

ABSTRACT
The interkingdom coevolution innovated the rhizobium-legume symbiosis. The application of this nitrogen-fixing system in sustainable agriculture is usually impeded by incompatible interactions between partners. However, the progressive evolution of rhizobium-legume compatibility remains elusive. In this work, deletions of rhcV encoding a structural component of the type three secretion system allow related Sinorhizobium strains to nodulate a previously incompatible soybean cultivar (Glycine max). These rhcV mutants show low to medium to high symbiotic efficiency on the same cultivated soybean while being indistinguishable on wild soybean plants (Glycine soja). The dual pantranscriptomics reveals nodule-specific activation of core symbiosis genes of Sinorhizobium and Glycine genes associated with genome duplication events along the chronogram. Unexpectedly, symbiotic efficiency is in line with lineage-dependent transcriptional profiles of core pathways which predate the diversification of Fabaceae and Sinorhizobium. This is supported by further physiological and biochemical experiments. Particularly, low-efficiency nodules show disordered antioxidant activity and low-energy status, which restrict nitrogen fixation activity. Collectively, the ancient core pathways play a crucial role in optimizing the function of later-evolved mutualistic arsenals in the rhizobium-legume coevolution.
IMPORTANCE Significant roles of complex extracellular microbiota in environmental adaptation of eukaryotes in ever-changing circumstances have been revealed. Given the intracellular infection ability, facultative endosymbionts can be considered pioneers within complex extracellular microbiota and are ideal organisms for understanding the early stage of interkingdom adaptation. This work reveals that the later innovation of key symbiotic arsenals and the lineage-specific network rewiring in ancient core pathways, predating the divergence of legumes and rhizobia, underline the progressive evolution of rhizobium-legume compatibility. This insight not only is significant for improving the application benefits of rhizobial inoculants in sustainable agriculture but also advances our general understanding of the interkingdom coevolution which is theoretically explored by all host-microbiota interactions.
KEYWORDS: adaptation, immunity, mutualism, pangenome, legume
INTRODUCTION
Despite significant roles of complex extracellular microbiota in environmental adaptation of eukaryotes in ever-changing circumstances (1, 2), rare microorganisms are indispensable for eukaryotes and vertically transmitted from parent to offspring (3, 4). Facultative intracellular microsymbionts are not strictly associated with their hosts but may confer adaptive benefits to hosts under particular conditions (5). Given the intracellular infection ability, facultative endosymbionts can be considered pioneers within complex extracellular microbiota and are ideal organisms for understanding the early stage of interkingdom adaptation.
Among diverse soil bacteria, more than 200 rhizobial species belonging to 18 genera of proteobacteria can induce and intracellularly infect nodules on diverse legumes (6). This is largely explained by the horizontal transfer of nodulation and nitrogen fixation genes among soil bacteria and subsequent recruitment of lineage-specific accessory genes (7–10). Despite the great diversity of rhizobia in the rhizosphere (11–13), the majority of nodules were individually infected by a single clone, a phenomenon called competitive nodulation (14–17). In most cases, legume roots exude flavonoids which specifically interact with a rhizobial transcriptional factor, NodD, to activate the expression of nodulation genes directing the synthesis of Nod factors (18). Specific perception of Nod factors by host receptors activates cascades of signal transduction and subsequent initiation of root hair curling, infection thread formation, and nodule organogenesis (19). The uptake of rhizobia into nodule cells leads to formation of symbiosome, consisting of differentiated rhizobial cell(s) called bacteroids which are surrounded by a host-derived membrane (20). Mature bacteroids are nongrowing and can reduce dinitrogen into ammonium that is exchanged for carbon and other nutrients from host cells (21–23), showing organelle-like features (23, 24). Great variations in nodulation and nitrogen fixation efficiency are commonly observed in agriculture practices (25). However, very few efforts have been undertaken to understand mechanisms underlying these variations (26–30).
Sinorhizobium strains are characterized by their multipartite genomes and are intensively studied as model facultative microsymbionts in mutualistic interactions and adaptive evolution of pangenome (23, 31–34). Diverse related Sinorhizobium strains have been isolated from soybean nodules in fields of alkaline-saline soils (11, 35, 36) and harbor an open pangenome (10). These strains exhibit notable variations in symbiotic compatibility (29), despite their conserved composition of Nod factors (37). We have recently discovered fast adaptive evolution of narrow-host-range strains into microsymbionts of previously incompatible soybean cultivars (29), and this process is efficiently mediated by parallel transpositions of insertion sequences into the T3SS (type three secretion system) gene cluster composed of genes encoding structural components of the T3SS (e.g., rhcV), the effector protein NopP, and the positive transcriptional regulator TtsI (29). Despite their improved nodulation ability, these newly evolved facultative microsymbionts differ in their symbiotic efficiency (29). This highlights a progressive evolution of Sinorhizobium compatibility with soybeans, though mechanisms underlying their variation of symbiotic efficiency remain unknown.
To get more evolutionary insights into this progressive interkingdom adaptation phenomenon, comparative transcriptomic analyses of symbiotic partners in soybean nodules were performed in a pangenome context, i.e., dual pantranscriptomics. To this end, we constructed rhcV mutants of five related Sinorhizobium strains including Sinorhizobium fredii CCBAU45436 (SF4), CCBAU25509 (SF2), and CCBAU83666 (SF8), Sinorhizobium sp. III CCBAU05631 (SS1), and Sinorhizobium sojae CCBAU05684 (SJ4). These rhcV mutants exhibited contrasting symbiotic performance on a test soybean cultivar (Glycine max cv. JD17) while being equally effective on a wild soybean accession (Glycine soja WSD). The dual transcriptome sequencing (RNA-seq) study in a pangenome context was used to analyze transcriptional profiles of core and accessory genes from both rhizobia and host cells in nodule samples. Active symbiotic roles of Sinorhizobium core symbiosis genes and host duplicated genes associated with key evolutionary events in the Fabaceae family were revealed. The symbiotic efficiency was, however, associated with lineage-specific rewiring patterns of core pathways predating the diversification of Fabaceae or Sinorhizobium. Core processes associated with these transcriptional profiles were further verified by physiological and biochemical experiments. Novel insights were obtained for the progressive evolution of symbiotic compatibility between legume hosts and their facultative microsymbionts, particularly highlighting a previously unknown mechanism mediated by lineage-specific rewiring of ancient core pathways.
RESULTS AND DISCUSSION
Bypassing standing variation of key symbiotic arsenals led to improved compatibility with lineage-dependent symbiotic efficiency.
Independent studies proposed a similar divergence time for Sinorhizobium-Rhizobium, as late as 203 million years ago (MYA) (38) or 201 MYA (39, 40), which is much earlier than 60 MYA, when legume (Fabaceae) evolved (41). With 203 to 336 MYA as a calibration point for the Sinorhizobium-Rhizobium split (38), the divergence between Sinorhizobium strains isolated from soybean nodules and those from Medicago nodules was estimated to take place 106 MYA (Fig. 1A), based on four housekeeping genes of strictly vertical evolutionary history in rhizobia (10, 42). A key difference between Glycine and Medicago microsymbionts is the presence of a functional T3SS in the former strains (Fig. 1B). In this work, the rhcV gene encoding an essential structural component of T3SS (43) (Fig. 1B) was deleted in a broad-host-range strain, SF4, and four narrow-host-range strains, SF2, SF8, SS1, and SJ4 (29, 44), resulting in SF4M, SF2M, SF8M, SS1M, and SJ4M, respectively (see Table S1 in the supplemental material). These rhcV mutants and their wild-type strains are all compatible with G. soja WSD and indistinguishable in symbiotic performance regarding leaf chlorophyll content, nodule number, and nitrogenase activity (Fig. 1C and Table S2). SF4 and five rhcV mutants formed nitrogen-fixing nodules on G. max cv. JD17 whereas SF8, SF2, SS1, and SJ4 induced pseudonodules on the same host (Fig. 1C). These phenotypes of rhcV mutants are similar to those of evolved compatible clones carrying insertion mutation in genes encoding T3SS structural components, TtsI or NopP, as reported previously (29). NopP of Sinorhizobium can be translocated into the host cell in a T3SS-dependent manner and can be phosphorylated by host kinases (45, 46). Variations in NopP of Sinorhizobium account for the restricted nodulation on rj2/Rfg1 soybeans but are not required for nodulation on Rj2/rfg1 soybeans (29, 47, 48). In this work, the Rj2/Rfg1 allelic genotypes of G. max cv. JD17 and G. soja WSD were determined as rj2/Rfg1 and Rj2/rfg1, respectively, thus supporting previous studies (29, 47). Although it remains unknown how NopP is recognized by different Rj2/Rfg1 soybeans (30, 47), the deletion of rhcV in five related Sinorhizobium strains allows bypassing the standing NopP variation.
FIG 1.

Lineage-specific symbiotic performance of related Sinorhizobium strains. (A) A chronogram based on four core genes of strictly vertical evolutionary history. Sinorhizobium strains associated with soybeans belong to cluster II. MYA, million years ago. (B) The cluster II strains have a conserved type three secretion system (T3SS). IM, inner membrane; OM, outer membrane. (C) Symbiotic performance of cluster II strains and their mutants lacking rhcV (SF4M, SF8M, SS1M, SF2M, and SJ4M), which encodes a structural component of T3SS. Error bars represent SD. Significant differences between means are indicated by different letters based on ANOVA followed by Tukey’s test (alpha = 0.05). Leaf chlorophyll content and nodule numbers were summarized from 18 to 30 scored plants from multiple independent experiments. Acetylene reduction activity was determined using nine plants from three independent experiments. Effective soybean nodules (halves) or pseudonodules/bumps and their representative thin sections (left, a section from pseudonodules induced by SF8; right, a section from effective nodules infected by SF4) are shown.
