

Abstract
The Australian backyard mosquito, Aedes notoscriptus, is a highly urbanised pest species that has invaded New Zealand and the USA. Importantly, Ae. notoscriptus has been implicated as a vector of Ross River virus, a common and arthritogenic arbovirus in Australia, and is a laboratory vector of numerous other pathogenic viruses, including West Nile, yellow fever, and Zika viruses. To further explore live viruses harboured by field populations of Ae. notoscriptus and, more specifically, assess the genetic diversity of its virome, we processed 495 pools, comprising a total of 6,674 female Ae. notoscriptus collected across fifteen suburbs in Brisbane, Australia, between January 2018 and May 2019. Nine virus isolates were recovered and characterised by metagenomic sequencing and phylogenetics. The principal viral family represented was Flaviviridae. Known viruses belonging to the genera Flavivirus, Orbivirus, Mesonivirus, and Nelorpivirus were identified together with two novel virus species, including a divergent Thogoto-like orthomyxovirus and an insect-specific flavivirus. Among these, we recovered three Stratford virus (STRV) isolates and an isolate of Wongorr virus (WGRV), which for these viral species is unprecedented for the geographical area of Brisbane. Thus, the documented geographical distribution of STRV and WGRV, both known for their respective medical and veterinary importance, has now been expanded to include this major urban centre. Phylogenies of the remaining five viruses, namely, Casuarina, Ngewotan, the novel Thogoto-like virus, and two new flavivirus species, suggested they are insect-specific viruses. None of these viruses have been previously associated with Ae. notoscriptus or been reported in Brisbane. These findings exemplify the rich genetic diversity and viral abundance within the Ae. notoscriptus virome and further highlight this species as a vector of concern with the potential to transmit viruses impacting human or animal health. Considering it is a common pest and vector in residential areas and is expanding its global distribution, ongoing surveillance, and ecological study of Ae. notoscriptus, together with mapping of its virome and phenotypic characterisation of isolated viruses, is clearly warranted. Immanently, these initiatives are essential for future understanding of both the mosquito virome and the evolution of individual viral species.
Keywords: Aedes notoscriptus, arbovirus, flavivirus, metagenomics, Stratford virus, thogotovirus
1. Introduction
Historically, there is no vector considered more important with regard to arthropod-borne virus (arbovirus) emergence or transmission than the mosquito. Describing the vast array and global distribution of arboviruses is complicated by the large number of mosquito species that can serve as vectors. There are also inherent difficulties in sampling in remote areas from where viruses may emerge or are introduced and potential limitations in adequate technologies and resources to support arbovirus discovery efforts. Whilst arboviral biodiversity remains largely underexplored, high-throughput metagenomic platforms such as next-generation sequencing (NGS) have revolutionised mosquito virome mapping from a variety of sample types and uncovered numerous novel viral families and species (Coffey et al., 2014; Bigot et al., 2018; Ramirez et al., 2020). Further to viruses of medical importance, numerous insect-specific viruses (ISVs) have also been identified, highlighting the complexity of arbovirus evolution and transmission dynamics (reviewed in Blitvich and Firth 2015; Hall et al., 2016; Vasilakis et al., 2013). Although restricted in their ability to infect vertebrate cells, the importance of ISVs in possible modulation of pathogenic virus transmission and their potential use in other innovative applications is only now being realised. Indeed, some ISVs have been used recently to produce chimeras incorporating key antigenic genes of pathogenic arboviruses, thereby becoming potential vehicles for non-infectious antigen delivery in diagnostic and vaccine applications (Hobson-Peters et al., 2019).
Holistic approaches encompassing investigations of both mosquito species and their respective viromes are needed to fully utilise the health benefits contained in the collective temporal and spatial mosquito distribution and virus data. In Australia, deep sequencing and metatranscriptomics have revealed new insights into virus ecology, diversity, and abundance within respective Aedes and Culex mosquito species, with one study reporting 19 new RNA virus species from a multitude of virus families (Shi et al., 2017). Viromes of Aedes aegypti from Cairns, Queensland (QLD; Zakrzewski et al., 2018) and other Aedes and Culex spp. from New South Wales (NSW; Coffey et al., 2014) were also investigated, yielding previously uncharacterised rhabdoviruses, bunyaviruses, and ISVs. Despite these concerted efforts, the viromes of many Australian mosquito species implicated in human health remain understudied, including that of Ae. notoscriptus, which to date has not been specifically investigated.
Aedes notoscriptus is a highly urbanised mosquito species distributed throughout much of the southwest Pacific region (Lee et al., 1982). It is considered an invasive species as it has expanded its global distribution, becoming established in New Zealand in the 1920s (Graham 1939), and was introduced into the USA in 2014 where it has since established in multiple counties in California (Peterson and Campbell 2015; Feiszli et al., 2020). Aedes notoscriptus is a common pest species in both residential and industrial areas of major cities and regional locations (Kay, Watson, and Ryan 2008; Strid 2008) and therefore has a high association with humans.
Aedes notoscriptus has been implicated as an urban vector of Ross River virus (RRV) (Doggett and Russell 1997; Ritchie et al., 1997; Ryan, Do, and Kay 2000), a prevalent arthritogenic alphavirus, which is endemic and enzootic in Australia and Papua New Guinea (Harley, Sleigh, and Ritchie 2001; Jansen et al., 2019). Within the laboratory, high transmission rates have been achieved with another endemic Australian alphavirus, Barmah Forest virus (BFV; Watson and Kay 1999), which suggest Ae. notoscriptus could also play a role in BFV transmission and subsequent human disease. Further laboratory studies have also demonstrated that Ae. notoscriptus can be infected with and transmit several other medically important arboviruses, including Zika virus (ZIKV), West Nile virus (WNV), Japanese encephalitis virus, chikungunya virus, and yellow fever virus (YFV) (van den Hurk et al., 2003, 2010, 2011; Jansen et al., 2008; Duchemin et al., 2017), albeit at rates generally lower than implicated vectors. There is also the possibility that Ae. notoscriptus could serve as a vector of medically important viruses in countries where it has become established, similar to what has occurred with Aedes japonicus (Kaufman and Fonseca 2014).
Given the close association of Ae. notoscriptus with humans, its ability to invade new ecosystems, and known incrimination in pathogenic arbovirus transmission, we investigated the virome of this mosquito species post isolation of viruses in cell culture using applied metagenomic sequencing and phylogenetics. Mosquitoes were sampled from Brisbane, the largest city and state capital of QLD, Australia, over an 18-month period. Herein, we describe the subsequent isolation of nine viruses from five different genera and their genetic and phylogenetic characterisation. Among these, we report the discovery of three novel arboviruses and their evolutionary relationships with respective viral groups.
2. Materials and methods
2.1. Mosquito collections
Adult mosquitoes were sampled fortnightly from fifteen suburbs in Brisbane, QLD, Australia, from 24 January 2018 to 29 May 2019. Mosquitoes were collected using CO2-baited light traps supplemented with 1-octen-3-ol deployed overnight from 16:00 to 07:00 (Ritchie and Kline 1995). Upon retrieval, trap collections were placed on a chill table and mosquitoes morphologically identified as Ae. notoscriptus (Lee et al., 1982) were placed into pools of ≤25 mosquitoes within 2-mL U-bottom microfuge tubes (Eppendorf AG, Hamburg, Germany) before being stored at −80°C. The remaining unsorted mosquitoes from each trap night and location were also stored at −80°C.
2.2. Virus isolation
A single 5-mm stainless steel ball and 1.8 ml of growth media (GM; Opti-MEM®; Gibco, Life Technologies Corporation, NY) containing 3 per cent foetal bovine serum, and antibiotics and antimycotics was added to each tube containing the pooled Ae. notoscriptus. The pools were homogenised for 3 min at 26 Hz in a TissueLyser II (Qiagen, Hilden, Germany) before centrifugation at 14,000 × g for 1 min. The supernatant was then filtered through a 0.8/0.2 μM Supor® membrane filter (Pall Corporation, Ann Arbour, MI) into three separate aliquots, two of which were stored at −80°C. The unfrozen aliquot was then used to inoculate confluent monolayers of C6/36 (American Type Culture Collection (ATCC), CRL-1660, Aedes albopictus) cells seeded on duplicate 96-well plates. For each plate, duplicate wells were each inoculated with 50 µl of the supernatant.
Plates were incubated at 28°C for 7 days before cells were examined for potential virus infection via observation of cell morphological changes and cytopathic effect (CPE). Fifty microlitres of the supernatant were then passaged onto confluent monolayers of C6/36 cells as described above. The supernatant from wells displaying CPE was retained for further analysis. All remaining supernatant from non-suspicious wells was discarded and all cells were fixed in 20 per cent acetone before plates were stored at −20°C. To further screen for prevalent flaviviruses and alphaviruses, fixed plates were subjected to a cell culture enzyme immunoassay (CC-EAI) utilising pan-reactive B10 anti-alphavirus and 4G2 anti-flavivirus monoclonal antibodies (mabs) (Broom et al., 1998).
The supernatant from wells containing cells exhibiting CPE or an aliquot from pools positive in the CC-EIA was further passaged up to three times in 3-ml flat-bottom culture tubes (Nunc™, ThermoFisher Scientific, Australia, catalogue number 156758) seeded with confluent monolayers of C6/36 cells grown in GM. Briefly, approximately 100 µl of supernatant obtained from combined duplicate wells of the mosquito pool microtitre plate inoculations were through a 13 mm, pore size 0.2 µM filter (Catalogue number 4602, Acrodisc, Bio-Strategy Pty Ltd, Tingalpa, QLD, Australia) before inoculation of C6/36 monolayers in the 3-ml culture tubes, followed by incubation at 1 h at 28°C. Cultures were re-fed with 2 ml of culture media (CM; Opti-MEM®; Gibco, Life Technologies Corporation, NY, supplemented with 0.2 per cent bovine serum albumin; Catalogue number A8412-100ML, Sigma-Aldrich, North Ryde, Australia) and incubated for 7 days at 28°C. Cells from these inoculated tubes were screened for flavivirus and alphavirus infection in an immunofluorescence assay (IFA) using a panel of mabs as previously described (Pyke et al., 2018).
Preliminary analysis suggested Isolate 208825 was related to potentially pathogenic orthomyxoviruses; therefore, it was investigated further by cell culture to determine its ability to infect vertebrate cell lines. The cell lines the isolate were characterised in were African green monkey kidney (Vero; ATCC, CRL-1586), human hepatocyte cellular carcinoma (Huh-7; obtained from Professor Paul Young, University of Queensland, Brisbane, Australia), human embryonic kidney (HEK) 293 (ATCC, CRL-1573), human lung (MRC-5; catalogue number 72-211, BioWhittaker, Walkersville, MD, USA), human muscle rhabdomyosarcoma (RD; ATCC CCL-136), human lung adenocarcinoma (A549; ATCC CCL-185), baby hamster kidney (BHK-21; ATCC CCL-10), buffalo green monkey kidney (BGMK; USA EPA, USA), rhesus monkey kidney (LLC-MK2; CSL, Australia), Madin-Darby canine kidney (MDCK; ATCC CCL-34), Madin-Darby bovine kidney (MDBK; CSL, Australia), human cervical cancer (HeLa; ATCC CCL-2), and mouse neuroblastoma (MNA; catalogue number 07-305T, BioWhittaker, Walkersville, MD, USA). The C6/36 cell supernatant from viral Passage three was first diluted 1:50 in CM and inoculated onto confluent cell monolayers grown in 3-ml flat-bottom culture tubes. Culture tubes were incubated for 1 h at 37°C, before re-feeding with 2 ml of CM (or Dulbecco’s modified eagle medium/F-12, GlutaMAX™ supplement; Catalogue number 10565018, ThermoFisher Scientific, Seventeen Mile Rocks, QLD, Australia, for MDCK and MRC-5 cells) and incubation for a total of seven days at 37°C. Cultures were passaged in each cell line a total of three times and were monitored for cell morphological changes and evidence of CPE.
