

Abstract
As a typical human pathogenic fungus, Cryptococcus neoformans is a life-threatening invasive fungal pathogen with a worldwide distribution causing ∼700,000 deaths annually. Cryptococcosis is not just an infection with multi-organ involvement, intracellular survival and extracellular multiplication of the fungus also play important roles in the pathogenesis of C. neoformans infections. Because adequate accumulation of drugs at target organs and cells is still difficult to achieve, an effective delivery strategy is desperately required to treat these infections. Here, we report a bioresponsive micro-to-nano (MTN) system that effectively clears the C. neoformans in vivo. This strategy is based on our in-depth study of the overexpression of matrix metalloproteinase 3 (MMP-3) in infectious microenvironments (IMEs) and secreted protein acidic and rich in cysteine (SPARC) in several associated target cells. In this MTN system, bovine serum albumin (BSA, a natural ligand of SPARC) was used for the preparation of nanoparticles (NPs), and then microspheres were constructed by conjugation with a special linker, which mainly consisted of a BSA-binding peptide and an MMP-3-responsive peptide. This MTN system was mechanically captured by the smallest capillaries of the lungs after intravenous injection, and then hydrolyzed into BSA NPs by MMP-3 in the IMEs. The NPs further targeted the lung tissue, brain and infected macrophages based on the overexpression of SPARC, reaching multiple targets and achieving efficient treatment. We have developed a size-tunable strategy where microspheres “shrink” to NPs in IMEs, which effectively combines active and passive targeting and may be especially powerful in the fight against complex fungal infections.
KEY WORDS: Albumin, SPARC, MMP-3, Size-tunable strategy, Microenvironment responsive, Complex fungal infection
Abbreviations: AmB, amphotericin B; BBB, blood‒brain barrier; BSA, bovine serum albumin; DDS, drug delivery system; IME, infectious microenvironment; MMP-3, matrix metalloproteinase 3; MTN, micro-to-nano; NP, nanoparticle; PEG, polyethylene glycol; PMVECs, pulmonary microvascular endothelial cells; RFP, red fluorescent protein; SPARC, secreted protein acidic and rich in cysteine
Graphical abstract
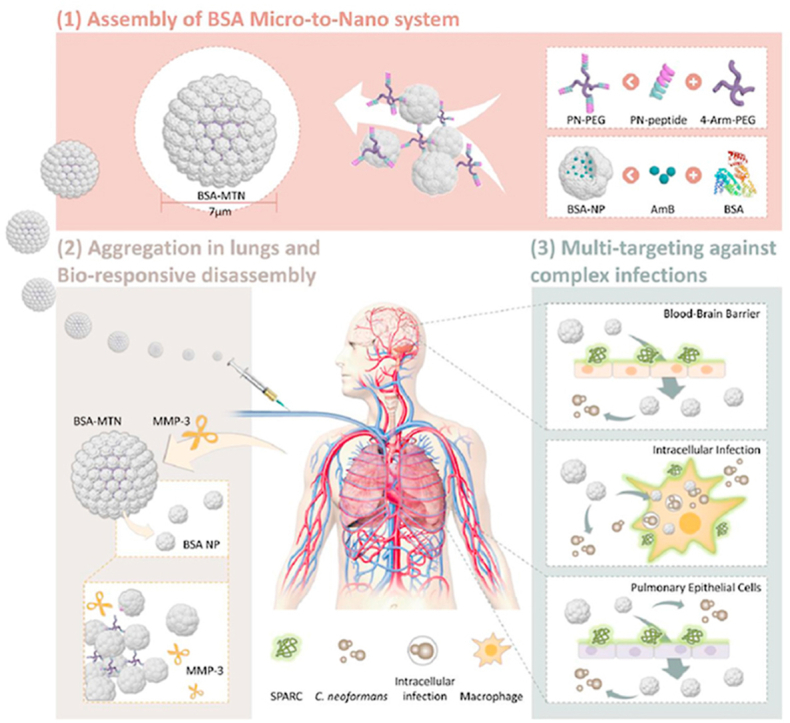
The bioresponsive micro-to-nano albumin-based system was accumulated in the lungs and responds to the upregulated MMP-3, then targeted the lung tissue, brain, and infected macrophages, reaching multiple targets and efficient treatment.
1. Introduction
In recent years, the incidence and mortality of invasive fungal infections (IFIs) have markedly increased worldwide. Patients with compromised immunity, such as those with acquired immunodeficiency syndrome (AIDS) and those who are undergoing chemotherapy, are vulnerable to fungal infections1. The high mortality of IFIs is associated with the lack of effective antifungal treatments. Clinically, there are only a few drugs for the treatment of IFIs, including amphotericin B (AmB), flucytosine, and fluconazole2. However, these antifungal drugs are limited by severe side effects, emerging drug resistance, and unavailability in countries with urgent demand. Thus, in addition to new drug development, an effective drug delivery system (DDS) will improve the therapeutic efficacy of existing antifungal agents and decrease toxicity. As a typical human pathogenic fungus, C. neoformans mainly invades through the respiratory tract, leading to pulmonary cryptococcosis3. If the pathogen spreads to the central nervous system, it will result in cryptococcal meningitis with over 700,000 deaths annually4. More seriously, C. neoformans is a facultative intracellular pathogen in pulmonary infections and can either lyse macrophages or escape from within them by a process called phagosomal extrusion, leading to disease recurrence5. This phenomenon is ubiquitous in all types of fungal infections. Therefore, effective elimination of infections caused by recalcitrant pathogens (such as C. neoformans) with antifungal drugs faces multiple physiological and pathological barriers. It is critically necessary to develop new IFI therapeutic agents with novel modes of action.
As a key characteristic of micro-/nano-DDSs, size greatly influences the efficiency of organ targeting in many ways, which include affecting the circulation, biodistribution, tissue accumulation and penetration, cellular uptake, and subcellular distribution6. Many studies have found that there is a close association between the treatment effect and size of a drug carrier7,8. Because of the structure and environment of tissues, there is a contradictory effect of nanoparticle (NP) size on drug delivery. Namely, large NPs tend to be more capable of retention in some tissue than small NPs9, 10, 11, 12. However, regarding permeability, the reverse is true, and smaller NPs can penetrate tissues to a greater extent. In recent years, an intelligent, size-tunable strategy has shown promise as a solution to the conundrum of the ideal drug carrier size13, 14, 15. Specifically, there are a series of stimulus-induced aggregation and shrinkage strategies for tumor-targeted drug delivery that can significantly increase the retention and penetration of nanodrugs simultaneously at tumor sites, thus promoting treatment efficacy. However, apart from in oncotherapy, the applications of size-tunable strategies to treat other diseases are scarce.