Strains, plasmids, and primers used in this study. Download Table S1, XLSX file, 0.01 MB (13.7KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Symbiotic performance of Sinorhizobium strains on G. max cv. JD17 and G. soja WSD. Download Table S2, XLSX file, 0.01 MB (11.4KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Notably, these rhcV mutants are not equally efficient on G. max cv. JD17 (Fig. 1C and Table S2). SJ4M showed the lowest nitrogenase activity as measured by acetylene reduction ability (analysis of variance [ANOVA] followed by Tukey’s test, alpha = 0.05). Nodulation ability sequentially increased in the order from SJ4M, SS1M-SF2M, SF8M-SF4M, to SF4, and significant variations in chlorophyll content of plants were also observed among treatments (ANOVA followed by Tukey’s test, alpha = 0.05). Collectively, these six strains can roughly be assigned into the high- (SF4, SF4M, and SF8M), medium- (SF2M and SS1M), and low-efficiency (SJ4M) groups (Fig. 1C). Therefore, conserved composition of the key symbiotic signal (Nod factor) of these related strains (37) and bypassing NopP variation are not enough to confer high-efficiency symbiotic performance on SF2M, SS1M, and SJ4M compared to SF8M and SF4M. Nodulation and nitrogen fixation genes are horizontally transferable and contribute to the great diversity of rhizobial germplasms (7, 8). An ongoing experimental evolution of the plant pathogen Ralstonia solanacearum into legume symbionts after receiving a symbiosis island has succeeded in obtaining clones with improved intracellular symbiosis with the Mimosa pudica legume, though no nitrogen-fixing clones have evolved yet (9, 49), possibly due to the limitation of gene content in this single ancestor strain. In this work, the newly generated symbiotic couples (rhcV mutants and G. max cv. JD17) with contrasting symbiotic performance may represent ideal materials for further investigation of the progressive evolution of interkingdom compatibility in a pangenome scenario.
Dual pantranscriptomes of nodules and T3SS-dependent common adaptation pathways.
The dual RNA-seq technique allows simultaneous investigation of interacting bacterium-host partners (50). Here, we extend its application in rhizobium-legume interactions in a pangenome context, i.e., dual pantranscriptomics. Dual RNA-seq was performed for 32 nodule samples from G. max cv. JD17 (G. max thereafter) and G. soja WSD (G. soja thereafter) plants inoculated with either wild type or rhcV mutants (Tables S3 to S5). Differentially expressed genes (DEGs) in nodules (|Log2R| > 1, adjusted P < 0.05) were determined between treatments of individual rhcV mutants and SF4 (Fig. S1). Both symbiotic performance and transcriptomes of the SF4M-G. soja couple are indistinguishable from the SF4-G. soja couple (7 and 0 DEGs in rhizobia and host, respectively). In contrast, the SF4M-G. max couple exhibits significant changes in transcriptome compared to SF4-G. max (694 and 3,644 DEGs), though no differences in their nitrogenase activity and leaf chlorophyll content were observed (Fig. 1C). In contrast to SF4M, the other test rhcV mutants induce various strain- and species-dependent transcriptional changes in G. soja nodules compared to either SF4 or their wild-type strains (Fig. S1 and S2), but rare DEGs are shared between multiple treatments. These rhcV mutants induce more DEGs in G. max than in G. soja (Fig. S1), in parallel with their significant difference in symbiotic performance on G. max while being indistinguishable on G. soja (Fig. 1C and Table S2).
Venn plot of differentially expressed genes revealed by dual RNA-seq. All samples from G. max cv. JD17 nodules (A) or G. soja WSD nodules (B) were compared to the treatment of SF4. No host DEGs were found in WSD nodules induced by SF4 and SF4M. Download FIG S1, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Venn plot of differentially expressed genes revealed by dual RNA-seq in G. soja WSD nodules. All samples from G. soja WSD nodules were compared to the individual treatments using corresponding wild-type strains. No host DEGs were found in WSD nodules induced by SF4 and SF4M. Download FIG S2, TIF file, 0.5 MB (525.5KB, tif) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Basic information on plant expression analysis within G. max cv. JD17 and G. soja WSD nodules. Download Table S3, XLSX file, 18.3 MB (18.3MB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In G. max nodules, there are 326 upregulated and 291 downregulated host DEGs, and 20 up- and 40 downregulated rhizobial DEGs that are common for all couples involving rhcV mutants compared to SF4-G. max (Fig. S1 and Tables S3 and S4), implying potential T3SS-dependent common adaptation pathways. In the functional enrichment analyses, the upregulated host DEGs are enriched in chromatin structure and dynamics (18 genes), cell cycle control and mitosis (10 genes), and cytoskeleton (8 genes) (P < 0.05, Fisher’s exact test) while downregulated ones are overrepresented by those belonging to carbohydrate metabolism and transport (14 genes), inorganic ion transport and metabolism (10 genes), and cell wall/membrane/envelope biogenesis (7 genes). Among the common downregulated genes within G. max nodules induced by rhcV mutants (Fig. S1 and Tables S3 and S4), Glyma.06G052000 encodes a zinc transporter homolog, ZIP (51, 52), and znuB encodes an essential component of the high-affinity zinc uptake system of S. fredii (53). The znuB mutant of SF4 forms many pseudonodules and few normal nodules on G. max cv. C08, and the inactivation of Znu together with accessory zinc uptake proteins leads to downregulation of T3SS genes in the presence of the symbiotic signal genistein (53). All of these defects can be rescued by supplying replete zinc (53). These findings imply a link between the activity of rhizobial T3SS and zinc homeostasis in symbiosis, though the underlying processes remain largely unknown. It has been reported that certain effector proteins of bacterial pathogens can target chromatin access in host cells through affecting histone-modifying enzymes (54, 55) and that the expression of many nodule-specific genes correlates with ploidy-dependent opening of the chromatin together with histone tail modifications in legume nodule cells (56). Direct interactions of pathogen T3SS effectors with host cytoskeletal arsenals (such as kinesin) and the consequent promotion of virulence have been reported (57, 58). In soybean nodules, cytoskeletal arrays differ between uninfected and infected nodule cells and between developing and mature nodule cells (59). Continued reorganization of actin cytoskeleton is associated with rhizobium release from infection threads and symbiosome development (60, 61). Notably, the key symbiotic phenotype associated with the inactivation of T3SS in incompatible Sinorhizobium strains is the shift from uninfected pseudonodules to well-infected nodules (Fig. 1C). Therefore, it is likely that effector proteins secreted by rhizobial T3SS can be directly or indirectly involved in regulating the above-mentioned host functions.
Basic information on bacterial expression analysis (versus SF4) within G. max cv. JD17 nodules and G. soja WSD nodules. Download Table S4, XLSX file, 12.4 MB (12.4MB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Evolutionary footprints of active pangenome members.
To investigate the relative contribution to symbiosis by pangenome members evolved along the evolutionary timeline of soybeans, a chronogram (Fig. 2A) was built for G. max, G. soja, and 14 reference genomes from both legume and nonlegume species within the nitrogen-fixing root nodule clade (62, 63). Common genes of G. max and G. soja were partitioned into seven sequential subsets of different conservation levels, 1 to 7, along the nodes of the chronogram (Fig. 2A and Table S3). Moreover, accessory orthologous gene clusters shared by 24 G. max accessions or 4 G. soja accessions excluding subsets 1 to 7 were defined as subset 8, and the remaining gene clusters were defined as subset 9 (Fig. 2A and Table S3). Based on RNA-seq data from different tissues of cultivated soybean (flower, pod, leaf, root, and nodule) in SoyBase (64), the subset 4 genes are particularly active in nodules compared to the other tissues (Fig. 2B). Independent of the efficiency of test Sinorhizobium strains (Fig. 1C), subset 4 genes, particularly those duplicated ones, are actively recruited by both G. max and G. soja (Fig. 2C). Duplicated genes belonging to subsets 5 and 7 and single-copy ones of subsets 4 and 7 are also explored by G. soja symbiosis (Fig. 2C and Fig. 3). In contrast, species-specific duplicated genes belonging to subsets 8 and 9 and single-copy ones of subsets 4, 5, 7, and 8 are explored by G. max symbiosis (Fig. 2C and Fig. 3). An ancestral-legume genome of 7 chromosomes has been proposed (65). A whole-genome duplication event happened around 58 MYA in the Papilionoideae leading to 14 chromosomes with evidence from early-diverging Papilionoideae lineages (66), and subsequent genome rearrangements led to a putative 9 chromosomes predating the split into Hologalegina (including Medicago, Cicer, and Lotus) and Milletioids (including Glycine, Vigna, Phaseolus, and Cajanus) around 54 MYA (66). The subset 4 genes are shared by Hologalegina and Milletioids (Fig. 2A), and symbiotic hemoglobins with 3 to 12 copies in Hologalegina and Milletioids belong to this subset and form a monophyletic group in parallel with nonsymbiotic hemoglobins present in both legumes and nonlegumes (Fig. S3). Indeed, all symbiotic hemoglobins essential for nitrogen fixation of G. max nodules (67) belong to this subset. Therefore, pangenome members associated with the 58-MYA duplication event innovated key symbiotic arsenals which have been further explored by Glycine and other members of Milletioids and Hologalegina for maintaining nodule function. However, duplicated genes belonging to subset 7 and subsequent accessory subsets 8 and 9, at least partially associated with the 13-MYA whole-genome duplication event in Glycine leading to 20 chromosomes (68), are differentially explored by G. max and G. soja, implying a strong domestication effect on symbiosis of cultivated soybeans.
FIG 2.

Pangenome members associated with the 58-MYA duplication event in the Papilionoids are actively transcribed in Glycine nodules. (A) A chronogram based on 20 single-copy genes with phylogenetic topology congruent with the phylogenomic species tree of test plants. The pangenome members of 24 Glycine max accessions and 4 Glycine soja accessions are divided into nine hierarchical subsets. The number of orthologous clusters for each subset is indicated. Subset 9 includes accessory genes of individual species. (B) Transcriptional levels of pangenome members in different tissues of G. max. (C) Transcriptional levels of single-copy genes or those with more than one copy in different pangenome subsets of G. max JD17 and G. soja WSD. Transcription levels are represented by reads per kilobase of transcript length per million reads (B) retrieved from RNA-seq data from https://soybase.org/soyseq or transcripts per million (C) obtained in this work. Proportions of DEGs belonging to individual pangenome subsets in all DEGs are shown in panel C. Confidence interval is shown for the average transcription levels in panels B and C. + and −, significant enrichment and depletion, respectively, of DEGs in individual pangenome subsets (Fisher’s exact test, alpha = 0.05).