2.3. RNA extraction and next-generation sequencing
Reagents and protocols for RNA extraction and NGS have been described previously (Pyke et al., 2020). Briefly, RNA was extracted from 140 µl of viral culture supernatant using the QIAamp viral RNA extraction kit (Qiagen, Chadstone, Australia) without carrier RNA. DNase treatment was performed to remove host and microbial DNA using the DNase Heat and Run kit (ArcticZymes, Scientifix, South Yarra, Victoria, Australia). First-strand (Protoscript II first-strand complementary DNA (cDNA) kit) cDNA was prepared from the RNA template followed by second-strand cDNA synthesis using an enzyme cocktail of Escherichia coli DNA ligase, DNA polymerase I, and RNase H (first- and second-strand cDNA synthesis reagents obtained from New England Biolabs, Notting Hill, Victoria, Australia). The cDNA libraries were constructed using the Nextera XT kit and individual indices (kit A) for barcoding (Illumina, San Diego, CA) according to manufacturer’s instructions. Paired-end (2 × 150 nucleotides) massive parallel sequencing was performed using the V2 mid-output kit on a NextSeq 500 machine (Illumina, San Diego, CA). Raw sequence reads were processed using Geneious R10 version 10.2.6 software (Kearse et al., 2012). Briefly, forward and reverse reads were paired before trimming at both ends with BBduk to remove low-quality reads. Near-complete genome sequences (non-segmented viruses) or sequences representing viral segments or individual genes (segmented viruses) were assembled using the SPAdes de novo assembler (Bankevich et al., 2012) within Geneious before prediction of respective open reading frames (ORFs). In order to determine viral genome identities, contigs were initially compared to the GenBank database using, blastn (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Where nucleotide sequence identity per cents were low or unable to be determined, ORFs of these unclassified viruses were translated within Geneious and the resulting protein sequences were subjected to blastp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) searches. Further sequence analyses and phylogenetics are described below.
2.4. Stratford virus RT-rtPCR
In addition to IFA, a reverse transcription real-time polymerase chain reaction (RT-rtPCR) was used to confirm the isolation of Stratford virus (STRV). Primers and a dual-labelled probe were designed using the STRV C338 reference sequence (GenBank accession number L48962) targeting the non-structural protein 5 (NS5) and Primer Express™ software (PE Applied Biosystems USA) as follows: forward primer (STRATfor) 5ʹ-316GGTGAAGATGCTGCCTGCTT335-3ʹ, reverse primer (STRATrev) 5ʹ-384GGCTCCATTGGTTGTTTATCCA363-3ʹ, and probe (STRATprobe) 5ʹ FAM-337CGGCTCAGGGTGATGCCCTGA357-TAMRA 3ʹ. Each reaction consisted of 0.4 µl of Superscript™ III RT/Platinum® Taq mix, 10.0 µl of 2× reaction mix, 500 nM primers, 200 nM dual-labelled probe, 50 nM ROX reference dye, and 5 µl of extracted viral RNA in a total of 20 µl. Amplification was performed on a Rotor-Gene™ Q machine (Qiagen, Australia) with cycling conditions of one cycle at 50°C for 5 min, one cycle at 95°C for 2 min, and 50 cycles at 95°C for 3 s and 60°C for 30 s.
2.5. Phylogenetic analyses
Phylogenetic analyses were performed on complete ORFs (non-segmented viruses) or ORFs of representative segments of segmented viruses. Methodologies are provided for each virus group in the following descriptions. Protein alignments were performed using MAFFT (Katoh and Standley 2013) version 7.450 and the E-INS-i algorithm before automative trimming using TrimAl (Capella-Gutierrez, Silla-Martinez, and Gabaldon 2009). To estimate the best-fitting model of evolution for the sequence alignments, jModelTest 2.1.6 software (Darriba et al., 2012) was used for nucleotide alignments and ProtTest 3.4 (Darriba et al., 2011) for amino acid alignments. A summary of all the viruses and respective sequences used in the study is provided in Supplementary Table S1.
2.5.1. Flaviviruses—Stratford virus
Nucleotide sequence alignments of complete STRV ORFs (14 taxa) or a 556-bp fragment of the NS5 gene were performed using ClustalW and MEGA software (Tamura et al., 2011) version 7.0.26. The latter NS5 alignment (50 taxa) enabled a larger number of STRV sequences which were available on GenBank to be compared. Bayesian phylogenetic trees were inferred using the MrBayes plugin version 2.2.4 within Geneious with four chains of 1,000,000 Markov chain Monte Carlo (MCMC) generations, using the GTR + I + G nucleotide substitution model, sampling every 200 generations and discarding the first 10 per cent as burn-in.
2.5.2. Flaviviruses—unclassified viruses
Blastn searches using complete nucleotide ORFs did not yield sufficiently high matches with the GenBank database, so amino acid sequence datasets representing major flavivirus groups were used for sequence pairwise distance (163 taxa) and phylogenetic evolutionary analyses (90 taxa), respectively. A Bayesian phylogenetic tree was derived as above with four chains of 10,000,000 MCMC generations, using the LG + I + G + F amino acid substitution model, sampling every 2,000 generations and a burn-in of 10 per cent.
2.5.3. Orthomyxovirus—unclassified virus
Complete ORFs were predicted from six viral RNA segments, namely PB2 polymerase subunit (Segment 1), PB1 polymerase subunit (Segment 2), PA polymerase subunit (Segment 3), glycoprotein (G, Segment 4), nucleoprotein (NP, Segment 5), and matrix-like protein (M, Segment 6). Maximum-likelihood (ML) trees were estimated for Segments 1–5 using Mega 7.0.26 with 1,000 replicate bootstrap support. The amino acid substitution model LG + I + G + F was used for all segments except for Segment 4 (G protein) where WAG + I + G + F was used. Protein sequence datasets were composed of most closely related (determined by blastp) and other representative orthomyxoviruses and included the following number of taxa per segment: 28 (PB2), 33 (PB1), 28 (PA), 28 (G), 28 (NP), and 26 (M).
2.5.4. Orbivirus—Wongorr virus
The near-complete ORF of the RNA-dependent RNA polymerase (RdRp) gene (viral protein 1 (VP1); Segment 1) and complete ORFs of structural genes encoding viral protein 2 (VP2; Segment 2) and viral protein 7 (VP7; Segment 8) were predicted following NGS of viral RNA of the Ae. notoscriptus Wongorr virus (WGRV) isolate 205783. Phylogenetic ML trees for each segment based on amino acid sequences were derived as stated above for the orthomyxoviruses using the LG + I + G + F amino acid substitution model and included the following number of taxa: 45 (VP1), 43 (VP2), and 41 (VP7).
2.5.5. Mesonivirus—Casuarina virus and Negevirus—Ngewotan virus
Near-complete genome mesonivirus sequences were determined by NGS, but due to the limited availability of similar length, related mesonivirus sequences in GenBank, a dataset of ORF1ab polyprotein sequences (twenty-two taxa) was used for ML tree construction as stated above using the LG + I + G + F amino acid substitution model. We further performed Bayesian phylogenetics (as described for STRV above) to compare the near-complete genome sequence (19,923 nucleotides) of Casuarina virus (CASV) isolate 205089, with other available CASV isolates including the prototype 2010 Darwin, Northern Territory (NT), Australia 0071 isolate and isolates collected from the military Shoalwater Bay Training Area (SWBTA) in QLD, Australia in 2019, namely, SWBTA-Mes-ann from Culex annulirostris mosquitoes and SWBTA-Mes-vigilax from Aedes vigilax mosquitoes (GenBank accession numbers MT522182 and MT522183, respectively). The sequence of a third CASV SWBTA isolate (GenBank accession number MT522184) obtained from Aedes procax mosquitoes (Newton et al., 2020) was not included in our analysis due to the presence of a large number of ambiguous bases.
We also determined a near-complete genome nucleotide sequence from the NGS data for the Ae. notoscriptus Ngewotan virus (NWTV) isolate 205737 (∼9.2 kb). An amino acid dataset of complete ORF1 sequences (24 taxa) was used to phylogenetically compare 205737 with other representative negeviruses and an ML tree was inferred using the LG + I + G + F amino acid substitution model.
3. Results
3.1. Initial viral screen
A total of 495 pools of female Ae. notoscriptus, representing 6,674 mosquitoes, were processed. Seven of these pools caused CPE in the inoculated C6/36 cell monolayers, whilst two pools reacted with the anti-flavivirus mab 4G2 in the CC-EIA but did not cause an observable CPE. These two pools subsequently reacted with the anti-STRV/KOKV mab, 8A2 (Pyke, A.T., unpublished data) in an IFA.
A total of nine virus isolates from four different Brisbane suburbs were recovered from the Ae. notoscriptus pools from known genera, namely Flavivirus (3 STRV isolates), Orbivirus (1 WGRV isolate), Mesonivirus (1 CASV isolate), and Nelorpivirus (1 NWTV isolate) and three new unclassified ISVs of which two were identified as insect-specific flaviviruses (ISFs) and one a thogoto-like orthomyxovirus (Fig. 1). Because the isolation of STRV in Brisbane was unprecedented and may be associated with potential public health implications, we processed the remaining mosquito species from the trap collection that yielded the first STRV isolate. Whilst no further STRV isolates were obtained, CASV isolates were recovered from single pools of Ae. procax and Cx. annulirostris mosquitoes.
Figure 1.
