

Abstract
Glucocorticoids have important anti-inflammatory and immunomodulatory activities. Dexamethasone (Dex), a synthetic glucocorticoid, induces insulin resistance, hyperglycemia, and hypertension. The hypertensive mechanisms of Dex are not well understood. Previously, we showed that exercise training prior to Dex treatment significantly decreases blood vessel loss and hypertension in rats. In this study, we examined whether the salutary effects of exercise are associated with an enhanced metabolic profile. Analysis of the NAD and ATP content in the tibialis anterior muscle of trained and non-trained animals indicated that exercise increases both NAD and ATP; however, Dex treatment had no effect on any of the experimental groups. Likewise, Dex did not change NAD and ATP in cultured endothelial cells following 24 h and 48 h of incubation with high concentrations. Reduced VEGF-stimulated NO production, however, was verified in endothelial cultured cells. Reduced NO was not associated with changes in survival or the BH4 to BH2 ratio. Moreover, Dex had no effect on bradykinin- or shear-stress-stimulated NO production, indicating that VEGF-stimulated eNOS phosphorylation is a target of Dex’s effects. The PTP1B inhibitor increased NO in Dex-treated cells in a dose-dependent fashion, an effect that was replicated by the glucocorticoid receptor inhibitor, RU486. In combination, these results indicate that Dex-induced endothelial dysfunction is mediated by glucocorticoid receptor and PTP1B activation. Moreover, since exercise reduces the expression of PTP1B and normalized insulin resistance in aging rats, our findings indicate that exercise training by reducing PTP1B activity counteracts Dex-induced hypertension in vivo.
Keywords: glucocorticoid receptor, endothelial dysfunction, nitric oxide, VEGF
Graphical Abstract
Introduction
Dexamethasone (Dex) is a synthetic glucocorticoid used for the treatment of inflammatory and allergic conditions and as an adjuvant in cancer therapies. The prolonged use of Dex can elicit several side effects such as hypertension, muscle atrophy, and insulin resistance, among others (1–3). Since Dex has negligible mineralocorticoid activity, drug-induced hypertension is thought to be mediated by central nervous system effects and alterations in vascular homeostasis (4,5). Recognizing that microcirculation plays a fundamental role in tissue oxygenation and perfusion and in the regulation of blood flow, some recent studies have examined the effect of Dex in microvessel homeostasis. Studies in Wistar rat models treated with Dex demonstrated a reduction in microvascular density in the cremaster and tibialis anterior, supporting a role for microvascular rarefaction in the hypertensive effects of the drug (3–6).
Blood vessel density stability depends on the balance between angiogenic and cell death mechanisms (7). Previous studies established that Dex causes skeletal muscle (tibialis anterior) microvascular rarefaction, which is accompanied by vascular endothelial growth factor receptor 2 (VEGF-R2) reduction and also a decreased in Bcl-2 and the Bcl-2:Bax ratio (3,4,8). These findings support the idea that Dex-induced hypertension is explained by the inhibition of angiogenic mechanisms coupled with increased endothelial cell death. A decrease in VEGF-dependent signaling is a well-known cause of endothelial dysfunction and apoptosis, which could mediate some of Dex’s actions. In vitro studies with endothelial cells, however, have indicated dissimilar mechanisms of Dex in apoptosis and inhibition of angiogenesis (9–11).
Dex treatment is known to impair insulin sensitivity transiently, increasing blood glucose and insulin levels; thus, some of the side effects of the drug have been connected with metabolic remodeling (1,12,13). The metabolic pathways and specific changes induced by glucocorticoids in endothelial cells, however, are not completely characterized (14). Both signaling and glucocorticoid receptor (GR) transcriptional mechanisms in the endothelium are implicated in several of the beneficial effects of Dex in preventing vascular wall disruption (15). Non-transcriptional effects of glucocorticoid actions to protect endothelial cells and improve function have been shown to enhance cerebral blood flow in vivo (16).
Exercise training is a well-known activator of striatal muscle metabolism via the PGC1α pathway and the control of blood glucose levels (17). Previously, we showed that young adult rats who were submitted to 8 weeks of aerobic exercise on a treadmill, at 60% of the maximal respiratory capacity, prior to Dex treatment significantly reduced the loss of blood vessels and hypertension as compared with non-trained control animals. We hypothesized that a possible link between exercise training and reduced Dex hypertensive side effects could be explained by the maintenance of endothelial cell bioenergetics via NAD/Sirt1 signaling and increase in endothelial nitric oxide synthase (eNOS) activity, oxidative metabolism, and mitochondrial function, resulting in enhanced endothelial cell survival. Thus, the effects of Dex on NAD and ATP levels in the rat skeletal muscle of trained versus non-trained animals and endothelial cell culture were examined to gain some insight on the metabolic mechanisms affecting endothelial cell function and hypertension. Our data indicate that although exercise improved metabolic function, the hypertensive effects of Dex are better explained by the non-transcriptional effects of GR on eNOS activity.
Materials and Methods
Materials.
All chemicals and high-performance liquid chromatography (HPLC) standards were of analytical grade and purchased from Sigma-Aldrich (Milwaukee, USA). Dexamethasone sodium phosphate (Decadron®) was obtained from Aché Laboratory, (Guarulhos, SP, Brazil), Protein tyrosine phosphatase 1B inhibitor KY-226 from Focus Biomolecules (Plymouth Meeting, PA).
Animal groups and training.
Wistar rats (10-week old males [250–300 g]) were obtained from the Center for Research and Production Facilities of UNESP (Botucatu, SP, Brazil). The animals were housed at the Animal Facility at UNESP (Bauru, SP, Brazil). Animals received water and food ad libitum. All methods used were approved by the Committee for Ethical Use of Animals (CEUA) at the São Paulo State University, UNESP (approved protocol #836/2016). After 2 weeks of running adaptation on a rodent treadmill (5–10 m/min, 5 min), all rats were subjected to the maximal exercise test (Tmax) that consisted of running on a treadmill with increments of 5 m/min every 3 min, with 0% grade elevation until the animal stopped running spontaneously (8). After completing this test, rats were assigned to one of four different groups, such that each group would have the same average Tmax performance. The total training time was 10 weeks, and Dex treatment began after week 8. The experimental groups were (1) Sedentary-Control (SC), (2) Sedentary-Dex (SD) (3) Trained-Control (TC), (4) Trained-Dex (TD). Dex was administered subcutaneously as a saline injection (50 µg/kg per day) for 14 days. Untreated animals received saline injections only. Dexamethasone treatment did not produce toxicity. Body weight was recorded daily during the time of the experiment in all groups. Also, at the end of the treatment, adrenal glands were weighed in all animals as control of Dex treatment. Tibia bone length was used to normalize adrenal weight changes.
Blood pressure measurements.
After treatment, rats were anesthetized, and a polyethylene catheter was inserted into the carotid artery for arterial pressure recording. After 24 h recovery, arterial pressure and heart rate were recorded during 30 min in a conscious animal using a pressure transducer (DPT100; Utah Medical Products Inc.; Midvale, UT, USA), amplifier (Quad Bridge Amp, AD-Instruments, Sidney, NSW, Australia), and acquisition board (PowerLab 4/35, ADInstruments, Sidney, NSW, Australia).
Skeletal muscle dissection.
After euthanasia, the left tibialis anterior muscle was removed, cleaned, and immediately embedded in neutral talc, fixed with Tissue Tech (Sakura Finetek, Torrance, CA, USA), snap-frozen in liquid nitrogen, and stored at −80°C for morphometric analyses. The right tibialis anterior muscles were also removed, immediately immersed in a cold solution of 5% perchloric acid, and snap-frozen in liquid nitrogen for nucleotide HPLC assays.
Morphometric analyses.
Skeletal muscle slices were stained with hematoxylin-eosin and analyzed by microscopy for capillary density and capillary-to-fiber ratio. All images from 6 slices for each muscle were digitalized and the analysis was conducted by a single observer blinded to rat identity.
Endothelial cells.
