

Abstract
R-loops are naturally occurring transcriptional intermediates containing RNA/DNA hybrids. Excessive R-loops cause genomic instability, DNA damage, and replication stress. Senataxin-associated exonuclease (San1) is a protein that interacts with Senataxin (SETX), a helicase resolving R-loops. It remains unknown if R-loops-induced DNA damage plays a role in the heart, especially in the proliferative neonatal cardiomyocytes (CMs). San1−/− mice were generated using the CRISPR/Cas9 technique. The newborn San1−/− mice show no overt phenotype, but their hearts were smaller with larger, yet fewer CMs. CM proliferation was impaired with reduced cell cycle-related transcripts and proteins. S9.6 staining revealed that excessive R-loops accumulated in the nucleus of neonatal San1−/− CMs. Increased γH2AX staining on newborn and adult heart sections exhibited increased DNA damage. Similarly, San1−/− AC16-cardiomyocytes showed cumulative R-loops and DNA damage, leading to the activation of cell cycle checkpoint kinase ATR and PARP1 hyperactivity, arresting G2/M cell-cycle and CM proliferation. Together, the present study uncovers an essential role of San1 in resolving excessive R-loops that lead to DNA damage and repressing CM proliferation, providing new insights into a novel biological function of San1 in the neonatal heart. San1 may serve as a novel therapeutic target for the treatment of hypoplastic cardiac disorders.
Keywords: San1, R-loops, DNA damage, Cardiomyocytes proliferation, Heart development
1. Introduction
Among all functional organs, the heart forms first in the embryo [1]. Cardiomyocytes (CMs) are responsible for ensuring the heart pumps blood efficiently throughout the body in an organized and rhythmic manner. CMs are relatively limited in renewal after the early developmental stage [2,3]. CMs in the mammalian heart possess regenerative capacities within 7 days after birth and then become terminally differentiated. DNA damage increases because of oxidative stress derived from the oxygen-rich postnatal environment during the perinatal period, which may trigger cell cycle exit and subsequently impair cardiac development [4–8].
R-loops are non-B DNA structures composed of an RNA/DNA hybrid that form during transcription when nascent transcript reanneals to the template DNA, thus displacing the non-template DNA strand [9,10]. However, excessive R-loops cause transcription-replication conflicts and replication fork stalling, which leads to DNA strand breaks and G2/M phase cell cycle arrest. Intriguingly, replication stress potentially further amplifies R-loop-associated DNA damage [11,12]. R-loop-associated DNA damage causes replication stress and genomic instability. Human diseases, such as neurodegenerative disease and cancer, have been linked to excessive R-loop accumulation [9,10]. However, the role of R-loops-induced DNA damage in neonatal CMs development is yet to be investigated.
San1 was initially identified and characterized as a transcriptional coactivator of peroxisome proliferator-activated receptor γ (PPARγ) to promote PPARγ-dependent adipogenesis [13]. Alex et al. [14] discovered San1 was a 5′ exonuclease, which shares a homologous N-terminal domain with conserved nuclease structure of the Flap Structure-Specific Endonuclease 1 (FEN1) and functioned to cleave the 5′ end of single-strand DNA (ssDNA). San1 participated in repairing DNA interstrand cross-links (ICLs) by interacting with SETX. SETX is a crucial RNA/DNA helicase and can specifically resolve unscheduled R-loops, thus promoting genomic stability [15]. Interestingly, the abundance of R-loops in San1−/− cells was slightly increased even though the San1 may not directly resolve. San1−/− 293T and HeLa cells did not exhibit increased sensitivity to replication stress and inhibitory proliferative activity.
Herein, we report that San1 deletion in CMs led to excessive R-loop accumulation, inhibiting CM proliferation during neonatal heart development. The resulted DNA lesions compromised cell proliferation profiles and eventually led to cardiac morphologic abnormalities and dysfunction. Further investigation of San1−/− AC16 human CM cell lines confirmed that San1 deficiency results in increased RNA/DNA hybrids, DNA damage response (DDR), and disturbed cellular proliferative activity. In San1−/− AC16 CMs, DNA damage-associated hyperactivation of PARP1 led to excessive degradation of X-Ray Repair Cross Complementing 1 (XRCC1), a DNA break repair protein [16], and SETX through poly(ADP-ribose)-dependent ubiquitination (PARdU), which further increased the aggregation of R-loops and DNA damage, forming a vicious circle.
2. Materials and methods
2.1. Human heart samples
Heart failure samples were obtained from patients diagnosed with dilated cardiomyopathy (DCM) who received heart transplants. Donor hearts were procured from age-matched healthy subjects suffering traffic accidents. The left ventricle samples from both donors and heart failure patients were collected, immediately stored in liquid nitrogen and kept at −80 °C. All study participants signed an informed consent form. All protocols involving human samples were approved by the Institutional Ethics Committee of Tongji Hospital of Huazhong University of Science and Technology and conducted in accordance with the Declaration of Helsinki.
2.2. Animal
All animal experimental procedures were performed according to the Guideline for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication No. 85–23, revised 1996), and approved by the Care of Experimental Animals Committee of Huazhong University of Science and Technology, Wuhan, China (IACUC Number: [2016] 737). CRISPR/Cas-mediated genome engineering was conducted to create a San1−/− knockout C57BL/6 mouse. Cas9 vector and guide-RNA for murine San1 exon 2 were generated by in vitro transcription, followed by injected into fertilized eggs for knockout mouse production. The positive founders were genotyped by PCR and DNA sequencing analysis. The primers for genotyping San1−/− mice were 5′-tctattgtctctgttaaagtcccactgtag-3′ (forward) and 5′-aatcacgacattgtggaccgag-3′ (reverse).
2.3. Establishment of the San1 gene knockout AC16 cell line
To construct the human San1 knockout AC16 cell line, San1 gene targeting guide RNAs were got from Andrews et al. [14]. The gRNA oligomers were synthesized, sequences were as follows: 5′-gcaggataagagagatgaat-3′ (gRNA-1) and 5′-gagaagctctgtgagagtct-3′ (gRNA-2). Both gRNAs are designed to create DNA double stranded breaks on exon 1 of San1 gene. The gRNA sequences were introduced into the px459 vector (Addgene #62988). A total of 1.2 × 105 AC16 cells were then transfected with 2 μg of px459-San1 at 37 °C for 48 h using Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific.) followed by screening in DMEM containing 1 μg/milliliter puromycin for two weeks. Single cell clones were obtained through limited dilution method. Colonies derived from single cells were separated and genotyped by PCR and subsequent sequencing.
2.4. Transfection and establishment of stable transfectants
AC16 cells were grown to ~80% confluency on 12-well plates, and then changed into fresh medium containing 10% FBS and antibiotics. pcDNA3.1-RNase H1 (1 μg), pcDNA3.1-San1 (1 μg) and empty vector (1 μg) were diluted in 50ul serum-free DMEM, and then mixed with 3ul polyJet (SignaGen Laboratories) diluted in 50ul serum-free DMEM. The PolyJet/DNA mixture was incubated for 12 min at room temperature, and then added onto the medium in each well. Remove the medium containing PolyJet/DNA mixture and replace with complete medium 12 h post-transfection. The cells were used for western blotting and fluorescent imaging 48 h post-transfection. For screening of stable transfectants, the culture medium was replaced with DMEM containing G-418 (500 μg/ml) and 10% FBS 48 h after transfection, and the medium was replaced on alternate days for 2 weeks. Stable transfected clones were confirmed by western blotting.
2.5. Preparation of cell lysates and western blotting
Collected cells were lysed in ice-cold RIPA buffer (50 mM Tris–HCl, pH 7.4, 100 mM NaCl, 50 mM NaF, 5 mM EDTA, 40 mM b-glycerophosphate, 1% Triton X-100) containing protease and phosphatase inhibitor cocktail. For heart tissue, whole-cell extracts from freezing heart tissue samples were manufactured in identical RIPA lysis buffer by homogenization using a glass homogenizer. Cell lysates were centrifuged at 12,000g for 10 min at 4 °C, to remove insoluble elements. Protein concentrations were determined using the BCA assay and samples were denatured (95 °C, 10 min) after DTT and bromophenol blue were added. 10–40 μg of proteins was loaded per lane, separated on 6–15% SDS–PAGE, and transferred onto PVDF membranes (Amersham, GE Healthcare). Membranes were blocked with 5% skimmed milk dissolved in TBST buffer and incubated with relevant primary antibody overnight at 4 °C followed by washed in TBST buffer. Membranes were incubated with HRP-conjugated secondary antibodies at RT for 2 h followed by washed in TBST buffer. Blots were detected by chemiluminescence using ECL reagent. The intensities of individual bands were analyzed by Gel-Pro analyzer.
Antibodies used for western blotting were commercially acquired: phosphor-ATM-Ser1981 (Abclonal AP0008), phospho-H2A.X (Ser139) (Millipore 05–636), FLAG M2 (Sigma-Aldrich), San1 (Abcam ab106455), XRCC1 (Boster BA3670), SETX (Novus NBP1–94712), phospho-CHK1-Ser345 (Cell Signaling Technologies 133D3), phosphor-RPA32-Ser33 (Bethyl Laboratories A300–246A), phosphor-KAP1-Ser824 (Bethyl Laboratories A300–767A), PCNA (Boster BM0104), PARP1 (Cell Signaling Technologies 9532T), Poly (ADP-Ribose) Polymer (Abcam ab14459), KI67 (NOVUS NB500–170), Cyclin D1 (ABclonal A19038), MCM2 (Abcam ab108935), MCM3 (ABclonal A11475), MCM4 (ABclonal A9251),GFP-tag (Abclonal AE011), GAPDH (Abcam ab8245), alpha-Tubulin (Abcam ab729).
2.6. Comet assay
DNA strand breaks were analyzed using single cell electrophoresis (comet assay) according to the manufacturer’s instructions (Trevigen 4250–050-K). Cells were gently trypsinized and detached from flask surface, washed, and resuspended with ice-cold PBS (Ca++ and Mg++ free) to achieve a cell density of 1 × 105/ml. Solid LMAgarose was melted in boiling water for 5 min, and the molten agar was placed in 37 °C water for 20 min. Cells at a concentration of 1 × 105/ml were mixed with molten agar at a volume ratio of 1:10. Immediately, 50 μL of the compound was pipetted onto FLARE Slides and incubated for 30 min at 4 °C in the dark. The slides were immersed in 4 °C Lysis Solution overnight at 4 °C. For alkaline comet assay, slides were immersed in Alkaline Unwinding/Electrophoresis Solution (200 mM NaOH, 1 mM EDTA, pH > 13) for 1 h at 4 °C. Slides were then electrophoresed at a constant voltage of 1 Volt/cm (Adjust volume of electrophoresis buffer until the current is approximately 300 mA) for 30 min. Slides were immersed twice in demineralized H2O for 5 min each, then 70% ethanol for 5 min, air-dried and stained with ethidium bromide (20 μg/ml, 20ul/slide) for 20 min at 25 °C, and followed with imaging with a fluorescent microscope. For neutral comet assay, slides were immersed in Neutral Electrophoresis Solution (0.1 M Tris Base, 0.3 M NaAc, PH = 9.0) for 30 min at 4 °C. Slides were then electrophoresed at a constant voltage of 1 Volt/cm (Adjust volume of electrophoresis buffer until the current is approximately 300 mA) for 45 min. Slides were immersed in DNA Precipitation Solution (1 M NH4Ac, 80% EtOH) for 30 min at 25 °C, then in 70% EtOH for 30 min at 25 °C, air-dried and stained with ethidium bromide (20 μg/ml, 20ul/slide) for 20 min at 25 °C, and followed with imaging with a fluorescent microscope. DNA damage was quantified by scoring the comet tail moment and percent DNA in comet tail, using comet-score software.