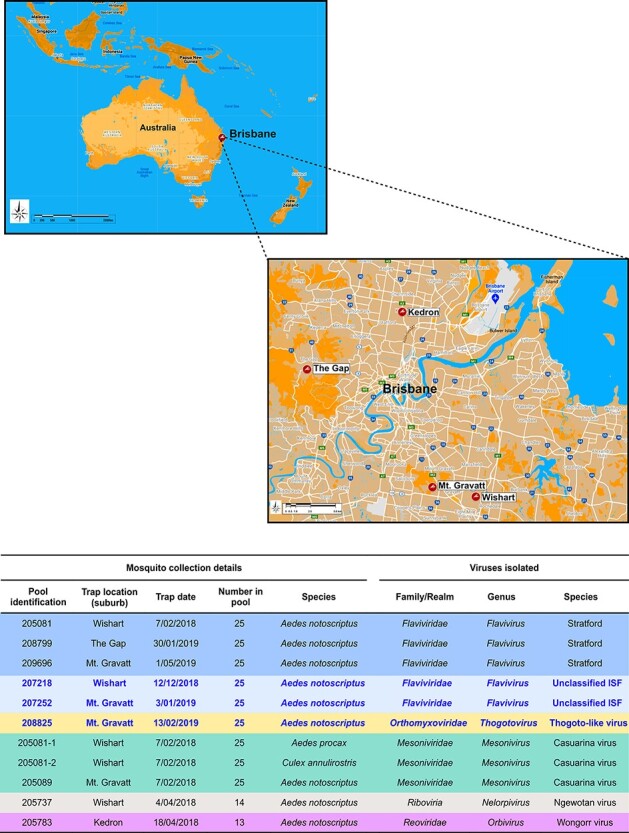
Summary of Ae. notoscriptus mosquito pool collections and viruses isolated. The maps show mosquito collection sites within the Brisbane urban area from which virus isolates were obtained. The table summarises details of the Ae. notoscriptus mosquito pool collections and virus isolates obtained. New, unclassified viruses and their respective mosquito pool collection data are highlighted in blue font. Two additional Casuarina virus isolates were detected in Ae. procax and Cx. annulirostris mosquito pools 205081–1 and 205081–2 respectively.
3.2. Phylogenetic analyses and virus characterisation
3.2.1. Stratford virus
Three isolates of STRV (205081, 209696, and 208799) were obtained from three different Brisbane suburbs from traps deployed 15 months apart. Infection with STRV or Kokobera virus (KOKV) was suspected based on the reaction to the anti-STRV/KOKV monoclonal antibody, 8A2 in the IFA (Fig. 2); the specific RT-rtPCR confirmed the identity of STRV.
Figure 2.

Isolation of STRV. Schematic summary showing the virus isolation process depicting images of Ae. notoscriptus (provided by Dr Cameron Webb), and STRV infected C6/36 cells stained in an immunofluorescence assay (IFA) using an anti-STRV/Kokobera virus monoclonal antibody, 8A2 (Pyke, A., unpublished data).
The complete ORF nucleotide sequences of the three Ae. notoscriptus STRV isolates 205081, 209696, and 208799 (GenBank accession numbers MZ358848 to MZ358850, respectively) were phylogenetically compared with two other available STRV sequences on GenBank (both of the C338 1961 prototype strain, GenBank accession numbers KF917540 and KM225263) and two new STRV sequences we had determined from Ae. vigilax mosquito isolates collected in 1995 (22911, GenBank accession number MZ358851) and 2006 (78701, GenBank accession numberMZ358852) from the state of NSW (Fig. 3A). These STRV sequences were also compared with representative sequences of six other closely related flaviviruses (Fig. 3A). Among the STRV isolates, the C338 prototype nucleotide sequences from 1961 were least similar to those of more recent isolates as shown by their separate grouping in the phylogenetic tree (Fig. 3A) and shared an average per cent nucleotide identity of between approximately 97 and 98 per cent with the other isolates. Among the recent STRV isolates, two of the Brisbane isolates, 205081 (MZ358848) and 208799 (MZ358850), were more closely related to NSW isolate 78701 (MZ358852), sharing 99.5 per cent pairwise nucleotide identity. Interestingly, Brisbane STRV isolate 209696 (MZ358849) did not group with the other two Brisbane STRV isolates, sharing only 98.4 per cent pairwise nucleotide identity with these viruses. This virus was more closely related to NSW STRV isolate 22911 (MZ358851) with which it shared 99.1 per cent nucleotide identity. Collectively, the STRV isolates, including the prototype C388 1961 strain (GenBank accession numbers KF917540 and KM225263), were most closely related to Torres virus (KM225265) which shared approximately 75 per cent mean nucleotide identity with STRV over the complete ORF region. Similar findings were apparent for the phylogenetic comparison of 50 STRV sequences for the 556-bp region of the NS5 gene (Fig. 3B) incorporating a larger number of available sequences from NSW, with the exception that STRV isolates 209696 (MZ358849) and 22911 (MZ358851) were more closely related to NSW isolates KU059146 and KU059132, respectively.
Figure 3.
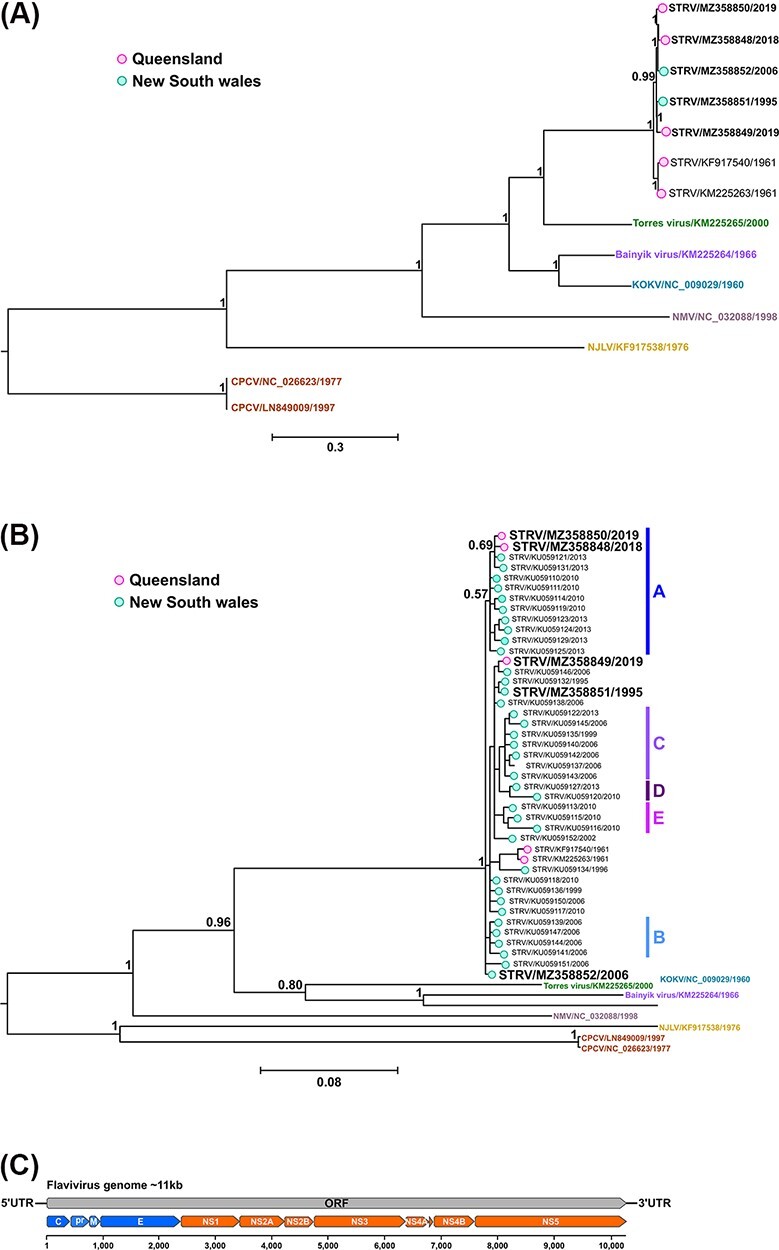
Bayesian phylogeny of nucleotide alignments comparing STRV isolates with other closely related flaviviruses and schematic of the flavivirus genome organisation. (A) Complete ORFs (14 taxa) and (B) a 556-bp region of the NS5 gene (50 taxa) were both analysed using the GTR + I + G substitution model. New STRV isolates are shown in bold font (including Ae. notoscriptus isolates, 205081, 209696, and 208799, GenBank accession numbers MZ358848 to MZ358850, respectively). The scale bars represent the substitution rates per site, and posterior probabilities of key nodes are included. For comparison only, the clade designations (A–E) previously determined for some STRV clusters (Toi et al., 2017) are also shown in (B). (C) Schematic of the flavivirus genome organisation.
3.2.2. Unclassified flaviviruses
Blastp searches on the translated amino acid ORFs of two unclassified Ae. notoscriptus isolates 207218 and 207252 (derived from near-complete genome nucleotide sequences, GenBank accession numbers MZ358856 and MZ358857, respectively) indicated the viruses were distantly related to other known flaviviruses, but were highly similar to each other, sharing 99.97 per cent pairwise amino acid identity (Fig. 4A). Indeed, only one amino acid change (valine to alanine at ORF position 459) was detected in the putative envelope (E) gene. Designated as Ae. notoscriptus flaviviruses (AenoFVs), a diagrammatic summary of the per cent pairwise amino acid identities and sliding-window analysis comparing the ORFs of 207218 (MZ358856) with 207252 (MZ358857) and 161 other flaviviruses is given in Fig. 4.
Figure 4.
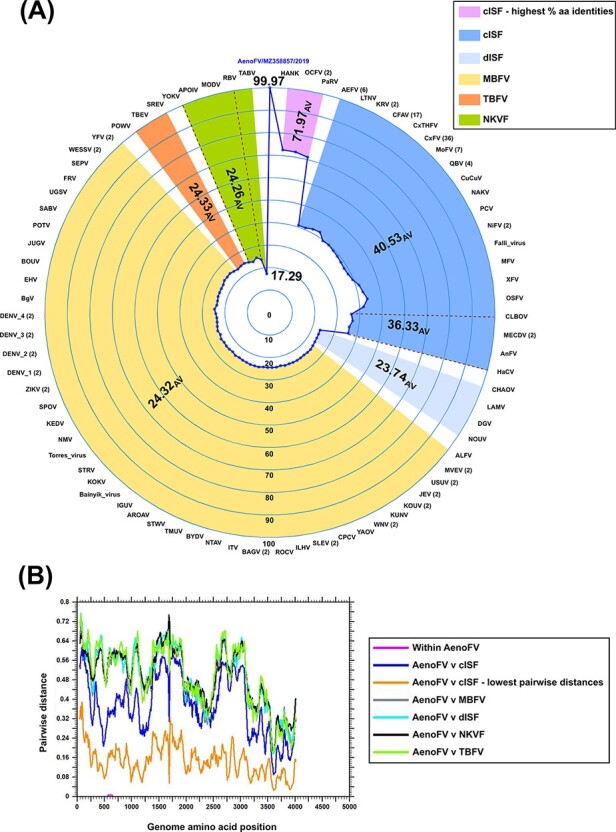
Pairwise amino acid analyses comparing unclassified Ae. notoscriptus flavivirus isolates with 161 known flaviviruses. Major flavivirus groups are shown, namely classical insect-specific flavivirus (cISF), dual-host associated insect-specific flavivirus (dISF), mosquito-borne flavivirus (MBFV), tick-borne flavivirus (TBFV), and no known vector flavivirus (NKVF). (A) Radar plot of per cent amino acid pairwise distances of complete flavivirus ORF sequences compared to Ae. notoscriptus flavivirus (AenoFV) 207218 (derived from the near-complete genome nucleotide sequence, GenBank accession number MZ358856) with number of sequences (if >1) used in the analysis included in parentheses. Average per cent (AV) pairwise values for each virus group are shown in bold. The cISFs, HANKV, OCFV, and PaRV sharing the highest per cent pairwise amino acid identities with AenoFVs 207218 (MZ358856) and 207252 (MZ358857) are also highlighted. (B) Sliding-window analysis using Simple Sequence Editor software (Simmonds 2012) comparing mean pairwise amino acid identities across the complete flavivirus ORF. Virus groupings and abbreviations as per Fig. 4A
Flaviviruses sharing the highest per cent pairwise amino acid identities with the two AenoFVs belonged to the classical insect-specific flavivirus (cISF) group and included Hanko virus (HANKV), Ochlerotatus caspius flavivirus (OCFV), and Parramatta River virus (PaRV) with 72.5 per cent, 72.3 per cent, and 71.0 per cent identities, respectively. Sliding-window analysis of mean pairwise distances (Fig. 4B) did not reveal any regions across the complete ORF of the AenoFVs that were disproportionate by comparison with other flaviviruses.