An ideal size-tunable system should be designed in response to the unique needs of a particular disease. For example, for lung diseases, both passive and active lung targeting have their own advantages and disadvantages. With passive targeting, microspheres of 7–30 μm can be rapidly concentrated in the lungs after intravenous administration due to mechanical filtration by the capillary beds. Drug carriers in this range of particle sizes can be quickly trapped and concentrated effectively in the pulmonary blood vessels, but drug efficacy is usually limited by the low permeability of these vessels16. When microspheres are deposited in the lung tissue, the loaded drugs are released and freely diffuse through the thin alveolar-capillary barrier into the alveolar space. However, the particle size and degradation rate of microspheres must be carefully regulated to ensure efficient clearance from the lung, avoiding embolization-related toxicity, such as inflammatory cell recruitment leading to local inflammation17,18. With active targeting ligand–receptor interactions could enhance penetration of the pulmonary vessels or increase the affinity of the preparation for lung lesions, enhancing the permeability of the pulmonary vessels through receptor-mediated endocytosis or by phagocytosis19. Nonetheless, the therapeutic effect is limited by the initial distribution of the drug in the lung. Therefore, the combination of these two targeting strategies may produce better targeting efficiency. This size-tunable strategy not only increases the drug concentration in the lungs by passive targeting, but also enhances the ability of the drug carriers to penetrate the pulmonary blood vessels through active targeting, thereby achieving better targeted delivery to the disease site.
Herein, we designed, constructed and tested a microenvironment-responsive micro-to-nano (MTN) albumin delivery system for complex fungal infections (see Fig. 1). 1) We found that matrix metalloproteinase 3 (MMP-3) is highly expressed in infectious microenvironments (IMEs), and secreted protein acidic and rich in cysteine (SPARC) is overexpressed in infected macrophages as well as lung and brain microvascular endothelial cells. 2) Since albumin is a good drug carrier material and natural ligand of SPARC, it was chosen as the carrier material, AmB was chosen as the model drug. Then, AmB-loaded bovine serum albumin (BSA) NPs were prepared. 3) To further assemble the NPs into microspheres, we constructed a special, dual-function linker (PN-PEG) that was able to bind BSA and was MMP-3 responsive. 4) The linker was incubated with the NPs, using particle size as an index, and the MTN system was successfully prepared by optimizing the NP/PN-PEG ratio. We expected that this responsive delivery system could be quickly trapped in the lungs through passive targeting, followed by cleavage of the connecting arm due to the upregulated MMP-3 in the IMEs. The NPs would then target cells or tissues that highly express SPARC, achieving multiplex targeting and efficient treatment. These expectations were tested both with in vitro cultured cells and in vivo mouse models.
Figure 1.

Schematic of IME-responsive construction of MTN albumin-based systems for targeted drug delivery against complex fungal infections. IME, infectious microenvironment; MTN, micro-to-nano; BSA, bovine serum albumin; PN, BSA-binding MMP-3-responsive peptide; PEG, polyethylene glycol; NP, nanoparticle; AmB, amphotericin B; MMP-3, matrix metalloproteinase 3; SPARC, secreted protein acidic and rich in cysteine; C. neoformans, Cryptococcus neoformans.
2. Materials and methods
2.1. Materials
4-arm-PEG10,000-MAL was purchased from Beijing JenKem Technology Co., Ltd. (Beijing, China). Amphotericin B (AmB) was purchased from Macklin Biochemical Co., Ltd. (Shanghai, China). Coumarin-6 (C6) was ordered from Aladdin Co., Ltd. (Shanghai, China). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbo cyanine iodide (DiR) was supplied by Invitrogen (Carlsbad, CA, USA). Rabbit anti-SPARC antibody, MMP-2 antibody, MMP-3 antibody, MMP-9 antibody, Alexor Fluo 488 labeled mouse anti-rabbit and HRP labeled goat anti-rabbit were obtained from Bioss biotechnology Co., Ltd. (Beijing, China). All other reagents were analytical grade.
2.2. Cell culture and animals
RAW 264.7 macrophage cells and bEnd3 cells were provided by Cell Bank of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; KeyGEN, China) containing 10% fetal bovine serum (FBS; Gibco, USA), 80 U/mL penicillin and 0.08 mg/mL streptomycin. All cells were kept at 37 °C in a humidified incubator with 5% CO2. C. neoformans var. grubii (serotype A) strain H99 was acquired from American Type Culture Collection (ATCC, USA) and grown in nutrient-rich yeast extract-peptone-dextrose (YPD) medium at 30 °C.
Female BALB/c mice (4 weeks, 18–22 g) and Sprague–Dawley (SD) rats (8 weeks, 180–220 g) were purchased from Chongqing Academy of Chinese Materia Medica (Chongqing, China) and raised in specific pathogen free (SPF) environment, housed on a 12 h light/dark cycle at ambient 22–24 °C with 30%–50% relative humidity. All animal experimental protocols (yxy201823) were reviewed and approved by the Experimental Animal Ethics Committee of College of Pharmaceutical Sciences, Southwest University.
2.3. Surface plasmon resonance (SPR) measurements
The interaction between the peptide and BSA was quantified by SPR (Nicoya Lifescience, Waterloo, Canada) at 25 °C with a constant flow rate of 20 μL/min. To immobilize BSA on the chip, 100 μL of 0.02 mol/L NHS and 0.1 mol/L EDC dissolved in activation buffer (Nicoya Lifescience) were injected to activate the carboxyl group, and BSA was suspended in HBS running buffer at 5 mg/mL. A pulse of 50 mmol/L cholamine was injected to quench redundant activated carboxyl groups. To validate the affinity between ligand and BSA, the peptide of different concentrations was injected at a flow rate of 20 μL/min with PBS. All concentrations were performed in triplicate and after each test the chips were regenerated by injection of 200 μL of 10 mmol/L HCl.
2.4. Preparation and characterization of BSA NP/AmB
NP was prepared by emulsion-solvent evaporation20,21. Briefly, 10 mg BSA was dissolved in 1 mL 0.9% NaCl, 0.5 mg AmB was dissolved in 25 μL dimethyl sulfoxide (DMSO) and 175 μL of a chloroform and methyl alcohol mixed solvent (4:1, v/v). Then, 1 mL of BSA was mixed with 200 μL AmB (0.5 mg) to sonicate for 5 min using a bath sonicator at a power of 400 W. The resulting NP was hardened during solvent evaporation for 3 h, and free AmB was removed by gel filtration on a Sephadex G-50 column.