Bovine aortic endothelial cells (BAEC) were obtained from American Type Culture Collection (Manassas, VA, USA). Cells were cultured in high-glucose DMEM (GIBCO, Thermo Fisher, Brookfield, WI, USA), supplemented with 10% (v/v) FBS, L-glutamine (2 mM), sodium pyruvate (1 mM), penicillin (100 units/mL), and streptomycin (100 units/mL). Culture dishes were kept at 37°C in a humidified air incubator with 5% CO2. All experiments used cells at 6–9 passages that were seed in 6-well plates at a density of 5×104 cell/ml for eNOS activity assays. BAECs were treated with Dex 1, 10 and 100 µM for 24h; untreated cells had DMSO as a control for drug-vehicle effects.
Cell viability.
Live/death assays were performed by the neutral red method as described (18). Values were normalized by protein content.
Pyrimidine nucleotide and adenine nucleotide quantification.
Briefly, adenine nucleotide (ATP, ADP) and oxidized forms of pyridine nucleotides (NAD) were extracted in cold 5% perchloric acid from tibialis anterior muscles (15 and 30 mg) or 100 mm confluent BAEC dishes. The extracts were neutralized with 1 M K2CO3. The supernatants were deproteinized using Amicon filters and combined with solvent A (0.1 M potassium phosphate and 4 mM tetrabutylammonium bisulfate, pH 6.0) for analysis. Protein concentrations were determined using a bicinchoninic acid assay in protein pellets resuspended in 0.5 N NaOH (200 µL). Samples were loaded onto a Supercosil LC-18T column (3 µm, 15 cm × 4.6 mm, Supelco) eluted with a gradient solvent A and solvent B (0.1 M potassium phosphate and 4 mM tetrabutylammonium bisulfate, pH 6.0), at a flow rate of 1 ml/min. Authentic NAD+, ATP, and ADP samples were used for calibration, sample concentrations of normalized per sample protein content.
Western blotting.
BAEC lysates were prepared using RIPA buffer with freshly added protease inhibitors and 20 µM PMSF. After clearing cell debris by centrifugation, proteins were resolved either on 12% or 8% SDS-PAGE gel and transferred onto a nitrocellulose membrane (Hybond-ECL, Amersham Biosciences, Piscataway, NJ, USA). After blocking membranes with 1% BSA/TBS solution, membranes were incubated with primary and secondary antibody and imaged using Immobilon Crescendo Western HRP substrate (MilliporeSigma, Burlington, MA) using a LICOR Biosciences scanning system (Lincoln, NE, USA). Primary antibodies are beta-actin, AKT, phospho-AKT (Cell signaling, Danvers, MA, USA), eNOS, p-eNOS (Upstate Technologies, Lake Placid, NY, USA).
Nitric oxide analysis.
Nitric oxide (NO) was analyzed by chemiluminescence using a NO-Analyzer (Sievers, GE, Boulder, CO). BAECS were stimulated with VEGF (50 µg/ml) in Hanks’ Balanced Salt solution (HBSS) supplemented with 1% FBS media and accumulated nitrite in the media (HBSS, 1% FBS) was analyzed by using triiodide reducing reagent (19). Calibration used authentic nitrite solutions (0.5–5 µM). Concentrations of nitrite were normalized to protein content.
Quantification of tetrahydrobiopterin by HPLC.
BAECs were lysed in 50 mM phosphate buffer, pH 2.6, and cleared supernatants were loaded onto a Synergi Polar-RP column (Phenomenex, Torrance, CA; 4 µ, 250×4.6 mm) eluted with argon-saturated 50 mM phosphate buffer, pH 2.6. Both tetrahydrobiopterin (BH4) and 7,8-dihydrobiopterin (BH2) detection were performed in 4-cell multielectrode, set potentials +0, 150, 280, 365 mV, equipped CoulArray system as described (20). Concentrations were estimated using the internal standard for extraction/recovery and authentic BH4/BH2 standards and normalized to protein content.
Statistical analysis.
Data are presented as mean ±SEM. Two-way or one-way analysis of variance was used. When interactions were found, Tukey’s post hoc test was adopted. The student’s t-test was used for comparisons between the 2 groups. Statistical significance was set as a p-value <0.05.
Results
Exercise training counteracts dexamethasone-induced high blood pressure and blood vessel rarefaction in the skeletal muscle of Wistar Rats.
To study the influence of NAD-dependent processes on the hypertensive mechanisms of Dex and reduction of blood vessels in skeletal muscle, a group of rats was subjected to the exercise protocol as previously described (4,21). Increased blood pressure in sedentary non-trained animals was verified, while blood pressure changes were attenuated in the trained group (Table 1). We also confirmed the loss of blood vessels in the tibialis anterior (left leg) of the sedentary treated groups, which was prevented by exercise training in the treated group (Table 1, Fig. 1). The pharmacological actions of Dex were also shown by the loss of body weight and adrenal gland atrophy (Table 1). The right leg tibialis anterior from the same animals was used to quantify NAD tissue levels, which were found significantly increased in exercise-trained animal control and Dex treated groups compared with non-trained sedentary groups (Fig. 2). Moreover, ATP but not ADP levels followed the same trend, showing that aerobic exercise training enhances muscle metabolic capacity. However, Dex did not alter NAD
Table 1.
Aerobic exercise training outcomes in rats receiving dexamethasone
| Experimental group | Bodyweight (g) | Body weight changec | Adrenal gland weightd (g) | Arterial pressuree (mmHg) | Heart rate (bpm) | Morphometric analysesf | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wistar Rats | Week-0 a | Week-8 b | (g) | SP | DP | MP | CD number/mm 2 | C:F ratio | ||
| Sedentary-Control | 203.6±9.7 | 438.0±14.1 | −2.3±2.5 | 10.9±0.7 | 106±6 | 91±3 | 96±6 | 426±12 | 192.6±16.3 | 0.90±0.02 |
| Sedentary-Dex | 203.2±6.4 | 204.2±6.4 | −52.2±3.4* | 6.2±0.2* | 129±4* | 119±3* | 122±4* | 429±26 | 152.7±8.8* | 0.70±0.02* |
| Trained-Control | 206.8±7.4 | 401±17.7 | +0.2±4.5 | 9.9±0.5 | 110±3 | 101±5 | 104±4 | 398±12 | 238.8±13.3┼ | 1.00±0.04┼ |
| Trained-Dex | 205.8±5.2 | 434.8±8.5 | −56.3±4.0* | 6.2±0.5* | 121±4 | 106±5┼ | 110±4┼ | 371±8┼ | 222.0±10.3┼ | 1.00±0.02┼ |
Wistar rats were trained as described in Materials and Methods.
Body weight was measured immediately after first Tmax and
after 8 weeks of training.
Difference in body weight at the end of 14 days of dexamethasone treatment.
Adrenal gland weight was measured at the end of the experiment.
Systolic (SP), diastolic (DP), and mean (MP) blood pressure.
Histological analysis of capillary density (CD) and capillary to fiber ratio (C:F ratio) in tibialis anterior muscle. The number of animals per group n=5–10. Values are mean±SEM,
p<0.05 for dexamethasone effects;
p<0.05 for training outcomes; statistical analysis was performed as described in methods.
Figure 1. Exercise training prevents skeletal muscle rarefaction induced by dexamethasone.