Bayesian phylogenetics of complete amino acid ORFs comparing the AenoFVs with 88 known flaviviruses is presented in Fig. 5. Both AenoFVs grouped as new distinct members of the Flaviviridae family in a monophyletic cluster with a high level of support (posterior probability of 1) within the cISF clade and were positioned basally to the cISFs HANKV and OCFV.
Figure 5.
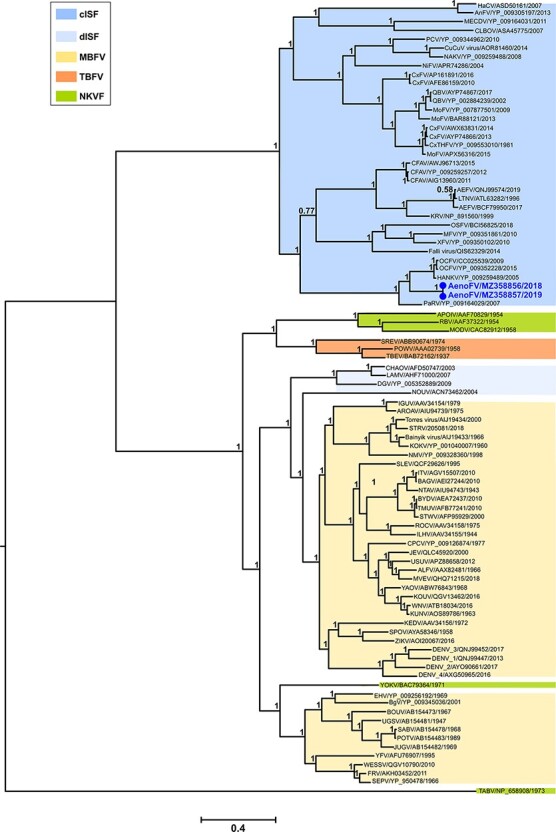
Bayesian phylogeny of amino acid alignments comparing unclassified Ae.notoscriptus flavivirus isolates with known flaviviruses. Complete ORFs (90 taxa) were analysed using the LG + I + G + F amino acid substitution model. The scale bar represents the substitution rates per site and support posterior probabilities are shown on key nodes. Flavivirus groupings and their abbreviations are as per those given in Fig. 4. New Ae. notoscriptus flavivirus (AenoFV) isolates, namely 207218 and 207252 (near-complete nucleotide sequence GenBank accession numbers MZ358856 and MZ358857, respectively) are highlighted in bold blue font.
3.2.3. Orthomyxovirus—unclassified virus
For the unclassified Ae. notoscriptus isolate 208825, blastp searches on the predicted translated ORFs of six RNA segments, later attributed to the PB2, PB1, PA, G, NP, and M segments (complete ORF nucleotide sequence GenBank accession numbers MZ363949 to MZ363954, respectively), produced highest per cent amino acid identities with other members of the Orthomyxoviridae virus family. Designated as Ae. notoscriptus orthomyxovirus virus (AenoOMV), the isolate was most closely related to viruses in the genus Thogotovirus (represented by major Thogotovirus (THOV) species) and to Varroa orthomyxovirus_1 (VOV_1) and Sinu virus (SINUV) (Table 1).
Table 1.
Genome segments of Ae. notoscriptus orthomyxovirus and summary of predicted orthomyxovirus ORFs with percentage pairwise amino acid sequence identities compared with cognate sequences of thogotovirus, Varroa orthomyxovirus_1 and Sinu virus.
| % Pairwise identity (aa) | |||||||
|---|---|---|---|---|---|---|---|
| Segment | Putative protein | ORF length (nt) | ORF length (aa) | THOVb (range) | VOV_1c | SINUVc | GenBank accession numbera |
| 1 | PB2 | 2,271 | 757 | 28.1–31.4 | 24.2 | 26.5 | MZ363949 |
| 2 | PB1 | 2,181 | 727 | 54.6–57.3 | 53.5 | 55.2 | MZ363950 |
| 3 | PA | 1,926 | 642 | 32.3–33.8 | 30.9 | 30.2 | MZ363951 |
| 4 | G | 1,272 | 424 | 19.0–22.9 | 16.1 | 18.3 | MZ363952 |
| 5 | NP | 1,320 | 440 | 32.5–41.5 | 37.6 | 34.5 | MZ363953 |
| 6 | Matrix-like | 819 | 273 | 13.7–18.1 | 16.0 | 10.3 | MZ363954 |
GenBank accession number of Ae. notoscriptus orthomyxovirus (AenoOMV) ORF nucleotide sequence from which amino acid sequences were derived.
Range of amino acid % pairwise identities with thogotovirus (THOV) sequences (10 representative sequences chosen from major THOV phylogenetic clades as shown in Figure 6).
GenBank accession numbers for Varroa orthomyxovirus_1 (VOV_1) and Sinu virus (SINUV) sequences are summarized in Supplementary Table 1.
Of all the AenoOMV segments, PB1 demonstrated the highest per cent amino acid identities (53.5–57.3 per cent) with THOV, VOV_1, and SINUV groups. In comparison with the same three virus groups, the AenoOMV PA and NP segments shared similar per cent amino acid identities (30.2–33.8 per cent and 32.5–41.5 per cent, respectively) and the G and M segments shared the lowest per cent amino acid identities (16.1–22.9 per cent and 10.3–18.1 per cent, respectively).
The ML phylogenies of five representative AenoOMV gene segments deduced from complete ORF amino acid sequences indicate that AenoOMV is a new divergent virus which groups independently in a monophyletic clade compared with other orthomyxoviruses (Fig. 6). Topologies of trees inferred from PB2, PB1, PA, and G proteins were similar with AenoOMV, SINUV, and VOV_1 each placed individually in basal positions relative to viruses of the Thogotovirus genus. For the NP, AenoOMV grouped within the THOV clade together with VOV_1 and SINUV, although both were still notably divergent and positioned in separate monophyletic clades.
Figure 6.
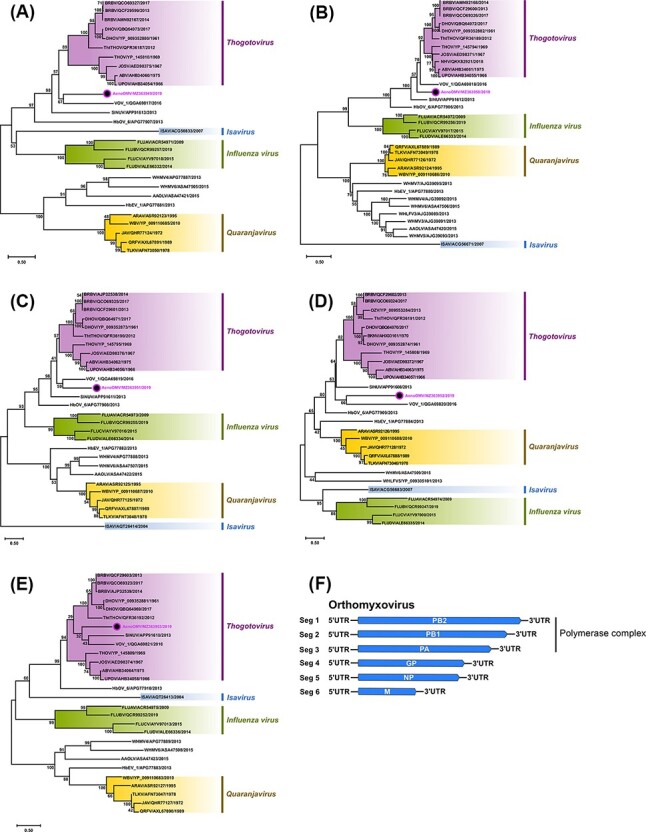
Phylogenetic analyses of five genes of unclassified AenoOMV based on amino acid sequences of predicted ORFs and orthomyxovirus genome segment summary. The midpoint rooted ML trees inferred from the full-length ORF sequences of (A) PB2, (B) PB1, (C) PA, (D) G, and (E) NP genes are shown with bootstrap support estimated from 1,000 replicates. Major orthomyxovirus groupings are named and highlighted (new Ae. notoscriptus isolate 208825 is identified by a coloured dot), and the scale bar represents the amino acid substitution rates per site. The LG + I + G + F amino acid substitution model was used for all analyses except for the G protein where WAG + I + G + F was used. (F) A schematic summary of the orthomyxovirus genome segments.
Recently, a newly identified THOV, Bourbon virus (BRBV), caused a fatal infection in a male from Bourbon County, Kansas, in the USA in 2014 (Kosoy et al., 2015). Given AenoOMV PB2, PB1, PA, NP, and G proteins shared some phylogenetic relatedness with corresponding proteins from members of the Thogotovirus genus, we assessed the ability for AenoOMV to infect vertebrate cells using 12 cell lines (Vero, BGMK, HuH-7, HEK293, MRC-5, RD, A549, HeLa, MDBK, MDCK, BHK-21, and LLCMK2). No cell morphological changes or CPE were observed for any of the cell lines following three passages, including the six human cell lines (HuH-7, HEK293, MRC-5, RD, A549, and HeLa).
3.2.4. Orbivirus—Wongorr virus
A near-complete ORF nucleotide sequence for VP1 (5ʹ-terminal M amino acid residue could not be resolved) and complete ORF sequences for VP2 and VP7 RNA segments were assembled from NGS data obtained from the Ae. notoscriptus isolate 205783 (GenBank accession numbers MZ358859, MZ358860, and MZ363936, respectively). Blastn (nucleotide sequences) and Blastp (amino acid sequences derived from nucleotide sequences) searches identified the isolate as the orbivirus WGRV, which shared high per cent sequence identities with the WGRV group incorporating the WGRV species WGRV, Paroo River virus (PARV), and Picola virus (PIAV) (Table 2). Whilst we were unable to definitively establish the isolation year of WGRV isolate MRM15174, another previously reported WGRV isolate, MRM13443, was originally isolated from Mitchell River, QLD, Australia, in 1970. Unfortunately, only partial VP1 nucleotide and VP3 sequences are publicly available for MRM1344 on GenBank and therefore were not used in this study.
Table 2.