2.5. In vitro interaction of NP with RAW264.7 cells
For in vitro interaction analysis, cells from the macrophage-like cell line RAW264.7 were diluted to 1 × 104 cells/mL and seeded into Cell Carrier Ultra 96-well culture plates (PerkinElmer) at a volume of 0.2 mL per well. Following overnight culture, C. neoformans were collected, washed twice with PBS, and opsonized with 20% mouse complement for 1 h at 37 °C and 5% CO222. Cryptococcal cells (1 × 105) were added to each well and cultured for 2 h. Extracellular cryptococcal cells were then removed by three washes with PBS. Next, 2.5 μg/mL coumarin-6-labeled NP was added and incubated for 2 h. Images were obtained for observation using an Operetta CLS high-content screen imaging system (HCS; PerkinElmer, USA).
2.6. Isolation of pulmonary microvascular endothelial cells (PMVECs)
The explant method was used to isolate and culture mouse PMVECs23. After anesthesia with 10% phenobarbital, the lungs of the mice were collected. Then, the peripheral lung tissue was cut into 1 mm3 pieces, rinsed with serum-free medium three times, and evenly placed into 25 cm2 plastic culture bottles. After incubation for 2 h, 2 mL of DMEM containing 20% fetal bovine serum, 90 U/mL heparin sodium, and 50 μg/mL endothelial cell growth supplement (ECGS) was added for static culture. The tissue pieces were carefully removed after 60 h, and 2 mL of culture medium was added to continue the culture. Half of the medium was changed the next day. Cell morphology was observed under a light microscope.
2.7. Cell protein level detection by immunofluorescence
Cells were infected as described above and fixed with 4% paraformaldehyde for 30 min, blocked with 5% BSA and incubated overnight at 4 °C with anti-SPARC antibody, then subsequently stained with Alexa Fluor 488-conjugated secondary antibody for 1 h at room temperature, and followed by DAPI (4ʹ,6-diamidino-2-phenylindole) staining. Imaging analysis was processed under an Operetta CLS HCS (PerkinElmer).
2.8. Conjugation of peptide with 4-arm-PEG10000-Mal
The amino acid sequences of the BSA-binding peptide and MMP-3-responsive peptide (NFF-3) were integrated into a new single peptide and selected as the linker, named the PN peptide. The PN peptide was conjugated to 4-arm-PEG10,000-MAL (PEG-Mal) by a nucleophilic substitution reaction to obtain the targeting compound PN-PEG. Briefly, the PN peptide and PEG-Mal (molar ratio 5:1) were dissolved in freshly distilled dimethyl sulfoxide (DMSO) and phosphate-buffered saline (PBS), respectively. After 18 h of incubation at room temperature, the reaction mixture was placed in a dialysis bag with a cut-off molecular weight (MW) of 8000–14,000 Da and dialyzed against deionized water for 48 h to remove the unreacted ligand24. Then, the solution was lyophilized and stored at −20 °C. The conjugation of the PN peptide with PEG-Mal was confirmed using a matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer (Bruker Daltonics, Germany). Acetonitrile:water (7:3) with 0.1% trifluoroacetate was used as the solvent, and 10 mg/mL of α-cyano-4-hydroxycinnamic acid was used as the matrix25.
2.9. Preparation and characterization of BSA MTN
To generate NP bound to PN-PEG, PN-PEG was first dissolved in 0.9% NaCl. Next, 4 mL of a 0.5 mg/mL NP solution was added to 1 mL of a PN-PEG (0.5 mg/mL) with stirring, incubated for 30 min at 37 °C. Size distribution was measured by Zeta sizer Nano ZS (Malvern Panalytical, UK) at 25 °C, and the samples were imaged using scanning electron microscopy (SEM).
2.10. Establishment of BALB/c mice model infected with C. neoformans
Cells of C. neoformans wild-type H99 strain or the RFP-expressing C. neoformans strain used for infection were cultured in YPD medium overnight at 30 °C. The medium was removed, and the cells were washed three times with PBS and diluted to 5 × 106 colony-forming units (CFU) per mL (CFU/mL). Then BALB/c mice were inoculated with 20 μL of the C. neoformans suspension via intranasal instillation to establish the infection model.
2.11. In vivo and ex vivo imaging of BSA MTN in infected mice
For in vivo and ex vivo real-time tracking, BSA NP, BSA MP, and BSA MTN were labeled with Near-infrared DiR fluorescent probe. After 48-h infection, the C. neoformans-bearing mice were injected with the preparations via the tail vein. In vivo images were monitored using an FX pro in vivo imaging system (Carestream, USA) at predetermined time points (1, 4, 8, 12, and 24 h). For ex vivo images, mice were sacrificed at 8 h post-administration, and the major organs (brain, heart, liver, spleen, lung, and kidney) were carefully collected, rinsed with cold PBS, and observed.
2.12. In vivo pharmacodynamics efficacy
2.12.1. Survival study
After 24-h infection, model mice were randomly divided into four groups and treated intravenously with saline, BSA NP/AmB, BSA MP/AmB, and BSA MTN/AmB respectively, with a single dose (AmB concentration 2 mg/kg). The survival rates for all mice were monitored for 42 days after treatment.
2.12.2. Tissue burden study
Mice were infected as described above for the infection model. After 24-h infection, infected mice were treated with saline, BSA NP/AmB, BSA MP/AmB, and BSA MTN/AmB at 2 mg/kg of AmB respectively. On Days 3 and 7 after treatment, mice were sacrificed, and then the lung and brain tissues from each group were carefully collected, weighed, and homogenized with sterile saline. The homogenate was continuously diluted by sterile saline, and then 50 μL of the suspension was inoculated onto YPD medium with 1% penicillin–streptomycin (1:1, v/v) and incubated at 30 °C for 48 h to determine CFU/g of lung tissue and CFU/g of brain tissue.
2.12.3. MRI assay
On the tenth day after treatment, animals were anaesthetized with isoflurane gas and magnetic resonance imaging (MRI) examinations were carried out at a 1.0 T MRI scanner (NM42-040H-I, Niumag, China), and the mean signal intensity in the lung area was measured by Image-Pro Plus.
