexation, and docked several candidates into the active site of β-lactamase in silico. Hit molecules were synthesized and evaluated, resulting in the discovery of vaborbactam, the first clinically approved boronic acid-containing SBLs inhibitor (Fig. 17A). In 2017, vaborbactam/meropenem (Vabomere) was approved to treat complex urinary tract infections10. This drug combination potently inhibits classes A and C SBLs. However, no MBLs inhibitor based on a boronic acid chemotype has yet been found.
Figure 17.

(A)–(D) Design strategies of vaborbactam, 41, taniborbactam and QPX7728, respectively. (B) Co-crystallization of 41 with VIM-2 (B1). The overlay compares the binding modes of 41 and hydrolysed cefuroxime complexed with NDM-1 (B2) (PDB ID: 4RL2). (C) Crystal structures of VNRX-5133 complexed with NDM-1 (PDB ID: 6RMF); chain A shows the major observed bicyclic form (C1), while chain B shows the tricyclic form (C2). (E) Activities of vaborbactam, 41, taniborbactam and QPX7728.
3.1.2. Broad-spectrum β-lactamase inhibitors (BLIs)
Brem et al.94 obtained cyclic boronate analogues showing nanomolar inhibition of SBLs and MBLs by applying a strategy of mimicking the common high-energy tetrahedral intermediate. As shown in Fig. 17B1, the binding modes of 41 with both BcII and VIM-2 are similar to those observed for complexes of MBLs with hydrolysed β-lactams, exemplified by hydrolysed cefuroxime. For instance, in the complex obtained by co-crystallization of 41 with VIM-2, the “endocyclic” boronate ester oxygen that corresponds to the cephalosporin dihydrothiazine ring nitrogen forms a dative bond with Zn, and two boron-bound exocyclic oxygen atoms chelate another zinc atom. Furthermore, the hydrogen bonding/electrostatic interaction with Lys224 (NDM-1 and BcII) or Arg228 (VIM-2) and hydrophobic interaction with conserved Trp87 and Phe61 residues are analogous to those with hydrolysed β-lactams (Fig. 17B2). Overall, therefore, the structure of the cyclic borate complex closely matches the tetrahedral transition state in MBL catalysis, and this may be the reason why 41 shows broad-spectrum β-lactamase-inhibitory activity.
Recognizing that the cyclic boronate scaffold might serve for the development of pan-spectrum β-lactamase inhibitors, Liu et al.27 introduced hydrophobic groups into the scaffold of 40b to enhance van der Waals interactions with hydrophobic residues at the active sites of SBL and MBL enzymes. Next, additional distal primary amino groups were also introduced to improve Gram-negative OM permeability. These studies led to compound 11 (VNRX-5133, taniborbactam), which is a potent inhibitor of all four Ambler classes of β-lactamase and a wide range of Gram-negative bacteria (Fig. 17C). Notably, taniborbactam was selective for bacterial enzymes, being nontoxic to mammalian cells. Currently, VNRX-5133/cefepime is in phase III clinical study for patients with complicated urinary tract infections. Crystallographic analysis of VNRX-5133 complexed with NDM-195 revealed a similar MBL inhibition mechanism to that 41 (Fig. 17C1), though an unanticipated tricyclic structure involving cyclization of the acylamino oxygen onto the boron of the bicyclic core was also observed (Fig. 17C2). The results further support the validity of the “high-energy intermediate” strategy for the development of boron analogues as broad-spectrum β-lactamase inhibitors.
Building on the above structures, Hecker et al.96 designed a series of methylthioacetamide analogues represented by 42 as potent inhibitors of classes A and C serine enzymes. Greatly improved potency against class B enzymes was obtained by replacing the amide linkage with a thio linkage, as exemplified by 43. Removal of the substituent next to boron resulted in remarkable activity against class D enzymes, but unfortunately the most promising compound 44 produced a potentially toxic long-lived metabolite via oxidative deboronation. Structural modifications to block this reaction while retaining the desired properties culminated in the discovery of 12 (QPX7728, Fig. 17D). QPX7728 shows a remarkably broad spectrum of inhibition (classes A, B, C and D enzymes), and is well tolerated in rats, with good oral bioavailability (F%) of 24%–53%, depending upon the dose. QPX7728 has advanced to phase I clinical trials18.
Acyclic boronates were also effective inhibitors of both SMLs and MBLs. Wang et al.97 obtained a new MBL/SBL inhibitor 45 by fusion of (2′S)-3′-mercapto-2′-methylpropanamido with acyclic α-amino boronic acid. The structures of VIM-2/45 and KPC-2/45 complexes revealed the dual MBL/SBL inhibition mode: a thiol bridges two active site zinc ions for MBL inhibition (Fig. 18A) and the boronic acid group covalently binds with the catalytic residue Ser70 in the active site for SBL inhibition (Fig. 18B). Further examination indicated that 45 did not occupy the subpocket consisting of the Trp105, Ser130, and Thr235 residues, suggesting that introduction of a substituent at the 1-position of 45 might result in enhanced potency. Indeed, this approach led to compound 46, which showed more potent inhibition of several tested MBL and SBL enzymes, including the clinically common carbapenemases VIM-2 (Ki = 0.44 μmol/L) and KPC-2 (Ki = 0.61 μmol/L).
Figure 18.

X-ray structure analyses of the VIM-2/45 (A) and KPC-2/45 (B) complexes, leading to the development of 46.
3.2. Targeting leucyl-tRNA synthetase (LeuRS) and extracellular glycans of Mtb
Benzoxaboroles (also known as oxaboroles) target fungal cytoplasmic LeuRS by an oxaborole tRNA-trapping (OBORT) mechanism, in which the boron atom forms a covalent adduct with the terminal nucleotide Ade76 of tRNA to capture the 3′ end of tRNALeu in the editing site, inhibiting leucylation and thus inhibiting protein synthesis (Fig. 19)23. Utilizing this mechanism to target M. tuberculosis LeuRS, Sonoiki and co-workers98 synthesized a series of 3-aminomethyl-benzoxaboranes, in which the boron atom is bonded to the cis-diols of AMP, a surrogates for Ade76, as seen in the X-ray co-crystal structure of LeuRS with compound 47. The most promising compound in this series, 48, displayed potent inhibition of Mtb LeuRS (IC50 = 0.056 μmol/L), but unfortunately there were potential toxicity issues arising from its inhibition of mammalian cytoplasmic LeuRS (IC50 = 38.8 μmol/L). Based on the co-crystal structure, modification studies focused on the solvent-exposed C-7, C-6, and C-6/C-7 sites were performed with the aim of increasing the selectivity99. Efficacy studies of these derivatives identified a clinical candidate, GSK3036656 (8), which was highly selective for Mtb LeuRS over human cytoplasmic LeuRS and HepG2 protein synthesis, even though its potency towards Mtb LeuRS was not significantly improved over that of the initial analogue 48 (Fig. 19). Importantly, GSK3036656 exhibited excellent PK profiles in mouse TB infection models with high tolerability. A phase II clinical study of GSK3036656 for TB was undergoing14.
Figure 19.

Mechanism of inhibition of LeuRS by tavaborole and X-ray cocrystal structure of LeuRS complexed with 47 (PDB ID: 5AGR).
The cell envelope of Mtb is a highly efficient permeability barrier, preventing entry of many antibiotics into cells, thus impeding anti-tubercular treatment. Boronic acids form bonds with cis-1,2- and 1,3-diols in carbohydrates and multiple boronic acids placed on a single scaffold provide a synergistic increase in binding affinity28. Guy et al.32 designed multimeric boronic acids to selectively target structurally unique Mtb cell envelope glycans, in order to overcome the Mtb cell envelope barrier (Fig. 20). The number of, and distance between, boronic acid units greatly influenced the binding selectivity and affinity. Among the synthesized compounds, two boronic acid units with shorter linkers were selective inhibitors of mycobacteria (MICs: 780–3100 μmol/L) over other strains of bacteria, and exhibited low cytotoxicity in a panel of human cell lines. BLI data revealed that multimeric boronic acids have intense and specific interactions with isolated Mtb glycans and whole integral Mtb cells versus other bacterial species or mammalian cells. Whole-cell proteomics identified a series of stress response instead of a single target, and this may facilitate the anti-resistance. This non-conventional approach means that the molecule does not need to cross the Mtb cell wall barrier.
Figure 20.

Design of multimeric boronic acids specifically targeting the extracellular Mtb cell-envelope glycans. The complex Mtb cell envelope and peptidoglycan layer are indicated by lines and hexagons, respectively.
3.3. Targeting S. aureus caseinolytic protease P (SaClpP)
Ju et al.100 screened 2632 molecules and selected a peptidomimetic boronate, MLN9708 (49) with an IC50 value of 5.7 μmol/L against SaClpP for further optimization. Based on the idea that boron binds to residue Ser98 in the SaClpP active site, they constructed a virtual library of boron-based compounds and modeled the binding to the active site of SaClpP. Privileged substituents were selected for micro-scale combinatorial synthesis and bioassay of SaClpP-inhibitory activity. This work led to 50 (Fig. 21), which showed an IC50 value of 0.9 μmol/L. In addition, the crystal structure of 50‒SaClpP complex indicated that compound 50 forms a reversible covalent bond with Ser98 while stabilizing the tetradecameric structure of SaClpP. This study offers an integrated strategy for peptidomimetic boronate optimization to develop covalently binding inhibitors. However, these compounds exhibit poor selectivity between 20S proteasome and the homologous hClpP and PaClpP1 from Pseudomonas aeruginosa, so further modification will be needed to increase the selectivity.
Figure 21.