Summary of Wongorr virus isolate 205783 VP1, VP2 and VP7 protein ORFs and percentage pairwise nucleotide and amino acid sequence identities compared to cognate sequences of Wongorr virus, Paroo River virus and Picola virus.
| % Pairwise identity (nt/aa) | |||||||
|---|---|---|---|---|---|---|---|
| Segment | Putative protein | ORF length (nt) | ORF length (aa) | WGRVc | PARVc | PIAVc | GenBank accession numberb |
| 1 | VP1 | 3,879a | 1,293a | 98.2/99.5 | 96.9/99.1 | 96.8/99.1 | MZ358859 |
| 2 | VP2 | 2,718 | 906 | 97.1/99.6 | 97.8/99.7 | 97.6/99.4 | MZ358860 |
| 8 | VP7 | 1,050 | 350 | 97.2/100 | 98.0/100 | 97.7/100 | MZ363936 |
The predicted 5 terminal methionine amino acid residue (ATG) for the VP1 ORF could not be resolved from the 205783 isolate NGS data.
GenBank accession number of the Ae. notoscriptus205783 Wongorr virus (WGRV) ORF nucleotide sequence.
The sequences of WGRV (MRM15174), Paroo River virus (PARV; GG668) and Picola virus (PIAV; PK886) isolates were used for the pairwise sequence analysis (GenBank accession numbers for each virus are summarized in Supplementary Table 1).
Consistent with the pairwise amino acid identity data, phylogenetic analyses of highly conserved VP1, VP2, and VP7 ORF amino acid sequences (Fig. 7) demonstrated grouping of the Ae. notoscriptus 205783 isolate with previously reported WGRV, PARV, and PIAV orbiviruses.
Figure 7.
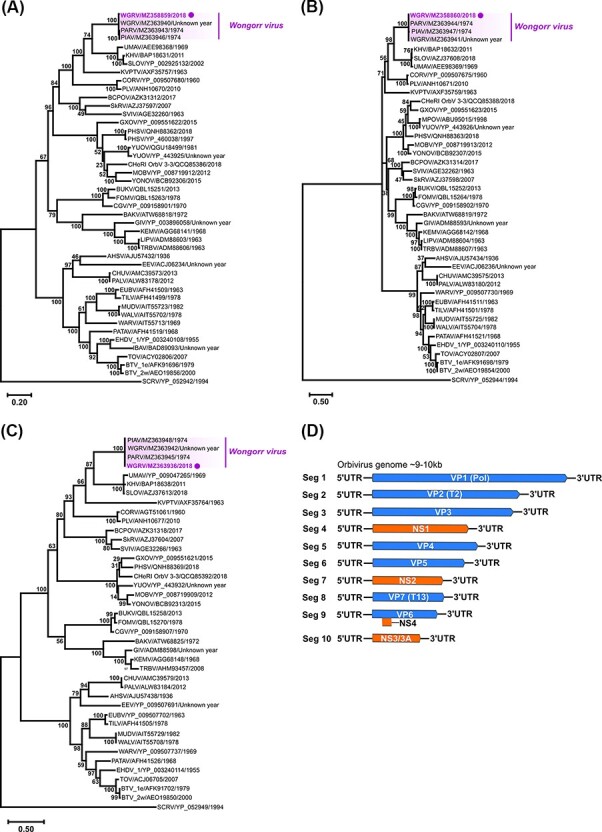
Phylogenetic analyses of orbivirus VP1, VP2, and VP7 genes based on ORF amino acid sequences and orbivirus genome segment summary. The midpoint rooted ML trees deduced from near-complete (A) VP1 (45 taxa) and complete (B) VP2 (43 taxa), and (C) VP7 (41 taxa) ORF sequences are shown with bootstrap support estimated from 1,000 replicates. The WGRV subgroup consisting of WGRV, PARV, and PIAV is highlighted (new Ae. notoscriptus isolate 205783 highlighted by a purple dot) and the scale bar represents the amino acid substitution rates per site. The LG + I + G + F amino acid substitution model was used for all analyses. (D) A schematic summary of the orbivirus genome segments.
The ORF sequences of the remaining seven orbivirus genes (VP3, VP4, VP5, VP6 (non-structural protein (NS) 4), NS1, NS2, and NS3) were also predicted following NGS of 205783 RNA (GenBank accession numbers MZ358861 to MZ358864 and MZ363937 to MZ363939, respectively) but further analyses were not performed due to the limited availability of representative WGRV group or other orbivirus sequences of these genes.
3.2.5. Mesonivirus—Casuarina virus
Near complete genome nucleotide sequences (∼20 kb) were assembled following NGS of Brisbane CASV isolates, 205081_1 (Ae. procax), 205081_2 (Cx. annulirolstris), and 205089 (Ae. notoscriptus) (GenBank accession numbers MZ358853 to MZ358855, respectively). Per cent pairwise nucleotide identities shared among these three isolates were extremely high (99.99 per cent), contrasting with lower per cent nucleotide identities when compared with cognate sequences (19,923 nucleotides) of the prototype 0071 (98.5 per cent), and the two SWBTA isolates (SWBTA-Mes-ann (95.0 per cent) and SWBTA-Mes-vigilax (96.7 per cent), respectively) (Table 3).
Table 3.
Percentage nucleotide and amino acid sequence identities of Casuarina virus isolate 205089 compared with sequences of other Casuarina virus isolates for functionally known ORFs.
| % Pairwise identity (nt/aa) | ||||||||
|---|---|---|---|---|---|---|---|---|
| ORF domain | Putative protein(s) | Length nucleotide (nt) | Length amino acid (aa) | 205081_1a | 205081_2a | 0071a | SWBTA-Mes-anna | SWBTA-Mes-vigilaxa |
| 1abb | Polyprotein incorporating 1a and 1b ORFs and RFS motif | 15,195 | 5,065 | 100/100 | 100/100 | 98.6/99.4 | 95.3/97.2 | 97.6/98.6 |
| 1a | 3C-like chymotrypsin-like protease (3CLpro) and flanking transmembrane domains | 7,434 | 2,478 | 100/100 | 100/100 | 98.6/99.4 | 95.5/97.7 | 99.8/100 |
| 1b | RNA-dependent RNA polymerase (RdRp) and other related replicase domains | 7,821 | 2,607 | 100/100 | 100/100 | 98.5/99.4 | 95.2/96.7 | 95.6/97.0 |
| 2a | Spike protein | 2,691 | 897 | 100/100 | 100/99.9 | 98.2/97.8 | 92.2/89.7 | 92.2/89.7 |
| 2b | Nucleocapsid protein | 630 | 210 | 100/100 | 100/100 | 97.5/98.6 | 90.2/91.0 | 90.3/91.0 |
| 3a | Membrane protein | 468 | 156 | 100/100 | 100/100 | 95.3/96.1 | 93.4/91.7 | 93.4/91.7 |
| 3b | Membrane protein | 345 | 115 | 100/100 | 100/100 | 96.8/94.8 | 95.9/94.8 | 95.9/94.8 |
GenBank accession numbers of near complete genome nucleotide Casuarina virus (CASV) sequences used: 205081_1 (MZ358853), 205081_2 (MZ358854) and 205089 (MZ358855), 0071 (KJ125489), SWBTA-Mes-ann (MT522182) and SWBTA-Mes-vigilax (MT522183).
Contains a transcriptional, slippage-prone ribosomal frameshift (RFS) motif GGAUUUU which can result in read-through of ORF1a and facilitate translation of the polyprotein 1ab (Vasilakis et al. 2014).
With the exception of Protein 1a, the pairwise sequence analyses of individual mesonivirus ORFs consistently indicated isolate 205089 shared similar and lower per cent nucleotide and amino acid identities with each of the SWBTA isolates, in particular, SWBTA-Mes-ann. Interestingly, the 1a protein of SWBTA-Mes-ann contained 56 amino acid changes compared with the 1a proteins of the three Brisbane CASV isolates with 35 of these (63 per cent) positioned in the 3ʹ-terminal, one-third of the protein sequence (amino acids 1652–2478). This region is essentially downstream from the putative 3C-like chymotrypsin-like protease (3CLpro) serine protease domain which is among several functional ORF 1ab domains that are highly conserved between mesoniviruses.
Phylogenetic trees were inferred from complete 1ab polyprotein ORF amino acid mesonivirus sequences and near-complete genome nucleotide CASV sequences (Fig. 8). In each tree, the Brisbane 2018 CASV isolates (GenBank accession numbers MZ358853 to MZ358855) clustered together within the CASV clade (subgenera Alphamesonivirus 4) and were more phylogenetically related to the prototype 0071 NT 2010 isolate (GenBank accession number KJ125489) than to either of the SWBTA 2019 isolates SWBTA-Mes-ann and SWBTA-Mes-vigilax (GenBank accession numbers MT522182 and MT522183, respectively) which formed an additional discrete cluster within the CASV clade.
Figure 8.
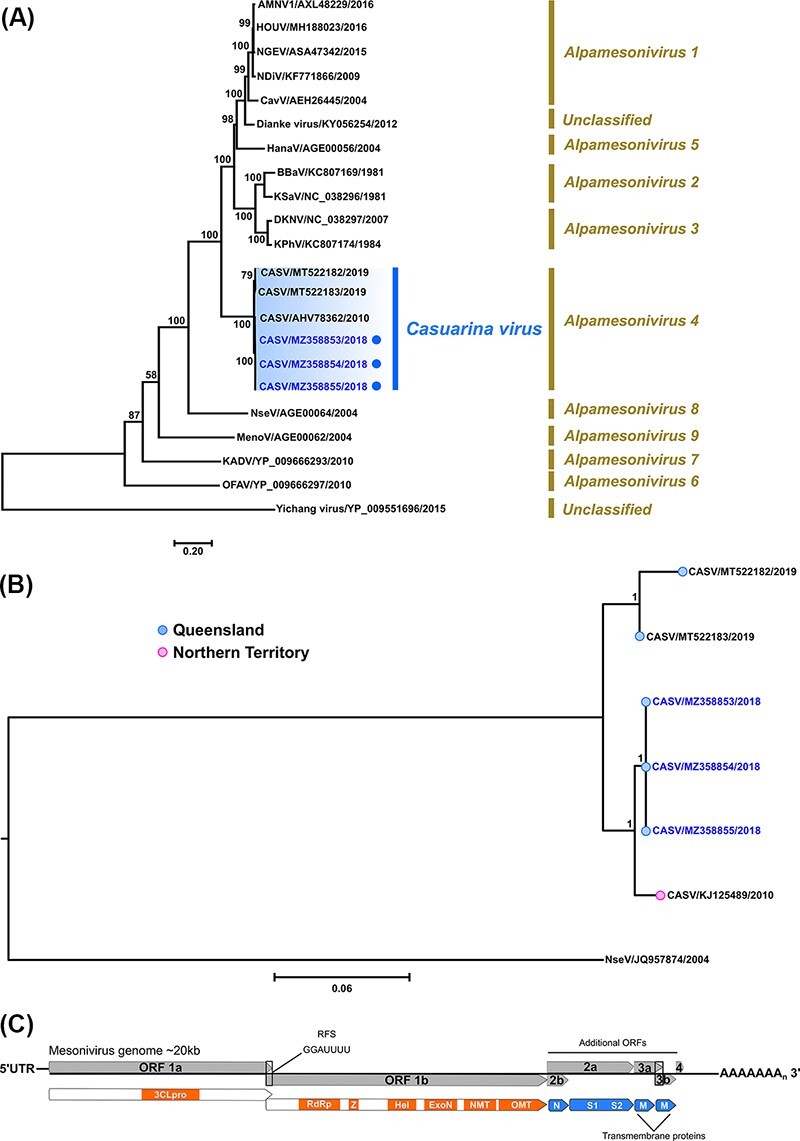
Phylogenetic analyses of complete mesonivirus 1ab polyprotein ORFs based on amino acid sequences and near-complete nucleotide CASV sequences. (A) Midpoint rooted ML tree inferred from complete 1ab polyprotein amino acid sequences (22 taxa) using the LG + I + G + F amino acid substitution model is shown (scale bar represents amino acid substitution rates per site) with bootstrap support estimated from 1,000 replicates. Corresponding mesonivirus subgenera (Alphamesonivirus) groupings are given and the CASV clade is highlighted (subgenera Alphamesonivirus 4), with the new Brisbane Ae. procax (205081–1), Cx. annulirostris (205081-2) and Ae. notoscriptus (205089) isolates (GenBank accession numbers MZ358853 to MZ358855, respectively) identified by blue. (B) Bayesian tree deduced from near-complete genome nucleotide CASV sequences (19,923 nucleotides) using the GTR + I + G substitution model which was rooted using the divergent Nsè virus as an outgroup. New CASV isolates are highlighted in blue font. Posterior probabilities are shown for key nodes and scale bar represents nucleotide substitution rates per site. (C) Graphic summary of the mesonivirus genome organisation.