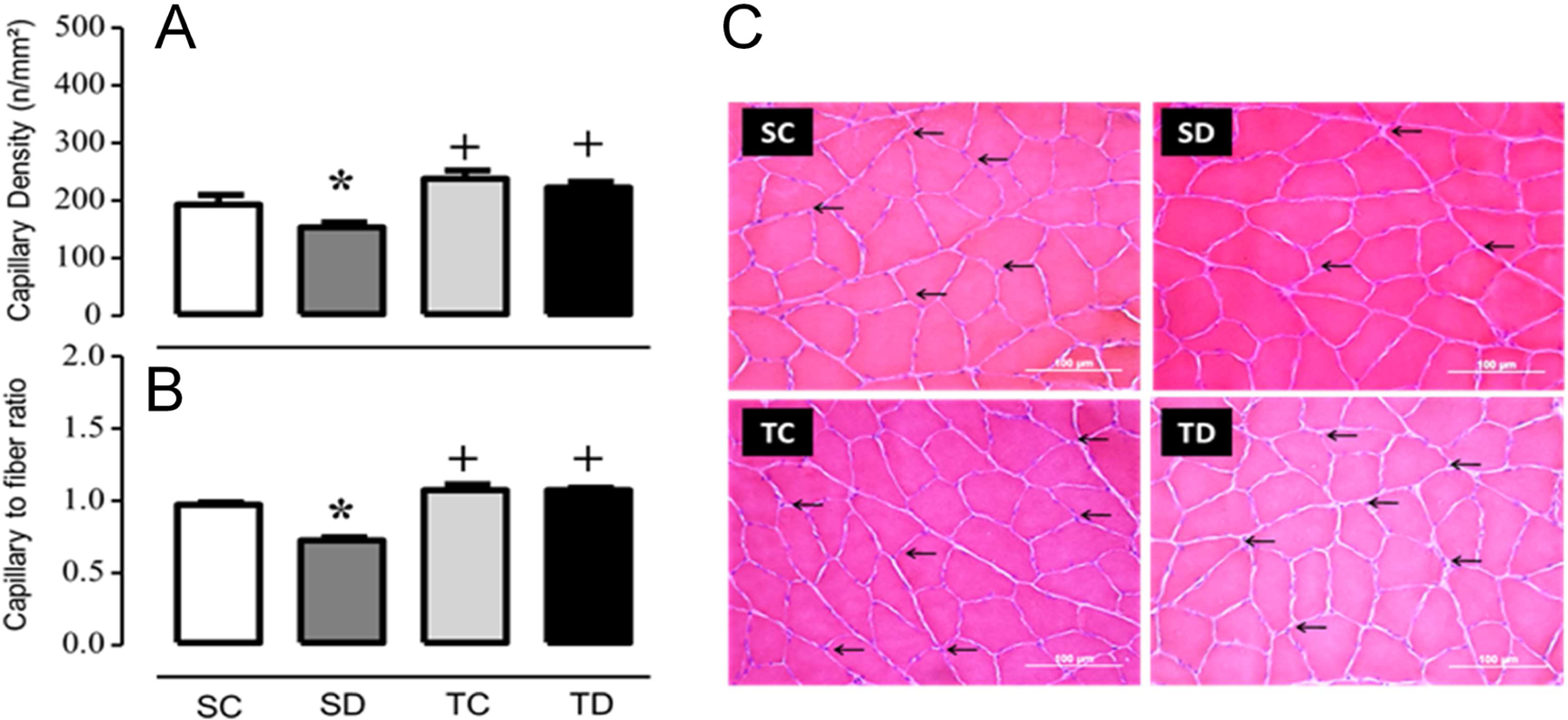
(A) capillary density, (B) capillary-to-fibers ratio, (C) histological sections from SC: sedentary control, SD: sedentary treated with DEX, TC: trained control and trained treated with DEX (TD) muscles stained with hematoxylin and eosin. The capillaries are marked by arrows (Scale bar= 100 m). Values represent the meanSEM from 6-histological slices obtained from 8–9 animals each group. +p<0.05 training; *p<0.05 dexamethasone effect.
Figure 2. Striatal muscle exercise training increases NAD, and ATP that is not changed by dexamethasone.
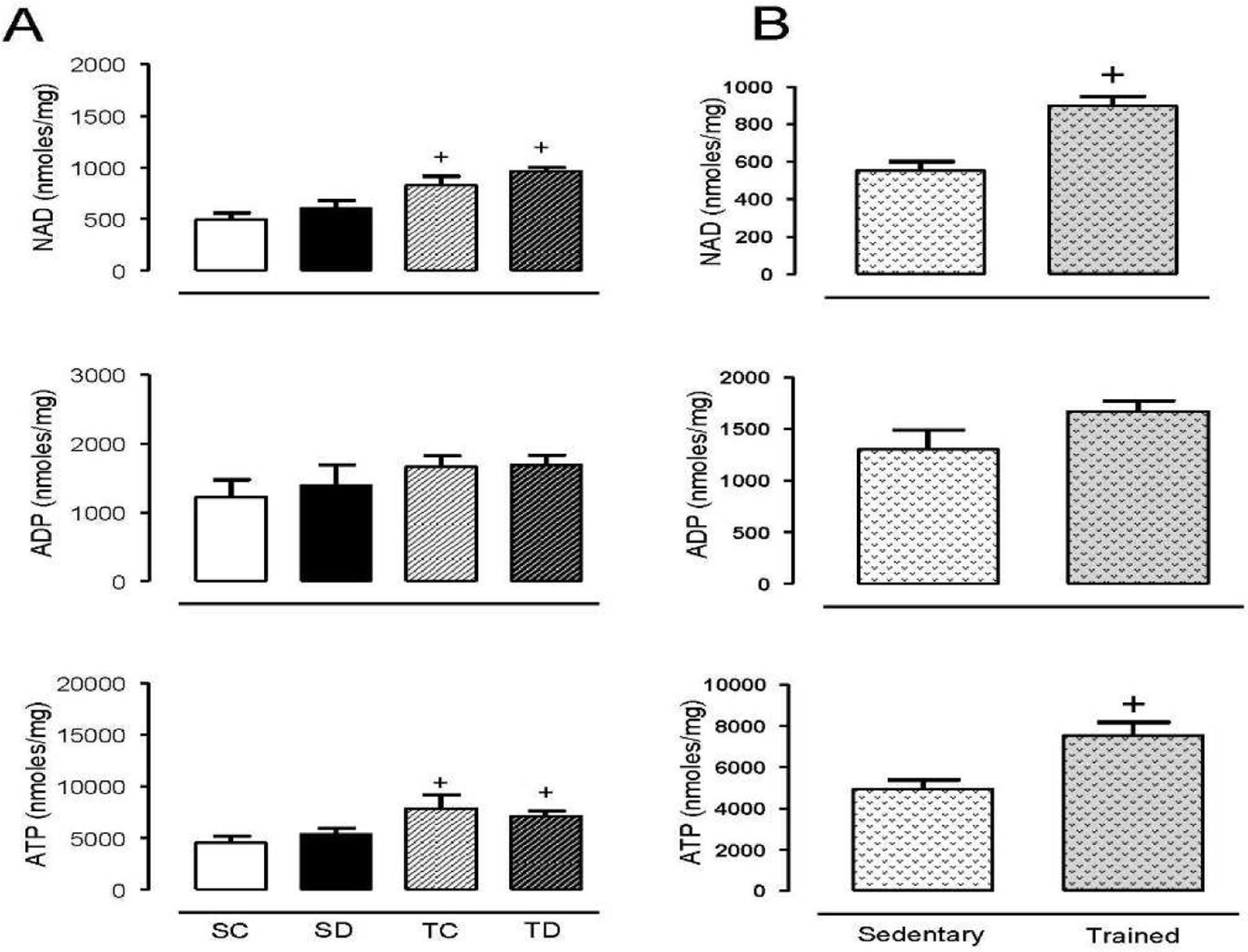
(A). Wistar rat tibialis anterior muscles from sedentary control (SC) and dexamethasone treated (SD) were compared to exercise trained control (TC) and dexamethasone treated (TD). (B) Direct comparison between sedentary and exercise groups regardless of treatment. All groups were exercised trained for 8 weeks prior to 14 days of treatment with dexamethasone (50 µg/kg/day, s.c. injections). Tibialis anterior muscles (30 mg) were collected and extracted with perchloric acid 5% and analyzed by HPLC as described in the Methods section. Values represent the mean±SEM from 5–10 samples in each group. +p<0.05 exercise training effect.
Dexamethasone effects on endothelial dysfunction.