Integrated strategies to optimize hit compound 49.
4. Boron-containing antiviral agents
Viral infections constitute a major threat to human health, as exemplified by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic101, 102, 103. Other major pathogens include influenza virus, Dengue virus, Ebola virus, Chikungunya virus, SARS-CoV, and Middle East respiratory syndrome-related coronavirus, some of which have high mortality rates and lack specific therapies. For example, the annual incidence of severe influenza is approximately 3–5 million worldwide, and there are around 290,000 to 650,000 deaths related to respiratory diseases each year104. The annual incidence of dengue fever worldwide is about 390 million cases105. Furthermore, it is very difficult to eradicate chronic infectious viruses such as human immunodeficiency virus (HIV), hepatitis B virus (HBV) or hepatitis C virus (HCV), which may infect large numbers of people. For example, about 40 million people worldwide are infected with HIV; in 2019 the incidence of new cases was estimated to be 1.7 million and the number of deaths from HIV-caused illnesses was about 700,000106. Similarly, about 71 million people worldwide are infected with HCV, and approximately 399,000 people died of it in 2016107. In antiviral research, boronic acids are expected to be useful to facilitate drug binding to the target protein even in the presence of resistance mutations, by forming multiple hydrogen bonds or a covalent adduct. The glycan-binding property of boronic acid derivatives can also be utilized to interfere with viral binding to the cell membrane and entry into the cell.
Windsor et al.25 inspected the interactions of darunavir (51) and its analogues (such as 52–55) with the S2′ subsite of HIV-1 protease. Considering potential hydrogen bonds that might be formed by a boronic acid group, they replaced the aniline moiety in darunavir with phenylboronic acid (PBA), obtaining compound 56 (Ki = 0.5 ± 0.3 pmol/L), which showed 20-fold increased affinity for HIV-1 WT protease. The analogues 52–55 showed only less than 2-fold increases in affinity compared with darunavir (Ki = 10 pmol/L). More importantly, compound 56 retained high affinity for the drug-resistant D30N variant (Ki = 0.4 ± 0.3 pmol/L). X-Ray crystallography demonstrated that the boronic acid group participates in three hydrogen bonds (Fig. 22). In particular, the hydrogen bond between the boronic acid hydroxyl group and the Asp30 (or Asn30) carbonyl group was short (rO···O = 2.2 Å) and density functional theory analysis revealed a high degree of covalency. These data highlight the ability of boronic acids to form multiple hydrogen bonds or robust hydrogen bonds with a high degree of covalency, thereby enhancing the affinity for both wild-type and drug-resistant HIV-1 proteases.
Figure 22.

Structures of compounds 51–56 and interactions between 56 and the S2ʹ subsite of HIV-1 protease.
Compound 57 is a non-nucleoside polymerase (NS5B) inhibitor of hepatitis C virus. Introduction of boronic acid moieties into compound 57 enhanced the in vitro potency towards wild-type HCV replicons and clinically relevant polymorphic and resistant HCV mutant replicons, resulting in a clinical candidate, GSK5852 (58)108. However, GSK5852 showed a short plasma half-life (t1/2 = 5 h) in human volunteers as a result of facile benzylic oxidation. To overcome this issue, GSK8175 (13) was designed by excising the benzylic methylene group and introducing N-phenyl sulfonamide analogues26. GSK8175 displayed a 60–63 h half-life and caused a robust decrease in viral RNA levels in HCV-infected patients. The co-crystal structure of GSK8175 bound to GT 1a 316Y protein revealed that boronic acid contributes a network of six hydrogen-bonding interactions mediated by water molecules (Fig. 23). Thus, boronic acid appears to promote the binding of NS5B by establishing multiple direct and through-water H-bond interactions. GSK8175 has completed phase I clinical trial for treating HCV infection19.
Figure 23.

The design of GSK8175 and the cocrystal structure of GSK8175 bound to GT 1a 316Y protein (PDB ID: 6MVO).
Influenza A virus (IAV) neuraminidase (NA) protein, a glycoprotein, can facilitate IAV attachment and entry into infected cells in the early period of viral life cycle109,110. Boronic acid derivatives possess good glycan-binding ability and reduce the cytotoxicity of some quindoline derivatives111, and Wang et al.112 utilized these features to design a novel series of novel boronic acid-modified quindoline derivatives as IAV inhibitors to interfere with viral binding and cell entry. Boronic acid-modified compounds, exemplified by 59, effectively prevented the entry of viral RNP into the nucleus and reduced the virus titer in IAV-infected cells. For example, in virus yield reduction assay, compound 59 possessed a broad antiviral spectrum against all strains tested including H1N1 strains A/California/04/2009 (Cal09) and A/Puerto Rico/8/34 (PR/8) and H3N2 strain A/swine/Minnesota/02719/2009 (Minnesota) with IC50 values of 4.3 ± 0.7, 2.5 ± 0.4, and 3.0 ± 0.4 μmol/L, respectively, being superior to boronic acid-free compound 60 (25.5 μmol/L, Fig. 24). More importantly, IAV-infected mice treated orally with compound 59 showed better survival rates than oseltamivir-treated mice. The results of NA inhibition assay and molecular docking indicated that boronic acid modification may enhance the interaction between NA protein and quindoline derivatives to block viral infection.
Figure 24.

Structures of compounds 59 and 60.
Introduction of a boronic acid moiety into C-terminal of dipeptidic inhibitors against Zika, West Nile, and dengue viral proteases achieved a thousand-fold affinity gain compared to the amide or carboxylic acid analogues (61 and 62)113. The most active compound 63 had Ki values of 0.051, 0.082 and 0.040 μmol/L, respectively. Crystallographic study showed that boronic acid can form a five- or six-membered ring with glycerol, suggesting that it would be able to take a variety of three-dimensional shapes to interact with surrounding amino acid residues in the target protein. Indeed, in the crystal structure of 63 in complex with ZIKV NS2B-NS3 protease114, a six-membered boronate structure was observed, in which the boron atom forms a covalent bond with Ser135 and two oxygen atoms are linked to Gly133 and His51 through hydrogen bonds (Fig. 25B). In the case of the West Nile virus113, the NS2B-NS3 protease recognizes a five-membered boronate structure with similar interactions (Fig. 25C). But, although the boronic acid moiety significantly contributes to the target affinity, the antiviral effect in cells is relatively weak. This is attributable to the high polarity of the boronic acid moiety and the basic side chain, which hinders passage through the cell membrane. To overcome this issue, the boronic acid was modified by esterification with diols to obtain prodrugs.
Figure 25.

(A) Structures of compounds 61–63. (B) Crystal structure of the ZIKV NS2B-NS3 in complex with compound 63 (PDB ID: 5LC0). (C) Schematic illustration of 63 in the substrate-binding site of WNV NS2B-NS3 protease.
5. Boron-containing antiparasitic agents
Malaria is a life-threatening disease, and Plasmodium falciparum and Plasmodium vivax pose the greatest threat among the five parasitic species that cause malaria in humans. In 2018, the incidence of malaria worldwide was about 228 million cases, and the estimated death toll was 405,000. Artemisinin combination therapies (ACTs) have become an indispensable tool in the control of malaria. However, the emergence of resistance to both artemisinin and partner drugs is threatening the efficacy of ACTs, especially in Southeast Asia115. New agents are needed urgently, preferably with novel mechanisms of action, and boron-containing compounds, especially benzoxaborole, seem to be promising candidates. Benzoxaborole exhibits good antimalarial activity against a variety of plasmodial species including P. falciparum strains, as well as high bioavailability. Further, it is easy to synthesize, with a low cost of production. Importantly, some boron-containing compounds appear to have a novel antimalarial target and new mechanism of action, and therefore may have potential for treating multidrug-resistant malaria in combination with other drugs.
In a search for new antimalarial compounds, Sonoiki et al.98 screened a benzoxaborole library rich in LeuRS inhibitors for potency against cultured P. falciparum. Two 3-aminomethyl compounds, AN6426 (64) and AN8432 (65), showed activity against cultured multidrug-resistant (W2 strain) P. falciparum (IC50 = 310 and 490 nmol/L, respectively) and efficacy against murine Plasmodium berghei infection when administered orally once daily for 4 days (ED90 = 7.4 and 16.2 mg/kg). Genetic and biochemical studies suggested that AN6426 and AN8432 have a novel antimalarial mechanism of action, i.e., inhibition of P. falciparum LeuRS. In the same year, Sonoiki et al.116 discovered AN3661 (compound 66 in Fig. 26) by screening a benzoxaborole library against cultured P. falciparum asexual blood stage parasites. AN3661 was effective against multiple plasmodial species, including P. falciparum strains (mean IC50: 32 nmol/L), Uganda field isolates (mean IC50: 64 nmol/L), and P. berghei and P. falciparum infections (ED90: 0.34 and 0.57 mg/kg, respectively). It was also highly effective when administered orally in mouse models of P. falciparum and P. berghei infection. Genetic and biochemical studies disclosed that AN3661 targets a homologue of mammalian cleavage and polyadenylation specificity factor subunit 3 (CPSF3).
Figure 26.