2.7. Immunostaining
Heart tissue samples were harvested at the indicated phases and immediately fixed in 4% paraformaldehyde (PFA) in PBS for 24 h at room temperature (RT), followed by paraffin-embedding and processing for serial sectioning (5 μm). Paraffin sections were deparaffinized with xylene and rehydrated with gradient alcohol. Antigens were retrieved by boiling in sodium citrate buffer (10 mM sodium citrate, 0.05% tween 20, PH 6) for 20 min. Slides were permeabilized with 0.3% Triton in PBS for 15 min, blocked with incubation buffer (10% normal goat serum dissolved in TBST) for 1 h at RT, and incubated overnight with the relevant primary antibody at 4 °C. After washed in PBS, sections were then incubated with applicable luciferase labeled secondary antibody (dissolved in incubated buffer) for 1 h at RT in the dark. After washed in PBS, the slides were stained with DAPI and mounted with mounting medium for imaging under a Zeiss (AXIO IMAGER A2) fluorescence microscope.
Antibodies used for immunofluorescence were commercially acquired: Cardiac Troponin T (Abclonal A4914; Abcam ab8295), Ki67 (Novus NB500–170), phospho-H2A.X (Ser139) (Millipore 05–636), PCNA (Boster BM0104), S9.6 (Sigma-Aldrich MABE1095), and Nucleolin (Abcam ab129200).
Immunofluorescence detection of RNA/DNA hybrids using S9.6 antibody was performed according to the previously described method [17]. Briefly, isolated neonatal mice cardiomyocytes were cultured on a 12-well plate. After washed with PBS, the CMs were fixed with ice-cold methanol for 10 min at −20 °C and were then permeabilized with ice-cold acetone for 1 min at 20 °C. The fixed cells were washed 3 times with PBST (PBS containing 0.1 Tween-20), and then incubated with blocking buffer (PBST containing 3% Bovine Serum Albumin (BSA)) for 1 h at room temperature, followed by incubation with the primary antibody (S9.6: 1:200; Nucleolin: 1:500) overnight at 4 °C. After the CMs were washed 3 times with PBST, they were then incubated with luciferase labeled secondary antibody (dissolved in blocking buffer) for 1 h at room temperature in the dark. After washing with PBST 3 times, the nucleus was stained with DAPI for 5 min. The slides were then mounted with mounting medium for imaging under a Zeiss (LSM710/780) confocal microscope.
2.8. Detection of cell cycle profile using flow cytometry
Cells were trypsinized, fixed with ice-cold 75% ethanol in PBS and incubated at 4 °C for 4 h. Cell suspension was centrifuged and the supernatant was discarded. The cell pellets were resuspended and washed with 1 ml pre-cooled PBS. Cells were then treated with 500ul dyeing working buffer (Meilunbio MA0334) (dyeing buffer: propidium iodide: RNase A = 100: 5: 2) for 30 min at 37 °C in the dark. The cell suspension was filtered through a 200 nylon mesh prior to examination by flow cytometry at 488 nm wavelength.
2.9. Cell growth curves
200,000 AC16 Human Cardiomyocytes (WT, San1−/−, San1−/− + San1, San1−/− + RNH1) were seeded into a 6-well plate (time 0). At the indicated times (24 h, 36 h, 48 h, 60 h), cells were trypsinized, resuspended with PBS, and counted with cell counting chamber. Growth curves were plotted using data from 4 independent biological replicates.
2.10. Real-time quantitative PCR
Total RNAs were extracted from cultured cells and heart tissue using TRIzol (Qiagen) reagent according to the manufacturer’s instructions. The concentration of RNA isolated was measured using an Ultramicro nucleic acid analyzer (Nano-200). Reverse transcription was carried out following particular temperature-time cycling procedure with 1 μg total RNA, RT buffer, primer mix(random primers and oligo dT primer) and primerscript enzyme mix, after digestion with gDNA Eraser at 42 °C for 2 min (TaKaRa). Real-time quantitative reverse transcriptase-PCR (qRT-PCR) was performed on 7900HT Fast Real-Time PCR System (Applied Biosystems) in 384-well plate format, after equal amounts of cDNA were mixed with TB Green (TaKaRa) and the specific primer pairs. The relative expression levels of target gene were measured by normalization to internal control reference genes (GAPDH and β-actin), using SDS2.4 software.
2.11. Echocardiography and hemodynamic monitoring
Cardiac structure and function were evaluated by Transthoracic echocardiography using a Visual Sonics Vevo 1100 Imaging System (Visual Sonic, Canada). After chests were shaved, the mice were anesthetized with isoflurane and placed in a supine position for detection. The parameters were calculated from 2-dimensional and M-mode images obtained in the long and short-axis views, according to built-in algorithms of Vevo 1100. The following parameters were measured: Left ventricular ejection fraction (EF), fractional shortening (FS), stroke volume (SV), diastolic volume (DV), LV end-systolic internal diameter (LVID, s), LV end-diastolic internal diameter (LVID, d), end-diastolic interventricular spectrum thickness (IVS, d) and end-diastolic left ventricular posterior wall thickness (LVPW, d).
Left ventricular hemodynamic measurements were performed using a Millar Catheter System (Millar 1.4F, SPR835, Millar Instruments Inc., USA). Maximal slope of systolic pressure increment (Max dP/dT) and minimal slope of diastolic pressure decrement (Min dP/dT), were measured.
2.12. DOT blot for RNA/DNA hybrids
DOT blot for RNA/DNA hybrids was performed as previously described [17]. Briefly, AC16 cells were cultured to 80% confluency in 10 cm dishes, trypsinized, and resuspended in 1.6 ml TE buffer (10 mM Tris, 1 mM EDTA, PH8). Add 50 μL 20% SDS and 5 μL 20 mg/mL proteinase K to cells-TE buffer mixture and incubate overnight at 37 °C. DNA was extracted with phenol/chloroform/isopropanol (25:24:1), precipitated with sodium acetate/ethyl alcohol, washed with 80% ethyl alcohol and dissolved in TE buffer. Genomic DNA was sonicated to 200–500 bp fragment using a Bioruptor apparatus (KONTES). For RNase H1-treated samples, 10 μg of DNA was treated with 20 U RNase H1 (Diamond B110070) for 6 h at 37 °C. 0.5 and 1 μg DNA was loaded to positively charged nylon membrane (Amersham, GE Healthcare). After UV crosslinking for 3 min, membranes were blocked with 5% skimmed milk dissolved in TBST wash buffer and incubated with S9.6 primary antibody (Sigma-Aldrich MABE1095) overnight at 4 °C. Membranes were incubated with HRP-conjugated secondary antibodies and blots were detected by chemiluminescence using ECL reagent.
2.13. DNA/RNA immunoprecipitation (DRIP)
DRIP assay was performed mainly as previously described [18]. The cell harvest and lysis, DNA extraction and fragmentation, and the RNase H-treated specificity control were similar to the description in DOT blot. Eight micrograms of the fragmentated nucleic acids (treated or not treated with RNase H1) were diluted with 500ul of TE buffer, leave 50ul for input, and the remaining 450ul for DRIP. The diluted DNA was added with 52ul of 10 × DRIP binding buffer (100 Mm Na3PO4, 1.4 M NaCl, 0.5% Triton-X100) and 10ul of S9.6 antibody (Sigma-Aldrich, MABE1095). Incubate the complex for 16 h at 4 °C while gently inverting on a mini-tube rotator (about 10 rpm per minute), followed by precipitation with protein A/G agarose beads. The precipitated DNA were eluted with 300ul of DRIP elution buffer (50 mM Tris, 10 mM EDTA, 0.5% SDS, PH 8) and 7ul of proteinase K (20 mg/ml), and incubated for 45 min at 55 °C. Immunoprecipitated DNA was then cleaned up by a phenol–chloroform extraction followed by ethanol precipitation, and resuspension with 10ul of TE buffer. qPCR was performed to analyze enrichment of DNA/RNA hybrids at actively transcribed genes. The relative abundance of DNA/RNA hybrid immunoprecipitated in each region was normalized to input values. Primer sequences used are shown below: RPL13A (positive loci): 5′-aggtgccttgctcacagagt-3′ (Forward) and ggttgcattgccctcattac (Reverse); EGR1: 5′-gccaagtcctccctctctactg-3′ (Forward) and 5′-ggaagtgggcagaaaggattg-3′ (Reverse); MALAT1: 5′-acgcagggagaattgcgtca-3′ (Forward) and 5′-ccttcccgtacttctgtcttcca-3′ (Reverse); SNRPN (negative loci): 5′-gccaaatgagtgaggatggt-3′ (Forward) and 5′-tcctctctgcctgactccat-3′ (Reverse).
2.14. Cardiomyocyte isolation and culture
The neonatal mice ventricular myocytes (NMVMs) were isolated from 1 to 2 day mouse pups of San1−/− and WT according to the previously described method. Briefly, pups were soaked in 75% alcohol and then the sheared hearts were placed in ice PBS. Ventricles were dissociated out, minced, and dissociated in 0.25% trypsin for 1 h at 4 °C. Then the CMs clumps were re-digested in digestive buffer (PBS containing 0.1% trypsin and 0.05% collagenase II) at 37 °C. Non-CMs were removed through differential adhesion method, and purified CMs were plated on slides within 12-well culture plates in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), penicillin and streptomycin. The CMs were stably adhered to the slide 48 h after plated, then replaced with fresh medium, and cultured for another 24 h to begin experiment.
2.15. Transcriptome sequencing
RNA-sequencing was performed to evaluate differentially expressed genes and enriched pathways. Briefly, the hearts of San1−/− and WT mice at postnatal day 1 and 12 weeks were respectively collected (n = 3), 1 μg RNA per sample was used as input material for the transcriptome sequencing. RNA sequencing was conducted by NovoGene (Beijing, China) using an instrument of Illumina (San Diego, California, USA) according to the manufacturer’s instructions. P (adj) < 0.05 and | log2 (fold change) | ≥ 0.5 between San1−/− and WT groups were considered as statistically significant.
2.16. Statistics
All results were expressed as mean ± standard error of measurement (SEM) from at least 3 independent experiments. Comparisons for independent data of normal distribution were performed using Student’s unpaired t-test, and multiple comparisons were determined by one-way analysis of variance (ANOVA). The statistical tests were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered statistically significant.