FIG 3.
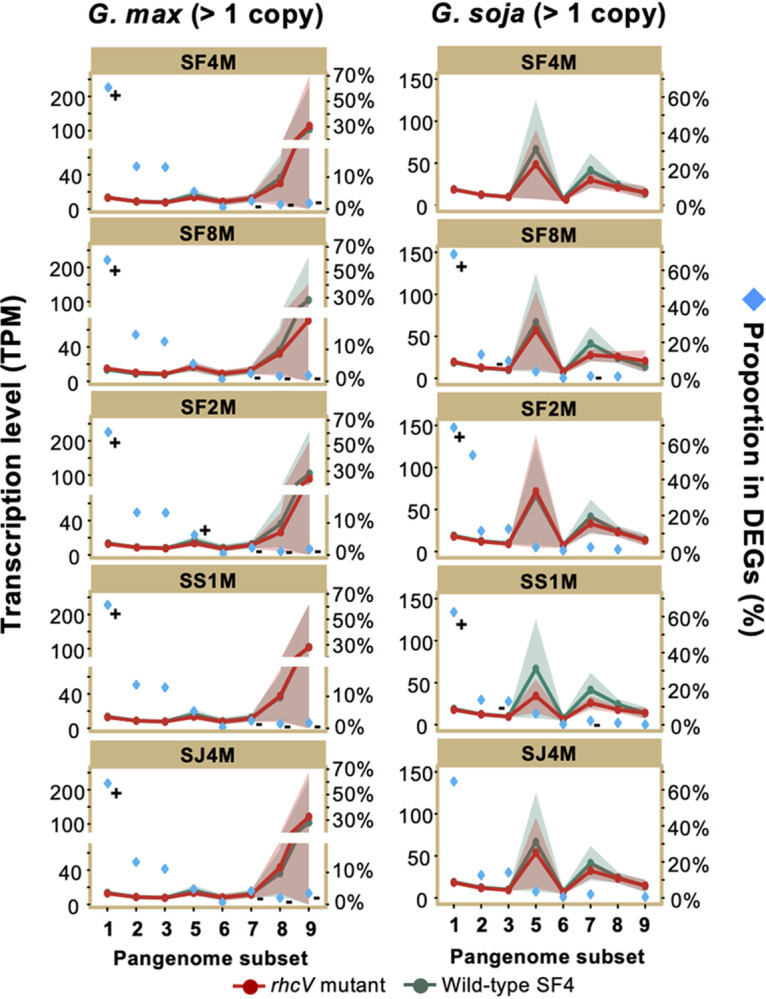
Lineage-specific pangenome members are explored by G. max symbiosis (left) and those associated with the 13-MYA duplication event are recruited by G. soja symbiosis (right). Transcriptional levels of genes with more than one copy in different pangenome subsets (except 4) of G. max JD17 and G. soja WSD. Transcription levels are represented by transcripts per million obtained. Proportions of DEGs belonging to individual pangenome subsets in all DEGs are shown. Confidence interval is shown for the average transcription levels. + and −, significant enrichment and depletion, respectively, of DEGs in individual pangenome subsets (Fisher’s exact test, alpha = 0.05).
The phylogenetic tree of phytoglobins in leguminous and nonleguminous plants forming nitrogen-fixing nodules. The protein sequences of Glycine soja and Lotus japonicus were extracted from SoyBase (□) and Kazusa DNA Research Institute (△), respectively. The protein sequences of other leguminous and nonleguminous plants were extracted from NCBI and GigaDB (*). Accession numbers are shown in brackets. Phytoglobin protein sequences were aligned with the ClustalW program, and the phylogenetic tree was constructed by the maximum-likelihood method using MEGA. Bootstrap values less than 75 are not shown. The phytoglobins of nonleguminous plants are highlighted in green. The red star indicates the putative duplication event generating symbiotic hemoglobins in the Papilionoids. Download FIG S3, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Despite more active roles of these pangenome members after whole-genome duplication events around 58 MYA and 13 MYA in soybean nodules, DEGs are significantly enriched in subset 1 (single- and multiple-copy genes) and subset 2 (single-copy genes) in G. max nodules induced by individual rhcV mutants compared to the treatment of SF4 (Fig. 2C and Fig. 3). This suggests a considerable role of network rewiring in the more conserved subsets in symbiotic optimization. This is supported by recent phylogenomics and transcriptomics studies indicating that ancient orthologs and duplication events in plants before the existence of legume might be preadapted for symbiosis (62, 69).
To investigate contributions to nodule function by rhizobial pangenome members of different conservation levels, transcriptional levels of core and accessory genes of test Sinorhizobium strains were analyzed regarding different replicons. Complete genome sequences of SF4 and SF2 have been obtained in our previous work (32), and those of SF8, SS1, and SJ4 were obtained in this study (Table S6). All five strains harbor chromosome, chromid, and symbiosis plasmid (pSymA), which are conserved replicons in Sinorhizobium (31). Moreover, 0 to 2 accessory plasmids are present in individual strains (Table S6). By including six Sinorhizobium strains associated with Medicago (Fig. 1A), a pangenome of SF4, SF2, SF8, SS1, and SJ4 was obtained and partitioned into four subsets of different conservation levels: subset I, genus core genes (2,649); subset II, genes shared by five test strains except genus core (1,238); subset III, genes shared by two to four test strains excluding subset I and subset II (809 to 2,093); and subset IV, strain-specific genes (521 to 1,054) (Fig. 4A). There are 83.4% of core genes within subsets I and II showing a consistent location at the replicon level (Fig. 4B). These pangenome features strengthen their relationships within a monophyletic group in the phylogenomic analysis (10) and the view that Sinorhizobium strains isolated from either Glycine or Medicago have an open pangenome (10, 70). Twenty reference transcriptomes of SF4, SF2, SF8, SS1, and SJ4 under free-living conditions from our previous work (29) were also included here for comparison. Subsets I to IV of symbiosis plasmids (pSymA) in test strains are generally upregulated in bacteroids within nodules compared to free-living cells (Fig. 4C), and the average transcription level of a pSymA gene in nodules generally showed a positive correlation with its conservation level. This is consistent with the view that the more conserved bacterial genes are more successfully integrated in the regulation network (32, 71), while the newer pangenome members (such as those of subset IV) are more likely silenced before integration (72). Indeed, nodulation genes responding to the soybean symbiotic signal genistein (29) and those nitrogen fixation genes active in nodules belong to the more conserved subsets I and II on the symbiosis plasmid pSymA (Fig. 4C and Tables S4 and S5), and these key symbiosis genes show a low diversity in a collection of 109 Sinorhizobium strains isolated from soybeans in different ecoregions (73). Even though subset I genes on chromosome are generally downregulated in nodules compared to free-living cells (Fig. 4C), in line with the nongrowing status of bacteroids (21, 74), these genus core genes (subset I) and the other genes shared by test strains (subset II) are still transcribed at a high level in nodules compared to those of less conserved ones in subset III or IV (Fig. 4C).
FIG 4.

Conserved pangenome members of Sinorhizobium are actively transcribed in nodules. (A) Hierarchical divisions of core/accessory subsets for the five test Sinorhizobium strains. Genus core = subset I; core genes shared by five strains = subset I + subset II; genes shared by two to four strains = subset I + subset II + subset III; strain-specific genes = subset IV. (B) Core genes localized on the same (left) or different (right) replicons in the multipartite genomes of five test strains. Lines in green, red, and yellow represent homologs on the chromosome (SF4c, SF2c, SF8c, SS1c, and SJ4c), chromid (SF4b, SF2b, SF8b, SS1b, and SJ4b), and symbiosis plasmid (SF4a, SF2a, SF8a, SS1a, and SJ4a), respectively. Replicons are shown in a linearized way with the length of fragments corresponding to sizes in the Mb scale. (C) Replicon-dependent transcriptional levels of pangenome subsets defined in panel A. C.I., confidence interval.
Basic information on bacterial expression analysis (versus wild type [WT]) within G. soja WSD nodules. Download Table S5, XLSX file, 3.0 MB (3MB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Overview of genome information for Sinorhizobium strains used in this work. Download Table S6, XLSX file, 0.01 MB (10.8KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DEGs are enriched in subsets I and II while depleted in subset III across chromosome and chromid in the treatment of SS1M and SJ4M compared to the treatment of SF4 in nodules of both G. max and G. soja (Fig. 5). This pattern is not observed for pSymA harboring key nodulation and nitrogen fixation genes (Fig. 5 and Tables S4 and S5). Notably, the divergence time between Sinorhizobium lineages associated with Glycine and Medicago, respectively, is around 106 MYA, which predates the existence of Fabaceae and the split of Papilionoideae into Milletioids (including Glycine) and Hologalegina (including Medicago) (Fig. 1A and Fig. 2A). These findings suggest a lineage-dependent network rewiring in nodules for the more conserved Sinorhizobium pangenome members predating the existence of legume nodulation.
FIG 5.

Enrichment of differentially expressed genes in conserved pangenome members predating the divergence of test Sinorhizobium strains. All samples from G. max cv. JD17 nodules (left) or G. soja WSD nodules (right) were compared to the treatment of SF4. Transcriptional levels are represented by transcripts per million (TPM). Proportions of DEGs belonging to individual pangenome subsets in all DEGs are shown. Confidence interval is shown for the average transcription levels. + and −, significant enrichment and depletion, respectively, of DEGs in individual pangenome subsets (Fisher’s exact test, alpha = 0.05).
Couple-dependent symbiotic optimization by conserved pathways predating the divergence of Fabaceae or Sinorhizobium.
To gain further functional insights into dual pantranscriptomes of nodules with contrasting symbiotic efficiency, KEGG enrichment analysis was performed (Fig. 6). Compared to G. max nodules induced by SF4, the treatments of rhcV mutants SF4M, SF8M, and SF2M derived from S. fredii strains showed a significant enrichment of upregulated host DEGs encoding various heat shock proteins and disease resistance NB-LRR (nucleotide binding site–leucine-rich repeat domain) proteins in the estrogen signaling pathway (Fig. 6A), while the low-efficiency G. max nodules induced by SJ4M are enriched with downregulated genes in the MAPK (mitogen-activated protein kinase) signaling pathway and glutathione metabolism (Fig. 6A). Notably, subset 1 genes are overrepresented in these three pathways (Fig. 6B), and the treatment of SJ4M exhibits the most divergent transcriptional profiles in these individual pathways. Moreover, the low- and medium-efficiency systems (SJ4M, SS1M, and SF2M) form a cluster with similar transcriptional profiles of glutathione metabolism genes.