Structures of antiparasitic agents 64–67.
Recent studies117 have identified the proteasome of P. falciparum as a promising drug target, and combinations of proteasome inhibitors with other drugs appear to have potential for treating multidrug-resistant malaria. Xie et al.118 screened a library of human proteasome inhibitors (peptidyl boronic acids) and identified selective anti-Plasmodium proteasome inhibitors. They obtained four hits showing excellent inhibitory activity and selectivity, as well as significant activity against both artemisinin-sensitive and -resistant parasites. Among them, 67 (Fig. 26) was 56 times more effective than HepG2 against P. falciparum cultures, showing 112-fold improvement in selectivity relative to bortezomib. Moreover, substrate profiling revealed that the P. falciparum 20S β2 site is more hydrophobic than the human 20S site, offering a path for researchers to design even more selective inhibitors.
Zhang et al.119,120 identified a hit compound (68) with an IC50 of 120 nmol/L against P. falciparum from a library screen. To improve the antimalarial activity, they designed and synthesized a series of 6-hetaryloxy benzoxaborole derivatives. Among them, compound 69 showed IC50 values of 1.4 and 1.9 nmol/L against P. falciparum W2 and 3D7 strains in a mouse model of infection. It also showed excellent in vivo efficacy against P. berghei (ED90 = 7.0 mg/kg). However, it had a poor oral PK profile (t1/2 = 1 h, F = 23%) in mice, which was attributed to facile hydrolysis of the methyl ester. Changing the ester to an amide and introducing a 7-Me substituent on the benzoxaborane core improved the PK properties, and the 7-methyl benzoxaborole pyrazine carboxamide scaffold was optimized to give compound 70. This compound exhibited a longer half-life (t1/2 = 5.2 h) and higher bioavailability (F = 96%) in mice. In addition, 70 demonstrated excellent in vivo efficacies against P. falciparum (ED90 = 0.85 mg/kg) and P. berghei (ED90 = 1.92 mg/kg) in infected mice (Fig. 27). Ames and genetic toxicity test results indicated that 70 is not mutagenic and does not damage micronuclei.
Figure 27.

Structures of antiparasitic agents 68–70.
6. Other boron-containing agents
Other activities of boron-containing compounds have also been reported. For example, boronic acid covalently inhibits transthyretin (TTR) to attenuate TTR amyloidosis and covalently inhibits carboxylesterase to block resistance to organophosphate (OPs) insecticides. A new approach to rheumatoid arthritis (RA) therapy has also been reported based on the use of PBA-containing prodrugs that are activated by H2O2 in an inflammatory environment. In addition, the coordination properties of boron with diols have been utilized to develop self-assembling dimer agents and have been applied for the purification and modification of proteins.
6.1. Boron moieties as functional groups for covalent complexation
The dissociation of TTR, a homotetrameric protein, into monomers is related to the pathogenic formation of fibrils in human amyloidogenic diseases. The binding of a ligand can enhance the conformational stability of TTR, so ligands that stabilize the TTR tetramer are candidates for the treatment of TTR amyloidosis. Smith et al.21 considered that TTR ligands binding in a covalent but reversible manner would be preferable, and developed boronic acid-based ligands for the T4-binding site of the TTR tetramer. These diboronic acids inhibit fibril formation by both wild-type TTR and a common disease-related variant, V30M TTR, in fibril formation assay. Compound 71 inhibited fibril formation as effectively as the clinically used agent tafamidis, both for wild-type TTR (3% versus 0% at 7.2 μmol/L) and for the V30M variant (8% versus 8% at 7.2 μmol/L). Co-crystallographic data of TTR with diboronic acid 71 revealed that the boronic acid moiety forms a boronic ester with Ser117 through reversible covalent bond formation (see Fig. 28).
Figure 28.

Planar structure of diboronic acid 71, and structure of the wild-type transthyretin complexed with 71 (PDB ID: 5u4f).
Pesticides and insecticides such as organophosphates (OPs) and carbamates are vital tools for increasing agricultural productivity. The most common resistance mechanism of insects to OPs and carbamates involves carboxylesterases (CEs). Therefore, CEs such as αE7 from the agricultural pest Lucilia cuprina are targets for inhibitors designed to eliminate resistance to insecticide. Correy et al.121 screened a virtual library of ∼23,000 boronic acid derivatives against the crystal structure of αE7 using DOCKovalent, an algorithm for screening covalent inhibitors. They identified covalent inhibitors of WT αE7 with IC50 values ranging 520 pmol/L to 25 nmol/L. Co-crystal structures revealed that boronic acids form both trigonal planar and tetrahedral adducts with Ser218 of LcαE7 (Fig. 29). Further structure–activity relationship analysis led to compound 72, which is a potent inhibitor of WT LcαE7 (IC50 = 0.9 nmol/L) and a common Gly137Asp variant (IC50 = 35 nmol/L). Compound 72 is highly selective for LcαE7 over other L. cuprina serine hydrolases and human AChE, and shows little or no toxicity to human cells and mice. These inhibitors act synergistically with the OPs diazinon and malathion, achieving an EC50 reduction by up to 16-fold and restoring the sensitivity of resistant strains to OPs. Moreover, the compounds significantly increase the efficacy of chlorpyrifos, another OP, against Myzus persicae (a common pest), indicating broad-spectrum potential for boronic acid compounds as a class of insecticide synergists to overcome OP resistance.
Figure 29.

Structures of tetrahedral and trigonal planar boronic acid adducts (A) and compound 72 (B).
6.2. Boronic acids as hydrogen peroxide-sensitive prodrugs
RA is an autoimmune disease whose pathogenesis is associated with oxidative stress. Under pathological inflammatory conditions, the extracellular concentration of H2O2 can reach 1.0 mmol/L, which is 100-fold higher than in healthy tissue122. In order to reduce the side effects of methotrexate (MTX, 73) and aminopterin (AMT, 74) RA therapy, Peiró Cadahía et al.123 designed and synthesized hydrogen peroxide-sensitive prodrugs of MTX and AMT based on a PBA group linked to MTX or AMT via a carbamate linkage or a direct C–N bond (Fig. 30). Pathological concentrations of H2O2 activate the prodrugs 75 and 76 to release the parent drug. Compared to the parent drugs, the prodrugs exhibited moderate to good solubility, high chemical and enzymatic stability, and comparable efficacy with lower toxicity in a collagen-induced arthritis (CIA) mouse model of RA.
Figure 30.

Chemical structures of prodrugs of AMT and MTX.
6.3. Boronic acids as linker components
One approach to bypass the poor membrane permeability and poor pharmacokinetics of many high-molecular-weight compounds is to utilize bioorthogonal reactions, which proceed under physiological conditions, and are unaffected by biological small molecules or proteins124. For example, monomers consisting of ligands and paired bioorthogonal linkers can be delivered to the cell, pass separately through the plasma membrane, and then spontaneously dimerize to form an active inhibitor (Fig. 31). This strategy is easily adaptable to a wide range of targets, especially challenging drug targets.
Figure 31.

Schematic representation of the self-assembling dimer approach.
Many bioorthogonal linkers can be utilized. For example, both copper-free click chemistry and Staudinger ligation exhibit bioorthogonality. However, the usefulness of the Staudinger ligation is limited by the slow reaction rate and the oxidation of reactants, while copper-free click chemistry also has limitations, though the reaction shows better kinetics (9.0 × 10−2 L/mol∙S) than the Staudinger ligation125,126. In contrast, aryl boronic acids react reversibly with vicinal hydroxyl groups to produce boronate esters in a rapid equilibrium, and the reaction between PBA and salicyl hydroxamic acid (SHA) shows extremely fast kinetics [(7.01 ± 2.04) × 106 L2/mol2∙S]. Multivalent bioactive inhibitors can be easily produced by using linkers containing multiple SHA moieties. Compared with the interaction between vicinal diol and boronic acid, SHA interacts with boronic acid through oxygen and nitrogen, which increase the resistance of the borate esters to hydrolysis (Fig. 32). Importantly, these boronic acid esters show pH-dependent hydrolysis, and are expected to be cleavable under cellular conditions. According to Shin et al.36,127, this reaction is not affected by common molecules such as amino acids, d-glucose, and ions in biological solutions. Thus, aryl boronic acids and paired diols are attractive bioorthogonal linkers that can be used to connect ligands in the intracellular environment, enabling the efficient delivery of potent and highly selective bivalent drugs in the form of small-molecular monomers.
Figure 32.