3.2.6. Negevirus—Ngewotan virus
A near-complete genome (∼9.2 kb) consisting of three ORFs which corresponded to NWTV virus ORFs (ORF1, ORF2, and ORF3) was assembled from the Ae. notoscriptus isolate 205737 NGS sequence data (near-complete genome nucleotide sequence, GenBank accession number MZ358858). For each ORF, 205737 consistently shared highest per cent pairwise nucleotide and amino acid identities (>98.0 per cent) with cognate ORF sequences of NWTV isolate 155762 (Fig. 9A), originally recovered from Cx. annulirostris mosquitoes collected in Cairns, QLD, Australia, in 2006 (Colmant et al., 2020).
Figure 9.
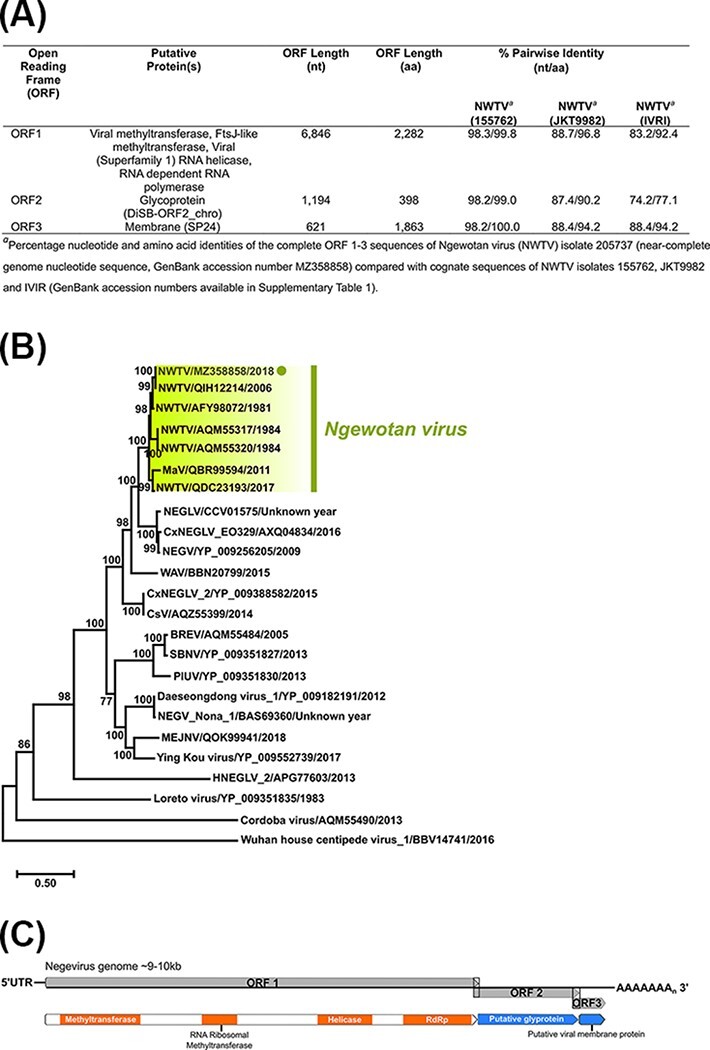
Per cent pairwise nucleotide and amino acid identities of complete Ngewotan ORF 1–3 sequences, ORF 1 phylogenetic analysis, and negevirus genome summary. (A) Per cent pairwise nucleotide and amino acid identities of Ae. notoscriptus NWTV isolate 205737 (near-complete genome nucleotide sequence, GenBank accession number MZ358858) complete ORF 1–3 sequences compared with cognate sequences of NWTV isolates 155762, JKT9982 and IVRI (GenBank accession numbers available in Supplementary Table S1). (B) Midpoint rooted ML tree deduced from complete ORF1 amino acid sequences (24 taxa) using the LG + I + G + F amino acid substitution model is shown (scale bar represents amino acid substitution rates per site) with bootstrap support estimated from 1,000 replicates. The Ae. notoscriptus NWTV isolate 205737 (MZ358858) is highlighted within the NWTV clade. (C) Schematic representation of the negevirus genome organisation.
Recently, NWTV was included in a new taxonomic group of Nelorpiviruses designated Negevirus along with Negev, Piura, Loreto, Dezidougou, and Santana viruses (Vasilakis et al., 2013). We deduced a ML tree from an amino acid dataset composed of 24 complete ORF1 sequences (Fig. 9B). The 205737 NWTV isolate (MZ358858) was most phylogenetically related to the NWTV isolate 155762 (QIH12214) and clustered with this virus within the NWTV clade.
4. Discussion
The rapid generation and extensive data afforded by high-throughput sequencing and metagenomic analysis of mosquito pools has facilitated the mapping of mosquito virome profiles (Shi et al., 2017; Sadeghi et al., 2018). We focused on culture techniques in C6/36 cells for the purposes of providing viral genomes for the sequencing and characterisation of live viruses carried by Ae. notoscriptus. Broader genotypic, phenotypic, and pathological investigations including a combination of these methods, rather than individually, provides enhanced capturing of both the existence and viability of viruses associated with this mosquito species. These could be further complemented by a comparison between viral sequences obtained directly from host samples and those acquired post isolation and culture to investigate any cell-induced, viral adaptive genomic changes. Inclusion of other host cell lines such as vertebrate cells for live virus isolation retrieval could also be considered; however, C6/36 cells are among the most sensitive cells available for isolation of potential arboviruses of medical significance.
Numerous arboviruses, such as RRV and BFV, are commonly reported from the Brisbane area. Isolation of STRV provides contemporary evidence of the presence of another potentially pathogenic arbovirus in Brisbane with implications for human health. Based on serological evidence, sporadic human infections with STRV have been reported in asymptomatic people in NSW (Hawkes et al., 1985) and symptomatic patients in NSW and QLD, with fever, arthritis, and lethargy listed among the symptoms (Phillips et al., 1993; Johansen, Maley, and Broom 2005). Although the extent and frequency of clinical and pathological manifestations resulting from STRV infections are largely unknown, between 2017 and 2020, the Queensland Department of Health reported a total of 49 specific anti-STRV immunoglobulin M (IgM) and 130 cross-reactive anti-STRV/KOKV IgM-positive cases which were temporally distributed across each of these years.
Within the Flavivirus genus, STRV is a member of the KOKV subgroup and was first isolated from Ae. vigilax mosquitoes collected in Cairns, QLD, Australia in 1961 (Doherty et al., 1963). Although limited to a comparison of five available contemporary STRV isolates, phylogenetic analysis of complete ORF nucleotide sequences demonstrated genetic diversity among each of the respective QLD and NSW isolate groups. Together with our detection of multiple isolates of STRV in Brisbane which occurred over 15 months, these findings support the serological evidence suggesting continual transmission of STRV in the Brisbane area. In our phylogenetic analysis, we also included a tentative, broader phylogenetic comparison of our virus isolates with other available NS5 STRV sequences (Toi et al., 2017) (Fig. 3B). However, only a relatively small (556 nt), conserved region could be analysed and a more robust whole-genome phylogenetic comparison with whole genomes, together with a review of previously reported clades, should be undertaken as more NGS data become available.
Although WGRV has not yet been associated with human disease, infections of cattle and marsupials have been reported based on serological evidence (Doherty et al., 1973; Belaganahalli et al., 2014). Other Australian orbiviruses, namely Wallal, Warrego, and Eubenangee viruses have also been isolated from marsupials and are casually linked with disease in these mammals (Rose et al., 2012; Belaganahalli et al., 2014). Our phylogenetic analysis and grouping of WGRV with PARV and PIAV based on VP1, VP2, and VP7 gene sequences correlates with previous studies using partial VP3 gene sequences (Parkes and Gould 1996). This WGRV subgroup was also most phylogenetically related to the orbiviruses Umatilla, Koyama Hill, and Stretch Lagoon viruses, the latter being reported from Australia in 2002 and 2007 (Cowled et al., 2009; Jansen et al., 2009), and the USA in 2018 (Tangudu et al., unpublished data, nucleotide sequence GenBank accession numbers MK100579, MK100580, and MK100585).
In the current study, we also isolated from Ae. notoscriptus viruses belonging to the genera Flavivirus, Mesonivirus and Nelorpivirus, thereby extending the range of mosquito species that ISVs infect. Because of their phylogenetic relatedness to other medically important flaviviruses, such as WNV, ZIKV, YFV, and dengue viruses, ISFs are increasingly being recognised as potential biological control, vaccine, or diagnostic delivery vehicles in either native or chimeric viral forms (Roundy et al., 2017; Hobson-Peters et al., 2019). The discovery and study of new ISFs, as presented here, assist these and future efforts to control and reduce the threat posed by pathogenic arboviruses. If AenoFV can lower mosquito susceptibility to pathogenic arboviruses, such has been observed for Palm Creek virus (Hobson-Peters et al., 2013; Hall-Mendelin et al., 2016), this ISF may have a role in modulating both enzootic transmission and subsequent spillover of harmful arboviruses to humans or animals.
A member of the Mesoniviridae family, CASV, belongs to the subgenera Alphamesonivirus 4 and was the first Australian mesonivirus identified (Warrilow et al., 2014). Now reported from both the NT and QLD, it is likely that the geographical distribution of the virus could be greater than is currently described. Interestingly, the phylogenetic analysis of both CASV 1ab amino acid sequences and near-complete nucleotide sequences (Fig. 8A, B, respectively) revealed QLD isolates grouped into two distinct CASV clades, characterised by spatial and temporal clustering. One clade, comprising the Brisbane 2018 CASV isolates, was found to be more phylogenetically related to the original CASV NT isolate sampled from Coquillettidia xanthogaster mosquitoes in 2010 (Warrilow et al., 2014) and was distinct from the other CASV clade which incorporated the SWBTA 2019 QLD CASV isolates recently discovered in Ae. vigilax and Cx. annulirostris mosquitoes (Newton et al., 2020). With so few isolates, it is difficult to infer if seeding of CASV from the NT into QLD has occurred, although there appears a tentative relationship between the Brisbane isolates and the NT 2010 prototype strain, with the latter positioned basally relative to the Brisbane viruses within the phylogenetic trees. Of note, the clustering of the Brisbane and SWBTA isolates into two distinct clades is not surprising given the high degree of diversity between the two respective geographical landscapes. Whilst Brisbane is a major urban centre, the SWBTA is a natural environment of 4,545 km2 characterised by a wide range of individual landscapes, including mountainous and coastal regions, with equally diverse populations of flora and fauna species. We also observed a higher degree of genetic diversity between the SWBTA isolates which shared 98.2 per cent nucleotide identity compared to the nucleotide identity shared between Brisbane isolates which was approximately 100.0 per cent. It is therefore plausible that there is higher degree of CASV evolution within the SWBTA indicative of more numerous and/or more complex modes of transmission.