2.13. Statistical analysis
Multiple group comparisons were conducted using one-way analysis of variance (ANOVA). All data were analyzed using IBM SPSS Statistics 20.0. All data are presented as the mean ± SD. Differences were considered significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
3. Results and discussion
3.1. Design and characterization of the MTN system
One of the major design considerations of the MTN system is the ability to transform between NPs and microspheres. The linker lies at the core of this system. This not only ensures that the albumin NPs form stable microspheres, but also allows the microspheres to respond quickly and transform into NPs in the microenvironment of the lesion. The linker consists of two parts, a ligand, which is a polypeptide fragment that binds to the albumin NPs and responds quickly to MMP-3, and a multi-armed PEG. In the ligand, the BSA-binding peptide (PGNLALRPDSNS) was identified by screening random peptide libraries. As displayed in Fig. 2A, this peptide specifically interacted with BSA at a binding constant (Kd) of 2.55 × 10−7 mol/L, while a scrambled peptide served as a negative control (Supporting Information Fig. S1). Further investigation through molecular docking indicated that the peptide favorably interacts with BSA. As shown in Fig. 2B and Supporting Information Fig. S2, the peptide occupies a wide cavity surrounded by the following amino acids: Asp111, Leu115, Lys114, Glu519, Lys523, Thr518, Glu424, and Arg458. And it was observed that it formed strong hydrogen-bond interactions with these residues. The other peptide was the MMP-3-responsive peptide NFF-3 (Arg-Pro-Lys-Pro-Val-Glu-Nva-Trp-Arg-Lys-NH2)26. The combination of (7-methoxycoumarin-4-yl)-acetyl (Mca) fluorophore and 2,4-dinitrophenyl (Dnp) quencher was used to fluorescently label NFF-3 (Mca-Arg-Pro-Lys-Pro-Val-Glu-Nva-Trp-Arg-Lys(Dnp)-NH2). We then used the specific fluorescent peptide Mca-Arg-Pro-Lys-Pro-Gln as a standard and measured the hydrolysis rate of the labeled NFF-3 in the presence of MMP-2, MMP-3, or MMP-9 by continuous fluorescence monitoring. Accordingly, the NFF-3 was rapidly hydrolyzed by MMP-3, but very slowly hydrolyzed by MMP-2 and MMP-9 (Fig. 2C). Based on these findings, we integrated the amino acid sequence of the BSA-binding and NFF-3 peptides into a new single peptide and named it PN (Supporting Information Fig. S3). Multi-armed PEG is a star-like structure carrying multi-hydroxyl or functional groups, which are nontoxic, non-immunogenic, non-antigenic and amphiphilic27. Initially, this technology was mainly used with macromolecular drugs, such as proteins and enzymes, and conjugation with multi-armed PEG has been approved by the FDA for the treatment of related diseases28. Among these multi-armed PEG, 4-arm-PEG is the most widely used. The successful use of 4-arm PEG macromolecular drugs has promoted the application of this technology to small molecule drugs to address therapy-related deficiencies, such as their potential poor solubility, high toxicity, undesirable pharmaceutical characteristics, and nonspecific biodistribution profiles. To date, several promising agents using 4-arm-PEG conjugated with small molecules have entered clinical trials, such as NKTR-102 (PEG-irinotecan), EZN-2208 (PEG-SN38), and NKTR-105 (PEG-docetaxel). In addition, 4-arm PEG can be combined with NPs for tissue engineering29. It is worth noting that the average molecular weight of PEG can impact the formation of the PEG micro-forms; PEG with an average molecular weight of 10 kDa can form microspheres with good shape and good mechanical properties30. In order to effectively assemble the NPs into microspheres, we chose 4-arm-PEG10,000-MAL (4-arm-PEG-Mal) as a structural carrier, and the successful attachment of the PN-PEG was verified by MALDI-TOF mass spectrometry (Fig. 2D).
Figure 2.

(A) Surface plasmon resonance (SPR) response analysis of the affinity between PN and BSA using BSA concentrations of 40–120 nmol/L. (B) Three-dimensional ligand–protein interaction mode for the binding site of BSA (PDB ID: 3V03) with the peptide. The active site residues are shown in cyan-colored stick form, the peptide is color-coded green, and the hydrogen-bond network with protein residues is represented as red dotted lines. (C) Continuous fluorometric assay of NFF-3 with (a) MMP-3, (b) MMP-2, and (c) MMP-9. (D) MALDI-TOF MS analysis of 4-arm-PEG10,000-MAL and PN-PEG (m/z). (E) Schematic representation of the construction of BSA MTN. MTN, micro-to-nano; BSA, bovine serum albumin; PN, BSA-binding MMP-3-responsive peptide; PEG, polyethylene glycol; NP, nanoparticle; AmB, amphotericin B; MMP, matrix metalloproteinase.
Since AmB is a preferred antifungal drug for the treatment of invasive fungal infections, it was chosen as the model drug. AmB-loaded BSA NPs (BSA NP/AmB) were prepared to be incubated with the special linker (Fig. 2E). Through optimizing the ratio of NP/PN-PEG, we found that the average particle size of the preparation changed from 115 nm to 7 μm when the ratio reached 4/1 (Supporting Information Figs. S4 and S5), and the preparations exhibited good stability (Supporting Information Fig. S6). More importantly, there was no apparent change in entrapment efficiency when the BSA NP/AmB was assembled into BSA MTN/AmB (91.13 ± 2.5% vs. 91.09 ± 1.3%), and the sustained release behavior of the BSA MTN/AmB was more favorable than that of the bare BSA NP/AmB (Supporting Information Fig. S7). These results clearly illustrate the successful preparation of the MTN system, in which the assembly process is efficient, gentle, and does not impact drug loading.
3.2. In vitro response and targeting of the MTN system
Having successfully constructed the MTN system, we first investigated the responsiveness of BSA MTN/AmB in vitro. After incubation with MMP-3 (0.15 μmol/L) at 37 °C, the average size of the BSA MTN/AmB decreased from 7 μm to 115 nm within 2 h, which was further confirmed by scanning electron microscope (SEM) analysis (Fig. 3B and Supporting Information Fig. S8). As a control, the BSA MP/AmB was prepared in the same way, except that the MMP-3-responsive peptide was replaced by a scrambled peptide. As shown in Supporting Information Fig. S9, the BSA MP/AmB could not be reduced in size by MMP-3. These results demonstrate that the size change triggered by MMP-3 occurred efficiently, and this responsiveness was specific to the MTN system.
Figure 3.

(A) Schematic representation of BSA MTN/AmB changes in the presence of MMP-3. (B) Dynamic light scattering and scanning electron microscopy visualization of the BSA MTN/AmB changes in the presence of MMP-3 at different time points. Scale bar = 5 μm. (C) Fluorescence imaging was used to observe the intracellular interaction between BSA NPs and normal/infected cells. RFP-tagged C. neoformans was used for intracellular infection. NPs were labeled with coumarin-6 (scale bar = 5 μm). (D) Analysis of the uptake of coumarin-6-loaded BSA NPs in normal/infected PMVECs. (E) Schematic illustration of the in vitro BBB model. (F) FACS analysis of uptake of normal/infected bEnd3 cells. RFP, red fluorescent protein; BBB, blood‒brain barrier; PMVECs, pulmonary microvascular endothelial cells; DAPI, 4′,6-diamidino-2-phenylindole.