FIG 6.

Symbiotic efficiency of Glycine max cv. JD17 nodules correlates with lineage-specific rewiring of core pathways predating the divergence of Fabaceae or Sinorhizobium. Function enrichment (A and C) and hierarchical clustering (B and D) analyses were performed for KEGG pathways harboring differentially expressed genes (DEGs) between treatments of individual rhcV mutants and that of wild-type SF4 [enrichment analysis: Fisher’s exact test, p.adjust < 0.05; hierarchical clustering analysis: red and green represent up- and downregulated genes, respectively, based on Log2R(TPMmutant/TPMSF4), and pangenome subset assignments of individual genes are indicated by different colors for both symbiotic partners].
Within the estrogen signaling pathway, two uncharacterized intracellular immune receptors (NB-LRR) are upregulated in the treatments of high-efficiency SF4M and SF8M, and Gm.14G197700, putatively involved in posttranscriptional RNA processing in NB-LRR-mediated immunity (75), is upregulated in the treatment of SF4M. The number of upregulated heat shock proteins sequentially increases in the treatment order from low- (SJ4M = 1) to medium- (SS1M = 4; SF2M = 4) to high-efficiency strains (SF4M = 7; SF8M = 11). More components at early steps of the MAPK pathway initiated by MAMP (microbe-associated molecular patterns) (76) are downregulated in the treatment of SJ4M, such as the cell surface immune receptor FLS2 recognizing bacterial flagellin (77, 78), seven RLCKs (receptor-like cytoplasmic kinases), and three MAPKs including a GmMEKK1 homolog (Gm.14G165700) negatively regulating defense responses (79). The thiol tripeptide glutathione is a well-known antioxidant and redox buffer (80), and glutathione S-transferase can conjugate target molecules to glutathione and act as a glutathione peroxidase to scavenge peroxides such as H2O2 (80). The number of downregulated glutathione S-transferases increased in the order from high- (SF4M = 2; SF8M = 3) to medium- (SF2M = 7; SS1M = 7) to low-efficiency strains (SJ4M = 10). In line with this transcriptional profile of glutathione S-transferase, it has been demonstrated that the downregulation of glutathione S-transferase in soybean nodules leads to a decrease in nitrogenase activity and an increase in oxidatively damaged proteins (81).
In bacteroids of G. max nodules, there is a general downshift of the oxidative phosphorylation pathway in less efficient strains (SJ4M, SS1M, and SF2M) (Fig. 6C and D and Table S4). This pathway is overrepresented by conserved subset I genes encoding respiratory components ATPase, Nuo, Cytcbb3, Cox, and Cytbd and those involved in various physiological steps producing NAD(P)H (Table S4) (23, 82). The energetically expensive processes including secretion systems T3SS and T4SS (overrepresented by subset II genes) and motility proteins (mainly subset I and II genes) are highly transcribed in SS1M and/or SJ4M (Fig. 6C). These findings indicate a low-energy status of bacteroids in these less efficient strains, though nitrogen fixation is an energetically high-cost reaction (23). In addition to these enriched pathways, notable transcriptional changes in individual conserved functional genes supporting nitrogen fixation were uncovered in low-efficiency bacteroids. The essential FeS assembly process (83) may be restricted in low-efficiency bacteroids due to the low transcription level of sufAE, paaD, and nifU genes (Table S4). However, the key regulator gene nifA; nitrogenase structural genes nifHDK; and nifBENQXW, fdxBN, and fixABC involved in FeMo-cofactor synthesis, nitrogenase maturation/stabilization, and electron donation (23, 83) are more actively transcribed in low-efficiency bacteroids than in those of SF4 (Table S4). In parallel with the disordered transcriptional profiles associated with host antioxidant functions, low-efficiency bacteroids also show a low transcription level in antioxidant functions including subset I members such as superoxide dismutase (SJ05684_c05970), peroxiredoxin (SJ05684_c05210), glutathione S-transferase (SJ05684_c36640), and thioredoxin reductase (SJ05684_c15680 and SJ05684_c05650) and a subset III member peptide-methionine (R)-S-oxide reductase (SJ05684_c06170). These functions are involved in the dismutation of superoxide radicals, H2O2 scavenging, and various detoxification processes (82, 84, 85).
As described above, the couples of different symbiotic efficiencies are characterized by distinct transcriptional files of genes involved in immunity response, antioxidant functions, and energy metabolism. Indeed, G. max nodules infected by SJ4M have fewer bacteroids (Fig. 7A). The MAPK kinase kinase GmMEKK1 homologs negatively regulating defense responses in soybean (79) are downregulated in the SJ4M treatment (Fig. 7B, qRT-PCR). An immunoblotting experiment using these nodule samples demonstrates highly active MPK3 and MPK6 (Fig. 7C), two well-studied MAPKs which are regulated by MEKK1 and respond to oxidative stress (79, 86, 87). Among diverse defense response marker genes (88), PR5 is transcribed at a significantly higher level in the treatment of SJ4M (Fig. 7D, qRT-PCR). All of these efficiency-related genes belong to subset 1 predating the divergence of Fabaceae.
FIG 7.

Defense response and energy status in G. max cv. JD17 nodules. (A) Thin sections of nodules induced by corresponding strains. (B) Relative transcription levels of MEKK1 homologs compared to an actin gene, Gm.15G05020. (C) Immunoblotting of defense kinases MPK6/MPK3 using antibody against phosphorylated MAPK. (D) Relative transcription levels of defense response gene PR5 compared to Gm.15G05020. (E) H2O2 production. (F to H) Energy status indicated by ratios of ADP/ATP (F), NADP+/NADPH (G), and NAD+/NADH (H). Error bars represent SD from biological triplicates. Significant differences between means are indicated by different letters based on ANOVA followed by Tukey’s test (alpha = 0.05).
In line with the elevated H2O2 in GmMEKK1-silenced soybean plants (79), the low-efficiency SJ4M treatment is characterized with the highest level of H2O2 (Fig. 7E). The medium-efficiency treatments SF2M and SS1M also exhibit a significantly higher level of H2O2 than high-efficiency SF4, SF4M, and SF8M (Fig. 7E). This phenomenon can be at least partially explained by the efficiency of rhizobial oxidative phosphorylation as measured by ratios of canonical energy currency molecules (Fig. 7F to H). As to ADP/ATP, NADP/NADPH, and NAD/NADH ratios, the low-efficiency SJ4M bacteroids are 86.3%, 523%, and 158% higher than that of SF4 bacteroids, respectively. The medium-efficiency treatments SF2M and SS1M show a significantly higher ratio of NAD/NADH than the high-efficiency treatments SF4, SF4M, and SF8M. These physiological and biochemical results provide strong evidence for the critical role of conserved pathways predating the divergence of Fabaceae and Sinorhizobium in optimizing symbiotic efficiency.
Conclusion.
With conserved compositions of Nod factors (37), five related Sinorhizobium strains are able to induce nodules/pseudonodules on cultivated and wild soybeans. The deletion of rhcV or fast adaptive evolution mediated by parallel transpositions of insertion sequences into the T3SS gene cluster (29) allows bypassing standing variations in effector proteins (47) and improving compatibility to different extents among test strains. Dual pantranscriptomics uncovers that the more actively transcribed genes in nodules are those conserved symbiosis genes in rhizobia and those associated with the whole-genome duplication event in the Papilionoideae (58 MYA) in soybeans. G. max and G. soja differentially explored pangenome members of different conservation levels, particularly those associated with the whole-genome duplication event in the Glycine genus (13 MYA). However, the variation in symbiotic efficiency correlates with lineage-dependent rewiring of ancient core pathways predating the divergence of Fabaceae or Sinorhizobium, which is supported by further physiological and biochemical evidences. Collectively, innovation of key symbiotic arsenals together with the largely unexplored network rewiring in existing ancient core pathways underlines the progressive evolution of rhizobium-legume compatibility. This insight not only is significant for improving the application benefits of rhizobial inoculants in sustainable agriculture but also advances our general understanding of the interkingdom coevolution which is theoretically explored by all host-microbiota interactions.
MATERIALS AND METHODS
Bacterial strains, plasmids, growth condition, and soybeans.
Strains, plasmids, and primers used in this work are listed in Table S1 in the supplemental material. Sinorhizobium strains and Escherichia coli strains were cultured as described previously (32). To generate rhcV mutants, the upstream and downstream DNA fragments of target rhcV genes in individual genomes were amplified by PCR and cloned into pCM351, carrying a cre-lox system (89). The resultant pCM351 derivatives were conjugated into Sinorhizobium strains with the helper pRK2013, and deletion mutants were screened on the TY (tryptone yeast)-agar plates supplied with 30 μg/ml nalidixic acid and 30 μg/ml gentamicin. Candidate clones were verified by colony PCR and Sanger sequencing. To determine Rj2/Rfg1 allelic genotypes of G. max cv. JD17 and G. soja WSD, RNA samples extracted from 1-week-old seedlings by using RNAiso Plus (TaKaRa) were used to synthesize cDNA with a reverse transcription kit (Genstar). Then, two pairs of primers (Table S1) were used to amplify two fragments corresponding to polymorphic regions 452 to 490 and 731 to 758 of Rj2/Rfg1 protein (28). PCR products were subject to Sanger sequencing. The resultant sequences were compared to those of different Rj2/Rfg1 genotypes reported previously (28).
Genome sequencing and functional annotation.
DNA of SF8, SS1, and SJ4 was extracted using the TIANamp bacterial DNA kit (Tiangen). The genomes were sequenced by PacBio and Illumina platforms and assembled at BGI-Shenzhen. Gaps were closed by Sanger sequencing of PCR products using primers listed in Table S1. Gene prediction and annotation were performed by using RAST (90) and eggNOG (91).
Pangenomic and phylogenetic analysis.