Schematic representation of the equilibria of boronic acid and diol/salicylhydroxamic acid that are utilized for dimer formation.
β-Tryptase, a homotetrameric serine protease with four identical active sites, is the most abundant protein in human mast cells, and its release is related to many inflammatory and allergic responses. Thus, it is an attractive target molecule for drug development128. Giardina et al.129 developed self-assembling dimeric inhibitors of tryptase by using boronic acid linkers and complementary catechols or cis-alkyl hydroxy partners to reversibly join two tryptase ligands. Co-dosing of successful combinations resulted in marked potency improvements over the monomers alone, and co-crystal structure analysis confirmed the presence of the dimer. For example, 77 and 78 formed a single stereoisomeric covalent boronate diester dimer (79), spanning approximately 27.3 Å and binding two adjacent subunits (Fig. 33), which exhibited an IC50 of 37 nmol/L, a 10-fold improvement over the more potent monomer. Further the heterodimer 82 formed from 80 and 81 exhibited a 35-fold improvement in IC50 in comparison to the more potent of the two monomers (22.1 nmol/L), and formed a phenolic-hydroxamate/boronate complex. The heterodimers showed more than 600-fold selectivity against related trypsin-family proteases. Plasma components had little effect on the synergy or efficacy of the compounds, and in the presence of the biological target, the boronic acids interact selectively with the target instead of naturally occurring diols.
Figure 33.

(A) Structures of boronic acid and partner molecules, and the self-assembled dimers. (B) X-ray crystallographic structure of the dimer of 77 and 78 bound to tryptase (PDB ID: 6P0P).
Recently, Pieszka et al.130 designed a peptide sequence that undergoes a multi-stage transformation, ultimately leading to the programmed death of cancer cells (Fig. 34). The first stage, based on dynamic formation of the PBA–SHA complex, involves linking the boronic acid-modified pro-assembling sequence to a transporter TAT (trans-activator of transcription) sequence that induces cellular uptake. After entering the cell, the boronic acid–salicylhydroxamate complex is cleaved under the acidic conditions, releasing the pro-assembling sequence. Endogenous or pathological hydrogen peroxide in cancer cells cleaves the boronic acid masking group to generate the isoleucine-serine-alanine (ISA) self-assembling motif that promotes the generation of fibrillar architectures. The formation of these superstructures, which can be imaged by fluorescence and electron microscopy, leads to programmed cell death.
Figure 34.

Intracellular co-assembly of peptides.
Boronic acids are also promising candidates for modifying and purifying proteins due to their small size, high biocompatibility, bioorthogonality, stability and reversibility. Zegota et al.131 reported the purification and reversible assembly of protein conjugates based on dynamic formation of the PBA-diol and PBA–SHA complexes (Fig. 35). Incorporation of the boronic acid tag into the protease (lysozyme) by reaction between disulfide bond of lysozyme and vinyl sulfone rebridging reagent bearing a PBA moiety was accomplished, and allowed the straightforward purification of BA-lysozyme by carbohydrate-based column chromatography. SHA-modified fluorescent dye (BODIPY FL) can be used for fast and efficient reversible functionalization of BA-lysozyme to obtain a stable biological conjugate using the interaction between boronic acid and SHA. The conjugate retains the original enzyme activity and remains intact in the cell, but can be cleaved by slight pH changes to allow further modification.
Figure 35.