Next, we explored the cellular uptake of the “shrinkable” BSA MTN in several potential target cells. C. neoformans is a facultative intracellular pathogen in pulmonary infections31, its interactions with macrophages are key events in determining the outcome of infection. However, phagocytosis is not always accompanied by fungal killing, since C. neoformans has a suite of traits that allow it to survive phagocytosis by macrophages and even proliferate within and escape from phagolysosomes32. There is an intracellular route through which macrophages can facilitate C. neoformans dissemination from the lungs and potentially cross the blood‒brain barrier (BBB) to cause devastating meningitis even with aggressive antifungal drug therapy33, resulting in high mortality and morbidity. The severity of this intracellular infection is the reason why current clinical treatment mainly involves increasing drug dosage. However, high-dose medication may cause toxic side effects and even lead to drug resistance34. More seriously, in immunosuppressed patients, reactivation of infection is usually caused by intracellular C. neoformans within host macrophages, complicating treatment. Therefore, we first evaluated whether BSA NPs released from “shrinkable” BSA MTN could specifically bind to infected macrophages. To visualize this interaction, RAW264.7 cells (a macrophage-like cell line) were infected with complement-opsonized red fluorescent protein (RFP)-tagged C. neoformans strain, followed by incubation with coumarin-6-labeled NPs. After a 2-h incubation, significantly stronger fluorescence signals were observed in the infected cells than in the normal cells (Fig. 3C), and quantitative fluorescence-activated cell sorting (FACS) revealed a similar trend (Supporting Information Fig. S10). Further, we found that this targeting preference was due to the significant upregulation of SPARC by macrophages after infection (Supporting Information Fig. S11); the uptake of BSA NPs was decreased when the infected cells were blocked with anti-SPARC antibody or silenced with SPARC siRNA (Supporting Information Figs. S12 and S13). This suggests that overexpressed SPARC is beneficial for the cell-specific delivery of BSA NPs. In addition, we investigated the intracellular distribution of NPs in the macrophage-like cells. As shown in Supporting Information Figs. S14 and S15, the NP endocytic pathway involved clathrin coating, which is consistent with the entry pathway of albumin NPs described in other reports35.
The lungs and brain are the major organs involved in C. neoformans infections. After intravenous injection, the penetration of NPs relies on the corresponding vascular endothelial cells of the lungs and brain. Therefore, we constructed in vitro models. After the PMVECs were extracted and identified (Supporting Information Fig. S16), we performed a C. neoformans infection experiment in the same way as we had done with macrophages. As shown in Supporting Information Fig. S17, we found high expression of SPARC in infected PMVECs by immunofluorescence analysis, and the uptake efficiency of the infected PMVECs was significantly higher than that of uninfected PMVECs (Fig. 3D and Supporting Information Fig. S18). Monolayer culture using immortalized mouse brain endothelial cells (bEnd3) is a commonly used in vitro BBB model to study the brain delivery of NPs36,37. The C. neoformans and bEnd3 cells were cocultured to develop the in vitro infected models (Fig. 3E). We then examined the BBB penetration ability of the NPs. As demonstrated in Fig. 3F and Supporting Information Fig. S19, BSA NP showed substantial penetration in the cell culture model, which may be attributed to the high level of SPARC expression on the infected bEnd3 cells (Supporting Information Fig. S20). These results indicate that BSA MTN has good responsiveness to MMP-3 in vitro. At the same time, after being reduced to BSA NPs, they are able to specifically recognize several potential target cells, providing a solid foundation for the responsiveness and targeting of BSA MTN in vivo.
3.3. In vivo distribution and targeting of BSA MTN
The lung has a unique feature that 7–30 μm microspheres can be quickly trapped and concentrated effectively in its blood vessels through passive targeting. To verify whether the BSA MTN can be specifically accumulated in the lungs, we used a qualitative imaging assay to investigate the in vivo and ex vivo targeting efficiency of BSA MTN/Dir. Compared to BSA NP/DiR, the BSA MTN/DiR exhibited specific accumulation in both normal (Supporting Information Fig. S21) and infected lungs (Fig. 4A and Supporting Information Fig. S22). Based on the live imaging, we then collected and analyzed major organs, and found a similar pattern (Fig. 4B and Supporting Information Fig. S22). Using a quantitative drug distribution assay, the concentration of AmB in different organs of normal and model mice was determined by high-performance liquid chromatography (HPLC). At all sampling time points, we found that the BSA MTN/AmB group showed significantly higher drug accumulation in lung tissue than the BSA NP/AmB group did, especially at 4 h post-administration. Over 4000 ng of AmB per gram of tissue from BSA MTN was found in both normal (Supporting Information Fig. S23) and infected lungs (Fig. 4C), while NP exhibited less than 2000 ng of AmB per gram of tissue. In addition, 24 h post-administration, the blood concentration of the BSA MTN/AmB group was 2-fold higher than that of the BSA NP/AmB group (Supporting Information Fig. S24).
Figure 4.

In vivo (A) and ex vivo (B) images of BSA NP, BSA MP, and BSA MTN in infected mouse models assessed by live imaging. Mice used in experiments were infected with C. neoformans for 48 h and then tail-vein injected with DiR-loaded preparations. Ex vivo images of various tissues from infected mice were collected after mice were euthanized 8 h after injection. (C) In vivo distribution of BSA NP/AmB, BSA MP/AmB, and BSA MTN/AmB in the brain, heart, lungs, liver, spleen, and kidneys of infected mice at different time points. Data are presented as mean ± SD (n = 3). ∗P < 0.05, ∗∗P < 0.01. (D) MMP-3 protein expression in the lungs of normal and infected mice determined by immunofluorescence. C. neoformans were labeled with RFP and shown as red, pulmonary tissues were labeled with MMP-3 (green) (scale bar = 20 μm). (E) Immunofluorescence of the colocalization of SPARC and comarin-6-labeled NPs in infected mice (Scale bar = 20 μm). (F) Fluorescence imaging was used to observe the intracellular interaction between BSA NP and normal/infected cells in vivo. RFP-tagged C. neoformans was used for intracellular infection. NPs were labeled with coumarin-6 (scale bar = 5 μm).