In contrast to CASV, analysis of NWTV sequences demonstrated that the two QLD isolates, 205737 (Ae. notoscriptus, 2018) and 155762 (Cx. annulirostris, 2006), shared high-per cent pairwise nucleotide and amino acid identities for all three ORF segments (Fig. 9A) and clustered together in the ORF 1 amino acid phylogenetic tree (Fig. 9B). NTWV is one of six ISV species in the recently proposed taxon, Negevirus, which has a broad geographic distribution (Vasilakis et al., 2013).
Amino acid phylogenetic analyses demonstrated that the new, previously unclassified ISV, AenoOMV, is a divergent orthomyxovirus most closely related to the ISVs VOV_1 and SINUV and other members of the Thogotovirus genus. VOV_1 was originally isolated from both Western honey bees (Apis mellifera) and the bee ectoparasite mite (Varroa destructor) (Levin et al., 2019), and SINUV was first isolated from mosquitoes collected in Columbia in 2013 (Contreras-Gutierrez et al., 2017). Thogotoviruses have been implicated as pathogens of human and veterinary importance, including Bourbon virus (BRBV), which caused the death of a male in 2014 following tick-bite exposure in Kansas in the USA (Lambert et al., 2015). Although we investigated the potential of AenoOMV to infect vertebrate cells, we did not find any evidence to support this in the cell lines used in this study, inferring that the virus is likely an ISV.
For each of the phylogenetic comparisons of orthomyxovirus genes, the AenoOMV isolate presented as a divergent, monophyletic clade in all ML trees generated. The basal phylogenetic positioning of the virus in most of the trees relative to the Thogotovirus genus clade also suggested that AenoOMV probably diverged earlier than the known thogotoviruses. These phylogenies are like those previously reported for SINUV and VOV_1, which were both reported as new orthomyxovirus species, most closely related to the Thogotovirus genus (Contreras-Gutierrez et al., 2017; Levin et al., 2019). However, we observed generally low bootstrap values for the positioning of the divergent AenoOMV within trees, hence there is currently only tentative support for its phylogenetic relatedness with the Thogotovirus genus. Interestingly, the analysis of the NP proteins of AenoOMV, VOV_1, and SINUV produced alternate phylogenetic variation whereby these viruses clustered within the Thogotovirus genus clade, albeit again with low bootstrap support. Although these phylogenies remain unclear, we did observe some regions sharing >50 per cent amino acid identity among AenoOMV, VOV_1, and SINUV species following comparison with Thailand tick, Bourbon, and Dhori thogotoviruses within the trimmed NP sequence alignment (final length, 366 residues; Supplementary Figure S1). Reassortment of gene segments occurs frequently in orthomyxoviruses and is a major factor contributing to their evolution and the emergence of new and occasionally more virulent strains (Markussen et al., 2008). It is possible that the consistent phylogenetic relatedness of AenoOMV, VOV_1, and SINUV is due to modes of viral transmission in instances where the temporal and spatial distributions of these diverse insect groups overlap. For example, both honey bees and mosquitoes consume floral nectaries (Foster 1995; Berenbaum and Calla 2021). Therefore, it is plausible that transmission may occur via exposure to virus-infected nectar, similar to that suggested by Newton et al., (2020) for mesoniviruses. Furthermore, it is also possible that each of these viruses have an even broader host range than the respective mosquito, bee, and mite species they were first isolated from, in which case, additional modes of transmission may also be involved.
Based on the results described herein and previous isolation of RRV (Ritchie et al., 1997), Edge Hill virus (Doggett et al., 2017), BFV (Doggett et al., 2011), STRV (Toi et al., 2017), Sindbis virus (Broom et al., 2002), and Nam Dinh virus (Newton et al., 2020), Ae. notoscriptus appears to harbour a genetically diverse virome. Further evidence for the diverse nature of the Ae. notoscriptus virome is the susceptibility of this species in laboratory studies to infection with pathogenic arboviruses from different viral families, including Flaviviridae (West Nile virus and Zika virus) (Jansen et al., 2008; Duchemin et al., 2017), Togaviridae (chikungunya virus) (van den Hurk et al., 2010), and Bunyaviridae (Rift Valley fever virus) (Turell and Kay 1998). Thus, it is likely that the diversity of virus species harboured by this mosquito may be even greater. Whilst consideration of any associated public health or veterinary implications of any of these viruses is of primary importance, many of these viruses require more specific characterisation and investigation with regard to their replication dynamics, host range, prevalence, and association with human and animal disease. Therefore, further mosquito sampling is warranted and should encompass a broader geographical and temporal range. This is important, because Ae. notoscriptus is a highly urbanised and invasive mosquito species, with the potential to follow in the footsteps of Ae. aegypti and Ae. albopictus and could become a vector of concern transmitting RRV, or other endemic or exotic pathogens, in newly colonised locations.
Supplementary Material
Acknowledgements
We sincerely thank Dr Son Nguyen, Forensic and Scientific Services, and Dr Sebastian Duchene, University of Melbourne, for assistance and advice regarding computational phylogenetic programmes and the Queensland Public Health and Infectious Diseases Reference Genomics for their assistance in running the NextSeq 500 sequencer. We are also grateful to Professor Roy Hall, University of Queensland for supply of B10 and 4G2 monoclonal antibodies and to Dr Stephen Doggett and John Haniotis, NSW Health Pathology, Westmead Hospital, Sydney, for provision of mosquito pools sampled from NSW from which STRV was subsequently isolated and Carmel Taylor, Forensic and Scientific Services, for retrieval of STRV patient data.
Contributor Information
Alyssa T Pyke, Department of Health, Public Health Virology Laboratory, Forensic and Scientific Services, Queensland Government, 39 Kessels Road, Coopers Plains, QLD 4108, Australia.
Martin A Shivas, Brisbane City Council, 20 Tradecoast Drive, Eagle Farm, Brisbane, QLD 4009, Australia.
Michael B Onn, Brisbane City Council, 20 Tradecoast Drive, Eagle Farm, Brisbane, QLD 4009, Australia; Metro North Public Health Unit, Queensland Health, Bryden Street, Windsor, QLD 4030, Australia.
Andrew Crunkhorn, Metro North Public Health Unit, Queensland Health, Bryden Street, Windsor, QLD 4030, Australia.
Ivan Montgomery, Brisbane City Council, 20 Tradecoast Drive, Eagle Farm, Brisbane, QLD 4009, Australia; Metro North Public Health Unit, Queensland Health, Bryden Street, Windsor, QLD 4030, Australia.
Cassie C Jansen, Communicable Diseases Branch, Queensland Health, 15 Butterfield Street, Herston, QLD 4006, Australia; Department of Health, Public Health Virology Laboratory, Forensic and Scientific Services, Queensland Government, 39 Kessels Road, Coopers Plains, QLD 4108, Australia.
Andrew F van den Hurk, Department of Health, Public Health Virology Laboratory, Forensic and Scientific Services, Queensland Government, 39 Kessels Road, Coopers Plains, QLD 4108, Australia.
Data availability
Virus sequences are available in the National Center for Biotechnology Information GenBank database at https://www.ncbi.nlm.nih.gov/genbank/.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This work was funded by the Mosquito and Arbovirus Research Committee.
Conflict of interest
None declared.
Human ethics statement
This work has received ethical clearance from the Forensic and Scientific Services Human Ethics Committee, approval reference HEC 21-17.