OrthoFinder (92) with default parameters (-og) was used to identify orthogroups. Briefly, gene length bias and phylogenetic distance were removed from length-normalized similarity score, and the lower limit of orthogroup sequence similarity was defined by using RBNHs (Reciprocal Best length-Normalized hit). Genes present in all Sinorhizobium strains were defined as subset I. Genes shared by 5 soybean microsymbionts excluding subset I were defined as subset II, and genes shared by 2 to 4 soybean microsymbionts but not present in subset II were defined as subset III. The remaining strain-specific genes were defined as subset IV. The OrthoFinder output file Orthogroups.GeneCount.tsv contains counts of the number of genes for each strain in each orthogroup; therefore, the orthogroups that contained fewer than two genes per strain were defined as single-copy genes, but the others were defined as multiple-copy genes. Similarly to the method of gene subset definition in Sinorhizobium, 9 pangenome subsets of Glycine were defined by analyzing 4 G. soja and 24 G. max genomes available in the public database (63). The protein sequences of phytoglobins from the leguminous and nonleguminous plants were extracted from the NCBI database and aligned with the ClustalW program (93), and the phylogenetic tree was constructed by the maximum-likelihood method using MEGA (94). Single-copy genes (1,290 genes) shared by 11 Sinorhizobium strains and an outgroup, Rhizobium leguminosarum bv. trifolii WSM1689, were used to construct a maximum-likelihood phylogenetic tree by RAxML (95) with the PROTGAMMAAUTO setting using 250 bootstrap replicates. This phylogenomic tree was used as the species tree. A species tree of test plants was inferred from 25,329 orthogroups shared by all 16 plants by using the STAG (species tree inference from all genes) algorithm (96) and rooted using STRIDE (97).
Divergence time estimation.
A Bayesian method implemented in BEAST2 (98) with an uncorrelated lognormal (UCLN) relaxed clock model was used to estimate divergence times for both bacteria and plant (parameters: Gamma Category Count = 4, Subset Model = GTR, Frequencies = Empirical). The selection of best substitution model was done with IQ-TREE (99). The Calibrated Yule Model with lognormal distribution was used for calibration with previous estimates of Sinorhizobium-Rhizobium divergence (203 to 346 MYA) (38) and the crown of Fabaceae (60 to 70 MYA) (41). To estimate intragenus-level divergence time of rhizobia, four high-resolution housekeeping genes (ctaE, rpoB, rplB, and glpK) (Table S7) with phylogenies congruent with the phylogenomic species tree were used. For plant, 20 single-copy core genes (Table S7) showing congruent phylogeny with the species tree were used.
Core genes used in chronogram construction. Download Table S7, XLSX file, 0.01 MB (10.2KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plant assays, nitrogenase activity assays, and nodule thin sections.
Seeds and rhizobial inoculants were prepared as described previously (32). Each seedling was inoculated with 1 ml rhizobial culture (optical density at 600 nm [OD600] = 0.2). Plants were grown at 24°C with 12-h day and night cycles for 30 days. Nitrogenase activity of nodules was measured by using the acetylene reduction method (100). Chlorophyll content was determined using a SPAD-502 meter (Konica Minolta) as described earlier (101, 102). More than three independent experiments were performed. To generate nodule thin sections, nodules were fixed in the fixative solution (90 ml 70% ethanol, 5 ml acetic acid, 5 ml 38% formaldehyde) at 4°C for 12 h. After dehydration and embedding of the nodule, 4-μm nodule slices were obtained by using a paraffin slicing machine (Leica, RM2016). Finally, nodule slices were dewaxed and stained in toluidine blue (1%) and further sealed by neutral balsam (60%).
RNA-seq and qRT-PCR.
Nodule total RNA was extracted by using RNAiso Plus (TaKaRa). Purified RNA was quantified by using NanoPhotometer Pearl (Implen), and quality was checked by using capillary electrophoresis (Bioanalyzer; Agilent). The resultant RNA samples were subjected to mRNA enrichment, library construction, and strand-specific RNA sequencing (PE150) on an Illumina HiSeq 2000 platform following the manufacturer’s instructions (Illumina) by Novogene (The Genome Analysis Centre, Beijing, China). Two sets of nodules from two independent experiments were used.
qRT-PCR was performed by using primers listed in Table S1. In detail, 8 μg nodule or root RNA was used for reverse transcription. The StarScript II first-strand cDNA kit with genomic DNA (gDNA) remover (Genstar) was used to synthesize cDNA. qRT-PCR was performed by using 2×RealStar Green Fast Mixture (Genstar) and a Roche LightCycler480 II system. Transcription levels were normalized to the expression of the control genes, including 16S rRNA gene of bacteria and an actin gene (Gm.15G05020) of soybean. Triplicates from three independent experiments were used.
Bioinformatics procedures in transcriptomic analysis.
RNA-seq reads of Sinorhizobium were mapped to their corresponding reference genomes with Bowtie 2 (103). RNA-seq reads from soybeans were mapped to genomes of G. max (Wm82.a2) and G. soja (PI483463.a1) obtained from SoyBase (https://soybase.org), with HISAT2 (104). Mapped reads for the protein-encoding gene in each sample were counted using featureCounts (105). Differentially expressed genes were identified by using DESeq2 (106) with an absolute fold change above 1 and adjusted P value of < 0.05. Hierarchical clustering was performed by using the standard R function hclust (hclust, method = average) (107). R package ClusterProfiler (108) was used in KEGG enrichment analysis. For all enrichment analyses, the annotation data set of individual genomes was used as background, and a P value below 0.05 indicates significant enrichment. R package ggplot2 (109) was used to visualize these enrichment results.
Immunoblot detection of phosphorylated MPK6 and MPK3.
Nodules (4 g) were ground into powder in liquid nitrogen, and protein was extracted by 5 ml extraction buffer (50 mM Tris-MES [morpholineethanesulfonic acid], pH 8, 0.5 M sucrose, 1 mM MgCl2, 10 mM EDTA, 5 mM dithiothreitol [DTT], and protease inhibitor cocktail). The extract was centrifuged at 10,000 × g at 4°C for 30 min. Protein concentration was determined by using the Bradford method (A53225; ThermoFisher). Then, 2 μg protein was separated by SDS-PAGE (15% acrylamide gel) and transferred to nitrocellulose filter membranes by wet electrotransfer (Bio-Rad). The membrane was blocked in 1× Tris-buffered saline (TBS) buffer containing 5% nonfat milk. The blocked membrane was further incubated with anti-phospho-p44/p42 MAPK (anti-pTEpY; Cell Signaling Technology) diluted at 1:2,000 overnight at 4°C and subsequently with secondary antibody diluted at 1:5,000 for 1 h. Finally, the bands were detected using chemiluminescent horseradish peroxidase (HRP) substrate, and the image was obtained by using Tanon 5200. Three independent biological replicates were used.
Determination of H2O2.
H2O2 concentration was determined by using hydrogen peroxide assay kit S0038 (Beyotime Biotech, China). Briefly, 2-g nodules were ground into powder in 2 ml lysis buffer S0038-3. Then, the mixture was centrifuged at 12,000 × g for 5 min at 4°C, and the supernatant was quickly chilled and stored at −80°C. Fifty microliters of supernatant was mixed with 100 μl test solution S0038-1 in a 96-well plate, at room temperature for 20 min, and measured immediately with a spectrometer (560 nm). Triplicates from three independent experiments were tested.
Determination of NAD+/NADH, NADP+/NADPH, and ADP/ATP.
To enrich bacteroids from nodules, 5-g nodules were ground by using a prechilled pestle in 15 ml extraction buffer (10 mM DTT, 300 mM sucrose, 10 mM phosphate buffer, pH 7.0, 2 mM MgCl2, and 0.33 g polyvinylpyrrolidone [PVP]), and the mixture was centrifuged at 400 × g for 10 min at 4°C to remove the majority of plant cell particles. The supernatant was centrifuged at 12,000 × g for 10 min at 4°C. The resultant pellet was suspended with 200 μl phosphate-buffered saline (PBS) buffer (pH 7.5) and stored at −80°C. The ratios of NAD+/NADH and NADP+/NADPH were determined by using the NAD+/NADH assay kit (S0175) and NADP+/NADPH assay kit (S0179), respectively, from Beyotime Biotech, China. Briefly, 20 μl supernatant and 90 μl working solution (S0175-6 for NAD+/NADH and S0179-5 for NADP+/NADPH) were added to a 96-well plate, placed at 37°C for 20 min, and measured immediately with a spectrometer (450 nm). Cellular ADP/ATP levels were measured by EnzyLight ATP assay (BioAssay Systems, Hayward, CA). Briefly, 10 μl supernatant was mixed with 90 μl ATP reagent in a 96-well plate. After 1 min, luminescence (RLU A) was detected on a luminometer. Then 5 μl ADP reagent was added to each well and mixed by tapping the plate. After 1 min, luminescence (RLU B) was detected on a luminometer. The ADP/ATP ratio was calculated as follows: (RLUB − RLUA)/RLUA. Three independent biological replicates were used in these experiments.
Data availability.
Raw sequence data from our RNA-seq analyses can be accessed via NCBI Sequence Read Archive (PRJNA652809). Complete genomes of five wild-type strains used in this study have been deposited in the GenBank database (BioProject no. PRJNA353922: SF8, SS1, and SJ4; BioProject no. PRJNA285929: SF4 and SF2).
ACKNOWLEDGMENTS
We thank the Shi-jiazhuang Branch of the National Soybean Improvement Center for providing seeds of JD17.
This work was supported by the National Key R&D Program of China (grant number 2019YFA0904700), the National Natural Science Foundation of China (grant numbers 32070078 and 31522003), the Innovative Project of the State Key Laboratory of Agrobiotechnology (grant numbers 2019SKLAB1-9 and 2020SKLAB1-5), and the Chinese Universities Scientific Fund (2019TC086).
C.-F.T. and Z.Z. planned and designed the research. W.-J.C., B.Z., R.Z., L.-X.L., and J.J. performed experiments and analyzed data. C.-F.T., W.-J.C., and B.Z. wrote the manuscript.
We declare that we have no competing interests.
Contributor Information
Ziding Zhang, Email: zidingzhang@cau.edu.cn.