To investigate whether Dex-induced hypertension in vivo is associated with increased endothelial cell dysfunction and death, cell survival and NO production were analyzed. Endothelial cell cultures treated with Dex for 24 h and 48 h at concentrations of 1–100 µM showed no changes in cell viability (Fig. 3). Also, no changes were found in NAD, ADP, or ATP levels after 24 h or 48 h treatments. To assess the effects of Dex on eNOS activity, cells were stimulated with VEGF, and the rate of nitrite accumulation in the media was quantified (Fig. 3). Dex at 100 µM but not 1–10 µM showed a significant decrease in NO production over time, which was not associated with a reduction in BH4 or the BH4:BH2 ratio. In combination, these data indicate that Dex induced endothelial dysfunction by mechanisms not related to NAD-dependent effects or eNOS uncoupling mechanisms.
Figure 3. Dexamethasone induces endothelial dysfunction that is not explained by changes in tetrahydrobiopterin or energy metabolism.
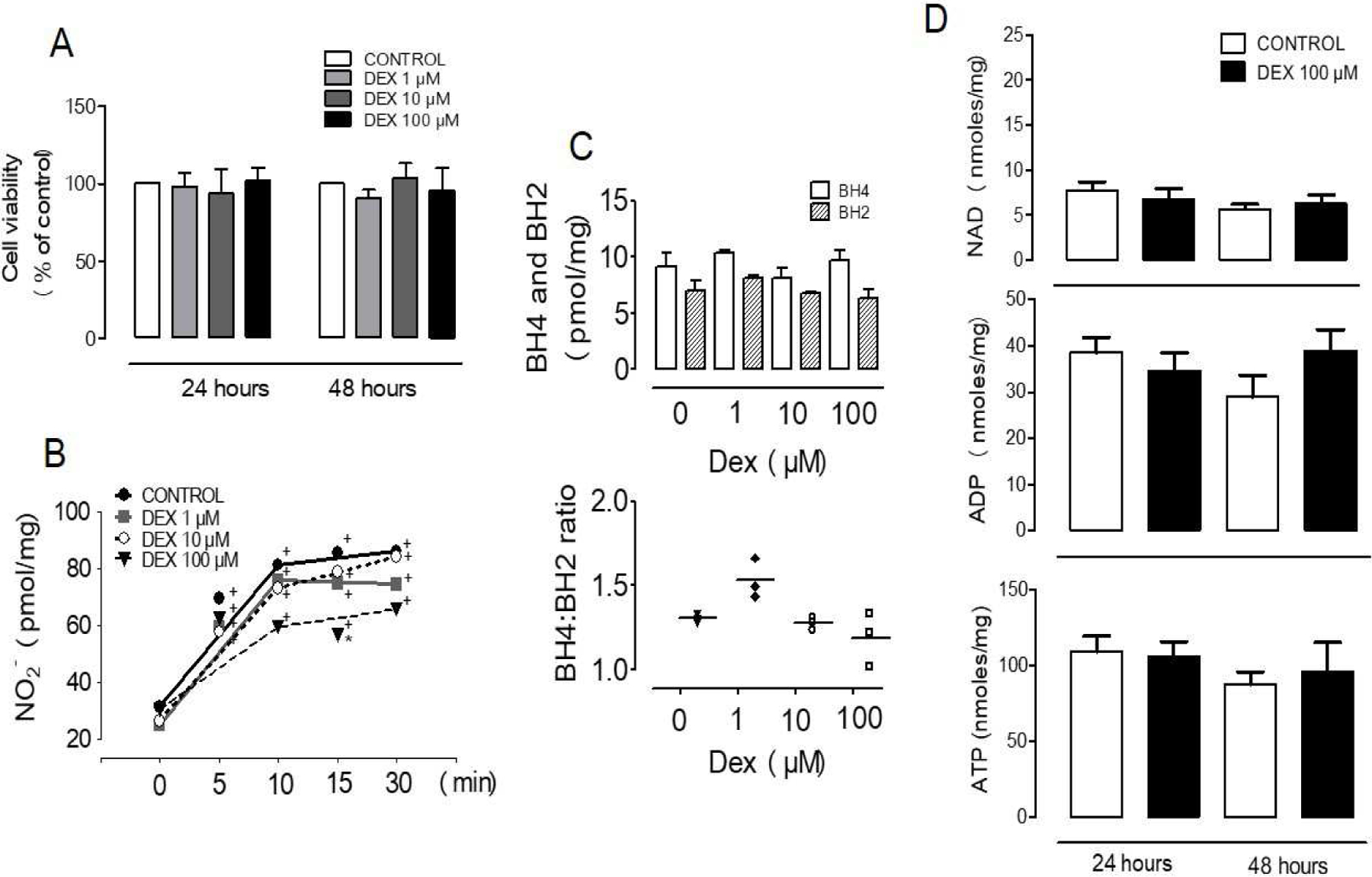
(A) BAECs viability was unaffected by 24 h or 48 h incubation with increasing concentrations of dexamethasone. (B) VEGF-stimulated NO production was decreased by 24 h Dex treatment. (C) BH4 and BH2 levels were not changed by 24 h dexamethasone treatment. (D) NAD, ADP, and ATP were not changed in cells treated for 24 h or 48 h with Dex. Values represent the mean±SEM from 3–10 independent replicates. +p<0.05 VEGF stimulation; *p<0.05 Dex effect.
Mechanisms of endothelial cell dysfunction by dexamethasone.
To better understand the mechanisms of Dex-induced endothelial dysfunction at high concentrations, we also examined the relationship of NO production induced by other eNOS activators. Bradykinin, a G-protein coupled receptor agonist, led to an acute increase in NO production that was unaffected by Dex treatments, indicating that Dex does not interfere with G-protein-coupled-receptor mediated mechanisms (Fig. 4A). Stimulation of endothelial cells by fluid shear stress is a well-established physiological mechanism increasing NO production. To model this stimulation in vitro, endothelial cells were exposed to a force of 18 dyne/cm2 for 4 h using a parallel disk viscometer. In this experimental setting, eNOS activation was ascertained by L-NMMA inhibition of NO production in the flow-stimulated cells. Basal NO detected in static cultures was significantly lower than flow-stimulated cells in the presence or absence of Dex, which was about 30% higher than that detected after a 30 min bolus addition of bradykinin (Fig. 4B). L-NMMA decreased NO accumulation to similar levels detected in static conditions (Fig 4B).
Figure 4. Mechanisms of endothelial cell dysfunction in dexamethasone-treated BAECs.