3. Results
3.1. San1 deletion in CMs leads to early cell cycle exit and limited CM proliferation in newborns
We first evaluated San1 expression in mouse CMs over a wide age range by immunoblotting. The results showed that San1 could be detected and persisted in newborn hearts 1–7 days post-birth (P1-P7) (Fig. 1A). San1 protein was decreased in the heart 7 days after birth (Fig. 1A). Furthermore, San1 was profoundly elevated in cardiac tissue from patients with end-stage heart failure (Fig. 1B). These results implicate a biological role of San1 in the neonatal and pathological heart. To define the in vivo role of San1, we generated a San1−/− mouse line through CRISPR-Cas9 technology. Immunoblotting confirms the deletion of San1 protein in San1−/− hearts (Fig. 1C). While the San1−/− mice were reproduced with a Mendelian pattern with no over embryonic and developmental phenotype, the heart weight to body weight (HW/BW) ratio of San1−/− hearts at P1 was slightly decreased compared with WT hearts (Fig. 1D). CM surface area evaluated on WGA staining heart sections was relatively increased in San1−/− CMs (Fig. 1E), suggesting that the San1−/− hearts are smaller because they have reduced CMs. To determine if the small San1−/− hearts with enlarged CMs are due to inhibition of CM proliferation, immunofluorescent Ki67 and PCNA staining revealed reduced positive staining on San1−/− relative to WT heart sections (Fig. 1F), indicating the impaired proliferation of the San1−/− CMs. RNAseq measurement on RNA samples extracted from neonatal San1−/− heart showed that cell cycle, mitosis, and DNA replication genes are consistently downregulated (Fig. 1G). Real-time PCR results supported that genes of the Cdc45-MCM2–7-GING (CMG) complex, indicators of DNA replication initiation, were down-regulated in neonatal hearts of San1−/− mice (Fig. 1H). Similarly, cell cycle genes, such as E2F1, RBL1, PCNA, CCND1, CCND2, and Mki67, were also downregulated in neonatal hearts of San1−/− mice (Fig. 1I). Western blot analysis also showed that cell cycle-related proteins, such as Ki67, Cyclin D1, MCM2, and MCM3, were all markedly reduced in the neonatal San1−/− compared with WT hearts (Fig. 1J). These results support the hypothesis that San1 deficiency leads to early cell cycle exit in CMs and impairing CM proliferation.
Fig. 1.
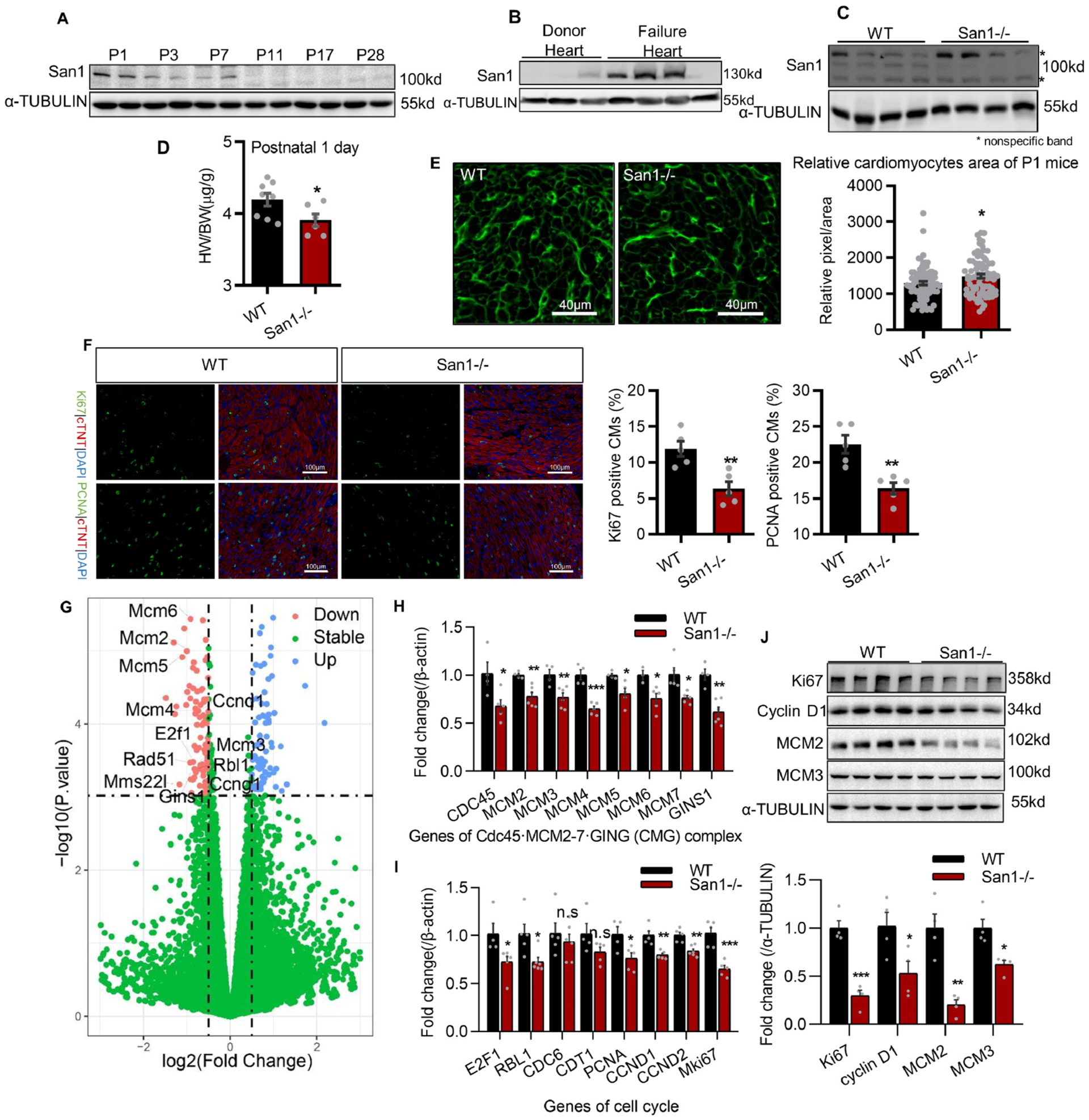
San1 deficient hearts revealed impaired CM proliferation in newborns. (A) San1 expression was monitored by immunoblotting in ventricular homogenates from mice of different ages. Relative protein levels were analyzed by densitometric methods and quantified by GraphPad Prism 8. P1–28 means postnatal 1–28 days. (B) San1 expression was detected by immunoblotting in hearts from the patients with end-stage heart failure and the normal adults. (C) Immunoblotting confirmed that San1 was completely broken in hearts of San1−/− mice. (D) In San1−/− and wild type (WT) mice of P1, the heart weight/body weight (HW/BW) ratio were determined (n = 8 in WT group, n = 6 in San1−/− groups). (E) Wheat germ agglutinin (WGA) staining of left ventricular sections was analyzed to indirectly determine the cross-sectional area (dot plot shows relative area per CMs from 6 different mice, scale bars, 40 μm). (F) Representative proliferative activity (labeled by Ki67 and PCNA) staining. Right panel: Quantification of cardiomyocytes proliferation activity from 5 different mice (scale bars, 100 μm). (G) volcano plot of RNA-sequencing data from hearts of WT and San1-deficiency mice at P1. (H and I) qRT-PCR analysis of DNA replication initiation and cell cycle genes in San1−/− and WT cardiomyocytes at P1 (n = 4–6). (J) Representative cell proliferation indicators (KI67, Cyclin D1) and DNA replication initiation helicases (MCM2, MCM3) were monitored in San1−/− and WT hearts of P1 mice by immunoblotting. Values are presented as mean ± SEM. n.s means not significant P > 0.05; *P < 0.05 vs. WT; **P < 0.01 vs. WT; ***P < 0.001 vs. WT. P values determined by unpaired Student’s t-test (2-tailed).
3.2. San1 deletion in mice leads to CM hypoplasia, cardiac dysfunction, CM hypertrophy, and fibrosis in adult hearts
We further assessed 3-month-old San1−/− mice to evaluate the function of their hypoplastic neonatal hearts. Series echocardiographic assessments (Fig. 2A) revealed that San1−/− mice exhibited decreased ejection fraction (Fig. 2B), stroke volume (Fig. 2C), left ventricular volume (Fig. 2D), and interventricular septal thickness (Fig. 2E). Cardiac catheterization measurement showed that both systolic (Max dP/dT) and diastolic (Min dP/dT) function were decreased in San1−/− hearts (Fig. 2F, 2G). The HW/BW ratios remained lower in San1−/− than WT mice (Fig. 2H). The cell surface area evaluated on H&E staining heart sections revealed that San1−/− CMs were slightly increased compared with WT CMs (Fig. 2I). WGA staining revealed that the adult San1−/− cardiomyocytes displayed disorganized arrangement (Fig. 2I), indicating that the cardiac development was incomplete and uncoordinated. Masson trichrome staining showed that cardiac fibrosis was increased in adult San1−/− hearts (Fig. 2I). Real-time PCR results showed elevated transcript levels of cardiac hypertrophic marks (ANP [atrial natriuretic peptide], BNP [brain natriuretic peptide], and MYH7 [myosin heavy chain 7]) in San1−/− relative to WT hearts (Fig. 2J). These results indicate that San1 deficiency in mice leads to CM hypoplasia, which shows weakened cardiac performance with CM hypertrophy and fibrosis when enduring the increased workload in an adult mouse. RNAseq measurement on cardiac RNA samples extracted from 3-month-old San1−/− mice showed a consistent change of cell cycle and replication genes (Fig. 2K). Genes reflecting cardiac hypertrophy, such as Nppa, Nppb, and Junb, were upregulated (Fig. 2K). Real-time PCR did confirm the downregulation of cell cycle genes, such as CDK1, CCNA2, CCNB1, BuB1, Mki67 and FOXM1, in 3-month-old San1−/− hearts compared with controls (Fig. 2L). Despite the cardiac disorders, survival rate of San1−/− mice was not significantly decreased compared with WT mice at least in the first nine months (65.2% vs. 83.3%, p = 0.14) (Fig. 2M). These results support the hypothesis that San1 deficiency impairs CM proliferation, leading to cardiac dysfunction and pathological development in the adult heart.
Fig. 2.