Chang-Fu Tian, Email: cftian@cau.edu.cn.
Rosie Alegado, University of Hawaii at Manoa.
REFERENCES
- 1.Mendes R, Garbeva P, Raaijmakers JM. 2013. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol Rev 37:634–663. doi: 10.1111/1574-6976.12028. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg E, Zilber-Rosenberg I. 2018. The hologenome concept of evolution after 10 years. Microbiome 6:78. doi: 10.1186/s40168-018-0457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiers ET, West SA. 2015. Evolving new organisms via symbiosis. Science 348:392–394. doi: 10.1126/science.aaa9605. [DOI] [PubMed] [Google Scholar]
- 4.Douglas GM, Bielawski JP, Langille MGI. 2020. Re-evaluating the relationship between missing heritability and the microbiome. Microbiome 8:87. doi: 10.1186/s40168-020-00839-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toft C, Andersson SGE. 2010. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet 11:465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- 6.de Lajudie PM, Andrews M, Ardley J, Eardly B, Jumas-Bilak E, Kuzmanović N, Lassalle F, Lindström K, Mhamdi R, Martínez-Romero E, Moulin L, Mousavi SA, Nesme X, Peix A, Puławska J, Steenkamp E, Stępkowski T, Tian C-F, Vinuesa P, Wei G, Willems A, Zilli J, Young P. 2019. Minimal standards for the description of new genera and species of rhizobia and agrobacteria. Int J Syst Evol Microbiol 69:1852–1863. doi: 10.1099/ijsem.0.003426. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan JT, Patrick HN, Lowther WL, Scott DB, Ronson CW. 1995. Nodulating strains of Rhizobium loti arise through chromosomal symbiotic gene transfer in the environment. Proc Natl Acad Sci U S A 92:8985–8989. doi: 10.1073/pnas.92.19.8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling J, Wang H, Wu P, Li T, Tang Y, Naseer N, Zheng H, Masson-Boivin C, Zhong Z, Zhu J. 2016. Plant nodulation inducers enhance horizontal gene transfer of Azorhizobium caulinodans symbiosis island. Proc Natl Acad Sci U S A 113:13875–13880. doi: 10.1073/pnas.1615121113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capela D, Marchetti M, Clérissi C, Perrier A, Guetta D, Gris C, Valls M, Jauneau A, Cruveiller S, Rocha EPC, Masson-Boivin C. 2017. Recruitment of a lineage-specific virulence regulatory pathway promotes intracellular infection by a plant pathogen experimentally evolved into a legume symbiont. Mol Biol Evol 34:2503–2521. doi: 10.1093/molbev/msx165. [DOI] [PubMed] [Google Scholar]
- 10.Tian CF, Zhou YJ, Zhang YM, Li QQ, Zhang YZ, Li DF, Wang S, Wang J, Gilbert LB, Li YR, Chen WX. 2012. Comparative genomics of rhizobia nodulating soybean suggests extensive recruitment of lineage-specific genes in adaptations. Proc Natl Acad Sci U S A 109:8629–8634. doi: 10.1073/pnas.1120436109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang XX, Guo HJ, Jiao J, Zhang P, Xiong HY, Chen WX, Tian CF. 2017. Pyrosequencing of rpoB uncovers a significant biogeographical pattern of rhizobial species in soybean rhizosphere. J Biogeogr 44:1491–1499. doi: 10.1111/jbi.12891. [DOI] [Google Scholar]
- 12.Xiong HY, Zhang XX, Guo HJ, Ji YY, Li Y, Wang XL, Zhao W, Mo FY, Chen CJ, Yang T, Zong X, Chen WX, Tian CF. 2017. The epidemicity of facultative microsymbionts in faba bean rhizosphere soils. Soil Biol Biochem 115:243–252. doi: 10.1016/j.soilbio.2017.08.032. [DOI] [Google Scholar]
- 13.Wang XL, Cui WJ, Feng XY, Zhong ZM, Li Y, Chen WX, Chen WF, Shao XM, Tian CF. 2018. Rhizobia inhabiting nodules and rhizosphere soils of alfalfa: a strong selection of facultative microsymbionts. Soil Biol Biochem 116:340–350. doi: 10.1016/j.soilbio.2017.10.033. [DOI] [Google Scholar]
- 14.Wang D, Wang YC, Wu LJ, Liu JX, Zhang P, Jiao J, Yan H, Liu T, Tian CF, Chen WX. 2016. Construction and pilot screening of a signature-tagged mutant library of Sinorhizobium fredii. Arch Microbiol 198:91–99. doi: 10.1007/s00203-015-1161-9. [DOI] [PubMed] [Google Scholar]
- 15.Mendoza-Suárez MA, Geddes BA, Sánchez-Cañizares C, Ramírez-González RH, Kirchhelle C, Jorrin B, Poole PS. 2020. Optimizing Rhizobium-legume symbioses by simultaneous measurement of rhizobial competitiveness and N2 fixation in nodules. Proc Natl Acad Sci U S A 117:9822–9831. doi: 10.1073/pnas.1921225117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pobigaylo N, Szymczak S, Nattkemper TW, Becker A. 2008. Identification of genes relevant to symbiosis and competitiveness in Sinorhizobium meliloti using signature-tagged mutants. Mol Plant Microbe Interact 21:219–231. doi: 10.1094/MPMI-21-2-0219. [DOI] [PubMed] [Google Scholar]
- 17.Shimoda Y, Mitsui H, Kamimatsuse H, Minamisawa K, Nishiyama E, Ohtsubo Y, Nagata Y, Tsuda M, Shinpo S, Watanabe A, Kohara M, Yamada M, Nakamura Y, Tabata S, Sato S. 2008. Construction of signature-tagged mutant library in Mesorhizobium loti as a powerful tool for functional genomics. DNA Res 15:297–308. doi: 10.1093/dnares/dsn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masson-Boivin C, Sachs JL. 2018. Symbiotic nitrogen fixation by rhizobia—the roots of a success story. Curr Opin Plant Biol 44:7–15. doi: 10.1016/j.pbi.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Roy S, Liu W, Nandety RS, Crook A, Mysore KS, Pislariu CI, Frugoli J, Dickstein R, Udvardi MK. 2020. Celebrating 20 years of genetic discoveries in legume nodulation and symbiotic nitrogen fixation. Plant Cell 32:15–41. doi: 10.1105/tpc.19.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parniske M. 2018. Uptake of bacteria into living plant cells, the unifying and distinct feature of the nitrogen-fixing root nodule symbiosis. Curr Opin Plant Biol 44:164–174. doi: 10.1016/j.pbi.2018.05.016. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Tian CF, Chen WF, Wang L, Sui XH, Chen WX. 2013. High-resolution transcriptomic analyses of Sinorhizobium sp. NGR234 bacteroids in determinate nodules of Vigna unguiculata and indeterminate nodules of Leucaena leucocephala. PLoS One 8:e70531. doi: 10.1371/journal.pone.0070531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiao J, Wu LJ, Zhang B, Hu Y, Li Y, Zhang XX, Guo HJ, Liu LX, Chen WX, Zhang Z, Tian CF. 2016. MucR is required for transcriptional activation of conserved ion transporters to support nitrogen fixation of Sinorhizobium fredii in soybean nodules. Mol Plant Microbe Interact 29:352–361. doi: 10.1094/MPMI-01-16-0019-R. [DOI] [PubMed] [Google Scholar]
- 23.Poole P, Ramachandran V, Terpolilli J. 2018. Rhizobia: from saprophytes to endosymbionts. Nat Rev Microbiol 16:291–303. doi: 10.1038/nrmicro.2017.171. [DOI] [PubMed] [Google Scholar]
- 24.De La Peña TC, Fedorova E, Pueyo JJ, Lucas M. 2018. The symbiosome: legume and rhizobia co-evolution toward a nitrogen-fixing organelle? Front Plant Sci 8:2229. doi: 10.3389/fpls.2017.02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brockwell J, Bottomley PJ. 1995. Recent advances in inoculant technology and prospects for the future. Soil Biol Biochem 27:683–697. doi: 10.1016/0038-0717(95)98649-9. [DOI] [Google Scholar]
- 26.Heath KD, Burke PV, Stinchcombe JR. 2012. Coevolutionary genetic variation in the legume-rhizobium transcriptome. Mol Ecol 21:4735–4747. doi: 10.1111/j.1365-294X.2012.05629.x. [DOI] [PubMed] [Google Scholar]
- 27.Burghardt LT, Guhlin J, Chun CL, Liu J, Sadowsky MJ, Stupar RM, Young ND, Tiffin P. 2017. Transcriptomic basis of genome by genome variation in a legume-rhizobia mutualism. Mol Ecol 26:6122–6135. doi: 10.1111/mec.14285. [DOI] [PubMed] [Google Scholar]
- 28.Yang S, Tang F, Gao M, Krishnan HB, Zhu H. 2010. R gene-controlled host specificity in the legume-rhizobia symbiosis. Proc Natl Acad Sci U S A 107:18735–18740. doi: 10.1073/pnas.1011957107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao R, Liu LX, Zhang YZ, Jiao J, Cui WJ, Zhang B, Wang XL, Li ML, Chen Y, Xiong ZQ, Chen WX, Tian CF. 2018. Adaptive evolution of rhizobial symbiotic compatibility mediated by co-evolved insertion sequences. ISME J 12:101–111. doi: 10.1038/ismej.2017.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugawara M, Takahashi S, Umehara Y, Iwano H, Tsurumaru H, Odake H, Suzuki Y, Kondo H, Konno Y, Yamakawa T, Sato S, Mitsui H, Minamisawa K. 2018. Variation in bradyrhizobial NopP effector determines symbiotic incompatibility with Rj2-soybeans via effector-triggered immunity. Nat Commun 9:6–10. doi: 10.1038/s41467-018-05663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J. 2001. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- 32.Jiao J, Ni M, Zhang B, Zhang Z, Young JPW, Chan T-F, Chen WX, Lam H-M, Tian CF. 2018. Coordinated regulation of core and accessory genes in the multipartite genome of Sinorhizobium fredii. PLoS Genet 14:e1007428. doi: 10.1371/journal.pgen.1007428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.López L, Lozano M, Lagares AJ, Fabre M, Draghi W, Del Papa M, Pistorio M, Becker A, Wibberg D, Schlüter A, Pühler A, Blom J, Goesmann A, Lagares A. 2019. Codon usage heterogeneity in the multipartite prokaryote location, expression level, and ancestry. mBio 10:e00505-19. doi: 10.1128/mBio.00505-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Batstone RT, O’Brien AM, Harrison TL, Frederickson ME. 2020. Experimental evolution makes microbes more cooperative with their local host. Science 370:476–478. doi: 10.1126/science.abb7222. [DOI] [PubMed] [Google Scholar]