“Tag and modify” protein conjugation with dynamic covalent chemistry.
7. Conclusions and perspectives
This article illustrates the diverse applications of boronic acids in the construction of therapeutically useful bioactive molecules. Briefly, boronic acid group is unique in several ways as follows. First, boron-containing compounds bind to biological targets through reversible covalent bonds, multiple hydrogen bonds or metal ion coordination bonds, facilitating biological activity as well as overcoming drug resistance. Second, such compounds can serve as prodrugs to improve drug targeting. Third, the boronic acid moiety is inaccessible to glucuronidation, and therefore can enhance bioavailability by blocking this metabolic pathway. Fourth, the boronic acid moiety can be designed to target unique bacterial glycoproteins or extracellular glycans. Fifth, bioorthogonal reactions involving boronic acid can be used to generate larger molecules intracellularly in order to overcome permeability issues. Sixth, boron nitride nanotubes and nanoparticles can be utilized for drug delivery. Seventh, boron cluster compounds can be used for boron neutron capture therapy.
In the past years, boron had always been ignored by medicinal chemists due to toxicity concerns in drug design. The investigation by Li et al.2 revealed that the toxicity concerns were derived from boric acid, a component of ant poisons and the toxicity of bortezomib. However, the LD of boric acid is 2660 mg/kg (rats, oral), which is equivalent to the LD value of table salt (3000 mg/kg rats, oral). And the toxicity of bortezomib is due to the mechanism of action rather than the presence of boron in the molecule. Moreover, recent research results show that the intake of boron is beneficial for bone growth and maintenance, brain function, and lower cancer risk132. Nevertheless, many challenges remain in extending the applicability of boron compounds. One significant challenge is the technical limitations of virtual screening, which has become an important tool in drug discovery. At present, most covalent docking methods are still too slow to conveniently screen thousands of compounds. Furthermore, during the calculation process, the side chain conformation of the reactive residue is usually assumed to be static, so that the result of the covalent docking depends greatly on the initially selected conformation. This assumption may be particularly problematic for boronic acid derivatives. Some covalent docking programs, such as the web server DOCKovalent developed by the Shoichet lab133 in 2014, are suitable for screening libraries of commercially available compounds, but no software is currently available for screening large user-defined libraries of covalent ligands.
Another problem is that boronic acids are less stable than aldehydes and acrylates, which are widely used as electrophiles, and thus may be degraded before binding to the target. They also show nonspecific reactivity with endogenous nucleophiles, limiting their specificity for nucleophilic residues at the active site of target enzymes and raising the possibility of off-target effects. It is also relevant that boronic acids are relatively difficult to prepare, especially when they contain C (sp3)‒B bonds134,135. To solve some of these problems, boronate esters and boron heterocycles such as benzoborazole and MIDA boronate have been developed. Compared with PBA, the boron–oxygen bond of benzoborazole is not easily hydrolyzed, and the boron–carbon bond (B–C) is stronger. Consequently, under reflux in 10% HCl or 15% NaOH, benzoborazole is more stable than PBA. Benzoborazole compounds also have higher water solubility than PBA136. Furthermore, the tertiary amine group in MIDA coordinates with the empty p orbital of boron and prevents unwanted reactions with external nucleophiles; therefore, the MIDA group contributes to increased stability and acts as a slow-release element in a biological milieu134. Indeed, the MIDA group has been found to improve the pharmacokinetic profile of dCTPase inhibitors.
The glycan-binding ability of boronic acid has also attracted increasing attention, but boronic acid has a wide specificity for glycoproteins. Therefore, it will be important to develop strategies to adjust the affinity of boronic acid for specific types of glycoproteins. For example, Lu et al.31 reported a pH adjustment strategy to fine-tune the boronate affinity in order to achieve specificity for two subclasses of glycoproteins.
Better characterization of the targets of boronic acids will be useful to guide the rational design of inhibitors that incorporate additional interactions in order to improve selectivity and to reduce the propensity for development of resistance. At present, the targets of some active compounds, especially antiparasitic and anti-inflammatory agents, are still unknown. For example, the cellular targets and mechanism of action of the anticancer and anti-malarial benzoxaborate remain unclear.
Lastly, boronic acids are commonly used to target serine residues. Therefore, there is considerable scope to expand their applicability to other nucleophiles such as arginine, tyrosine and threonine. In this context, it is interesting that multivalent boronic acids targeting the extracellular glycans of Mtb have a different mechanism of action from monomeric boronic acid. We believe there is still enormous scope to extend the role of boron in drug discovery and development to obtain new agents for treating a range of diseases.
Acknowledgments
Financial support from the National Natural Science Foundation of China (No. 81973181, to Xinyong Liu, China), Shandong Provincial Key Research and Development Project (No. 2019JZZY021011, to Peng Zhan, China), National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (2019ZX09301126, to Xinyong Liu, China), Outstanding Youth Fund of Shandong Province (ZR2020JQ31, to Peng Zhan, China), Foreign Cultural and Educational Experts Project (GXL20200015001, to Xinyong Liu, China), the Program for Outstanding Ph.D. Candidate of Shandong University (to Lin Sun, China), Qilu Young Scholars Program of Shandong University (to Peng Zhan, China) and the Taishan Scholar Program at Shandong Province (to Xinyong Liu, China) is gratefully acknowledged.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Vasanthanathan Poongavanam, Email: nobelvasanth@gmail.com.
Peng Zhan, Email: zhanpeng1982@sdu.edu.cn.
Xinyong Liu, Email: xinyongl@sdu.edu.cn.
Author contributions
Xinyong Liu and Peng Zhan were responsible for the conception and design of the review. Shu Song wrote the paper and made all the figures. Peng Zhan, Ping Gao, Lin Sun, Dongwei Kang, Jacob Kongsted and Vasanthanathan Poongavanam were in charge of checking and revision.
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.Albertoa R., Abramb U. International year of the periodic table 2019: elements important for life sciences. Chimia. 2019;73:207–209. doi: 10.2533/chimia.2019.207. [DOI] [PubMed] [Google Scholar]
- 2.Baker S.J., Ding C.Z., Akama T., Zhang Y.K., Hernandez V., Hernandez V. Therapeutic potential of boron-containing compounds. Future Med Chem. 2009;1:1275–1288. doi: 10.4155/fmc.09.71. [DOI] [PubMed] [Google Scholar]
- 3.Das B.C., Thapa P., Karki R., Schinke C., Das S., Kambhampati S. Boron chemicals in diagnosis and therapeutics. Future Med Chem. 2013;5:653–676. doi: 10.4155/fmc.13.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandes G.F.S., Denny W.A., Santos J.L.D. Boron in drug design: Recent advances in the development of new therapeutic agents. Eur J Med Chem. 2019;179:791–804. doi: 10.1016/j.ejmech.2019.06.092. [DOI] [PubMed] [Google Scholar]
- 5.Kahlert J., Austin C.J.D., Kassiou M., Rendina L.M. The fifth element in drug design: Boron in medicinal chemistry. ChemInform. 2013;66:1118–1123. [Google Scholar]
- 6.Richardson P.G., Hideshima T., Anderson K.C. Bortezomib (PS-341): A novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Contr. 2003;10:361–369. doi: 10.1177/107327480301000502. [DOI] [PubMed] [Google Scholar]
- 7.Gupta A.K., Versteeg S.G. Tavaborole—a treatment for onychomycosis of the toenails. Expet Rev Clin Pharmacol. 2016;9:1145–1152. doi: 10.1080/17512433.2016.1206467. [DOI] [PubMed] [Google Scholar]
- 8.Richardson P.G., Zweegman S., O'Donnell E.K., Laubach J.P., Raje N., Voorhees P. Ixazomib for the treatment of multiple myeloma. Expet Opin Pharmacother. 2018;19:1949–1968. doi: 10.1080/14656566.2018.1528229. [DOI] [PubMed] [Google Scholar]
- 9.Jarnagin K., Chanda S., Coronado D., Ciaravino V., Zane L.T., Guttman-Yassky E. Crisaborole topical ointment, 2%: A nonsteroidal, topical, anti-inflammatory phosphodiesterase 4 inhibitor in clinical development for the treatment of atopic dermatitis. J Drugs Dermatol JDD. 2016;15:390–396. [PubMed] [Google Scholar]
- 10.Burgos R.M., Biagi M.J., Rodvold K.A., Danziger L.H. Pharmacokinetic evaluation of meropenem and vaborbactam for the treatment of urinary tract infection. Expet Opin Drug Metabol Toxicol. 2018;14:1007–1021. doi: 10.1080/17425255.2018.1511702. [DOI] [PubMed] [Google Scholar]
- 11.ChEMBL database. December 17, 2020. https://www.ebi.ac.uk/chembl/g/#search_results/all/query=boron Available from: [Google Scholar]
- 12.M.D. Anderson Cancer Center . November 20, 2019. Talabostat and pembrolizumab for the treatment of advanced solid cancers. ClinicalTrials.gov. Identifier: NCT04171219.https://clinicaltrials.gov/ct2/show/NCT04171219?term=TALABOSTAT&draw=2&rank=2 Available from: [Google Scholar]
- 13.Recardio, Inc . April 3, 2018. Study of dutogliptin in combination with filgrastim in post-myocardial infarction. ClinicalTrials.gov. Identifier: NCT03486080.https://clinicaltrials.gov/ct2/show/NCT03486080?term=Dutogliptin&draw=2&rank=1 Available from: [Google Scholar]
- 14.GlaxoSmithKline . June 15, 2018. An early bactericidal activity, safety and tolerability of GSK3036656 in subjects with drug-sensitive pulmonary tuberculosis. ClinicalTrials.gov. Identifier: NCT03557281.https://clinicaltrials.gov/ct2/show/NCT03557281?term=GSK3036656&draw=2&rank=1 Available from: [Google Scholar]