Unlike the distribution in normal mouse lungs, the BSA MTN had good responsiveness in the lung microenvironment of infected mice. Because of the complexity of deep C. neoformans infections, we expected that the MTN system would not only accumulate in the lungs through passive targeting, but would also penetrate into the major involved tissues. Accordingly, we found that the BSA MTN distributed faster at different time points than did BSA MP (Supporting Information Fig. S25). The BSA MTN was no longer observed as micron-sized particles and completely disappeared from the pulmonary microvessels and penetrated into the tissues of infected mice 8 h after injection (Supporting Information Fig. S26). This may be explained by the high expression levels of MMP-3 in infected lungs (Fig. 4D and Supporting Information Fig. S27).
The “shrinkable” BSA MTN also showed good lung penetration and brain targeting. In addition to the passive targeting that played a major role in the mechanism responsible for preferential accumulation in the lungs, active-targeting mechanisms were also involved for albumin-based lung penetration and brain delivery, as demonstrated by the above mentioned cellular uptake study. We further explored the in vivo targeting behavior of the BSA MTN that “shrinks” to NPs via MMP-3 activity in IMEs. We found that both the lung and brain blood vessels highly expressed SPARC, which was demonstrated by clear colocalization of CD31 and SPARC (Supporting Information Fig. S28). More importantly, BSA NPs showed a significantly enhanced penetration in the infected lung and brain, and SPARC overlapped with the distribution of the NPs, demonstrating the importance of the SPARC-mediated uptake mechanism (Fig. 4E).
The “shrinkable” BSA MTN can even target infected macrophages. We extracted normal and infected mouse pulmonary alveolar macrophages (PAM) from bronchoalveolar lavage fluid (BALF). The results showed that the coumarin-6 fluorescence from the BSA NPs colocalized with the infected macrophages (Supporting Information Fig. S29), which could be attributed to high expression of SPARC (Fig. 4F). Thus, the MTN strategy is based on IMEs, in which MMP-3 is highly expressed. SPARC is overexpressed in macrophages, lung, and brain microvascular endothelial cells, indicating that SPARC-mediated delivery is an important mechanism for targeting IMEs. By combining passive and active targeting, this MMP-3-responsive MTN system can target multiple sites, which is consistent with our in vitro MTN studies.
3.4. Therapeutic efficacy of BSA MTN/AmB
The in vitro antifungal results demonstrated that the effect of BSA NP/AmB was superior to that of free AmB; free AmB did not show effective inhibition until its concentration exceeded 2 μg/mL, which was twice the concentration needed for the BSA NP/AmB (Fig. 5A). In addition, in vivo experiments demonstrated reduced C. neoformans infection as well as prolonged survival time with BSA MTN/AmB treatment. Four groups of infected mice (n = 10 per group) received two doses of saline solution or 2 mg/kg of preparation loaded with AmB. Within the 42-day observation period, we found that the survival rate of the mice treated with BSA MTN/AmB was significantly higher than that of the other treatment groups (Fig. 5B). Notably, the untreated negative control mice all died within 24 days, which is in stark contrast to the 80% survival rate of the mice treated with BSA MTN/AmB. As summarized in Fig. 5C and Supporting Information Figs. S30 and S31, compared with the BSA NP/AmB group, the colony-forming units (CFU) in the lungs and brain of the model animals were significantly reduced after BSA MTN/AmB treatment for 3 and 7 days. Magnetic resonance imaging (MRI) was performed to visualize the lung infection in vivo. As shown in Fig. 5D and Supporting Information Fig. S32, in the pulmonary lesions, we found that the untreated infected mice exhibited high signal intensities (white) on MRI due to inflammation, whereas the BSA MTN/AmB group exhibited no inflammatory signals. This implies a complete recovery in the latter group, which was superior to the results obtained from the BSA NP/AmB and BSA MP/AmB groups. Semi-quantitative measurements of the MRI signal intensities in the lung regions are shown in Supporting Information Fig. S33.
Figure 5.

(A) In vitro antifungal effect of free AmB, BSA NP/AmB, BSA MP/AmB and BSA MTN/AmB.Data are presented as mean ± SD (n = 3). (B) Survival rate of mice bearing C. neoformans after different treatments. ∗∗∗P < 0.001. (C) Colony-forming units (CFU) on Days 3 and 7 in the lungs of infected mouse models. Data are presented as mean ± SD (n = 3). ∗∗P < 0.01. (D) In vivo assessment by MRI of infection signals after different treatments. The infected part of the lungs is highlighted by a red rectangle. (E) PAS-stained slides prepared from cross-sections of lungs at Day 10 post-infection and observed by light microscopy. Cryptococci are indicated by black arrows (scale bar = 50 μm). (F) In vivo therapeutic effects of BSA NP/AmB, BSA MP/AmB, and BSA MTN/AmB treatments on intracellular C. neoformans. C. neoformans are shown in red due to RFP fluorescence (scale bar = 10 μm).
In addition, histopathological examination revealed that the lung tissue from untreated mice was heavily infiltrated by inflammatory cells with widened alveolar interstitial spaces and patchy hemorrhages, but the lungs from the mice treated with BSA MTN/AmB showed clear indications of recovery (Fig. 5E). To further investigate the antifungal effect of the preparations on the presence of intracellular C. neoformans, the free PAM of mice were collected from BALF and observed using fluorescence microscopy. As shown in Fig. 5F and Supporting Information Fig. S34, compared with that of the other groups, the BSA MTN/AmB showed an excellent therapeutic effect.
Moreover, the main physiological indexes and hematoxylin and eosin staining results demonstrated good compatibility of AmB therapy with the BSA MTN system (Supporting Information Figs. S35 and S36). We also measured insulin clearance rate to assess renal function of the mice 24 h post-poisoning38. BSA MTN/AmB showed no obvious renal toxicity when compared with the normal mice (Supporting Information Fig. S37). These results indicate that BSA MTN/AmB is highly biocompatible and safe, making it suitable for targeted drug delivery to treat complex C. neoformans infections.
Due to its high water solubility, low immunogenicity, nontoxicity, and biodegradability, albumin is emerging as a versatile protein carrier for targeted drug delivery. As a case in point, some albumin binding proteins are overexpressed on tumor cells and tumor vascular endothelial cells39,40, and these albumin-binding proteins can be exploited for efficient targeted drug delivery to tumors by simple formation of drug–albumin conjugates. Although albumin plays an increasing role as a drug carrier in clinical settings, the use of albumin as a versatile drug carrier has been underutilized. In our study, albumin served as both a drug carrier and targeting ligand. First, the AmB-loaded BSA NPs were assembled into microspheres through a special linker, thus increasing the concentration of the drug in the lungs by passive targeting. Then, the enzyme-responsive BSA MTN system was reduced to NPs in the IMEs, and upregulated SPARC on the target cells and tissues allowed BSA NP penetration through the pulmonary blood vessels and BBB, achieving multi-targeted drug delivery and efficacious treatment.