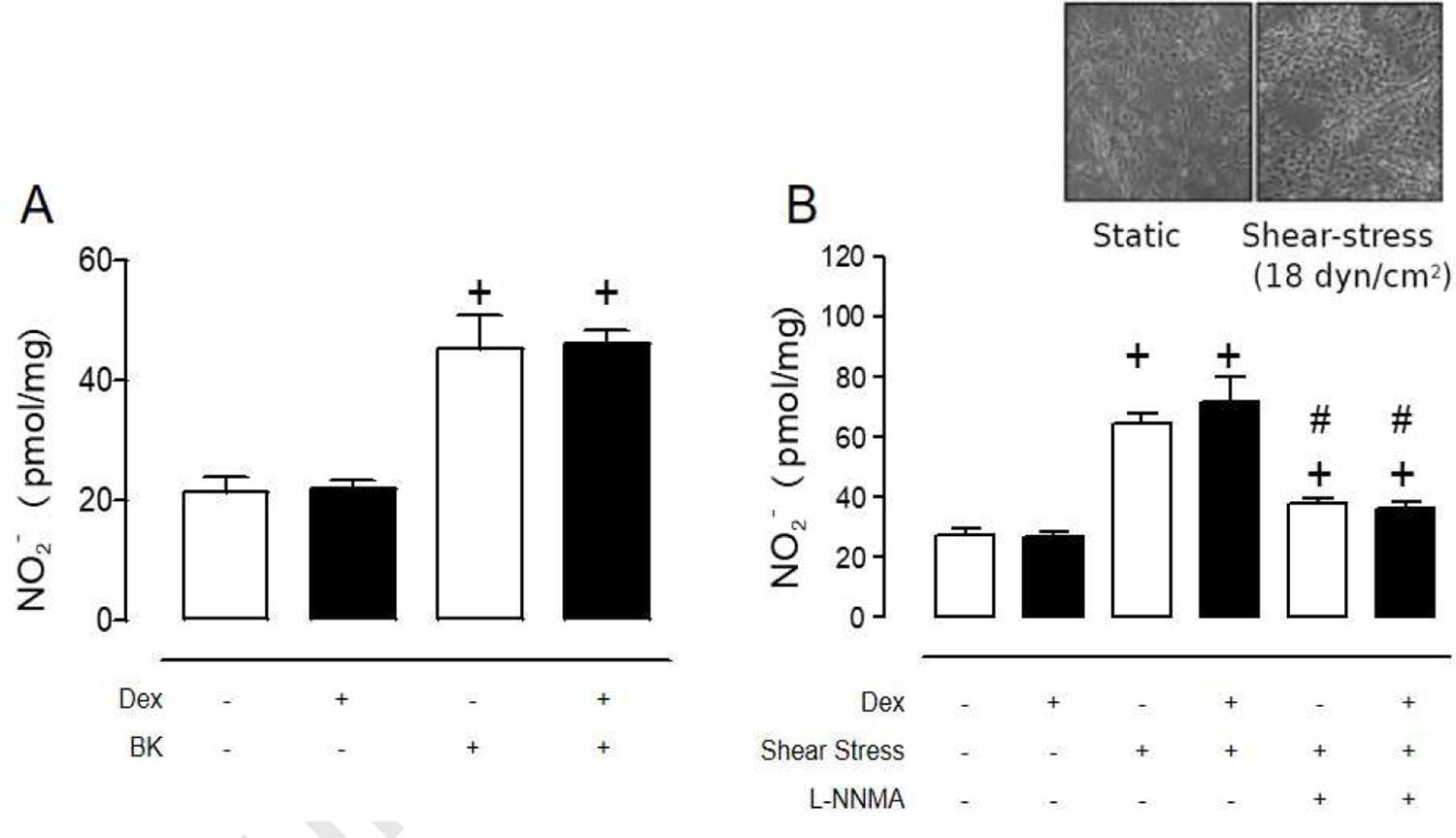
(A) Dex (100 µM, 24 h) did not modify bradykinin-stimulated NO release from BAECs. (B) Dex treatment (100 µM, 24 h) have no effect in NO release from shear stress-stimulated BAECs (18 dyen/cm2;4 h). Values represent the mean±SEM from 3–6 independent replicates. +p<0.05 bradykinin or shear stress stimulation; #p<0.05 L-NNMA effect.
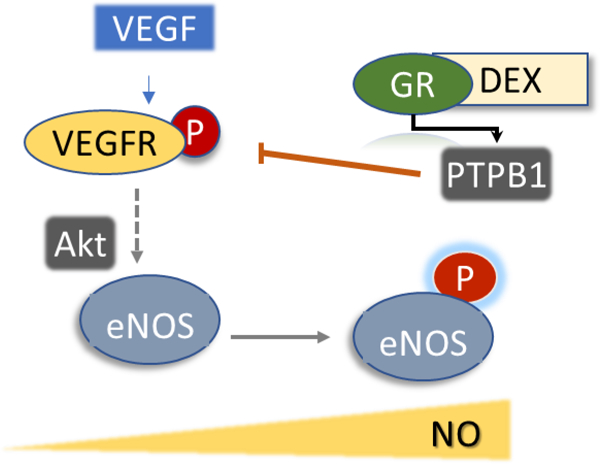
To ascertain the mechanisms by which Dex alters the rates of NO production in VEGF-simulated cells (Fig. 3B), control experiments confirmed the intermediacy of Akt and eNOS phosphorylation, which was enhanced by co-treatment with the protein tyrosine phosphatase 1B (PTP1B) inhibitor, KY226 (Fig. 5). There is evidence that PTP1B dephosphorylates VEGFR2 leading to attenuated ERK signaling. The PTP1B deficiency leads to decreased angiogenesis, which is also thought to be resultant of VEGF-Akt-eNOS pathway inhibition (22,23).
Figure 5. Effects of dexamethasone on VEGF-stimulated eNOS phosphorylation.
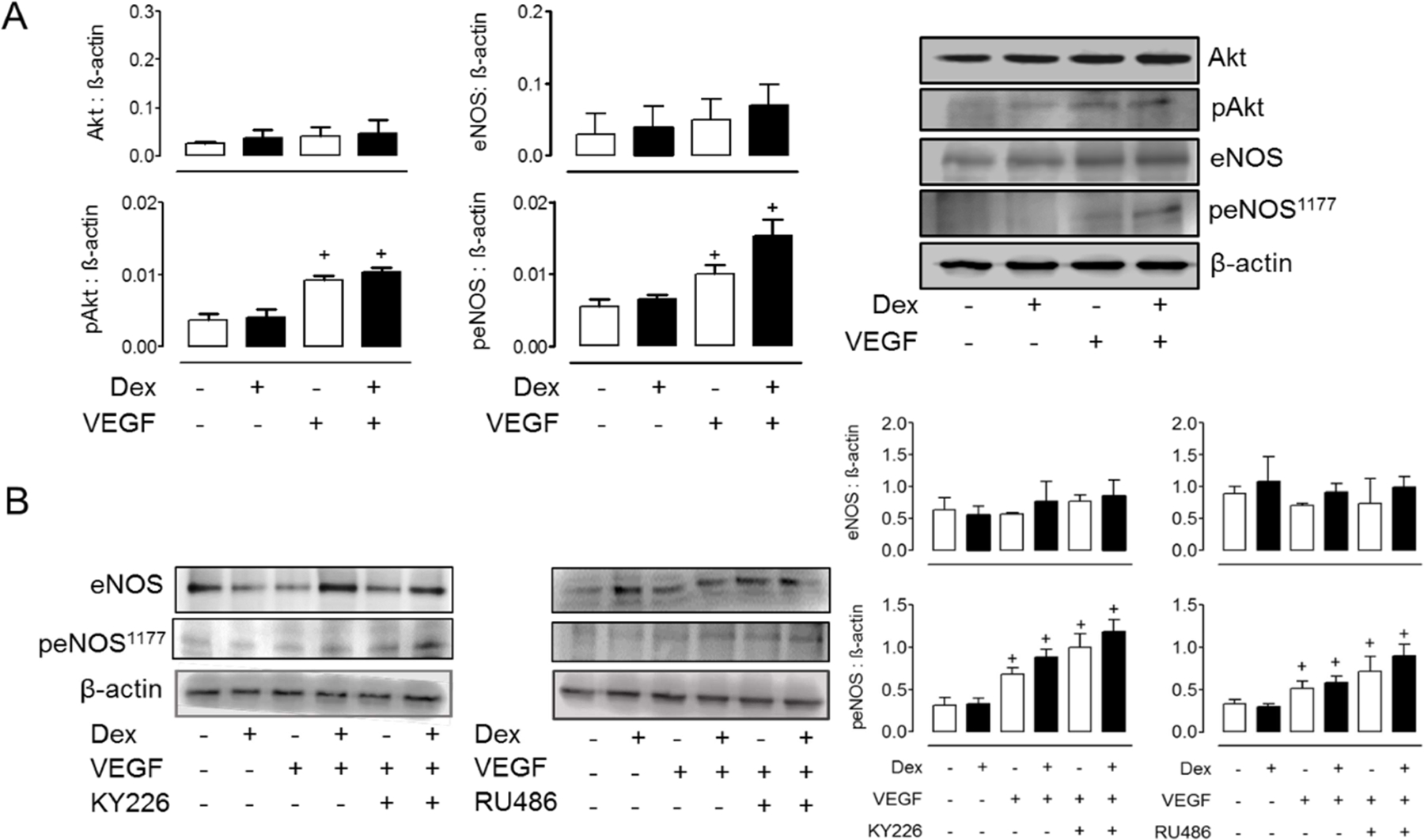
(A) Dex (100 µM, 24 h) did not modify VEGF-induced Akt and eNOS phosphorylation. Basal levels of Akt and eNOS were confirmed in duplicate experiments (B) PTP1B inhibitor (KY226), glucocorticoid receptor inhibitor (RU486), and Dex (100 µM, 24 h) enhance VEGF-induced eNOS phosphorylation. Values represent the mean±SEM from 3 independent replicates. +p<0.05 VEGF stimulation.