- 15.Drugs for Neglected Diseases . March 23, 2017. Prospective study on efficacy and safety of acoziborole (SCYX-7158) in patients infected by human African trypanosomiasis due to T.b. Gambiense (OXA002). ClinicalTrials.gov. Identifier: NCT03087955.https://clinicaltrials.gov/ct2/show/NCT03087955?term=SCYX-7158&draw=2&rank=1 Available from: [Google Scholar]
- 16.Pfizer . February 23, 2011. Efficacy and safety of AN2898 and AN2728 topical ointments to treat mild-to-moderate atopic dermatitis. ClinicalTrials.gov. Identifier: NCT01301508.https://clinicaltrials.gov/ct2/show/NCT01301508?term=AN2898&draw=2&rank=1 Available from: [Google Scholar]
- 17.VenatoRx Pharmaceuticals, Inc . February 15, 2019. Safety and efficacy study of cefepime/VNRX-5133 in patients with complicated urinary tract infections. ClinicalTrials.gov. Identifier: NCT03840148.https://clinicaltrials.gov/ct2/show/NCT03840148?term=Taniborbactam&draw=2&rank=1 Available from: [Google Scholar]
- 18.Qpex Biopharma, Inc . May 8, 2020. P1 single and multiple ascending dose (SAD/MAD) study of IV QPX7728 alone and combined with QPX2014 in NHV. ClinicalTrials.gov. Identifier: NCT04380207.https://clinicaltrials.gov/ct2/show/NCT04380207?term=QPX7728&draw=2&rank=1 Available from: [Google Scholar]
- 19.GlaxoSmithKline . December 18, 2013. Study to assess the safety, pharmacokinetics, and antiviral activity of repeat doses of GSK2878175 in subjects with chronic hepatitis C. ClinicalTrials.gov. Identifier: NCT02014571.https://clinicaltrials.gov/ct2/show/NCT02014571?term=GSK2878175&draw=2&rank=1 Available from: [Google Scholar]
- 20.Diaz D.B., Yudin A.K. The versatility of boron in biological target engagement. Nat Chem. 2017;9:731–742. doi: 10.1038/nchem.2814. [DOI] [PubMed] [Google Scholar]
- 21.Smith T.P., Windsor I.W., Forest K.T., Raines R.T. Stilbene boronic acids form a covalent bond with human transthyretin and inhibit its aggregation. J Med Chem. 2017;60:7820–7834. doi: 10.1021/acs.jmedchem.7b00952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groll M., Berkers C.R., Ploegh H.L., Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Rock F.L., Mao W., Yaremchuk A., Tukalo M., Crépin T., Zhou H. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 24.Bandyopadhyay A., McCarthy K.A., Kelly M.A., Gao J. Targeting bacteria via iminoboronate chemistry of amine-presenting lipids. Nat Commun. 2015;6:6561. doi: 10.1038/ncomms7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Windsor I.W., Palte M.J., Lukesh J.C., 3rd, Gold B., Forest K.T., Raines R.T. Sub-picomolar inhibition of HIV-1 protease with a boronic acid. J Am Chem Soc. 2018;140:14015–14018. doi: 10.1021/jacs.8b07366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong P.Y., Shotwell J.B., Miller J., Price D.J., Maynard A., Voitenleitner C. Design of N-benzoxaborole benzofuran GSK8175-optimization of human pharmacokinetics inspired by metabolites of a failed clinical HCV inhibitor. J Med Chem. 2019;62:3254–3267. doi: 10.1021/acs.jmedchem.8b01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu B., Trout R.E.L., Chu G.H., McGarry D., Jackson R.W., Hamrick J.C. Discovery of taniborbactam (VNRX-5133): A broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. J Med Chem. 2020;63:2789–2801. doi: 10.1021/acs.jmedchem.9b01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnaud J., Audfray A., Imberty A. Binding sugars: From natural lectins to synthetic receptors and engineered neolectins. Chem Soc Rev. 2013;42:4798–4813. doi: 10.1039/c2cs35435g. [DOI] [PubMed] [Google Scholar]
- 29.James T.D., Sandanayake K.R.A.S., Shinkai S. Saccharide sensing with molecular receptors based on boronic acid. Angew Chem Int Ed Engl. 1996;35:1910–1922. [Google Scholar]
- 30.Kuivila H.G., Keough A.H., Soboczenski E.J. Areneboronates from diols and polyols. J Org Chem. 1954;19:780–783. [Google Scholar]
- 31.Lu Y., Bie Z., Liu Y., Liu Z. Fine-tuning the specificity of boronate affinity monoliths toward glycoproteins through pH manipulation. Analyst. 2013;138:290–298. doi: 10.1039/c2an36048a. [DOI] [PubMed] [Google Scholar]
- 32.Guy C.S., Gibson M.I., Fullam E. Targeting extracellular glycans: Tuning multimeric boronic acids for pathogen-selective killing of Mycobacterium tuberculosis. Chem Sci. 2019;10:5935–5942. doi: 10.1039/c9sc00415g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J., Zheng S., Akerstrom V.L., Yuan C., Ma Y., Zhong Q. Fulvestrant-3 boronic acid (ZB716): An orally bioavailable selective estrogen receptor downregulator (SERD) J Med Chem. 2016;59:8134–8140. doi: 10.1021/acs.jmedchem.6b00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C., Zhong Q., Zhang Q., Zheng S., Miele L., Wang G. Boronic prodrug of endoxifen as an effective hormone therapy for breast cancer. Breast Cancer Res Treat. 2015;152:283–291. doi: 10.1007/s10549-015-3461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong Q., Zhang C., Zhang Q., Miele L., Zheng S., Wang G. Boronic prodrug of 4-hydroxytamoxifen is more efficacious than tamoxifen with enhanced bioavailability independent of CYP2D6 status. BMC Cancer. 2015;15:625. doi: 10.1186/s12885-015-1621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin S.B.Y., Almeida R.D., Gerona-Navarro G., Bracken C., Jaffrey S.R. Assembling ligands in situ using bioorthogonal boronate ester synthesis. Chem Biol. 2010;17:1171–1176. doi: 10.1016/j.chembiol.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Demetriades M., Leung I.K.H., Chowdhury R., Chan M.C., McDonough M.A., Yeoh K.K. Dynamic combinatorial chemistry employing boronic acids/boronate esters leads to potent oxygenase inhibitors. Angew Chem Int Ed Engl. 2012;51:6672–6675. doi: 10.1002/anie.201202000. [DOI] [PubMed] [Google Scholar]
- 38.Wanner J., Romashko D., Werner D.S., May E.W., Peng Y., Schulz R. Reversible linkage of two distinct small molecule inhibitors of Myc generates a dimeric inhibitor with improved potency that is active in Myc over-expressing cancer cell lines. PLoS One. 2015;10 doi: 10.1371/journal.pone.0121793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuivila H.G., Armour A.G. Electrophilic Displacement Reactions. IX. Effects of substituents on rates of reactions between hydrogen peroxide and benzeneboronic acid1-3. J Am Chem Soc. 1957;79:5659–5662. [Google Scholar]
- 40.Zheng S., Guo S., Zhong Q., Zhang C., Liu J., Yang L. Biocompatible boron-containing prodrugs of belinostat for the potential treatment of solid tumors. ACS Med Chem Lett. 2018;9:149–154. doi: 10.1021/acsmedchemlett.7b00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marzenell P., Hagen H., Sellner L., Zenz T., Grinyte R., Pavlov V. Aminoferrocene-based prodrugs and their effects on human normal and cancer cells as well as bacterial cells. J Med Chem. 2013;56:6935–6944. doi: 10.1021/jm400754c. [DOI] [PubMed] [Google Scholar]
- 42.Leśnikowski Z.J. Challenges and opportunities for the application of boron clusters in drug design. J Med Chem. 2016;59:7738–7758. doi: 10.1021/acs.jmedchem.5b01932. [DOI] [PubMed] [Google Scholar]
- 43.Ocampo-Néstor A.L., Trujillo-Ferrara J.G., Abad-García A., Reyes-López C., Geninatti-Crich S., Soriano-Ursúa M.A. Boron's journey: Advances in the study and application of pharmacokinetics. Expert Opin Ther Pat. 2017;27:203–215. doi: 10.1080/13543776.2017.1252750. [DOI] [PubMed] [Google Scholar]
- 44.Genady A.R. Promising carboranylquinazolines for boron neutron capture therapy: Synthesis, characterization, and in vitro toxicity evaluation. Eur J Med Chem. 2009;44:409–416. doi: 10.1016/j.ejmech.2008.02.037. [DOI] [PubMed] [Google Scholar]
- 45.Ferlay J.A.O., Colombet M., Soerjomataram I., Mathers C., Parkin D.M., Piñeros M. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- 46.Kuczynski E.A., Sargent D.F., Grothey A., Kerbel R.S. Drug rechallenge and treatment beyond progression—implications for drug resistance. Nat Rev Clin Oncol. 2013;10:571–587. doi: 10.1038/nrclinonc.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holohan C., Schaeybroeck S.V., Longley D.B., Johnston P.G. Cancer drug resistance: An evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 48.Murray M.Y., Auger M.J., Bowles K.M. Overcoming bortezomib resistance in multiple myeloma. Biochem Soc Trans. 2014;42:804–808. doi: 10.1042/BST20140126. [DOI] [PubMed] [Google Scholar]
- 49.Kikuchi J., Wada T., Shimizu R., Izumi T., Akutsu M., Mitsunaga K. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 2010;116:406–417. doi: 10.1182/blood-2009-07-235663. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Y., Liu X., Xue J., Liu L., Liang T., Li W. Discovery of peptide boronate derivatives as histone deacetylase and proteasome dual inhibitors for overcoming bortezomib resistance of multiple myeloma. J Med Chem. 2020;63:4701–4715. doi: 10.1021/acs.jmedchem.9b02161. [DOI] [PubMed] [Google Scholar]
- 51.Song F.F., Xia L.L., Ji P., Tang Y.B., Huang Z.M., Zhu L. Human dCTP pyrophosphatase 1 promotes breast cancer cell growth and stemness through the modulation on 5-methyl-dCTP metabolism and global hypomethylation. Oncogenesis. 2015;4:e159. doi: 10.1038/oncsis.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Llona-Minguez S., Höglund A., Jacques S.A., Johansson L., Calderón-Montaño J.M., Claesson M. Discovery of the first potent and selective inhibitors of human dCTP pyrophosphatase 1. J Med Chem. 2016;59:1140–1148. doi: 10.1021/acs.jmedchem.5b01741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhandari V., Wong K.S., Zhou J.L., Mabanglo M.F., Batey R.A., Houry W.A. The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem Biol. 2018;13:1413–1425. doi: 10.1021/acschembio.8b00124. [DOI] [PubMed] [Google Scholar]
- 54.Cole A., Wang Z., Coyaud E., Voisin V., Gronda M., Jitkova Y. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2015;27:864–876. doi: 10.1016/j.ccell.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akopian T., Kandror O., Tsu C., Lai J.H., Wu W., Liu Y. Cleavage specificity of Mycobacterium tuberculosis ClpP1P2 protease and identification of novel peptide substrates and boronate inhibitors with anti-bacterial activity. J Biol Chem. 2015;290:11008–11020. doi: 10.1074/jbc.M114.625640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan J., Grouleff J.J., Jitkova Y., Diaz D.B., Griffith E.C., Shao W. De novo design of boron-based peptidomimetics as potent inhibitors of human ClpP in the presence of human ClpX. J Med Chem. 2019;62:6377–6390. doi: 10.1021/acs.jmedchem.9b00878. [DOI] [PubMed] [Google Scholar]