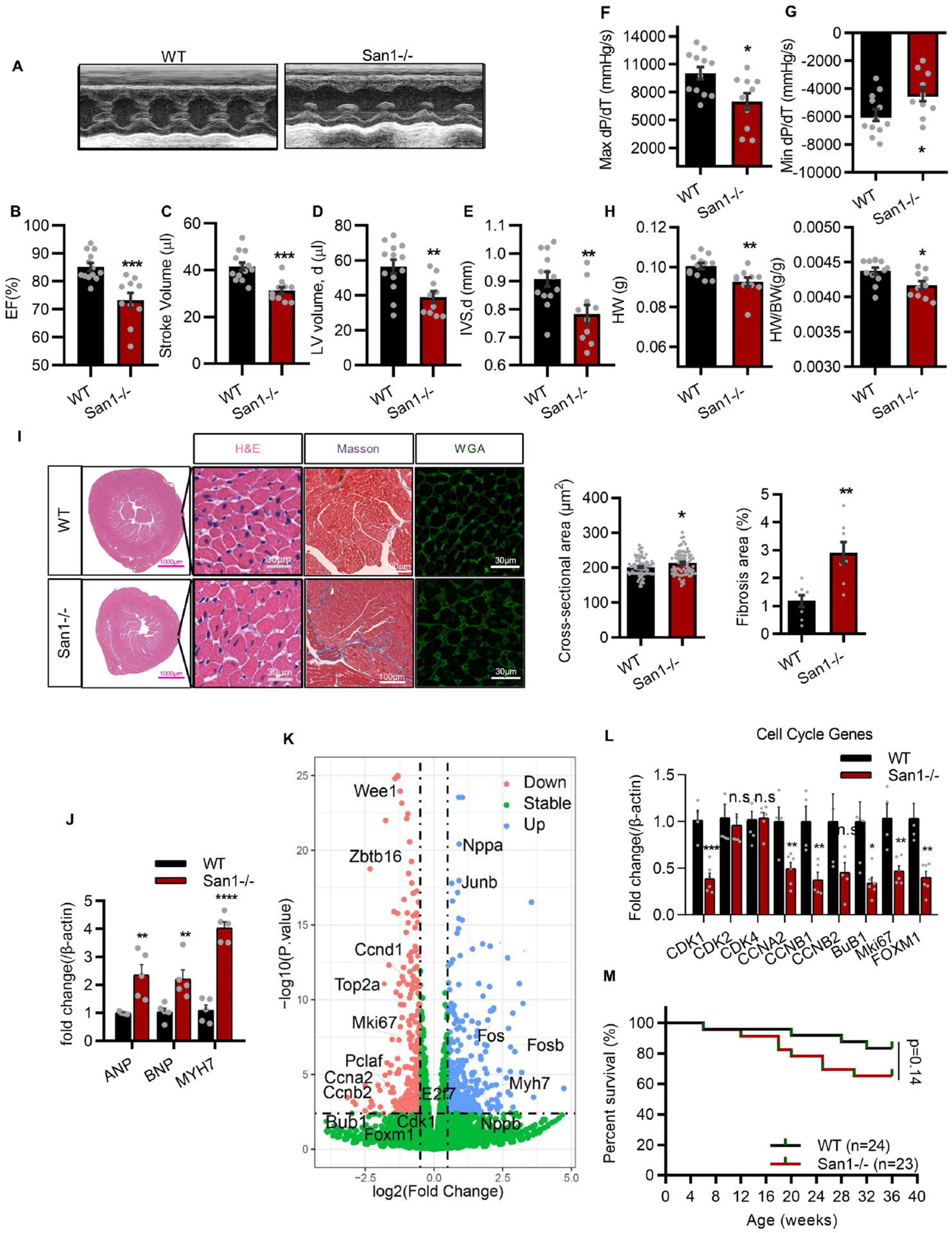
CM hypoplasia, CM hypertrophy, cardiac dysfunction, and fibrosis in 12-week-old San1−/− mice. (A-E) San1−/− and WT mice at 12 weeks were subjected to transthoracic echocardiographic examination (A), left ventricular ejection fraction (EF, %) (B), stroke volume (μl) (C), diastolic volume (μl) (D) and interventricular septal thickness at diastole (IVSd, mm) (E) were recorded (n = 10–13). (F and G) Maximal slope of systolic pressure increment (Max dP/dT, mmHg/s), and minimal slope of diastolic pressure decrement (Min dP/dT, mmHg/s) of San1−/− and WT mice were evaluated through cardiac catheterization method (n = 10–12). (H) The heart weight and the HW/BW ratio (n = 10–12). (I) Representative images of surface area of cardiomyocytes in San1−/− and WT mice were evaluated by wheat germ agglutinin (WGA) staining (scale bars, 30 μm) and hematoxylin/eosin (HE) staining (scale bars, 30 μm). Fibrosis level was staining by Masson trichrome (scale bars, 100 μm). Right panel: Quantification of cross-sectional area per CMs and relative fibrosis area from 6 different mice in the left panel. (J) Real-time quantitative PCR analysis of cardiac hypertrophic genes in San1−/− and WT hearts (n = 5). (K) volcano plot of RNA-sequencing data in San1−/− and WT hearts of mice at 12 weeks. (L) Real-time quantitative PCR analysis of cardiac proliferation gene in San1−/− and WT hearts (n = 6). (M) Survival curve of WT and San1−/− mice. p = 0.14, determined by log-rank (Mantel–Cox) testing (n = 24 in WT group, n = 23 in San1−/− groups). n.s means not significant P > 0.05; *P < 0.05 vs. WT; **P < 0.01 vs. WT; ***P < 0.001 vs. WT; ****P < 0.0001 vs. WT. P values determined by unpaired Student’s t-test (2-tailed).
3.3. San1 deficiency in cultured CMs results in checkpoint activation, cell cycle aggregating in S/G2 phase and down-regulated cell proliferation
To further investigate the potential role of San1 in cellular proliferation, we generated a San1-knockout AC16 (human cardiomyocyte) cell line utilizing CRISPR-Cas9 editing technology. Immunoblotting confirmed the complete deletion of San1 proteins in cultured AC16 CMs (Fig. 3A). The phosphorylation levels of checkpoint kinases RPA32 (S33) and p-CHK1 (S345) were increased in San1−/− AC16 CMs (Fig. 3B), suggesting collapsed replication forks and enhanced replication stress. The worsening replication stress got relieved in San1−/− AC16 cells with transfection of Flag-San1 (Fig. 3B). To further evaluate the role of San1 in the checkpoint pathway, we treated San1−/− and WT AC16 CMs with camptothecin, a topoisomerase inhibitor, to induce moderate replication stress [19]. We found that San1−/− cells displayed defective phosphorylation of CHK1 after camptothecin treatment (Fig. 3C). These results implied that San1 may participate in mediating the ATR-CHK1 pathway. Furthermore, immunoblotting also showed decreased expression of indicators for cell proliferation and DNA synthesis (PCNA, Ki67 and cyclin D1), and DNA replication licensors (MCM2, MCM3 and MCM4) in San1−/− AC16 CMs (Fig. 3D). Cell cycle profiling indicated that the proportion of cells stagnant in S and G2/M phase were significantly increased (66.2% vs. 50.3%, San1−/− vs. WT) (Fig. 3E). Cell counting also revealed defective proliferative activity in San1−/− cells (Fig. 3F). To further determine maladjusted cell proliferation, we measured the expression of several mitosis-promoting genes in San1−/− and WT cells and found that the expression of cell cycle-relevant genes declined sharply (Fig. 3G). Therefore, these results demonstrate that San1 participates in cell cycle regulation.
Fig. 3.
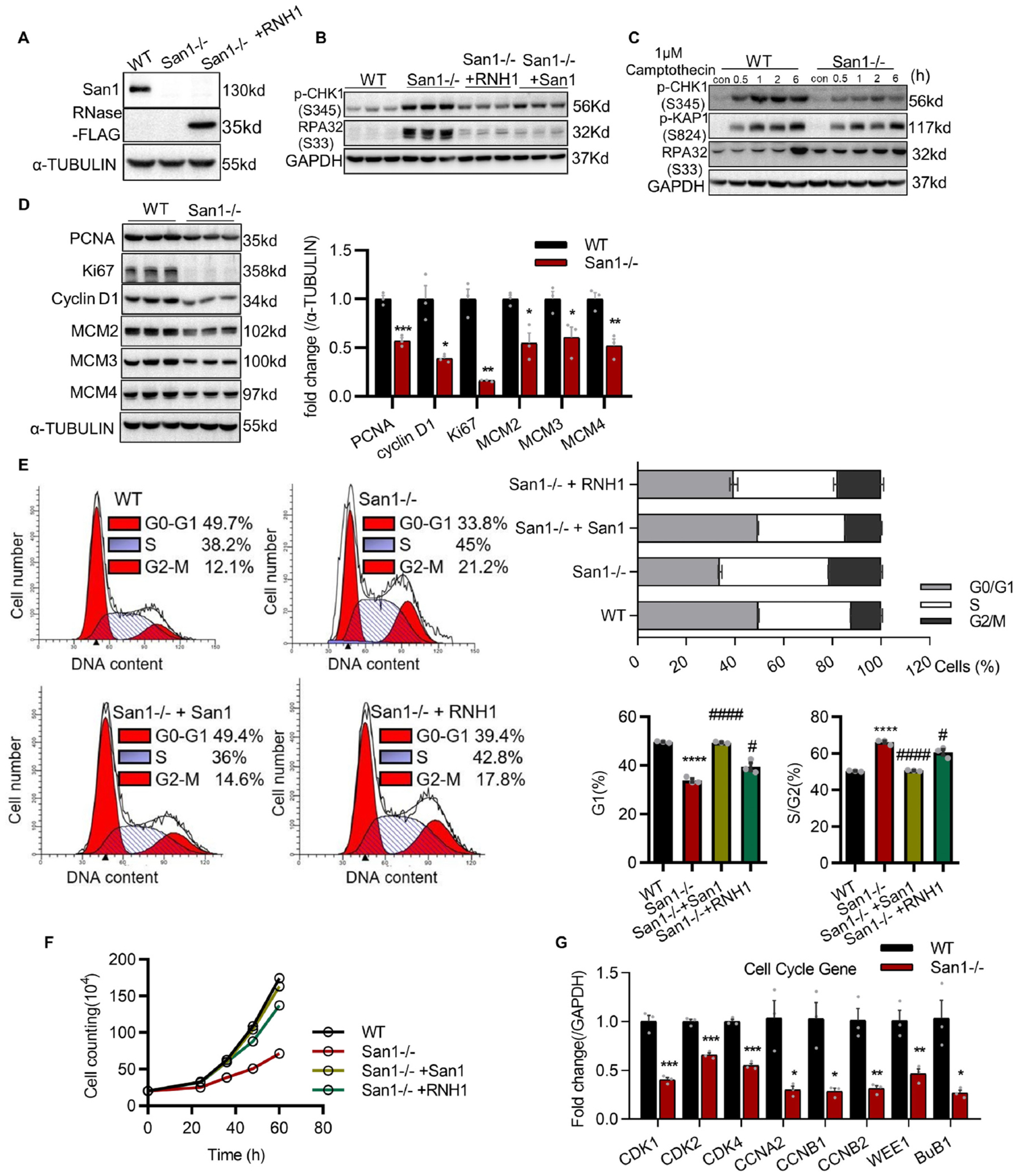
Loss of San1 causes checkpoint activation, cell cycle accumulating in S/G2 phase and cell proliferation down regulated. (A) Verification of complete deletion of San1 in AC16 cells. Verification of RNase H1 overexpression in San1 null cells. (B) Basal levels of p-CHK1 (S345) and RPA32 (S33), representative replication stress indicators, in San1−/−, San1−/− + RNH1, San1−/− + San1 and WT cells were monitored by immunoblotting. (C) Immunoblotting analysis of CHK1, KAP1 and RPA32 activity in total extracts of San1−/− and WT cells exposed or not to 1 μM Camptothecin for indicated time. (D) Basal levels of PCNA, Ki67, cyclin D1, MCM2–4, indicating DNA replication activity, in total extracts of San1−/− and WT cells were monitored by immunoblotting (n = 3). (E) Cell cycle profiles of San1−/−, San1−/− + RNH1, San1−/− + San1 and WT cells were evaluated by Propidium Iodide (PI) staining through flow cytometry under basal conditions. The cell cycle distribution was presented in the right panel (n = 3). (F) Proliferation curves of San1−/−, San1−/− + RNH1, San1−/− + San1 and WT cells were traced through cell counting. (G) qRT-PCR analysis of cell cycle genes in San1−/− and WT cells. *P < 0.05 vs. WT; **P < 0.01 vs. WT; ***P < 0.001 vs. WT; #P < 0.05 vs. San1−/−; # # # # P < 0.0001 vs. San1−/−. P values determined by unpaired Student’s t-test (2-tailed) for D and G; 1-way ANOVA for E.