Also, PTP1B inhibition normalized the NO production rates in a dose-dependent fashion when ECs were co-treated with KY226 prior to activation with VEGF, suggesting that activation of PTP1B by Dex is the likely mechanism of reduced NO activity (Fig. 6A). Whether the actions of Dex are mediated by the glucocorticoid receptor (GR), the GR-inhibitor RU486 (Fig. 6B) was used. Pre-treatment with RU486 did not affect eNOS phosphorylation upon VEGF stimulation; however, RU486 rescued the NO production in stimulated cells to the same levels of non-Dex treated cells (Fig. 6B). The same NO rescue effect was seen with the PTP1B inhibitor, indicating that Dex likely disrupts eNOS signaling by GR-mediated activity on PTP1B (Fig. 6).
Figure 6. Inhibition of PTP1B by KY226 or glucocorticoid receptor-inhibitor (RU486) ameliorate dexamethasone-induced endothelial dysfunction.
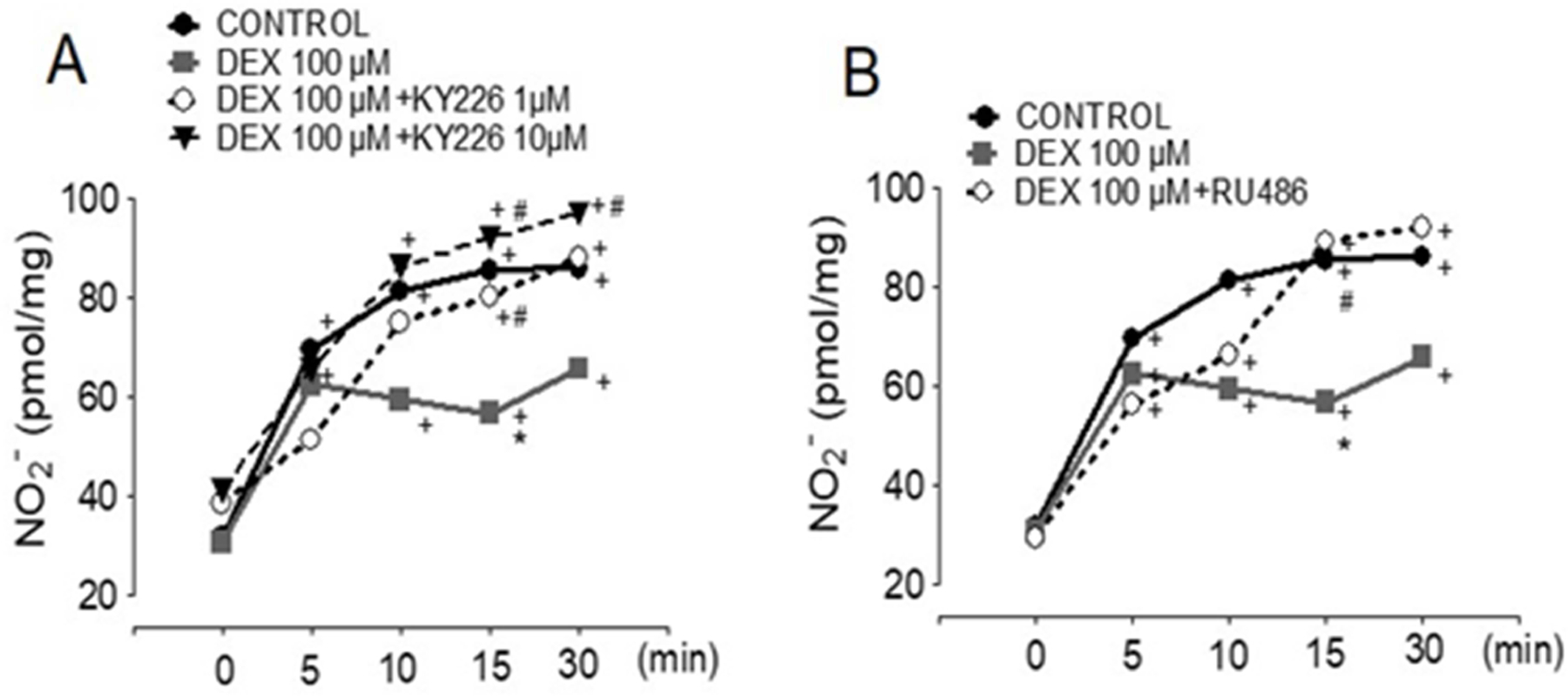
Controls are VEGF-stimulated BAECs. (A) KY226 dose-dependently rescued Dex (100 µM, 24 h) dependent NO reduction (B) The GR inhibitor (RU486, 10 µM) re-established NO release in Dex (100 µM, 24 h) treated cells. Values represent the mean±SEM from 3‒10 independent replicates. +p<0.05 for VEGF stimulation, *p<0.05, VEFG+Dex, #p<0.05 for KY226+VEGF+Dex or RU486+VEGF+Dex.
Discussion
Understanding the mechanisms and determinant factors associated with Dex-induced hypertension is important to ameliorate clinical outcomes. Genetic factors such as polymorphisms of the GR are considered to play a role, but a close look at the different GR mutations revealed divergent physiological responses that preclude a simple analysis of the effects of GR mutations on Dex responses, including hypertension (24,25). Inbred animal models with identical genetic makeup eliminate GR genetic variations as a factor in the hypertensive mechanisms, making them suitable for use establishing the biochemistry of Dex-induced vascular effects. We and others have shown that Wistar rats develop hypertension following long-term (14 days) Dex treatment. We also showed that aerobic exercise training ameliorates hypertension and the loss of small blood vessels, which could explain the ameliorating effects on blood pressure (3–5).
In rats, Dex induces body weight loss (Table 1), which is the opposite of its effect in humans; however, like humans, rats have significant muscle waste (26). Aerobic exercise training prior to Dex treatment did not prevent muscle waste; however, it improved muscle capillary density in Dex-treated rats (1), suggesting that these events are governed by different mechanisms. To evaluate the possibility that exercise training improved microvascular metabolic function under the control of NAD-signaling pathways and mitochondrial function (27,28), in this study NAD and ATP were quantified in the tibialis anterior skeletal muscle in both trained and non-trained rats (sedentary). Although exercise-trained animals showed a positive change, this response was not influenced by Dex, suggesting that improved metabolic function by exercise training alone plays a role in maintaining capillary density. We speculated that the Dex-sedentary treated group would show significant loss of NAD and ATP, which would correlate with a decreased capillary density (Table 1); however, no changes in nucleotides were found between treated and non-treated animals (Fig. 1). Assessment of the direct effects of Dex on cultured endothelial cell metabolism also showed no changes in NAD or ATP (Fig. 2).