With the unprecedented progress in biomedical nanotechnology over the past few decades, conventional DDSs have evolved into smart DDSs with stimuli-responsive characteristics. In order to provide smart properties, some developed smart DDSs were designed with sophisticated structures and formulations, which are difficult to scale up for industrial production41. Therefore, design simplicity is still one of the key points for successful translation of these smart drug carriers. In our delivery system, albumin can serve as a drug carrier and targeting ligand, making drug delivery simple and efficient. In addition, our assembly process is efficient and gentle; thus, the constructed delivery system possesses good drug-loading capacity and the formulation is relatively stable, laying a solid foundation for the formulation's application. Moreover, intracellular invasion and pathogen survival in phagocytic cells are known to contribute to potential antibiotic resistance and the treatment failure of pathogen infections, such as those involving Mycobacterium tuberculosis42 and Staphylococcus aureus43. Therefore, clearing intracellular infections is also important. After pathogens invade macrophages, the infected macrophages are activated, and cytokine secretion and the expression of extracellular receptors changes dramatically. These changes can provide mechanistic directions for the design of preparations targeting intracellular infections. Our strategy can also be applied to treat intracellular and extracellular infections of these pathogens, achieving effective treatment and avoiding disease recurrence.
4. Conclusions
We successfully designed a new DDS, BSA MTN, which is responsive to MMP-3 in IMEs and combines passive and active targeting strategies against complex fungal infections. This system is based on our in-depth study of IMEs, where MMP-3 is highly expressed, and several associated target cells that overexpress SPARC. In addition, the full use of albumin plays an essential role, in which it can serve as both a drug carrier and targeting ligand, imbuing the constructed delivery system with good drug-loading capacity and efficiency. As a result, the DDS is well-balanced in terms of multi-targeting and structural complexity. In view of the fact that the lung is the core invasion organ for many infectious diseases, this study is expected to provide useful insights for treating many lung diseases, especially intracellular infections.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 82073789, and 81673376), the project for Innovative Research Group at Higher Educational Institutions in Chongqing (CXQT20006, China), Chongqing Postgraduate Research and Innovation Project (CYB20106, China) and CAS Interdiscipliary Innovation Team (China).
Author contributions
Chong Li and Liting Cheng designed the research. Liting Cheng, Miao-Miao Niu, Tong Yan, Zhongyi Ma, Kexin Huang, Ling Yang and Xin Zhong carried out the experiments and performed data analysis. Liting Cheng and Chong Li wrote and revised the manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.04.020.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Decote-Ricardo D., LaRocque-de-Freitas I.F., Rocha J.D.B., Nascimento D.O., Nunes M.P., Morrot A. Immunomodulatory role of capsular polysaccharides constituents of Cryptococcus neoformans. Front Med. 2019;6:129. doi: 10.3389/fmed.2019.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aversa F., Busca A., Candoni A., Cesaro S., Girmenia C., Luppi M. Liposomal amphotericin B (AmBisome) at beginning of its third decade of clinical use. J Chemother. 2017;29:131–143. doi: 10.1080/1120009X.2017.1306183. [DOI] [PubMed] [Google Scholar]
- 3.May R.C., Stone N.R., Wiesner D.L., Bicanic T., Nielsen K. Cryptococcus: from environmental saprophyte to global pathogen. Nat Rev Microbiol. 2016;14:106–117. doi: 10.1038/nrmicro.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z., Liu N., Tu J., Ji C., Han G., Sheng C. Discovery of simplified sampangine derivatives with potent antifungal activities against cryptococcal meningitis. ACS Infect Dis. 2019;5:1376–1384. doi: 10.1021/acsinfecdis.9b00086. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez M., Casadevall A. Cell-to-cell spread and massive vacuole formation after Cryptococcus neoformans infection of murine macrophages. BMC Immunol. 2007;8:16. doi: 10.1186/1471-2172-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu W., Liu R., Zhou Y., Gao H. Size-tunable strategies for a tumor targeted drug delivery system. ACS Cent Sci. 2020;6:100–116. doi: 10.1021/acscentsci.9b01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carnovale C., Bryant G., Shukla R., Bansal V. Size, shape and surface chemistry of nano-gold dictate its cellular interactions, uptake and toxicity. Prog Mater Sci. 2016;83:152–190. [Google Scholar]
- 8.Talamini L., Violatto M.B., Cai Q., Monopoli M.P., Kantner K., Krpetic Z. Influence of size and shape on the anatomical distribution of endotoxin-free gold nanoparticles. ACS Nano. 2017;11:5519–5529. doi: 10.1021/acsnano.7b00497. [DOI] [PubMed] [Google Scholar]
- 9.Sykes E.A., Chen J., Zheng G., Chan W.C.W. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8:5696–5706. doi: 10.1021/nn500299p. [DOI] [PubMed] [Google Scholar]
- 10.She W., Luo K., Zhang C., Wang G., Geng Y., Li L. The potential of self-assembled, pH-responsive nanoparticles of mPEGylated peptide dendron–doxorubicin conjugates for cancer therapy. Biomaterials. 2013;34:1613–1623. doi: 10.1016/j.biomaterials.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Perrault S.D., Walkey C., Jennings T., Fischer H.C., Chan W.C.W. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9:1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 12.Hou X., Shou C., He M., Xu J., Gao F. A combination of lighton gene expression system and tumor microenvironment-responsive nanoparticle delivery system for targeted breast cancer therapy. Acta Pharm Sin B. 2020;10:1741–1753. doi: 10.1016/j.apsb.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C.Y., Gao J., Wang Z. Bioresponsive nanoparticles targeted to infectious microenvironments for sepsis management. Adv Mater. 2018;30 doi: 10.1002/adma.201803618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Q., Sun W., Lu Y., Bomba H.N., Ye Y., Jiang T. Tumor microenvironment-mediated construction and deconstruction of extracellular drug-delivery depots. Nano Lett. 2016;16:1118–1126. doi: 10.1021/acs.nanolett.5b04343. [DOI] [PubMed] [Google Scholar]
- 15.Wy A., Ms C., Xc B., Hg A. Advances in aggregatable nanoparticles for tumor-targeted drug delivery. Chin Chem Lett. 2020;31:1366–1374. [Google Scholar]