3.4. Increased DNA damage in San1−/− CMs attributes to the impaired CM proliferation
To determine if DNA damage contributes to the impaired CM proliferation, we conducted immunofluorescent obtaining on heart sections from San1−/− and WT mice. We found remarkably increased double-strand breaks (DSBs) as indicated by accumulated γH2AX in the San1-deleted CMs of P1 mice (Fig. 4A). Immunofluorescent staining on heart sections from 12-week-old mice also revealed increased γH2AX in San1−/− compared with WT hearts (Fig. 4B). Western blots also revealed that the first responder of DNA damage, PARP1, was upregulated in cardiac tissue of San1−/− mice (Fig. 4C). In cultured AC16 CMs, alkaline and Neutral comet assays demonstrated increased tail moment and percent DNA in the tail in San1−/− relative to WT AC16 CMs (Fig. 4D), implicating the upregulation of both single-strand breaks (SSBs) and DSBs. Also, San1 deficiency led to upregulated γH2AX and phosphorylated Ataxia telangiectasia mutated (p-ATM, Ser1981), an early response to DNA damage, in cultured AC16 CMs (Fig. 4E). Furthermore, the protein abundances of XRCC1 and SETX were down-regulated in cultured San1−/− AC16 CMs, and overexpression of San1 can at least partly recover the expression of these proteins (Fig. 4E).
Fig. 4.
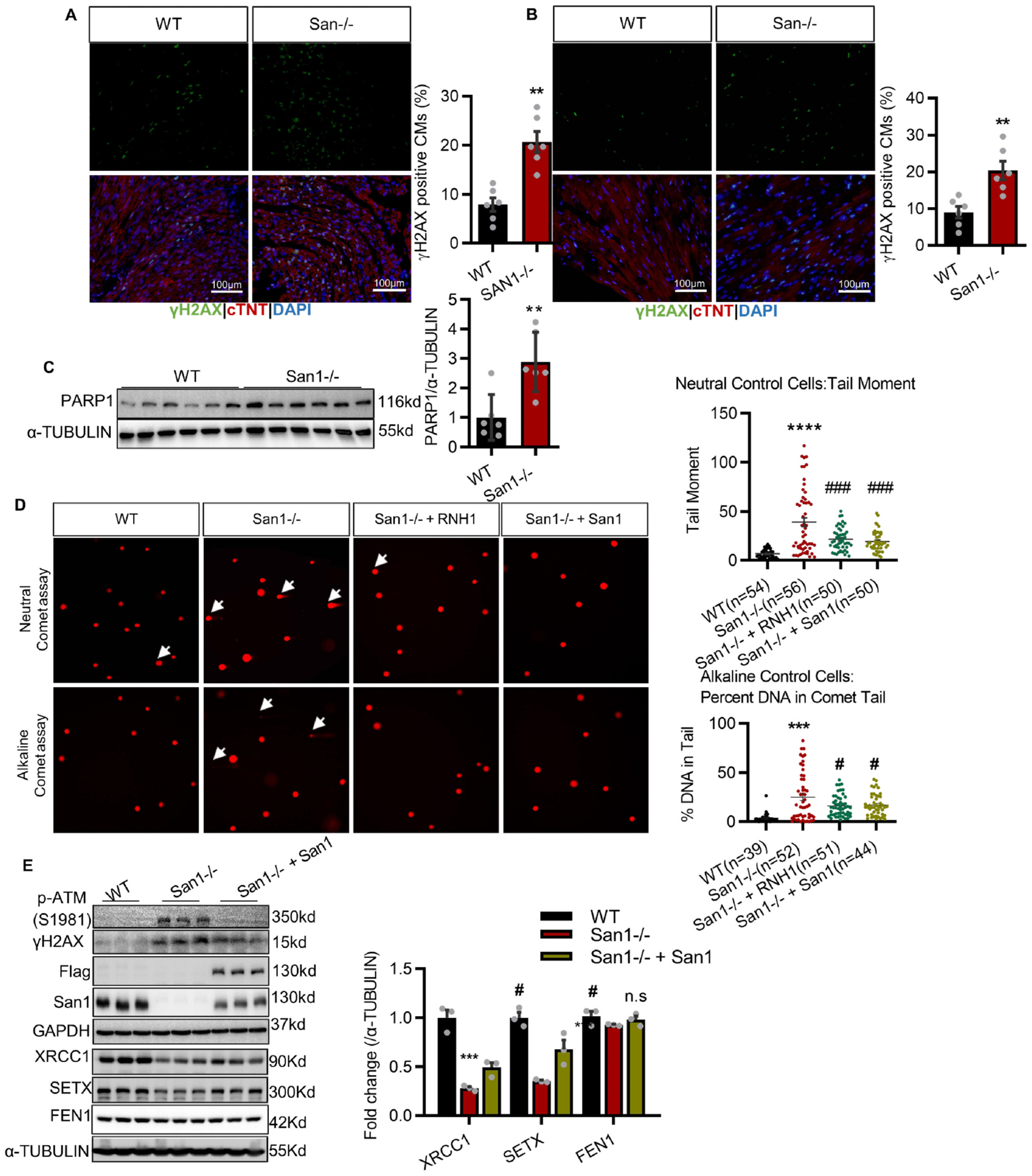
San1 deficient hearts exhibit DNA damage. (A and B) Representative DNA damage indicator γH2AX staining in the San1−/− and WT CMs of P1 mice (A) and 12-weeks (B) mice (n = 6, scale bars, 100 μm). (C) Immunoblotting analysis of PARP1 in San1−/− and WT hearts (n = 6). (D) Representative comet tail images from single-cell electrophoresis (Alkaline assay & Neutral assay) in San1−/−, San1−/− + RNH1, San1−/− + San1 and WT cells (White arrow indicates longer tails). The right panel showed quantification of tail moment and tail percentage (n = 39–56 from 3 independent experiments). (E) DDR indicators (p-ATM (S1981) and γH2AX) and crucial genomic repair and processing proteins (XRCC1, SETX and FEN1), in San1−/−, San1−/− + San1 and WT cells, were monitored by immunoblotting (n = 3). n.s means not significant P > 0.05, **P < 0.01 vs. WT, ***P < 0.001 vs. WT, ****P < 0.0001 vs. WT; # P < 0.05 vs. San1−/−, ### P < 0.001 vs. San1−/−. P values determined by unpaired Student’s t-test (2-tailed) for A-C; 1-way ANOVA for D-E.
To investigate if San1-related DNA damage would trigger DNA damage response (DDR) signaling, we conducted immunoblotting to measure PARP1 autoPARylation and hyperactivity in San1−/− and control AC16 CMs. After treated with PARP1 agonist camptothecin, the San1−/− AC16 CMs displayed increased PARylation of substrate proteins (Fig. 5A). Western blots revealed that XRCC1 and SETX were reduced under basal conditions in San1−/− relative to WT AC16 CMs, which were more pronounced when treated with protein synthesis inhibitor cycloheximide (CHX) treatment (Fig. 5B). Proteasome inhibitor MG132 almost completely inhibited their degradation (Fig. 5C), demonstrating that the degradations were mainly mediated by the ubiquitin-proteasome system (UPS). PARP inhibitor Olaparib partially delayed the proteolysis rates of XRCC1 and SETX in San1−/− AC16 CMs (Fig. 5D). Consistently, Western blots showed that XRCC1 and SETX were decreased in WT cells with PARP1-GFP overexpression (Fig. 5E). Besides, Olaparib treatment in San1−/− AC16 CMs reduced PAR and recovered the protein expression of XRCC1 and SETX in a dose-dependent manner (Fig. 5F). However, Olaparib treatment appeared insufficient to correct the replication stress in San1−/− AC16 CMs despite reducing DDR (p-ATM and γH2AX) (Fig. 5G). Collectively, the DNA lesions caused by San1 deficiency led to hyperactivation of PARP1, which resulted in accelerated SETX and XRCC1 degradation through PARdU, further aggravating DNA lesions, forming a vicious circle.
Fig. 5.