Previous studies by others have indicated that Dex treatment in a dose-dependent fashion reduces NO production from endothelial cells (29,30). A decline in the eNOS expression levels and tetrahydrobiopterin as well as enhanced caveolin-1 expression have been proposed to mediate the loss of NO production (30,31). In this study, Dex (1–100 µM) showed no effect on BAEC survival or on BH4 and BH2 levels, confirming that eNOS uncoupling and enhanced ROS production are not mechanisms of Dex-induced endothelial cell dysfunction. Reduced VEGF-stimulated eNOS activity, however, was shown only in cells treated with high concentrations of Dex. Other studies have found responses at low concentrations of Dex but using low serum conditions which may sensitize cells to Dex, or using other cell types (30,31). When bradykinin or shear stress was used as a stimulus, eNOS activity was not altered by Dex, indicating specific mechanisms of interference of Dex with the VEGF pathway. Additional experiments ruled out an effect of Dex on eNOS protein levels and confirmed increased Akt/eNOS phosphorylation in Dex-stimulated cells. Furthermore, we demonstrated that the GR inhibitor increases VEGF-stimulated eNOS phosphorylation and eNOS activity, in agreement with the notion that Dex interferes with downstream eNOS-activation pathways. We observed that inhibition of the GR receptor reproduced the rescue of NO production observed with the PTP1B inhibitor, which increases eNOS activity to control levels (Fig. 7). Prolonged NF-kB signaling has been shown in GR knockdown HUVECs, demonstrating that endothelial GR is an important regulator of protein phosphorylation with significant physiological functions, including, as shown here, endothelial dysfunction (32).
Figure 7.
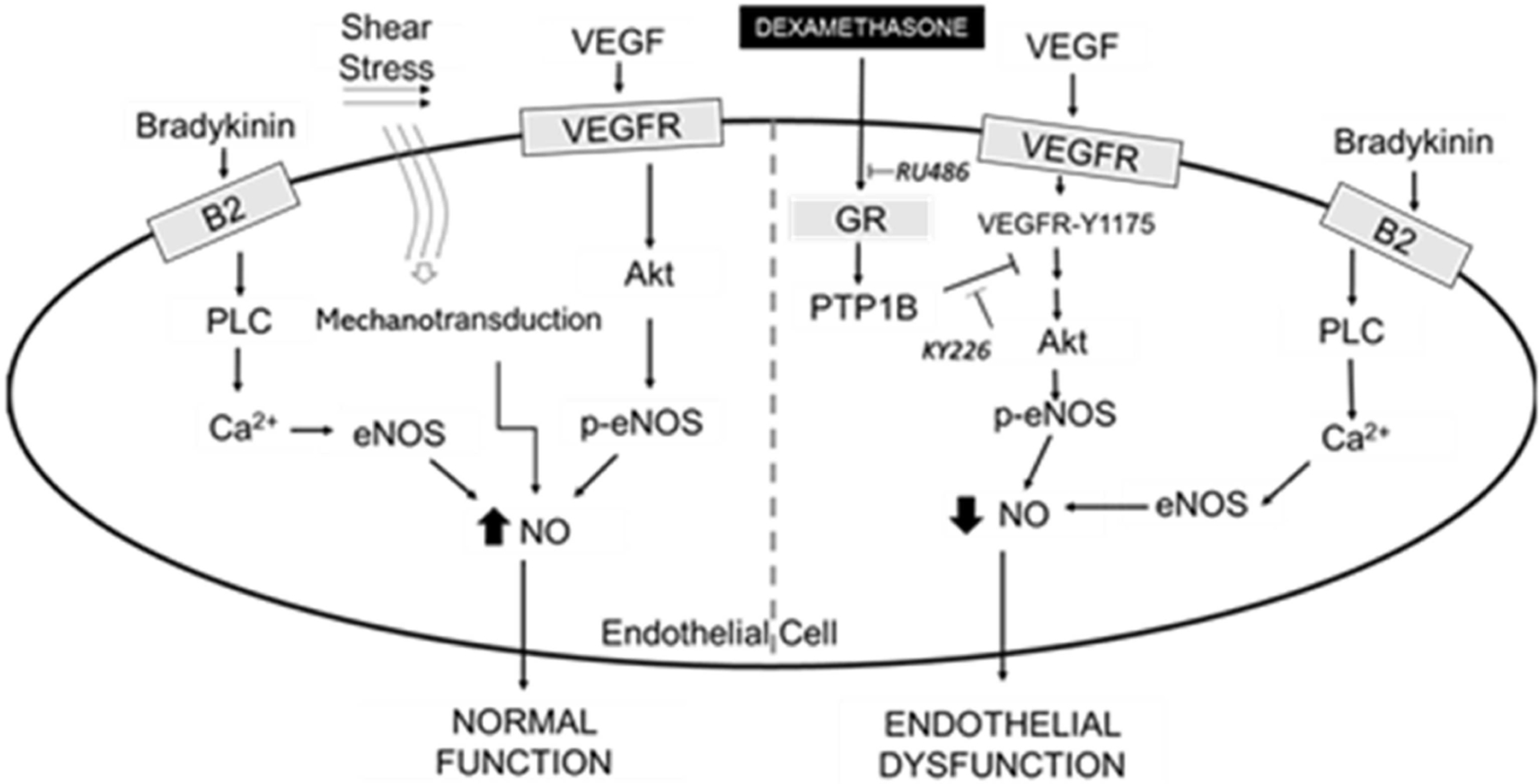
Glucocorticoid receptor mediates dexamethasone-induced endothelial dysfunction.
The key role of eNOS in the hypertensive effects of Dex has been supported by the observation that eNOS knockdown animals do not develop high blood pressure when receiving Dex (33). Our data indicate that Dex decreases NO production from VEGF but not bradykinin stimulated cells. This decrease is not transcriptionally dependent as no changes in eNOS or Akt protein levels were found. A sustained eNOS activation in Dex treatments was verified when blocking the GR receptor, which only occurs in the presence of Dex. Crosstalk between GR and PTP1B is a likely mechanism by which Dex diminishes NO since PTP1B inhibition also restores NO-release to control levels.
Because sustained NO production is a plausible mechanism preventing blood vessel rarefaction and enhancing angiogenesis (34), it would be interesting to establish the effects of endothelial-specific GR-knockdown on capillary density as well as examine the specific effects of exercise training ion GR activity, which could explain its positive hemodynamic effects. Recent evidence favoring this possibility comes from studies on the effects of acute exercise training, which has been shown to improve insulin sensitivity in the liver of aging Wistar rats by decreasing PTP1B protein level and enhancing insulin receptor signaling (35).
Highlights.
Dexamethasone (Dex) induces hypertension that is ameliorated by exercise training
Dex causes skeletal muscle (tibialis anterior) microvascular rarefaction
Dex does not modify NAD or ATP levels in vivo or in cultured endothelial cells
Dex alters VEGF but not bradykinin or shear stress-mediated NO activation
Glucocorticoid receptor or PTPB1 inhibition improves NO signaling in Dex-treatments
Acknowledgments
Funding statement
This study was supported by São Paulo Research Foundation, FAPESP #2017/00509–1 to SLA, FAPESP #2018/06998–7 and 2016/12532–5 to NAH, and FAPESP #2017/14405–3 and CAPES to FD, as well as by the National Institutes of Health under award R01 NS081936 to JVV.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Data availability
All data used to support the findings of this study are included in the article.