- 57.Hackl M.W., Lakemeyer M., Dahmen M., Glaser M., Pahl A., Lorenz-Baath K. Phenyl esters are potent inhibitors of caseinolytic protease P and reveal a stereogenic switch for deoligomerization. J Am Chem Soc. 2015;137:8475–8483. doi: 10.1021/jacs.5b03084. [DOI] [PubMed] [Google Scholar]
- 58.Akçay G., Belmonte M.A., Aquila B., Chuaqui C., Hird A.W., Lamb M.L. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol. 2016;12:931–936. doi: 10.1038/nchembio.2174. [DOI] [PubMed] [Google Scholar]
- 59.Yik-Sham Chung C., Timblin G.A., Saijo K., Chang C.J. Versatile histochemical approach to detection of hydrogen peroxide in cells and tissues based on puromycin staining. J Am Chem Soc. 2018;140:6109–6121. doi: 10.1021/jacs.8b02279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Narayanaswamy N., Narra S., Nair R.R., Saini D.K., Kondaiah P., Govindaraju T. Stimuli-responsive colorimetric and NIR fluorescence combination probe for selective reporting of cellular hydrogen peroxide. Chem Sci. 2016;7:2832–2841. doi: 10.1039/c5sc03488d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maslah H., Skarbek C., Pethe S., Labruère R. Anticancer boron-containing prodrugs responsive to oxidative stress from the tumor microenvironmol/Lent. Eur J Med Chem. 2020;207:112670. doi: 10.1016/j.ejmech.2020.112670. [DOI] [PubMed] [Google Scholar]
- 62.Chen W., Balakrishnan K., Kuang Y., Han Y., Fu M., Gandhi V. Reactive oxygen species (ROS) inducible DNA cross-linking agents and their effect on cancer cells and normal lymphocytes. J Med Chem. 2014;57:4498–4510. doi: 10.1021/jm401349g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen W., Fan H., Balakrishnan K., Wang Y., Sun H., Fan Y. Discovery and optimization of novel hydrogen peroxide activated aromatic nitrogen mustard derivatives as highly potent anticancer agents. J Med Chem. 2018;61:9132–9145. doi: 10.1021/acs.jmedchem.8b00559. [DOI] [PubMed] [Google Scholar]
- 64.Huang G., Chen H., Dong Y., Luo X., Yu H., Moore Z. Superparamagnetic iron oxide nanoparticles: Amplifying ROS stress to improve anticancer drug efficacy. Theranostics. 2013;3:116–126. doi: 10.7150/thno.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagen H., Marzenell P., Jentzsch E., Wenz F., Veldwijk M.R., Mokhir A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J Med Chem. 2012;55:924–934. doi: 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- 67.Han D.C., Lee M.Y., Shin K.D., Jeon S.B., Kim J.M. 2′-Benzoyloxycinnamaldehyde induces apoptosis in human carcinoma via reactive oxygen species. J Biol Chem. 2004;279:6911–6920. doi: 10.1074/jbc.M309708200. [DOI] [PubMed] [Google Scholar]
- 68.Noh J., Kwon B., Han E., Park M., Yang W., Cho W. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat Commun. 2015;6:6907. doi: 10.1038/ncomms7907. [DOI] [PubMed] [Google Scholar]
- 69.Miller E.W., Tulyathan O., Isacoff E., Chang C.J. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat Chem Biol. 2017;5:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- 70.Govan J.M., McIver A.I., Riggsbee C., Deiters A. Hydrogen peroxide induced activation of gene expression in mammalian cells using boronate estrone derivatives. Angew Chem Int Ed Engl. 2012;51:9066–9070. doi: 10.1002/anie.201203222. [DOI] [PubMed] [Google Scholar]
- 71.Chou D.H., Webber M.J., Tang B.C., Lin A.B., Thapa L.S., Deng D. Glucose-responsive insulin activity by covalent modification with aliphatic phenylboronic acid conjugates. Proc Natl Acad Sci U S A. 2015;112:2401–2406. doi: 10.1073/pnas.1424684112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J., Zhang J., Hao G., Xin W., Yang F., Zhu M. Design, synthesis, and structure‒activity relationship of 7-propanamide benzoxaboroles as potent anticancer agents. J Med Chem. 2019;62:6765–6784. doi: 10.1021/acs.jmedchem.9b00736. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki M. Boron neutron capture therapy (BNCT): A unique role in radiotherapy with a view to entering the accelerator-based BNCT era. Int J Clin Oncol. 2020;25:43–50. doi: 10.1007/s10147-019-01480-4. [DOI] [PubMed] [Google Scholar]
- 74.Kankaanranta L., Saarilahti K., Mäkitie A., Välimäki P., Tenhunen M., Joensuu H. Boron neutron capture therapy (BNCT) followed by intensity modulated chemoradiotherapy as primary treatment of large head and neck cancer with intracranial involvement. Radiother Oncol. 2011;99:98–99. doi: 10.1016/j.radonc.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 75.Kankaanranta L., Seppälä T., Koivunoro H., Saarilahti K., Atula T., Collan J. Boron neutron capture therapy in the treatment of locally recurred head-and-neck cancer: final analysis of a phase I/II trial. Int J Radiat Oncol Biol Phys. 2012;82:e67–75. doi: 10.1016/j.ijrobp.2010.09.057. [DOI] [PubMed] [Google Scholar]
- 76.Santa Cruz G.A., González S.J., Bertotti J., Marín J. First application of dynamic infrared imaging in boron neutron capture therapy for cutaneous malignant melanoma. Med Phys. 2009;36:4519–4529. doi: 10.1118/1.3218760. [DOI] [PubMed] [Google Scholar]
- 77.Ariyoshi Y., Miyatake S., Kimura Y., Shimahara T., Kawabata S., Nagata K. Boron neuron capture therapy using epithermal neutrons for recurrent cancer in the oral cavity and cervical lymph node metastasis. Oncol Rep. 2007;18:861–866. [PubMed] [Google Scholar]
- 78.Matović J., Järvinen J., Bland H.C., Sokka I.K., Imlimthan S., Mateu Ferrando R. Addressing the biochemical foundations of a glucose-based "trojan horse"-strategy to boron neutron capture therapy: From chemical synthesis to in vitro assessment. Mol Pharm. 2020;17:3885–3899. doi: 10.1021/acs.molpharmaceut.0c00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kreiner A.J., Bergueiro J., Cartelli D., Baldo M., Castell W., Asoia J.G. Present status of accelerator-based BNCT. Rep Practical Oncol Radiother. 2016;21:95–101. doi: 10.1016/j.rpor.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barth R.F., Grecula J.C. Boron neutron capture therapy at the crossroads—where do we go from here? Appl Radiat Isot. 2020;160:109029. doi: 10.1016/j.apradiso.2019.109029. [DOI] [PubMed] [Google Scholar]
- 81.Barth R.F., Mi P., Yang W. Boron delivery agents for neutron capture therapy of cancer. Cancer Commun (Lond) 2018;38:35. doi: 10.1186/s40880-018-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferreira T.H., Marino A., Rocca A., Liakos I., Nitti S., Athanassiou A. Folate-grafted boron nitride nanotubes: possible exploitation in cancer therapy. Int J Pharm. 2015;481:56–63. doi: 10.1016/j.ijpharm.2015.01.048. [DOI] [PubMed] [Google Scholar]
- 83.Jampilek J. Design and discovery of new antibacterial agents: Advances, perspectives, challenges. Curr Med Chem. 2018;25:4972–5006. doi: 10.2174/0929867324666170918122633. [DOI] [PubMed] [Google Scholar]
- 84.WHO . September 03, 2020. Global tuberculosis report 2019.https://www.who.int/tb/publications/global_report/en/ Available from: [Google Scholar]
- 85.Krajnc A., Lang P.A., Panduwawala T.D., Brem J., Schofield C.J. Will morphing boron-based inhibitors beat the β-lactamases? Curr Opin Chem Biol. 2019;50:101–110. doi: 10.1016/j.cbpa.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bush K. Proliferation and significance of clinically relevant β-lactamases. Ann N Y Acad Sci. 2013;1277:84–90. doi: 10.1111/nyas.12023. [DOI] [PubMed] [Google Scholar]
- 87.Cahill S.T., Tyrrell J.M., Navratilova I.H., Calvopiña K., Robinson S.W., Lohans C.T. Studies on the inhibition of AmpC and other β-lactamases by cyclic boronates. Biochim Biophys Acta Gen Subj. 2019;1863:742–748. doi: 10.1016/j.bbagen.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 88.Morandi F., Caselli E., Morandi S., Focia P.J., Blázquez J., Shoichet B.K. Nanomolar inhibitors of AmpC beta-lactamase. J Am Chem Soc. 2003;125:685–695. doi: 10.1021/ja0288338. [DOI] [PubMed] [Google Scholar]
- 89.Caselli E., Romagnoli C., Vahabi R., Taracila M.A., Bonomo R.A., Prati F. Click chemistry in lead optimization of boronic acids as β-lactamase inhibitors. J Med Chem. 2015;58:5445–5458. doi: 10.1021/acs.jmedchem.5b00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wong D., Nielsen T.B., Bonomo R.A., Pantapalangkoor P., Luna B., Spellberg B. Clinical and pathophysiological overview of acinetobacter infections: A century of challenges. Clin Microbiol Rev. 2017;30:409–447. doi: 10.1128/CMR.00058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Caselli E., Fini F., Introvigne M.L., Stucchi M., Taracila M.A., Fish E.R. 1,2,3-Triazolylmethaneboronate: a structure activity relationship study of a class of β-lactamase inhibitors against Acinetobacter baumannii cephalosporinase. ACS Infect Dis. 2020;6:1965–1975. doi: 10.1021/acsinfecdis.0c00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ness S., Martin R., Kindler A.M., Paetzel M., Gold M., Jensen S.E. Structure-based design guides the improved efficacy of deacylation transition state analogue inhibitors of TEM-1 beta-lactamase. Biochemistry. 2000;39:5312–5321. doi: 10.1021/bi992505b. [DOI] [PubMed] [Google Scholar]
- 93.Hecker S.J., Reddy K.R., Totrov M., Hirst G.C., Lomovskaya O., Griffith D.C. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J Med Chem. 2015;58:3682–3692. doi: 10.1021/acs.jmedchem.5b00127. [DOI] [PubMed] [Google Scholar]