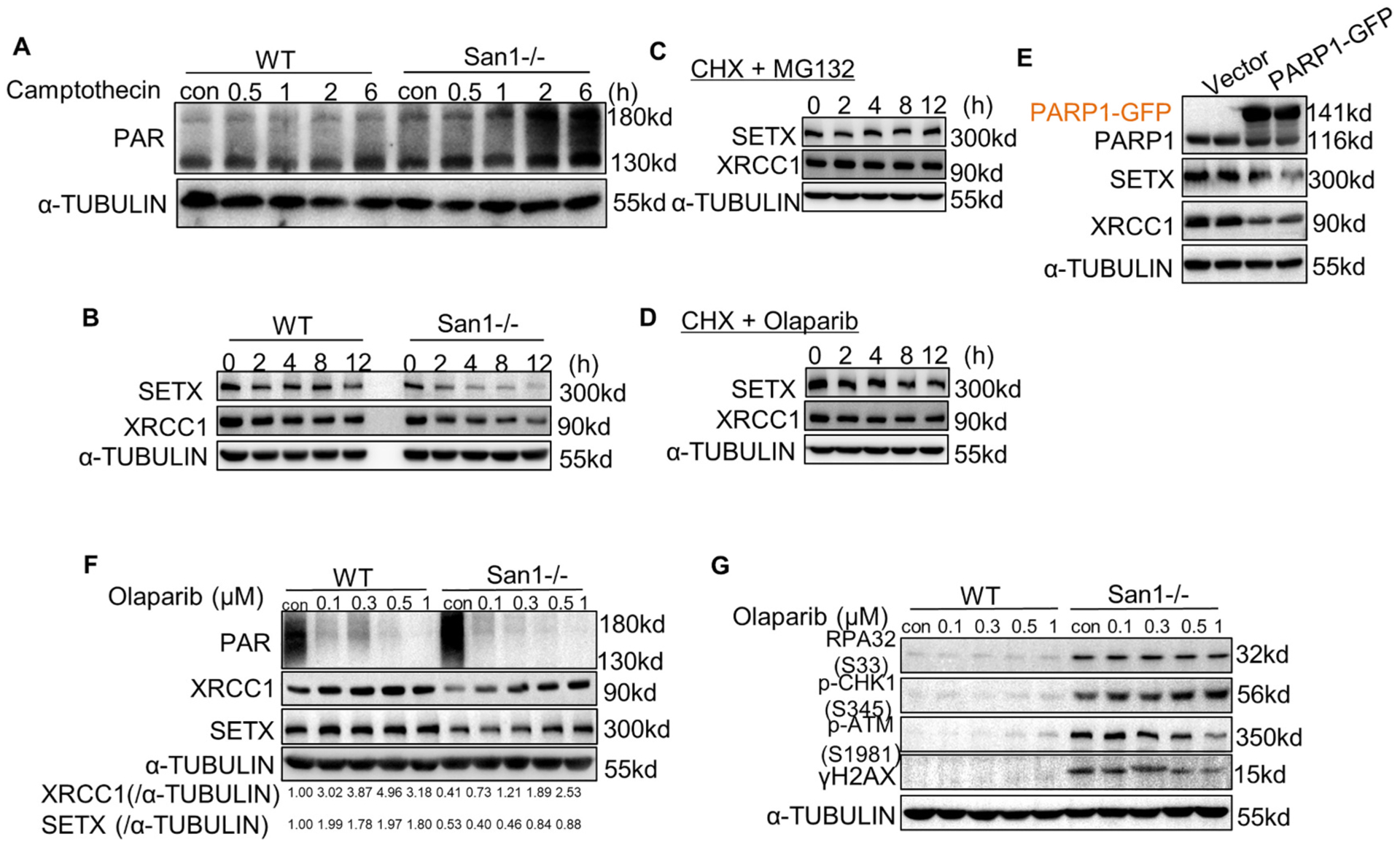
DDR forms a vicious circle with PARP1 overactivation, and Olaparib partially terminated this cycle. (A) San1−/− and WT cells were incubated with 1 μM camptothecin for indicated time. ADP-ribosylated proteins were determined by immunoblotting. (B) San1−/− and WT cells were incubated with 50 μM cycloheximide (CHX) for indicated time. SETX and XRCC1 were monitored by immunoblotting. (C) San1−/− cells were incubated with 50 μM cycloheximide (CHX) and 10 μM MG132 for indicated time. SETX and XRCC1 were detected by immunoblotting. (D) San1−/− cells were incubated with 50 μM cycloheximide (CHX) and 0.5 μM Olaparib for indicated time. SETX and XRCC1 were detected by immunoblotting. (E) WT cells were transfected with PARP1-GFP expression plasmid for 48 h. SETX and XRCC1 were detected by immunoblotting. (F and G) San1−/− and WT cells were incubated with Olaparib of different concentration gradients for 24 h. Indicators of DDR (p-ATM (S1981), γH2AX), replication stress (p-CHK1(S345), RPA32 (S33) and genomic processing proteins were monitored by immunoblotting.
3.5. Accumulation of R-loops is responsible for DNA damage and genomic instability in San1-deleted cells
Since San1 is associated with SETX, which is a known protein that resolves R-loop forming [20], we next investigated if San1 deficiency would change R-loop levels in CMs. We found that San1−/− hearts showed substantially more pronounced S9.6 staining in the CM nucleus (Fig. 6A), suggesting markedly more RNA/DNA hybrids (R-loops) accumulation in the San1−/− CMs. Consistent with those of the primary CMs from neonatal hearts, S9.6 staining revealed that San1 deficiency led to increased RNA/DNA hybrids, which are eliminated by RNase H1-treatment (Fig. 6B). We also confirmed R-loop accumulation in San1−/− cells by RNA/DNA hybrids immunoprecipitation (DRIP)-qPCR using the anti-S9.6 antibody (Fig. 6C) at various actively transcribed genes (RPL13A, EGR1, MALAT1 and SPRPN), previously reported to naturally form R-loops structures [18,21,22]. San1−/− AC16 CMs with stable flag-tagged-RNase H1 expression (San1−/− + RNH1) largely eliminated R-loops aggregation (Fig. 6D). Notably, ectopic RNase H1 strongly suppressed the upregulation of γH2AX, p-ATM, and p-KAP1 in San1−/− AC16 CMs (Fig. 6E), suggesting that R-loops was primarily responsible for DNA strand breaks. Interestingly, overexpression of RNase H1 effectively reduced the upregulation of PAR San1−/− AC16 CMs (Fig. 6F). Essentially, RNase H1 suppressed the upregulation of p-CHK1 and RPA32 (Fig. 3B), demonstrating that DNA damage-dependent checkpoint activation was mainly mediated by R-loops. Similarly, RNase H1 greatly collected the blocked cell cycle profiles (Fig. 3E) and disturbed proliferative activity (Figs. 3F and 6G). Together, these data demonstrated that deletion of San1 leads to R-loops-dependent-genomic instability and replication stress in cultured AC16 CMs. Therefore, these results support that R-Loop accumulation is responsible for genomic instability in San1-deleted cells.
Fig. 6.
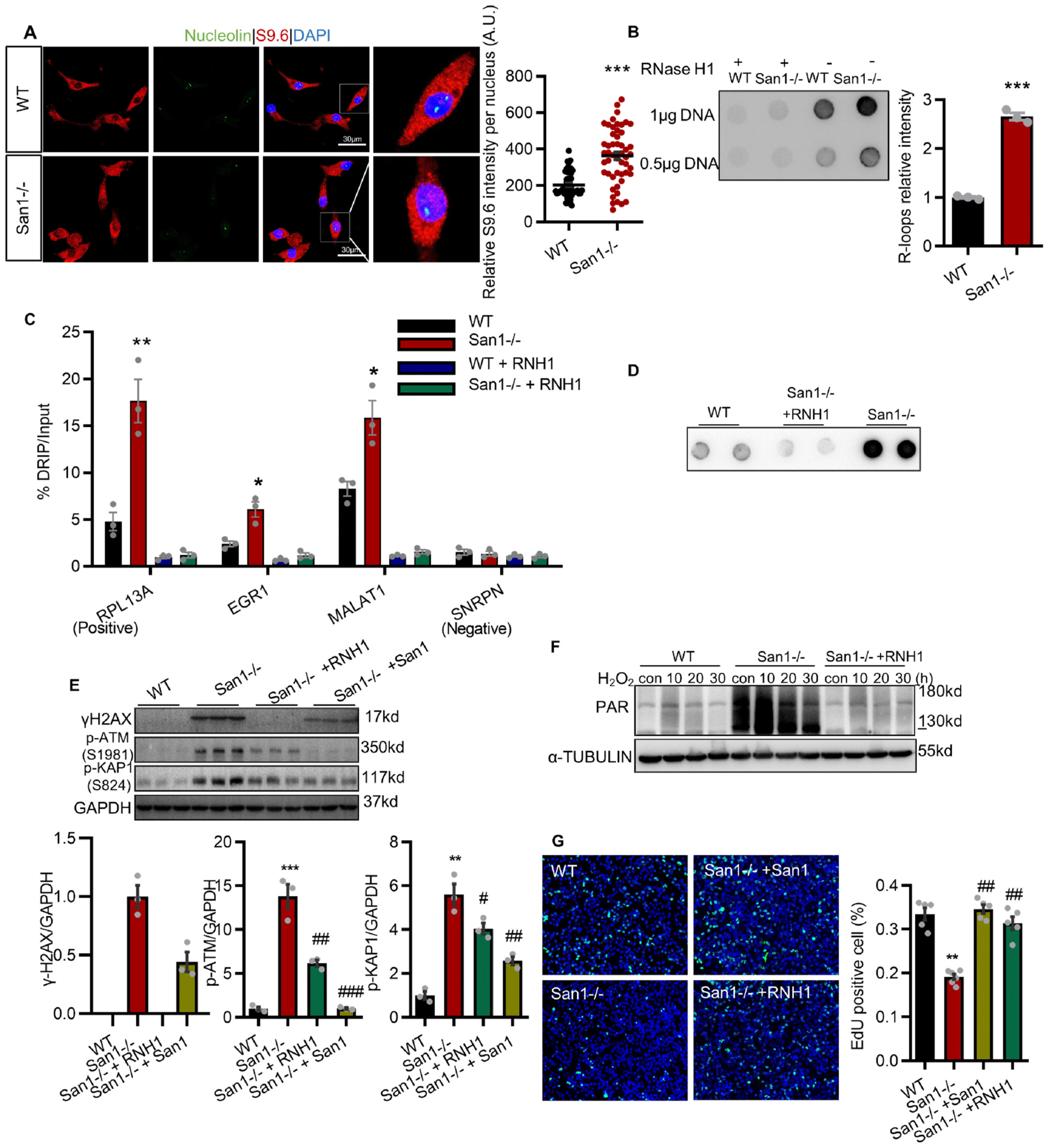
In San1−/− cells, accumulated R-loops are responsible for genomic instability and dysregulation of proliferation. (A) RNA/DNA hybrids (R-loops) were detected by immunostaining with anti-S9.6 and nucleolin antibodies in isolated neonatal CMs from San1−/− and WT P1 mice. Representative images are given (scale bars, 30 μm). Right panel: Quantification of relative S9.6 intensity per nucleus from 3 independent experiment described in the left panel. The intensity of nuclear S9.6 staining was determined by subtracting from nucleolin signal. (B) Dot blots with S9.6 antibodies to detect RNA/DNA hybrids in San1−/− and WT genomic DNA samples treated or not with RNase H1 from 3 independent experiment. (C) San1−/− and WT AC16 cells were subjected to DRIP-qPCR analysis with anti-S9.6 antibody at RPL13A (positive loci), EGR1, MALAT1 and SNRPN (negative loci) genes, with or without RNase H1 treatment (negative control) from 3 independent experiment. The relative abundance of R-loops immunoprecipitated in each genomic region was normalized to input values. (D) Dot blots with S9.6 antibodies to detect DNA/RNA hybrids in San1−/−, San1−/− + RNH1 and WT cells under basal conditions. (E) Basal levels of γH2AX, p-ATM (S1981) and p-KAP1 (S824), representative DDR indicators, in San1−/−, San1−/− + RNH1, San1−/− + San1 and WT cells, were monitored by immunoblotting (n = 3). (F) San1−/−, San1−/− + RNH1, and WT cells were incubated with 400 μM hydrogen peroxide for 10 min, 20 min and 30 min. ADP-ribosylated proteins were monitored by western blotting. (G) DNA synthesis activity were analyzed using EdU click assay in San1−/−, San1−/− + RNH1, San1−/− + San1 and WT cells (n = 5–6). *P < 0.05 vs. WT; **P < 0.01 vs. WT; ***P < 0.001 vs. WT; #P < 0.05 vs. San1−/−; # # P < 0.01 vs. San1−/−; # # # P < 0.001 vs. San1−/−. P values determined by unpaired Student’s t-test (2-tailed) for A and B; One-way ANOVA for D and F.