- 35.Wu LJ, Wang HQ, Wang ET, Chen WX, Tian CF, Chen WX. 2011. Genetic diversity of nodulating and non-nodulating rhizobia associated with wild soybean (Glycine soja Sieb. & Zucc.) in different ecoregions of China. FEMS Microbiol Ecol 76:439–450. doi: 10.1111/j.1574-6941.2011.01064.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhang YM, Li Y, Jr, Chen WF, Wang ET, Tian CF, Li QQ, Zhang YZ, Sui XH, Chen WX. 2011. Biodiversity and biogeography of rhizobia associated with soybean plants grown in the North China Plain. Appl Environ Microbiol 77:6331–6342. doi: 10.1128/AEM.00542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, Couderc F, Tian CF, Gu W, Liu LX, Poinsot V. 2018. Conserved composition of Nod factors and exopolysaccharides produced by different phylogenetic lineage Sinorhizobium strains nodulating soybean. Front Microbiol 9:2852. doi: 10.3389/fmicb.2018.02852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner SL, Young JPW. 2000. The glutamine synthetases of rhizobia: phylogenetics and evolutionary implications. Mol Biol Evol 17:309–319. doi: 10.1093/oxfordjournals.molbev.a026311. [DOI] [PubMed] [Google Scholar]
- 39.Kumar S, Stecher G, Suleski M, Hedges SB. 2017. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol 34:1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- 40.Chriki-Adeeb R, Chriki A. 2016. Estimating divergence times and substitution rates in Rhizobia. Evol Bioinform Online 12:87–97. doi: 10.4137/EBO.S39070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lavin M, Herendeen PS, Wojciechowski MF. 2005. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol 54:575–594. doi: 10.1080/10635150590947131. [DOI] [PubMed] [Google Scholar]
- 42.Zhang YM, Tian CF, Sui XH, Chen WF, Chen WX. 2012. Robust markers reflecting phylogeny and taxonomy of rhizobia. PLoS One 7:e44936. doi: 10.1371/journal.pone.0044936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tampakaki AP. 2014. Commonalities and differences of T3SSs in rhizobia and plant pathogenic bacteria. Front Plant Sci 5:114. doi: 10.3389/fpls.2014.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu LX, Li QQ, Zhang YZ, Hu Y, Jiao J, Guo HJ, Zhang XX, Zhang B, Chen WX, Tian CF. 2017. The nitrate-reduction gene cluster components exert lineage-dependent contributions to optimization of Sinorhizobium symbiosis with soybeans. Environ Microbiol 19:4926–4938. doi: 10.1111/1462-2920.13948. [DOI] [PubMed] [Google Scholar]
- 45.Skorpil P, Saad MM, Boukli NM, Kobayashi H, Ares-Orpel F, Broughton WJ, Deakin WJ. 2005. NopP, a phosphorylated effector of Rhizobium sp strain NGR234, is a major determinant of nodulation of the tropical legumes Flemingia congesta and Tephrosia vogelii. Mol Microbiol 57:1304–1317. doi: 10.1111/j.1365-2958.2005.04768.x. [DOI] [PubMed] [Google Scholar]
- 46.Schechter LM, Guenther J, Olcay EA, Jang SC, Krishnan HB. 2010. Translocation of NopP by Sinorhizobium fredii USDA257 into Vigna unguiculata root nodules. Appl Environ Microbiol 76:3758–3761. doi: 10.1128/AEM.03122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rehman HM, Cheung W-L, Wong K-S, Xie M, Luk C-Y, Wong F-L, Li M-W, Tsai S-N, To W-T, Chan L-Y, Lam H-M. 2019. High-throughput mass spectrometric analysis of the whole proteome and secretome from Sinorhizobium fredii strains CCBAU25509 and CCBAU45436. Front Microbiol 10:2569. doi: 10.3389/fmicb.2019.02569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan Y, Liu J, Lyu S, Wang Q, Yang S, Zhu H. 2017. The soybean Rfg1 gene restricts nodulation by Sinorhizobium fredii USDA193. Front Plant Sci 8:1548. doi: 10.3389/fpls.2017.01548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchetti M, Capela D, Glew M, Cruveiller S, Chane-Woon-Ming B, Gris C, Timmers T, Poinsot V, Gilbert LB, Heeb P, Medigue C, Batut J, Masson-Boivin C. 2010. Experimental evolution of a plant pathogen into a legume symbiont. PLoS Biol 8:e1000280. doi: 10.1371/journal.pbio.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolf T, Kämmer P, Brunke S, Linde J. 2018. Two’s company: studying interspecies relationships with dual RNA-seq. Curr Opin Microbiol 42:7–12. doi: 10.1016/j.mib.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Moreau S, Thomson RM, Kaiser BN, Trevaskis B, Guerinot M, Udvardi MK, Puppo A, Day DA. 2002. GmZIP1 encodes a symbiosis-specific zinc transporter in soybean. J Biol Chem 277:4738–4746. doi: 10.1074/jbc.M106754200. [DOI] [PubMed] [Google Scholar]
- 52.Abreu I, Saéz Á, Castro-Rodríguez R, Escudero V, Rodríguez-Haas B, Senovilla M, Larue C, Grolimund D, Tejada-Jiménez M, Imperial J, González-Guerrero M. 2017. Medicago truncatula Zinc-Iron Permease6 provides zinc to rhizobia-infected nodule cells. Plant Cell Environ 40:2706–2719. doi: 10.1111/pce.13035. [DOI] [PubMed] [Google Scholar]
- 53.Zhang P, Zhang B, Jiao J, Dai S-Q, Chen W-X, Tian C-F. 2020. Modulation of symbiotic compatibility by rhizobial zinc starvation machinery. mBio 11:e03193-19. doi: 10.1128/mBio.03193-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, Sansonetti PJ. 2007. An injected bacterial effector targets chromatin access for transcription factor NF-κB to alter transcription of host genes involved in immune responses. Nat Immunol 8:47–56. doi: 10.1038/ni1423. [DOI] [PubMed] [Google Scholar]
- 55.Dong W, Hamon MA. 2020. Revealing eukaryotic histone-modifying mechanisms through bacterial infection. Semin Immunopathol 42:201–213. doi: 10.1007/s00281-019-00778-9. [DOI] [PubMed] [Google Scholar]
- 56.Nagymihály M, Veluchamy A, Györgypál Z, Ariel F, Jégu T, Benhamed M, Szűcs A, Kereszt A, Mergaert P, Kondorosi É. 2017. Ploidy-dependent changes in the epigenome of symbiotic cells correlate with specific patterns of gene expression. Proc Natl Acad Sci U S A 114:4543–4548. doi: 10.1073/pnas.1704211114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang Y, Jelenska J, Cecchini NM, Li Y, Lee MW, Kovar DR, Greenberg JT. 2014. HopW1 from Pseudomonas syringae disrupts the actin cytoskeleton to promote virulence in Arabidopsis. PLoS Pathog 10:e1004232. doi: 10.1371/journal.ppat.1004232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimono M, Lu YJ, Porter K, Kvitko BH, Henty-Ridilla J, Creason A, He SY, Chang JH, Staiger CJ, Day B. 2016. The Pseudomonas syringae type III effector HopG1 induces actin remodeling to promote symptom development and susceptibility during infection. Plant Physiol 171:2239–2255. doi: 10.1104/pp.16.01593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whitehead LF, Day DA, Hardham AR. 1998. Cytoskeletal arrays in the cells of soybean root nodules: the role of actin microfilaments in the organisation of symbiosomes. Protoplasma 203:194–205. doi: 10.1007/BF01279476. [DOI] [Google Scholar]
- 60.Zhang X, Han L, Wang Q, Zhang C, Yu Y, Tian J, Kong Z. 2019. The host actin cytoskeleton channels rhizobia release and facilitates symbiosome accommodation during nodulation in Medicago truncatula. New Phytol 221:1049–1059. doi: 10.1111/nph.15423. [DOI] [PubMed] [Google Scholar]
- 61.Genre A, Timmers T. 2019. The symbiotic role of the actin filament cytoskeleton. New Phytol 221:611–613. doi: 10.1111/nph.15506. [DOI] [PubMed] [Google Scholar]
- 62.Griesmann M, Chang Y, Liu X, Song Y, Haberer G, Crook MB, Billault-Penneteau B, Lauressergues D, Keller J, Imanishi L, Roswanjaya YP, Kohlen W, Pujic P, Battenberg K, Alloisio N, Liang Y, Hilhorst H, Salgado MG, Hocher V, Gherbi H, Svistoonoff S, Doyle JJ, He S, Xu Y, Xu S, Qu J, Gao Q, Fang X, Fu Y, Normand P, Berry AM, Wall LG, Ané JM, Pawlowski K, Xu X, Yang H, Spannagl M, Mayer KFX, Wong GKS, Parniske M, Delaux PM, Cheng S. 2018. Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science 361:eaat1743. doi: 10.1126/science.aat1743. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Du H, Li P, Shen Y, Peng H, Liu S, Zhou GA, Zhang H, Liu Z, Shi M, Huang X, Li Y, Zhang M, Wang Z, Zhu B, Han B, Liang C, Tian Z. 2020. Pan-genome of wild and cultivated soybeans. Cell 182:162–176.e13. doi: 10.1016/j.cell.2020.05.023. [DOI] [PubMed] [Google Scholar]
- 64.Severin AJ, Woody JL, Bolon Y-T, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC. 2010. RNA-Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol 10:160. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cannon SB, McKain MR, Harkess A, Nelson MN, Dash S, Deyholos MK, Peng Y, Joyce B, Stewart CN, Rolf M, Kutchan T, Tan X, Chen C, Zhang Y, Carpenter E, Wong GKS, Doyle JJ, Leebens-Mack J. 2015. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol Biol Evol 32:193–210. doi: 10.1093/molbev/msu296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren L, Huang W, Cannon SB. 2019. Reconstruction of ancestral genome reveals chromosome evolution history for selected legume species. New Phytol 223:2090–2103. doi: 10.1111/nph.15770. [DOI] [PubMed] [Google Scholar]