- 16.Lu B., Zhang J.Q., Yang H. Lung-targeting microspheres of carboplatin. Int J Pharm. 2003;265:1–11. doi: 10.1016/s0378-5173(03)00332-6. [DOI] [PubMed] [Google Scholar]
- 17.Zagorski J., Debelak J., Gellar M., Watts J.A., Kline J.A. Chemokines accumulate in the lungs of rats with severe pulmonary embolism induced by polystyrene microspheres. J Immunol. 2003;171:5529–5536. doi: 10.4049/jimmunol.171.10.5529. [DOI] [PubMed] [Google Scholar]
- 18.Minami K., Okamoto K., Doi K., Harano K., Noiri E., Nakamura E. siRNA delivery targeting to the lung via agglutination-induced accumulation and clearance of cationic tetraamino fullerene. Sci Rep. 2014;4:4916. doi: 10.1038/srep04916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao P., Yin W., Wu A., Tang Y., Wang J., Pan Z. Dual-targeting to cancer cells and M2 macrophages via biomimetic delivery of mannosylated albumin nanoparticles for drug-resistant cancer therapy. Adv Funct Mater. 2017;27:1700403. [Google Scholar]
- 20.Lu C., Zahedi P., Forman A., Allen C. Multi-arm PEG/silica hydrogel for sustained ocular drug delivery. J Pharmacol Sci. 2014;103:216–226. doi: 10.1002/jps.23777. [DOI] [PubMed] [Google Scholar]
- 21.Phaechamud T., Tuntarawongsa S. Transformation of eutectic emulsion to nanosuspension fabricating with solvent evaporation and ultrasonication technique. Int J Nanomed. 2016;11:2855–2865. doi: 10.2147/IJN.S108355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang W., Liao G., Baker G.M., Wang Y., Lau R., Paderu P. Lipid flippase subunit CDC50 mediates drug resistance and virulence in Cryptococcus neoformans. mBio. 2016;7 doi: 10.1128/mBio.00478-16. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arora S., Olszewski M.A., Tsang T.M., McDonald R.A., Toews G.B., Huffnagle G.B. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect Immun. 2011;79:1915–1926. doi: 10.1128/IAI.01270-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z., Yu Y., Dai W., Lu J., Cui J., Wu H. The use of a tumor metastasis targeting peptide to deliver doxorubicin-containing liposomes to highly metastatic cancer. Biomaterials. 2012;33:8451–8460. doi: 10.1016/j.biomaterials.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 25.Roberts M.J., Bentley M.D., Harris J.M. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2012;64:116–127. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 26.Nagase H., Fields C.G., Fields G.B. Design and characterization of a fluorogenic substrate selectively hydrolyzed by stromelysin 1 (matrix metalloproteinase-3) J Biol Chem. 1994;269:20952–20957. [PubMed] [Google Scholar]
- 27.Brocchini S., Godwin A., Balan S., Choi J.W., Shaunak S. Disulfide bridge based PEGylation of proteins. Adv Drug Deliv Rev. 2008;60:3–12. doi: 10.1016/j.addr.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Li W., Zhan P., Clercq E.D. Current drug research on PEGylation with small molecular agents. Prog Polym Sci. 2013;38:421–444. [Google Scholar]
- 29.Fernandes-Cunha G.M., Chen K.M., Chen F., Le P., Han J.H., Mahajan L.A. In situ-forming collagen hydrogel crosslinked via multi-functional PEG as a matrix therapy for corneal defects. Sci Rep. 2020;10:312–319. doi: 10.1038/s41598-020-72978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuang Y., Shen H., Yang F., Wang X., Wu D. Synthesis and characterization of PLGA nanoparticle/4-arm-PEG hybrid hydrogels with controlled porous structures. RSC Adv. 2016;6:53804–53812. [Google Scholar]
- 31.Feldmesser M., Kress Y., Novikoff P., Casadevall A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect Immun. 2000;68:4225–4237. doi: 10.1128/iai.68.7.4225-4237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Leon-Rodriguez C.M., Rossi D.C.P., Fu M.S., Dragotakes Q., Coelho C., Guerrero Ros I. The outcome of the Cryptococcus neoformans‒macrophage interaction depends on phagolysosomal membrane integrity. J Immunol. 2018;201:583–603. doi: 10.4049/jimmunol.1700958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steven D., Jessica B. Mechanisms of pulmonary escape and dissemination by Cryptococcus neoformans. J Fungi. 2018;4:25. doi: 10.3390/jof4010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longo D., Fauci A., Kasper D., Hauser S., Jameson J., Loscalzo J. Harrison's principles of internal medicine. Practitioner. 2011;204:831–837. 18th Edition. [Google Scholar]
- 35.Wagner S., Zensi A., Wien S.L., Tschickardt S.E., Maier W., Vogel T. Uptake mechanism of ApoE-modified nanoparticles on brain capillary endothelial cells as a blood‒brain barrier model. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang X., Xin H., Ren Q., Gu J., Zhu L., Du F. Nanoparticles of 2-deoxy-d-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials. 2014;35:518–529. doi: 10.1016/j.biomaterials.2013.09.094. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y., Ran R., Chen J., Kuang Q., Tang J., Mei L. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials. 2014;35:4835–4847. doi: 10.1016/j.biomaterials.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 38.Li C., Xie Z., Chen Q., Zhang Y., Chu Y., Guo Q. Supramolecular hunter stationed on red blood cells for detoxification based on specific molecular recognition. ACS Nano. 2020;14:4950–4962. doi: 10.1021/acsnano.0c01119. [DOI] [PubMed] [Google Scholar]
- 39.Merlot A.M., Kalinowski D.S., Richardson D.R. Unraveling the mysteries of serum albumin-more than just a serum protein. Front Physiol. 2014;5:299. doi: 10.3389/fphys.2014.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin T., Zhao P., Jiang Y., Tang Y., Jin H., Pan Z. Blood–brain-barrier-penetrating albumin nanoparticles for biomimetic drug delivery via albumin-binding protein pathways for antiglioma therapy. ACS Nano. 2016;10:9999–10012. doi: 10.1021/acsnano.6b04268. [DOI] [PubMed] [Google Scholar]
- 41.Dong L., Fang Y., Fei X., Ning G. The smart drug delivery system and its clinical potential. Theranostics. 2016;6:1306–1323. doi: 10.7150/thno.14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Machelart A., Salzano G., Li X., Demars A., Debrie A.S., Menendez-Miranda M. Intrinsic antibacterial activity of nanoparticles made of β-cyclodextrins potentiates their effect as drug nanocarriers against tuberculosis. ACS Nano. 2019;13:3992–4007. doi: 10.1021/acsnano.8b07902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hussain S., Joo J., Kang J., Kim B., Braun G.B., She Z.G. Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy. Nat Biomed Eng. 2018;2:95–103. doi: 10.1038/s41551-017-0187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.