4. Discussion
Our findings demonstrate that San1 deficiency leads to excessive R-loops, increased DNA damage, and cell cycle arrest at the G2/M phase, impairing CM proliferation. As a result, San1−/− mice showed hypoplasia cardiac dystrophy in newborns and cardiac dysfunction in adults. The present study uncovers a novel biological function of San1 in resolving excessive R-loops that lead to DNA damage and reducing CM proliferation, thus leading to CM dysplasia and cardiomyopathy (Fig. 7).
Fig. 7.
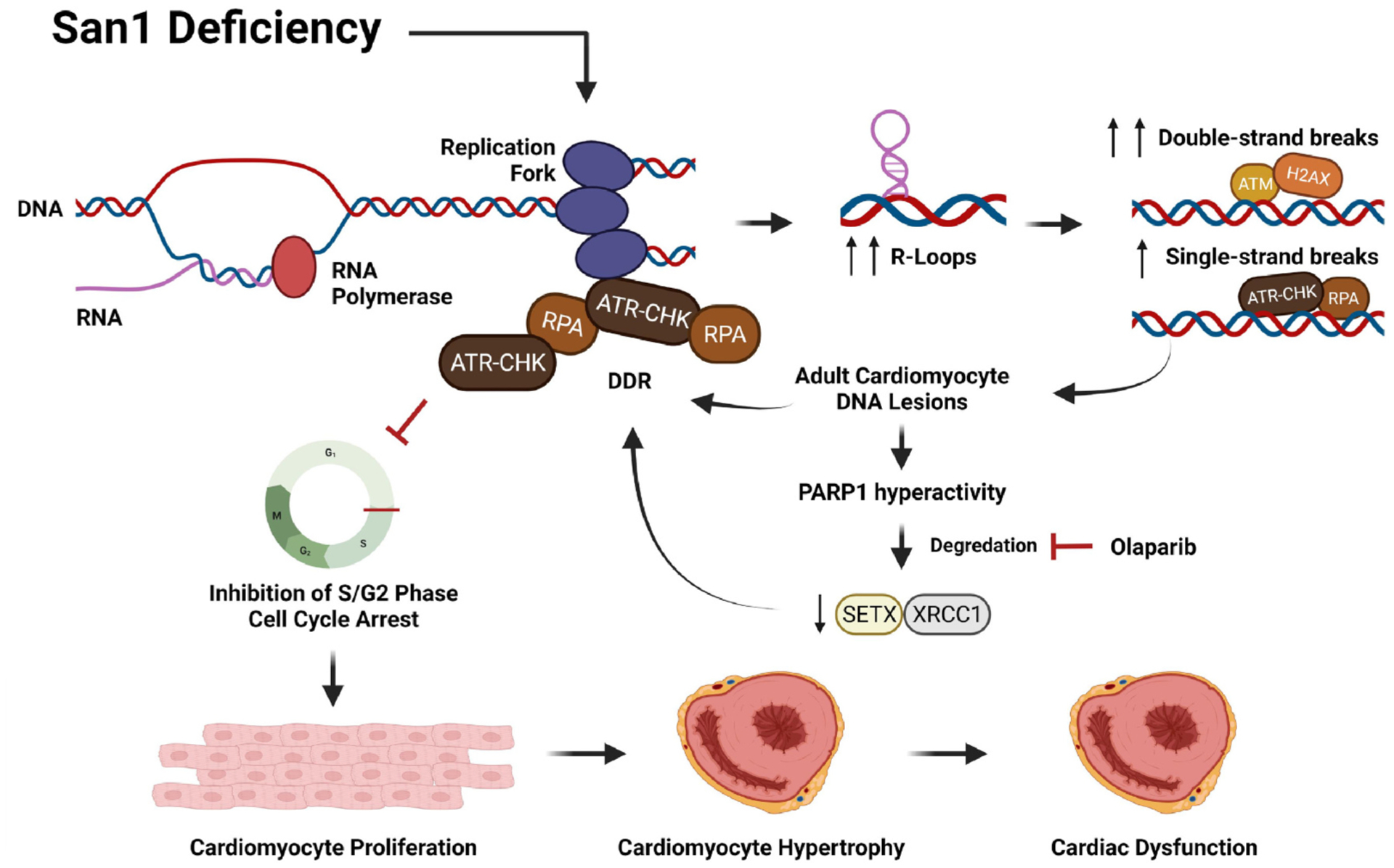
Schematic representation of the working hypothesis. This image was created using Biorender.com.
R-loops are naturally occurring transcriptional intermediates containing RNA/DNA hybrids, which have been linked to the regulation of fundamental biological processes, such as transcription, replication, and genome stability [10,23]. Excessive formation of R-loops results in DNA damage, genomic instability, and replication stress, mainly in proliferating cells [23]. During the development of mammalian CMs, replication and transcriptional activity is vigorous [24]. Little is known about the role of R-loops in fully proliferated CMs. As a crucial transcriptional intermediate, R-loops likely contribute to the modulation of chromatin status, gene expression, and other crucial genomic processes [9,10] in cardiac development. Given the newly defined function of San1 in regulating DNA damage via interacting with SETX [14], a helicase that resolves R-loops [25], it is not a surprise that we found San1 deficiency leads to elevated R-loops in primary and AC-16 CMs. S9.6 immunofluorescent staining is a commonly used method to identify R-loops in cultured cells, but it also binds dsRNA with a weaker affinity that leads to false-positive staining [26]. RNase H1 is a ribonuclease that specifically recognizes and binds to RNA/DNA hybrids to digest RNA within R-loops [27–29]. Supporting the role of San1 in resolving R-loops, RNase H1 treatment or RNase H1 overexpression in cultured AC-16 CMs can effectively eliminate the accumulated R loops. Therefore, this is the first evidence that San1 deficiency leads to excessive R-loops accumulation in CMs. This suggests that exonuclease San1 contributes to R-loops resolution, potentially via interacting with other R-loops resolving factors, such as SETX or DHX9, in collaboration with other nucleases, helicases and polymerases.
Excessive R-loops are linked to elevated DNA damage, especially DSBs. DNA damage-induced cell cycle exit at least partly attributes the losing proliferative potential in CMs after 1–2 weeks [5,7,30–32]. We confirmed that R-loops and DNA strand breaks, especially DSBs, were apparently aggregated in the nucleus of neonatal San1−/− CMs, indicated by the increased positive Comet assay and increased γH2AX protein expression. Furthermore, DDR was elevated in response to the increased DNA damage. Activated ATR protects genomic stability from R-loops by restraining transcription-replication conflicts, inhibiting immoderate cleavage of forks, and compelling a G2/M cell-cycle termination [11,12]. ATM activation limits the accumulation of R-loop-associated genomic instability when mild replication stress occurs [33]. The ATM signals (p-ATM, γH2AX, p-KAP1) and ATR signals (p-CHK1, RPA32) in San1−/− AC16 CMs were enhanced, consistent with the characteristics of R-loops-associated-DDR-dependent-replication stress [11,12,33]. Additionally, after intervention with camptothecin (a Topoisomerase I inhibitor which induces R-loops formation and DNA replication stress [19]), the San1−/− AC16 CMs showed defective and compromised phosphorylation of CHK1, which implicates that San1-related DNA damage interferes with the checkpoint process. The present study, however, will not exclude that San1 deficiency may also be activating other DNA repair mechanisms, such as the DNA-PKcs pathway, a signaling molecule sensing DSBs to initiate non-homologous end joining (NHEJ) [34,35]. Future studies are needed to further define the down-stream changes in response to San1 deficiency.
Our studies demonstrated that San1 was abundantly expressed in neonatal hearts but was reduced in adult mouse hearts. Additionally, the number of CMs and the heart size were significantly decreased in San1−/− neonatal hearts, suggesting that San1 may play a crucial role in neonatal heart development. Consistently, the crucial proliferative genes (Ki67 and cyclin D1) were prominently reduced, supporting the San1−/− neonatal CM proliferation activity was distinctly restrained. The transcriptome sequencing results further confirmed that the proliferative activity of San1−/− CMs was affected. RNA-sequencing showed that DNA replication initiation-related genes, especially genes of Cdc45-MCM2–7-GING (CMG) complex, were prominently down-regulated in San1−/− hearts of mice postnatal day 1. Both mRNA and protein levels revealed that the MCMs pathway was down-regulated. CMG proteins form a highly conserved heterohexameric complex, functioning as a replicative DNA helicase, which is essential for replication origin licensing [36]. Chronic replication stress down-regulates MCM2–7 expression[37], which can lead to genetic perturbations and cell proliferation disturbances [38]. DNA damage insults in the neonatal stage of CM development cause premature termination of cell cycle, resulting in CMs dysplasia, cardiac dysfunction, and even cardiogenic death [4,7,8]. Excessive R-loops can lead to DNA damage and genome instability [39,40]. Many DNA and RNA processing/regulatory factors prevent R-loop formation, while dysregulation of those modulators induces R-loop accumulation, causing R-loop-dependent DNA damage [41–46]. However, the role and regulation of R-loops in CMs are not completely understood. The cardiac phenotype in the young adult San1 heart could originate from CM hypoplasia during the neonatal stage, but could also attribute to DNA damage and cell cycle arrest in the adult heart. Further studies using the conditional knockout strategy should help solve this puzzle.
PARP1 principally senses and binds to SSBs where it becomes catalytically active, undergoes autoPARylation, and subsequently recruits and PARylates other nucleic acid metabolic proteins to damaged genomic regions [47–49]. PARP1 was hyperactive in San1−/− cells, while RNase H1 almost completely eliminated this state, indicating that PARP1 over-activation was mediated via R-loops-associated DNA damage. Many studies reported that PARylated protein could be degraded by the ubiquitin-proteasome system (UPS) [50,51]. Ubiquitylation of XRCC1 promotes its degradation through UPS [52]. Also, SUMOylation and ubiquitylation were thought to regulate the stability of SETX [25]. We found that hyperactivity of PARP1 in San1−/− cells negatively regulated the stability of SETX and XRCC1 through poly(ADP-ribose)-dependent ubiquitination (PARdU [51]). Importantly, Olaparib alleviated DDR; although it did not relieve replication stress, it had a strong correlation with the recovery of SETX and XRCC1. These results suggest that immoderate PARylation of XRCC1 and SETX induced their degradation by proteasome via PARdU and Olaparib potentially inhibited the aberrant proteolytic process. In the present study, we focus on the effects of PARP1 over-activation on DNA breakage, but we cannot exclude that non-DNA damage-related effects of PARP1 may also contribute to the detrimental cardiac effects of San1 deficiency.