- 94.Brem J., Cain R., Cahill S., McDonough M.A., Clifton I.J., Jiménez-Castellanos J.C. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat Commun. 2016;8:12406. doi: 10.1038/ncomms12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krajnc A., Brem J., Hinchliffe P., Calvopiña K., Panduwawala T.D., Lang P. Bicyclic boronate VNRX-5133 inhibits metallo- and serine-β-lactamases. J Med Chem. 2019;62:8544–8556. doi: 10.1021/acs.jmedchem.9b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hecker S.J., Reddy K.R., Lomovskaya O., Griffith D.C., Rubio-Aparicio D., Nelson K. Discovery of cyclic boronic acid QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases. J Med Chem. 2020;63:7491–7507. doi: 10.1021/acs.jmedchem.9b01976. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y.L., Liu S., Yu Z.J., Lei Y., Huang M.Y., Yan Y.H. Structure-based development of (1-(3′-mercaptopropanamido)methyl)boronic acid derived broad-spectrum, dual-action inhibitors of metallo- and serine-β-lactamases. J Med Chem. 2019;61:7160–7184. doi: 10.1021/acs.jmedchem.9b00735. [DOI] [PubMed] [Google Scholar]
- 98.Sonoiki E., Palencia A., Guo D., Ahyong V., Dong C., Li X. Antimalarial benzoxaboroles target plasmodium falciparum leucyl-tRNA synthetase. Antimicrob Agents Chemother. 2016;60:4886–4895. doi: 10.1128/AAC.00820-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li X., Hernandez V., Rock F.L., Choi W., Mak Y.S.L., Mohan M. Discovery of a potent and specific M. tuberculosis leucyl-tRNA synthetase inhibitor: (S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656) J Med Chem. 2017;60:8011–8026. doi: 10.1021/acs.jmedchem.7b00631. [DOI] [PubMed] [Google Scholar]
- 100.Ju Y., He L., Zhou Y., Yang T., Sun K., Song R. Discovery of novel peptidomimetic boronate ClpP inhibitors with noncanonical enzyme mechanism as potent virulence blockers in vitro and in vivo. J Med Chem. 2020;63:3104–3119. doi: 10.1021/acs.jmedchem.9b01746. [DOI] [PubMed] [Google Scholar]
- 101.Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S. Inhibitors of SARS-CoV-2 entry: Current and future opportunities. J Med Chem. 2020;63:12256–12274. doi: 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xian Y., Zhang J., Bian Z., Zhou H., Zhang Z., Lin Z. Bioactive natural compounds against human coronaviruses: A review and perspective. Acta Pharm Sin B. 2020;10:1163–1174. doi: 10.1016/j.apsb.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ni L., Chen L., Huang X., Han C., Xu J., Zhang H. Combating COVID-19 with integrated traditional Chinese and Western medicine in China. Acta Pharm Sin B. 2020;10:1149–1162. doi: 10.1016/j.apsb.2020.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paget J., Spreeuwenberg P., Charu V., Taylor R.J., Iuliano A.D., Bresee J. Global mortality associated with seasonal influenza epidemics: New burden estimates and predictors from the GLaMOR Project. J Glob Health. 2019;9 doi: 10.7189/jogh.09.020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bhatt S., Gething P.W., Brady O.J., Messina J.P., Farlow A.W., Moyes C.L. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.WHO . September 02, 2020. Summary of the global HIV epidemic, 2019.https://www.who.int/hiv/data/2019_summary-global-hiv-epi.png?ua=1 Available from: [Google Scholar]
- 107.GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990‒2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maynard A., Crosby R.M., Ellis B., Hamatake R., Hong Z., Johns B.A. Discovery of a potent boronic acid derived inhibitor of the HCV RNA-dependent RNA polymerase. J Med Chem. 2014;57:1902–1913. doi: 10.1021/jm400317w. [DOI] [PubMed] [Google Scholar]
- 109.Ohuchi M., Asaoka N., Sakai T., Ohuchi R. Roles of neuraminidase in the initial stage of influenza virus infection. Microb Infect. 2006;8:1287–1293. doi: 10.1016/j.micinf.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 110.Matsuoka Y., Swayne D.E., Thomas C., Rameix-Welti M.A., Naffakh N., Warnes C. Neuraminidase stalk length and additional glycosylation of the hemagglutinin influence the virulence of influenza H5N1 viruses for mice. J Virol. 2009;83:4704–4708. doi: 10.1128/JVI.01987-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yin R., Zhang M., Hao C., Wang W., Qiu P., Wan S. Different cytotoxicities and cellular localizations of novel quindoline derivatives with or without boronic acid modifications in cancer cells. Chem Commun (Camb) 2013;49:8516–8518. doi: 10.1039/c3cc45203d. [DOI] [PubMed] [Google Scholar]
- 112.Wang W., Yin R., Zhang M., Yu R., Hao G., Zhang L. Boronic acid modifications enhance the anti-influenza A virus activities of novel quindoline derivatives. J Med Chem. 2017;60:2840–2852. doi: 10.1021/acs.jmedchem.6b00326. [DOI] [PubMed] [Google Scholar]
- 113.Nitsche C., Zhang L., Weigel L.F., Schilz J., Graf D., Bartenschlager R. Peptide-boronic acid inhibitors of flaviviral proteases: medicinal chemistry and structural biology. J Med Chem. 2017;60:511–516. doi: 10.1021/acs.jmedchem.6b01021. [DOI] [PubMed] [Google Scholar]
- 114.Lei J., Hansen G., Nitsche C., Klein C.D., Zhang L., Hilgenfeld R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science. 2016;353:503–505. doi: 10.1126/science.aag2419. [DOI] [PubMed] [Google Scholar]
- 115.WHO . June 05, 2020. World malaria report, 2019.https://www.who.int/malaria/publications/world-malaria-report-2019/en/ Available from: [Google Scholar]
- 116.Sonoiki E., Ng C.L., Lee M.C., Guo D., Zhang Y.K., Zhou Y. A potent antimalarial benzoxaborole targets a plasmodium falciparum cleavage and polyadenylation specificity factor homologue. Nat Commun. 2017;8:14574. doi: 10.1038/ncomms14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stokes B.H., Yoo E., Murith J.M., Luth M.R., Afanasyev P., da Fonseca P.C.A. Covalent plasmodium falciparum-selective proteasome inhibitors exhibit a low propensity for generating resistance in vitro and synergize with multiple antimalarial agents. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xie S.C., Gillett D.L., Spillman N.J., Tsu C., Luth M.R., Ottilie S. Target validation and identification of novel boronate inhibitors of the plasmodium falciparum proteasome. J Med Chem. 2018;61:10053–10066. doi: 10.1021/acs.jmedchem.8b01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Y.K., Plattner J.J., Easom E.E., Jacobs R.T., Guo D., Sanders V. Benzoxaborole antimalarial agents. Part 4. Discovery of potent 6-(2-(alkoxycarbonyl)pyrazinyl-5-oxy)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles. J Med Chem. 2015;98:5344–5354. doi: 10.1021/acs.jmedchem.5b00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang Y.K., Plattner J.J., Easom E.E., Jacobs R.T., Guo D., Freund Y.R. Benzoxaborole antimalarial agents. Part 5. Lead optimization of novel amide pyrazinyloxy benzoxaboroles and identification of a preclinical candidate. J Med Chem. 2017;60:5889–5908. doi: 10.1021/acs.jmedchem.7b00621. [DOI] [PubMed] [Google Scholar]
- 121.Correy G.J., Zaidman D., Harmelin A., Carvalho S., Mabbitt P.D., Calaora V. Overcoming insecticide resistance through computational inhibitor design. Proc Natl Acad Sci U S A. 2019;116:21012–21021. doi: 10.1073/pnas.1909130116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Perez C., Monserrat J.P., Chen Y., Cohen S.M. Exploring hydrogen peroxide responsive thiazolidinone-based prodrugs. Chem Commun (Camb) 2015;51:7116–7119. doi: 10.1039/c4cc09921d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Peiró Cadahía J., Bondebjerg J., Hansen C.A., Previtali V., Hansen A.E., Andresen T.L. Synthesis and evaluation of hydrogen peroxide sensitive prodrugs of methotrexate and aminopterin for the treatment of rheumatoid arthritis. J Med Chem. 2018;61:3503–3515. doi: 10.1021/acs.jmedchem.7b01775. [DOI] [PubMed] [Google Scholar]
- 124.Prescher J.A., Bertozzi C.R. Chemistry in living systems. Nat Chem Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 125.Baskin J.M., Prescher J.A., Laughlin S.T., Agard N.J., Chang P.V., Miller I.A. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Codelli J.A., Baskin J.M., Agard N.J., Bertozzi C.R. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J Am Chem Soc. 2008;130:11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stolowitz M.L., Ahlem C., Hughes K.A., Kaiser R.J., Kesicki E.A., Li G. Phenylboronic acid-salicylhydroxamic acid bioconjugates. 1. A novel boronic acid complex for protein immobilization. Bioconjugate Chem. 2001;12:229–239. doi: 10.1021/bc0000942. [DOI] [PubMed] [Google Scholar]
- 128.Reber L.L., Frossard N. Targeting mast cells in inflammatory diseases. Pharmacol Ther. 2014;142:416–435. doi: 10.1016/j.pharmthera.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 129.Giardina S.F., Werner D.S., Pingle M., Feinberg P.B., Foreman K.W., Bergstrom D.E. Novel, self-assembling dimeric inhibitors of human β tryptase. J Med Chem. 2020;63:3004–3027. doi: 10.1021/acs.jmedchem.9b01689. [DOI] [PubMed] [Google Scholar]
- 130.Pieszka M., Han S., Volkmann C., Graf R., Lieberwirth I., Landfester K. Controlled supramolecular assembly inside living cells by sequential multi-staged chemical reactions. J Am Chem Soc. 2020;142:15780–15789. doi: 10.1021/jacs.0c05261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zegota M.M., Wang T., Seidler C., Wah Ng D.Y., Kuan S.L., Weil T. "Tag and Modify" protein conjugation with dynamic covalent chemistry. Bioconjugate Chem. 2018;29:2665–2670. doi: 10.1021/acs.bioconjchem.8b00358. [DOI] [PubMed] [Google Scholar]
- 132.Nielsen F.H. Update on human health effects of boron. J Trace Elem Med Biol. 2014;28:383–387. doi: 10.1016/j.jtemb.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 133.London N., Miller R.M., Krishnan S., Uchida K., Irwin J.J., Eidam O. Covalent docking of large libraries for the discovery of chemical probes. Nat Chem Biol. 2014;10:1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Adachi S., Cognetta A.B., Niphakis M.J., He Z., Zajdlik A., St Denis J.D. Facile synthesis of borofragments and their evaluation in activity-based protein profiling. Chem Commun (Camb) 2015;51:3608–3611. doi: 10.1039/c4cc09107h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.António J.P.M., Russo R., Carvalho C.P., Cal P.M.S.D., Gois P.M.P. Boronic acids as building blocks for the construction of therapeutically useful bioconjugates. Chem Soc Rev. 2019;48:3513–3536. doi: 10.1039/c9cs00184k. [DOI] [PubMed] [Google Scholar]
- 136.Adamczyk-Woźniak A., Borys K.M., Sporzyński A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem Rev. 2015;115:5224–5247. doi: 10.1021/cr500642d. [DOI] [PubMed] [Google Scholar]