References
- Bankevich A. et al. (2012) ‘SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-cell Sequencing’, Journal of Computational Biology, 19: 455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaganahalli M. N. et al. (2014) ‘Full Genome Characterization of the Culicoides-borne Marsupial Orbiviruses: Wallal Virus, Mudjinbarry Virus and Warrego Viruses’, PLoS One, 9: e108379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum M. R., and Calla B. (2021) ‘Honey as a Functional Food for Apis Mellifera’, Annual Review of Entomology, 66: 185–208. [DOI] [PubMed] [Google Scholar]
- Bigot D. et al. (2018) ‘Discovery of Culex Pipiens Associated Tunisia Virus: A New ssRNA(+) Virus Representing a New Insect Associated Virus Family’, Virus Evolution, 4: vex040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitvich B. J., and Firth A. E. (2015) ‘Insect-specific Flaviviruses: A Systematic Review of Their Discovery, Host Range, Mode of Transmission, Superinfection Exclusion Potential and Genomic Organization’, Viruses, 7: 1927–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broom A. K. et al. (1998) ‘Identification of Australian Arboviruses in Inoculated Cell Cultures Using Monoclonal Antibodies in ELISA’, Pathology, 30: 286–8. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2002) ‘Investigation of the Southern Limits of Murray Valley Encephalitis Activity in Western Australia during the 2000 Wet Season’, Vector-Borne andZoonotic Diseases, 2: 87–95. [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S., Silla-Martinez J. M., and Gabaldon T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey L. L. et al. (2014) ‘Enhanced Arbovirus Surveillance with Deep Sequencing: Identification of Novel Rhabdoviruses and Bunyaviruses in Australian Mosquitoes’, Virology, 448: 146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmant A. M. G. et al. (2020) ‘Novel monoclonal antibodies against Australian strains of negeviruses and insights into virus structure, replication and host -restriction’, Journal of General Virology, 101: 440–52. [DOI] [PubMed] [Google Scholar]
- Contreras-Gutierrez M. A. et al. (2017) ‘Sinu Virus, a Novel and Divergent Orthomyxovirus Related to Members of the Genus Thogotovirus Isolated from Mosquitoes in Colombia’, Virology, 501: 166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowled C. et al. (2009) ‘Genetic and Epidemiological Characterization of Stretch Lagoon Orbivirus, a Novel Orbivirus Isolated from Culex and Aedes Mosquitoes in Northern Australia’, Journal of General Virology, 90: 1433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D. et al. (2011) ‘ProtTest 3: Fast Selection of Best-fit Models of Protein Evolution’, Bioinformatics, 27: 1164–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2012) ‘jModelTest 2: More Models, New Heuristics and Parallel Computing’, NatureMethods, 9: 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett S., et al. (2011) ‘The New South Wales Arbovirus Surveillance and Mosquito Monitoring Program 2010-2011 Annual Report’, (Westmead: Department of Medical Entomology, ICPMR, Westmead Hospital; ), p. 37. [Google Scholar]
- ——— et al. (2017) ‘The New South Wales Arbovirus Surveillance and Mosquito Monitoring Program 2016-2017 Annual Report’. (Westmead: Department of Medical Entomology, ICPMR, Westmead Hospital; ), p. 40. [Google Scholar]
- Doggett S., and Russell R. (1997) ‘Aedes Notoscriptus Can Transmit Inland and Coastal Isolates of Ross River and Barmah Forest Viruses from New South Wales’, Arbovirus Research in Australia, 7: 79–81. [Google Scholar]
- Doherty R. L. et al. (1963) ‘Studies of Arthropod-borne Virus Infections in Queensland’, Australian Journal of Experimental Biology and Medical Science, 41: 17–40. [DOI] [PubMed] [Google Scholar]
- ——— et al. (1973) ‘Isolation of Arboviruses from Mosquitoes, Biting Midges, Sandflies and Vertebrates Collected in Queensland, 1969 and 1970’, Transactions of the Royal Society of Tropical Medicine and Hygiene, 67: 536–43. [DOI] [PubMed] [Google Scholar]
- Duchemin J. B. et al. (2017) ‘Zika Vector Transmission Risk in Temperate Australia: A Vector Competence Study’, Virology Journal, 14: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiszli T., et al. (2020) ‘Mosquito-borne Diseases’, Vector-Borne Disease Section Annual Report, 2019, (Sacramento, CA: California Department of Public Health; ), pp. 13–20. [Google Scholar]
- Foster W. A. (1995) ‘Mosquito Sugar Feeding and Reproductive Energetics’, Annual Review of Entomology, 40: 443–74. [DOI] [PubMed] [Google Scholar]
- Graham D. W. (1939) ‘Mosquito Life in the Auckland District’, Transactions and Proceedings of the Royal Society of New Zealand, 69: 210–54. [Google Scholar]
- Hall R. A. et al. (2016) ‘Commensal Viruses of Mosquitoes: Host Restriction, Transmission, and Interaction with Arboviral Pathogens’, Evolutionary Bioinformatics Online, 12: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Mendelin S. et al. (2016) ‘The Insect-specific Palm Creek Virus Modulates West Nile Virus Infection in and Transmission by Australian Mosquitoes’, Parasites &Vectors, 9: 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley D., Sleigh A., and Ritchie S. (2001) ‘Ross River Virus Transmission, Infection, and Disease: A Cross- Disciplinary Review’, Clinical Microbiology Reviews, 14: 909–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes R. A. et al. (1985) ‘Arbovirus Infections of Humans in New South Wales. Seroepidemiology of the Flavivirus Group of Togaviruses’, Medical Journal of Australia, 143: 555–61. [PubMed] [Google Scholar]
- Hobson-Peters J. et al. (2013) ‘A New Insect-specific Flavivirus from Northern Australia Suppresses Replication of West Nile Virus and Murray Valley Encephalitis Virus in Co-infected Mosquito Cells’, PLoS One, 8: e56534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2019) ‘A Recombinant Platform for Flavivirus Vaccines and Diagnostics Using Chimeras of A New Insect-specific Virus’, Science Translational Medicine, 11: eaax7888. [DOI] [PubMed] [Google Scholar]
- Jansen C. C. et al. (2008) ‘Vector Competence of Australian Mosquito Species for a North American Strain of West Nile Virus’, Vector-Borne andZoonotic Diseases, 8: 805–11. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2009) ‘Arboviruses Isolated from Mosquitoes Collected from Urban and Peri-urban Areas of Eastern Australia’, Journal of the American MosquitoControl Association, 25: 272–8. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2019) ‘Epidemiologic, Entomologic, and Virologic Factors of the 2014-15 Ross River Virus Outbreak, Queensland, Australia’, Emerging Infectious Diseases, 25: 2243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen C. A., Maley F. M., and Broom A. K. (2005) ‘Isolation of Stratford Virus from Mosquitoes Collected in the Southwest of Western Australia’, Arbovirus Research in Australia, 9: 164–6. [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman M. G., and Fonseca D. M. (2014) ‘Invasion Biology of Aedes japonicus japonicus (Diptera: Culicidae)’, Annual Review of Entomology, 59: 31–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay B. H., Watson T. M., and Ryan P. A. (2008) ‘Definition of Productive Aedes Notoscriptus (Diptera: Culicidae) Habitats in Western Brisbane, and a Strategy for Their Control’, Australian Journal of Entomology, 47: 142–8. [Google Scholar]
- Kearse M. et al. (2012) ‘Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data’, Bioinformatics, 28: 1647–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosoy O. I. et al. (2015) ‘Novel Thogotovirus Associated with Febrile Illness and Death, United States, 2014’, Emerging Infectious Diseases, 21: 760–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert A. J. et al. (2015) ‘Molecular, Serological and in Vitro Culture-based Characterization of Bourbon Virus, a Newly Described Human Pathogen of the Genus Thogotovirus’, Journal of Clinical Virology, 73: 127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. J. et al. (1982) The Culicidae of the Australasian Region. Entomology Monograph No.2, p. 2. Canberra: Australian Government Publishing Service Press. [Google Scholar]
- Levin S. et al. (2019) ‘New Viruses from the Ectoparasite Mite Varroa Destructor Infesting Apis Mellifera and Apis Cerana’, Viruses, 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markussen T. et al. (2008) ‘Evolutionary Mechanisms Involved in the Virulence of Infectious Salmon Anaemia Virus (ISAV), a Piscine Orthomyxovirus’, Virology, 374: 515–27. [DOI] [PubMed] [Google Scholar]
- Newton N. D. et al. (2020) ‘Genetic, Morphological and Antigenic Relationships between Mesonivirus Isolates from Australian Mosquitoes and Evidence for Their Horizontal Transmission’, Viruses, 12: 1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes H., and Gould A. R. (1996) ‘Characterisation of Wongorr Virus, an Australian Orbivirus’, Virus Research, 44: 111–22. [DOI] [PubMed] [Google Scholar]
- Peterson A. T., and Campbell L. P. (2015) ‘Global Potential Distribution of the Mosquito Aedes Notoscriptus, a New Alien Species in the United States’, Journal ofVector Ecology, 40: 191–4. [DOI] [PubMed] [Google Scholar]
- Phillips D. A. et al. (1993) ‘Epidemiology of Arbovirus Infection in Queensland, 1989-1992’, Arbovirus Research in Australia, 6: 245–8. [Google Scholar]
- Pyke A. T. (1995) ‘Production of Stratford Virus Monoclonal Antibodies’ (unpublished data), Queensland Health: Brisbane, Queensland. [Google Scholar]
- Pyke A. T. et al. (2018) ‘On the Home Front: Specialised Reference Testing for Dengue in the Australasian Region’, Tropical Medicine Infectious Diseases, 3: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Genome Sequences of Chikungunya Virus Strains from Bangladesh and Thailand’, Microbiology Resource Announcements, 9: e01452–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A. L. et al. (2020) ‘Metagenomic Analysis of the Virome of Mosquito Excreta’, mSphere, 5: e00587–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie S. A. et al. (1997) ‘Ross River Virus in Mosquitoes (Diptera: Culicidae) during the 1994 Epidemic around Brisbane, Australia’, Journal of Medical Entomology, 34: 156–9. [DOI] [PubMed] [Google Scholar]
- Ritchie S. A., and Kline D. L. (1995) ‘Comparison of CDC and EVS Light Traps Baited with Carbon Dioxide and Octenol for Trapping Mosquitoes in Brisbane, Queensland (Diptera: Culicidae)’, Journal of the Australian Entomological Society, 34: 215–8. [Google Scholar]
- Rose K. A. et al. (2012) ‘Epizootics of Sudden Death in Tammar Wallabies (Macropus Eugenii) Associated with an Orbivirus Infection’, Australian Veterinary Journal, 90: 505–9. [DOI] [PubMed] [Google Scholar]
- Roundy C. M. et al. (2017) ‘Insect-Specific Viruses: A Historical Overview and Recent Developments’, Advances in Virus Research, 98: 119–46. [DOI] [PubMed] [Google Scholar]
- Ryan P. A., Do K. A., and Kay B. H. (2000) ‘Definition of Ross River Virus Vectors at Maroochy Shire, Australia’, Journal of Medical Entomology, 37: 146–52. [DOI] [PubMed] [Google Scholar]
- Sadeghi M. et al. (2018) ‘Virome of >12 Thousand Culex Mosquitoes from Throughout California’, Virology, 523: 74–88. [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2017) ‘High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia’, Journal of Virology, 91: e00680–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. (2012) ‘SSE: A Nucleotide and Amino Acid Sequence Analysis Platform’, BMC Research Notes, 5: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strid G. S. M. (2008) ‘Mosquitoes (Diptera: Culicidae) of City of Ryde, New South Wales’, General and Applied Entomology, 37: 37–41. [Google Scholar]
- Tamura K. et al. (2011) ‘MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods’, Molecular Biology and Evolution, 28: 2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangudu C. S. et al. (2019) ‘Stretch Lagoon virus, isolate SLOV-IA08’ (unpublished data), Veterinary Microbiology and Preventive Medicine, Iowa State University, Veterinary Medicine: Ames. [Google Scholar]
- Toi C. S. et al. (2017) ‘Seasonal Activity, Vector Relationships and Genetic Analysis of Mosquito-borne Stratford Virus’, PLoS One, 12: e0173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turell M. J., and Kay B. H. (1998) ‘Susceptibility of Selected Strains of Australian Mosquitoes (Diptera: Culicidae) to Rift Valley Fever Virus’, Journal of Medical Entomology, 35: 132–5. [DOI] [PubMed] [Google Scholar]
- van den Hurk A. F. et al. (2003) ‘Vector Competence of Australian Mosquitoes (Diptera: Culicidae) for Japanese Encephalitis Virus’, Journal of Medical Entomology, 40: 82–90. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2010) ‘Vector Competence of Australian Mosquitoes for Chikungunya Virus’, Vector-borne and Zoonotic Diseases, 10: 489–95. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2011) ‘Vector Competence of Australian Mosquitoes for Yellow Fever Virus’, The American Journal of Tropical Medicine and Hygiene, 85: 446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilakis N. et al. (2013) ‘Negevirus: A Proposed New Taxon of Insect-specific Viruses with Wide Geographic Distribution’, Journal of Virology, 87: 2475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2014) ‘Mesoniviruses are Mosquito-specific Viruses with Extensive Geographic Distribution and Host Range’, Virology Journal, 11: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrilow D. et al. (2014) ‘A New Species of Mesonivirus from the Northern Territory, Australia’, PLoS One, 9: e91103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson T. M., and Kay B. H. (1999) ‘Vector Competence of Aedes Notoscriptus (Diptera: Culicidae) for Barmah Forest Virus and of This Species and Aedes Aegypti (Diptera: Culicidae) for Dengue 1-4 Viruses’, Journal of Medical Entomology, 36: 508–14. [DOI] [PubMed] [Google Scholar]
- Zakrzewski M. et al. (2018) ‘Mapping the Virome in Wild-caught Aedes Aegypti from Cairns and Bangkok’, Scientific Reports, 8: 4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Virus sequences are available in the National Center for Biotechnology Information GenBank database at https://www.ncbi.nlm.nih.gov/genbank/.