While the present study provides strong evidence supporting that lack of San1 in the neonatal heart impedes the development of CM proliferation due to R-loop build-up-related DNA damages that suppress cell cycle, the role of San1 in adult CM proliferation is not as definitive. However, we did observe elevated DNA damage, DDR with repressed cell cycle signal and DNA replication. Further investigation using a mouse model of conditional loss of San1 function in the adult heart should provide definitive insights. In addition, San1 deletion obstructed cell cycle progression or premature cell cycle arrest, which led to CM hypoplasia. The fewer CMs of the San1−/− newborn hearts were functionally compensated. The San1−/− hearts endured an increasing workload as the animal entered young adulthood, and the CMs in the smaller heart become hypertrophied in response to the increasing wall stress. The increased fibrosis and cardiac hypertrophy gene expression further demonstrated that San1−/− deficiency leads to cardiomyopathy, potentially due to early CM hypoplasia-related disorders. However, to confirm the specificity and function of San1 in cardiac development and pathology, further investigations on conditional San1−/− hearts, including the gain-of-function or rescuing effect via San1 reintroduction, are warranted.
In summary, the above results indicate that R-loop build-up during the neonatal period of San1−/− cardiomyocytes lead to increased DNA damage, early cell cycle exit, and CM hypoplasia. This process, in turn, affects the successful proliferation and development of cardiomyocytes, leading to cardiac dysfunction. This study, for the first time, indicates that excessive R-loops are detrimental to neonatal heart development, and San1 contributes to R-loop resolution, which prevents CM hypoplasia-related cardiac disorders.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81500312) to QL and grants from National Institutes of Health (NIH) (R01 HL135336) and American Diabetes Association (ADA) (1-17-IBS-184) to QY.
Footnotes
Ethics statement
The animal study was reviewed and approved by the Committee on the Ethics of Animal Experiments of Tongji Medical College, Huazhong Science and Technology University, China.
CRediT authorship contribution statement
ZL designed the study, performed the laboratory experiments, analyzed, interpreted the data, and drafted the paper. XG, ZZ, YY, CZ and ZW helped to perform in vivo experiments. SWK and HL conducts RANseq data analysis, DW, XR and DW reviewed and revised the draft. QL and QY designed, analyzed, and revised the manuscript. All authors read and approved the final manuscript.
Declaration of competing interest
The authors have no conflicts of interest to declare.
References
- [1].Meilhac SM, Buckingham ME, The deployment of cell lineages that form the mammalian heart, Nat. Rev. Cardiol 15 (2018) 705–724. [DOI] [PubMed] [Google Scholar]
- [2].Steinhauser ML, Lee RT, Regeneration of the heart, EMBO Mol. Med 3 (2011) 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Walsh S, Ponten A, Fleischmann BK, Jovinge S, Cardiomyocyte cell cycle control and growth estimation in vivo–an analysis based on cardiomyocyte nuclei, Cardiovasc. Res 86 (2010) 365–373. [DOI] [PubMed] [Google Scholar]
- [4].Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA, The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response, Cell 157 (2014) 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yuan X, Braun T, Multimodal regulation of cardiac myocyte proliferation, Circ. Res 121 (2017) 293–309. [DOI] [PubMed] [Google Scholar]
- [6].Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, Foley A, Magnitsky S, Srivastava D, Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration, Cell 173 (2018) 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang D, Li Y, Heims-Waldron D, Bezzerides V, Guatimosim S, Guo Y, Gu F, Zhou P, Lin Z, Ma Q, Liu J, Wang DZ, Pu WT, Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation, Circ. Res 122 (2018) 74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou L, Dai H, Wu J, Zhou M, Yuan H, Du J, Yang L, Wu X, Xu H, Hua Y, Xu J, Zheng L, Shen B, Role of FEN1 S187 phosphorylation in counteracting oxygen-induced stress and regulating postnatal heart development, FASEB J. 31 (2016) 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Crossley MP, Bocek M, Cimprich KA, R-loops as cellular regulators and genomic threats, Mol. Cell 73 (2019) 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Santos-Pereira JM, Aguilera A, R loops: new modulators of genome dynamics and function, Nat. Rev. Genet 16 (2015) 583–597. [DOI] [PubMed] [Google Scholar]
- [11].Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, Maiorano D, An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism, EMBO J. 36 (2017) 1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Matos DA, Zhang JM, Ouyang J, Nguyen HD, Genois MM, Zou L, ATR protects the genome against R loops through a MUS81-triggered feedback loop, Mol Cell 77 (2020) 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li D, Kang Q, Wang DM, Constitutive coactivator of peroxisome proliferator-activated receptor (PPARgamma), a novel coactivator of PPARgamma that promotes adipogenesis, Mol. Endocrinol 21 (2007) 2320–2333. [DOI] [PubMed] [Google Scholar]
- [14].Andrews AM, McCartney HJ, Errington TM, D’Andrea AD, Macara IG, A senataxin-associated exonuclease SAN1 is required for resistance to DNA interstrand cross-links, Nat. Commun 9 (2018) 2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Groh M, Albulescu LO, Cristini A, Gromak N, Senataxin: genome guardian at the interface of transcription and neurodegeneration, J. Mol. Biol 429 (2017) 3181–3195. [DOI] [PubMed] [Google Scholar]
- [16].Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA, Staras K, Mancini GM, McKinnon PJ, Wang ZQ, Wagner JD, C. Care4Rare Canada, G. Yoon, K.W. Caldecott, XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia, Nature 541 (2017) 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA, Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability, Mol. Cell 56 (2014) 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sanz LA, Chedin F, High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing, Nat. Protoc 14 (2019) 1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tuduri S, Crabbé L, Conti C, Tourrìere H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, Pommier Y, Tazi J, Coquelle A, Pasero P, Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription, Nat. Cell Biol 11 (2009) 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cohen S, Puget N, Lin YL, Clouaire T, Aguirrebengoa M, Rocher V, Pasero P, Canitrot Y, Legube G, Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations, Nat. Commun 9 (2018) 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Salas-Armenteros I, Perez-Calero C, Bayona-Feliu A, Tumini E, Luna R, Aguilera A, Human THO-Sin3A interaction reveals new mechanisms to prevent R-loops that cause genome instability, EMBO J. 36 (2017) 3532–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mersaoui SY, Yu Z, Coulombe Y, Karam M, Busatto FF, Masson JY, Richard S, Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids, EMBO J. 38 (2019), e100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Garcia-Muse T, Aguilera A, Loops R, From physiological to pathological roles, Cell 179 (2019) 604–618. [DOI] [PubMed] [Google Scholar]
- [24].O’Meara CC, Wamstad JA, Gladstone RA, Fomovsky GM, Butty VL, Shrikumar A, Gannon JB, Boyer LA, Lee RT, Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration, Circ. Res 116 (2015) 804–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bennett CL, La Spada AR, Senataxin A novel helicase at the interface of RNA transcriptome regulation and neurobiology: from normal function to pathological roles in motor neuron disease and cerebellar degeneration, Adv. Neurobiol, 20 (2018) 265–281. [DOI] [PubMed] [Google Scholar]
- [26].Phillips DD, Garboczi DN, Singh K, Hu Z, Leppla SH, Leysath CE, The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6, J. Mol. Recognit 26 (2013) 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hyjek M, Figiel M, Nowotny M, RNases H: structure and mechanism, DNA Repair (Amst) 84 (2019), 102672. [DOI] [PubMed] [Google Scholar]
- [28].Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, Chedin F, Prevalent, Dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals, Mol. Cell, 63 (2016) 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F, R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters, Mol. Cell 45 (2012) 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Muralidhar SA, Sadek HA, Meis1 regulates postnatal cardiomyocyte cell cycle arrest, in: Nakanishi T, Markwald RR, Baldwin HS, Keller BB, Srivastava D, Yamagishi H(Eds.) Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology, Tokyo, 2016, pp. 93–101. [PubMed] [Google Scholar]
- [31].Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA, Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA, Transient regenerative potential of the neonatal mouse heart, Science 331 (2011) 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Marabitti V, Lillo G, Malacaria E, Palermo V, Sanchez M, Pichierri P, Franchitto A, ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells, Nucleic Acids Res. 47 (2019) 3485–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhou H, Toan S, Zhu P, Wang J, Ren J, Zhang Y, DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis, Basic Res. Cardiol 115 (2020) 11. [DOI] [PubMed] [Google Scholar]
- [35].Kannan A, Bhatia K, Branzei D, Gangwani L, Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy, Nucleic Acids Res. 46 (2018) 8326–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ishimi Y, Regulation of MCM2–7 function, Genes Genet. Syst 93 (2018) 125–133. [DOI] [PubMed] [Google Scholar]
- [37].Bai G, Smolka MB, Schimenti JC, Chronic DNA replication stress reduces replicative lifespan of cells by TRP53-dependent, microRNA-assisted MCM2–7 downregulation, PLoS Genet. 12 (2016), e1005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chuang CH, Wallace MD, Abratte C, Southard T, Schimenti JC, Incremental genetic perturbations to MCM2–7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress, PLoS Genet. 6 (2010), e1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rinaldi C, Pizzul P, Longhese MP, Bonetti D, Sensing R-loop-associated DNA damage to safeguard genome stability, Front. Cell Dev. Biol 8 (2020), 618157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Niehrs C, Luke B, Regulatory R-loops as facilitators of gene expression and genome stability, Nat. Rev. Mol. Cell Biol 21 (2020) 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M, Liberi G, Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes, Cell 151 (2012) 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cristini A, Groh M, Kristiansen MS, Gromak N, RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage, Cell Rep. 23 (2018) 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morales JC, Richard P, Patidar PL, Motea EA, Dang TT, Manley JL, Boothman DA, XRN2 links transcription termination to DNA damage and replication stress, PLoS Genet. 12 (2016), e1006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Garcia-Rubio ML, Perez-Calero C, Barroso SI, Tumini E, Herrera-Moyano E, Rosado IV, Aguilera A, The fanconi anemia pathway protects genome integrity from R-loops, PLoS Genet. 11 (2015), e1005674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Stirling PC, Hieter P, Canonical DNA repair pathways influence R-loop-driven genome instability, J. Mol. Biol 429 (2017) 3132–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W, The fanconi anemia pathway maintains genome stability by coordinating replication and transcription, Mol. Cell 60 (2015) 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Alemasova EE, Lavrik OI, Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins, Nucleic Acids Res. 47 (2019) 3811–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Krishnakumar R, Kraus WL, The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets, Mol. Cell 39 (2010) 8–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Azarm K, Smith S, Nuclear PARPs and genome integrity, Genes Dev. 34 (2020) 285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hu K, Wu W, Li Y, Lin L, Chen D, Yan H, Xiao X, Chen H, Chen Z, Zhang Y, Xu S, Guo Y, Koeffler HP, Song E, Yin D, Poly(ADP-ribosyl)ation of BRD7 by PARP1 confers resistance to DNA-damaging chemotherapeutic agents, EMBO Rep. 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vivelo CA, Ayyappan V, Leung AKL, Poly(ADP-ribose)-dependent ubiquitination and its clinical implications, Biochem. Pharmacol 167 (2019) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yousafzai NA, Zhou Q, Xu W, Shi Q, Xu J, Feng L, Chen H, Shin VY, Jin H, Wang X, SIRT1 deacetylated and stabilized XRCC1 to promote chemoresistance in lung cancer, Cell Death Dis. 10 (2019) 363. [DOI] [PMC free article] [PubMed] [Google Scholar]