References
- 1.Dionisio TJ, Louzada JC, Viscelli BA, Dionisio EJ, Martuscelli AM, Barel M, Perez OA, Bosqueiro JR, Brozoski DT, Santos CF, and Amaral SL (2014) Aerobic training prevents dexamethasone-induced peripheral insulin resistance. Horm Metab Res 46, 484–489 [DOI] [PubMed] [Google Scholar]
- 2.Macedo AG, Krug AL, Herrera NA, Zago AS, Rush JW, and Amaral SL (2014) Low-intensity resistance training attenuates dexamethasone-induced atrophy in the flexor hallucis longus muscle. The Journal of steroid biochemistry and molecular biology 143, 357–364 [DOI] [PubMed] [Google Scholar]
- 3.Herrera NA, Jesus I, Shinohara AL, Dionisio TJ, Santos CF, and Amaral SL (2016) Exercise training attenuates dexamethasone-induced hypertension by improving autonomic balance to the heart, sympathetic vascular modulation and skeletal muscle microcirculation. J Hypertens 34, 1967–1976 [DOI] [PubMed] [Google Scholar]
- 4.Herrera NA, Jesus I, Dionisio EJ, Dionisio TJ, Santos CF, and Amaral SL (2017) Exercise Training Prevents Dexamethasone-induced Rarefaction. J Cardiovasc Pharmacol 70, 194–201 [DOI] [PubMed] [Google Scholar]
- 5.Constantino PB, Dionisio TJ, Duchatsch F, Herrera NA, Duarte JO, Santos CF, Crestani CC, and Amaral SL (2017) Exercise attenuates dexamethasone-induced hypertension through an improvement of baroreflex activity independently of the renin-angiotensin system. Steroids 128, 147–154 [DOI] [PubMed] [Google Scholar]
- 6.Vogt CJ, and Schmid-Schonbein GW (2001) Microvascular endothelial cell death and rarefaction in the glucocorticoid-induced hypertensive rat. Microcirculation (New York, N.Y. : 1994) 8, 129–139 [PubMed] [Google Scholar]
- 7.Gerber HP, Dixit V, and Ferrara N (1998) Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem 273, 13313–13316 [DOI] [PubMed] [Google Scholar]
- 8.Barel M, Perez OA, Giozzet VA, Rafacho A, Bosqueiro JR, and do Amaral SL (2010) Exercise training prevents hyperinsulinemia, muscular glycogen loss and muscle atrophy induced by dexamethasone treatment. European journal of applied physiology 108, 999–1007 [DOI] [PubMed] [Google Scholar]
- 9.Chen WL, Lin CT, Yao CC, Huang YH, Chou YB, Yin HS, and Hu FR (2006) In-vitro effects of dexamethasone on cellular proliferation, apoptosis, and Na+-K+-ATPase activity of bovine corneal endothelial cells. Ocular immunology and inflammation 14, 215–223 [DOI] [PubMed] [Google Scholar]
- 10.Logie JJ, Ali S, Marshall KM, Heck MM, Walker BR, and Hadoke PW (2010) Glucocorticoid-mediated inhibition of angiogenic changes in human endothelial cells is not caused by reductions in cell proliferation or migration. PLoS One 5, e14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Yin J, Ding H, Zhang C, and Gao YS (2016) Vitamin K2 Ameliorates Damage of Blood Vessels by Glucocorticoid: a Potential Mechanism for Its Protective Effects in Glucocorticoid-induced Osteonecrosis of the Femoral Head in a Rat Model. International journal of biological sciences 12, 776–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi D, Pulinilkunnil T, An D, Ghosh S, Abrahani A, Pospisilik JA, Brownsey R, Wambolt R, Allard M, and Rodrigues B (2004) Single-Dose Dexamethasone Induces Whole-Body Insulin Resistance and Alters Both Cardiac Fatty Acid and Carbohydrate Metabolism. Diabetes 53, 1790. [DOI] [PubMed] [Google Scholar]
- 13.Alabbood M, Ling M, and Ho K (2019) Effect of high-dose dexamethasone on patients without diabetes during elective neurosurgery: a prospective study. Diabetology international 10, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasieka AM, and Rafacho A (2016) Impact of Glucocorticoid Excess on Glucose Tolerance: Clinical and Preclinical Evidence. Metabolites 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zielińska KA, Van Moortel L, Opdenakker G, De Bosscher K, and Van den Steen PE (2016) Endothelial Response to Glucocorticoids in Inflammatory Diseases. Frontiers in Immunology 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, Schutz G, Moskowitz MA, and Liao JK (2002) Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest 110, 1729–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finck BN, and Kelly DP (2006) PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. The Journal of clinical investigation 116, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Repetto G, del Peso A, and Zurita JL (2008) Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nature protocols 3, 1125–1131 [DOI] [PubMed] [Google Scholar]
- 19.Piknova B, Park JW, Cassel KS, Gilliard CN, and Schechter AN (2016) Measuring Nitrite and Nitrate, Metabolites in the Nitric Oxide Pathway, in Biological Materials using the Chemiluminescence Method. Journal of visualized experiments : JoVE [DOI] [PMC free article] [PubMed]
- 20.Whitsett J, Picklo MJ Sr., and Vasquez-Vivar J (2007) 4-Hydroxy-2-nonenal increases superoxide anion radical in endothelial cells via stimulated GTP cyclohydrolase proteasomal degradation. Arterioscler Thromb Vasc Biol 27, 2340–2347 [DOI] [PubMed] [Google Scholar]
- 21.Silva GJ, Brum PC, Negrao CE, and Krieger EM (1997) Acute and chronic effects of exercise on baroreflexes in spontaneously hypertensive rats. Hypertension 30, 714–719 [DOI] [PubMed] [Google Scholar]
- 22.Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, and Simons M (2014) PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 130, 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Li Q, Youn JY, and Cai H (2017) Protein Phosphotyrosine Phosphatase 1B (PTP1B) in Calpain-dependent Feedback Regulation of Vascular Endothelial Growth Factor Receptor (VEGFR2) in Endothelial Cells: IMPLICATIONS IN VEGF-DEPENDENT ANGIOGENESIS AND DIABETIC WOUND HEALING. J Biol Chem 292, 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vitellius G, Trabado S, Bouligand J, Delemer B, and Lombes M (2018) Pathophysiology of Glucocorticoid Signaling. Annales d’endocrinologie 79, 98–106 [DOI] [PubMed] [Google Scholar]
- 25.Koper JW, van Rossum EFC, and van den Akker ELT (2014) Glucocorticoid receptor polymorphisms and haplotypes and their expression in health and disease. Steroids 92, 62–73 [DOI] [PubMed] [Google Scholar]
- 26.Salehian B, and Kejriwal K (1999) Glucocorticoid-induced muscle atrophy: mechanisms and therapeutic strategies. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists 5, 277–281 [DOI] [PubMed] [Google Scholar]
- 27.Yan Z, Lira VA, and Greene NP (2012) Exercise training-induced regulation of mitochondrial quality. Exercise and sport sciences reviews 40, 159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ziaaldini MM, Hosseini SR, and Fathi M (2017) Mitochondrial adaptations in aged skeletal muscle: effect of exercise training. Physiological research 66, 1–14 [DOI] [PubMed] [Google Scholar]
- 29.Limbourg FP, Huang Z, Plumier J-C, Simoncini T, Fujioka M, Tuckermann J, Schütz G, Moskowitz MA, and Liao JK (2002) Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. The Journal of clinical investigation 110, 1729–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tobias S, Habermeier A, Siuda D, Reifenberg G, Xia N, Closs EI, Forstermann U, and Li H (2015) Dexamethasone, tetrahydrobiopterin and uncoupling of endothelial nitric oxide synthase. Journal of geriatric cardiology : JGC 12, 528–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Igarashi J, Hashimoto T, Shoji K, Yoneda K, Tsukamoto I, Moriue T, Kubota Y, and Kosaka H (2013) Dexamethasone induces caveolin-1 in vascular endothelial cells: implications for attenuated responses to VEGF. Am J Physiol Cell Physiol 304, C790–800 [DOI] [PubMed] [Google Scholar]
- 32.Goodwin JE, Feng Y, Velazquez H, and Sessa WC (2013) Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A 110, 306–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallerath T, Godecke A, Molojavyi A, Li H, Schrader J, and Forstermann U (2004) Dexamethasone lacks effect on blood pressure in mice with a disrupted endothelial NO synthase gene. Nitric Oxide 10, 36–41 [DOI] [PubMed] [Google Scholar]
- 34.Gogiraju R, Schroeter MR, Bochenek ML, Hubert A, Munzel T, Hasenfuss G, and Schafer K (2016) Endothelial deletion of protein tyrosine phosphatase-1B protects against pressure overload-induced heart failure in mice. Cardiovasc Res 111, 204–216 [DOI] [PubMed] [Google Scholar]
- 35.de Moura LP, Souza Pauli LS, Cintra DE, de Souza CT, da Silva ASR, Marinho R, de Melo MAR, Ropelle ER, and Pauli JR (2013) Acute exercise decreases PTP-1B protein level and improves insulin signaling in the liver of old rats. Immunity & Ageing 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used to support the findings of this study are included in the article.