- 67.Du M, Gao Z, Li X, Liao H. 2020. Excess nitrate induces nodule greening and reduces transcript and protein expression levels of soybean leghaemoglobins. Ann Bot 126:61–72. doi: 10.1093/aob/mcaa002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang X-C, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA. 2010. Genome sequence of the palaeopolyploid soybean. Nature 463:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 69.Wu Z, Wang M, Yang S, Chen S, Chen X, Liu C, Wang S, Wang H, Zhang B, Liu H, Qin R, Wang X. 2019. A global coexpression network of soybean genes gives insights into the evolution of nodulation in nonlegumes and legumes. New Phytol 223:2104–2119. doi: 10.1111/nph.15845. [DOI] [PubMed] [Google Scholar]
- 70.Sugawara M, Epstein B, Badgley BD, Unno T, Xu L, Reese J, Gyaneshwar P, Denny R, Mudge J, Bharti AK, Farmer AD, May GD, Woodward JE, Médigue C, Vallenet D, Lajus A, Rouy Z, Martinez-Vaz B, Tiffin P, Young ND, Sadowsky MJ. 2013. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol 14:R17. doi: 10.1186/gb-2013-14-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Golicz AA, Bayer PE, Bhalla PL, Batley J, Edwards D. 2020. Pangenomics comes of age: from bacteria to plant and animal applications. Trends Genet 36:132–145. doi: 10.1016/j.tig.2019.11.006. [DOI] [PubMed] [Google Scholar]
- 72.Singh K, Milstein JN, Navarre WW. 2016. Xenogeneic silencing and its impact on bacterial genomes. Annu Rev Microbiol 70:199–213. doi: 10.1146/annurev-micro-102215-095301. [DOI] [PubMed] [Google Scholar]
- 73.Guo HJ, Wang ET, Zhang XX, Li QQ, Zhang YM, Tian CF, Chen WX. 2014. Replicon-dependent differentiation of symbiosis-related genes in Sinorhizobium nodulating Glycine max. Appl Environ Microbiol 80:1245–1255. doi: 10.1128/AEM.03037-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DiCenzo GC, Benedict AB, Fondi M, Walker GC, Finan TM, Mengoni A, Griffitts JS. 2018. Robustness encoded across essential and accessory replicons in an ecologically versatile bacterium. PLoS Genet 14:e1007357. doi: 10.1371/journal.pgen.1007357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dong OX, Meteignier LV, Plourde MB, Ahmed B, Wang M, Jensen C, Jin H, Moffett P, Li X, Germain H. 2016. Arabidopsis TAF15b localizes to RNA processing bodies and contributes to snc1-mediated autoimmunity. Mol Plant Microbe Interact 29:247–257. doi: 10.1094/MPMI-11-15-0246-R. [DOI] [PubMed] [Google Scholar]
- 76.Kanyuka K, Rudd JJ. 2019. Cell surface immune receptors: the guardians of the plant’s extracellular spaces. Curr Opin Plant Biol 50:1–8. doi: 10.1016/j.pbi.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zipfel C, Oldroyd GED. 2017. Plant signalling in symbiosis and immunity. Nature 543:328–336. doi: 10.1038/nature22009. [DOI] [PubMed] [Google Scholar]
- 78.Wei Y, Balaceanu A, Rufian JS, Segonzac C, Zhao A, Morcillo RJL, Macho AP. 2020. An immune receptor complex evolved in soybean to perceive a polymorphic bacterial flagellin. Nat Commun 11:3763. doi: 10.1038/s41467-020-17573-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu HY, Zhang C, Li ZC, Wang ZR, Jiang XX, Shi YF, Tian SN, Braun E, Mei Y, Qiu WL, Li S, Wang B, Xu J, Navarre D, Ren D, Cheng N, Nakata PA, Graham MA, Whitham SA, Liu JZ. 2018. The MAPK kinase kinase GMMEKK1 regulates cell death and defense responses. Plant Physiol 178:907–922. doi: 10.1104/pp.18.00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar S, Trivedi PK. 2018. Glutathione S-transferases: role in combating abiotic stresses including arsenic detoxification in plants. Front Plant Sci 9:751. doi: 10.3389/fpls.2018.00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dalton DA, Boniface C, Turner Z, Lindahl A, Kim HJ, Jelinek L, Govindarajulu M, Finger RE, Taylor CG. 2009. Physiological roles of glutathione s-transferases in soybean root nodules. Plant Physiol 150:521–530. doi: 10.1104/pp.109.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bueno E, Mesa S, Bedmar EJ, Richardson DJ, Delgado MJ. 2012. Bacterial adaptation of respiration from oxic to microoxic and anoxic conditions: redox control. Antioxid Redox Signal 16:819–852. doi: 10.1089/ars.2011.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Curatti L, Rubio LM. 2014. Challenges to develop nitrogen-fixing cereals by direct nif-gene transfer. Plant Sci 225:130–137. doi: 10.1016/j.plantsci.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 84.Raghunath A, Sundarraj K, Nagarajan R, Arfuso F, Bian J, Kumar AP, Sethi G, Perumal E. 2018. Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biol 17:297–314. doi: 10.1016/j.redox.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dalton DA, Becana M, Matamoros MA, Udvardi M. 2010. Recent insights into antioxidant defenses of legume root nodules. New Phytol 188:960–976. doi: 10.1111/j.1469-8137.2010.03512.x. [DOI] [PubMed] [Google Scholar]
- 86.Cao Y, Halane MK, Gassmann W, Stacey G. 2017. The role of plant innate immunity in the legume-rhizobium symbiosis. Annu Rev Plant Biol 68:535–561. doi: 10.1146/annurev-arplant-042916-041030. [DOI] [PubMed] [Google Scholar]
- 87.Gourion B, Berrabah F, Ratet P, Stacey G. 2015. Rhizobium–legume symbioses: the crucial role of plant immunity. Trends Plant Sci 20:186–194. doi: 10.1016/j.tplants.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 88.Van Loon LC, Rep M, Pieterse CMJ. 2006. Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol 44:135–162. doi: 10.1146/annurev.phyto.44.070505.143425. [DOI] [PubMed] [Google Scholar]
- 89.Marx CJ, Lidstrom ME. 2002. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques 33:1062–1067. doi: 10.2144/02335rr01. [DOI] [PubMed] [Google Scholar]
- 90.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T, Bork P. 2008. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res 36:D250–D254. doi: 10.1093/nar/gkm796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Emms DM, Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20:238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 96.Emms D, Kelly S. 2018. STAG: Species Tree Inference from All Genes. https://github.com/davidemms/STAG.
- 97.Emms DM, Kelly S. 2017. STRIDE: species tree root inference from gene duplication events. Mol Biol Evol 34:3267–3278. doi: 10.1093/molbev/msx259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Buendia-Claveria AM, Romero F, Cubo T, Perez-Silva J, Ruiz-Sainz JE. 1989. Inter and intraspecific transfer of a Rhizobium-fredii symbiotic plasmid - expression and incompatibility of symbiotic plasmids. Syst Appl Microbiol 12:210–215. doi: 10.1016/S0723-2020(89)80016-5. [DOI] [Google Scholar]
- 101.Ling Q, Huang W, Jarvis P. 2011. Use of a SPAD-502 meter to measure leaf chlorophyll concentration in Arabidopsis thaliana. Photosynth Res 107:209–214. doi: 10.1007/s11120-010-9606-0. [DOI] [PubMed] [Google Scholar]
- 102.Li YZ, Wang D, Feng XY, Jiao J, Chen WX, Tian CF. 2016. Genetic analysis reveals the essential role of nitrogen phosphotransferase system components in Sinorhizobium fredii CCBAU 45436 symbioses with soybean and pigeonpea plants. Appl Environ Microbiol 82:1305–1315. doi: 10.1128/AEM.03454-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liao Y, Smyth GK, Shi W. 2014. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 106.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaufman L, Rousseeuw PJ. 1990. Finding groups in data: an introduction to cluster analysis. John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 108.Yu G, Wang LG, Han Y, He QY. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wickham H. 2011. ggplot2. WIREs Comput Stat 3:180–185. doi: 10.1002/wics.147. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains, plasmids, and primers used in this study. Download Table S1, XLSX file, 0.01 MB (13.7KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Symbiotic performance of Sinorhizobium strains on G. max cv. JD17 and G. soja WSD. Download Table S2, XLSX file, 0.01 MB (11.4KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Venn plot of differentially expressed genes revealed by dual RNA-seq. All samples from G. max cv. JD17 nodules (A) or G. soja WSD nodules (B) were compared to the treatment of SF4. No host DEGs were found in WSD nodules induced by SF4 and SF4M. Download FIG S1, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Venn plot of differentially expressed genes revealed by dual RNA-seq in G. soja WSD nodules. All samples from G. soja WSD nodules were compared to the individual treatments using corresponding wild-type strains. No host DEGs were found in WSD nodules induced by SF4 and SF4M. Download FIG S2, TIF file, 0.5 MB (525.5KB, tif) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Basic information on plant expression analysis within G. max cv. JD17 and G. soja WSD nodules. Download Table S3, XLSX file, 18.3 MB (18.3MB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Basic information on bacterial expression analysis (versus SF4) within G. max cv. JD17 nodules and G. soja WSD nodules. Download Table S4, XLSX file, 12.4 MB (12.4MB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The phylogenetic tree of phytoglobins in leguminous and nonleguminous plants forming nitrogen-fixing nodules. The protein sequences of Glycine soja and Lotus japonicus were extracted from SoyBase (□) and Kazusa DNA Research Institute (△), respectively. The protein sequences of other leguminous and nonleguminous plants were extracted from NCBI and GigaDB (*). Accession numbers are shown in brackets. Phytoglobin protein sequences were aligned with the ClustalW program, and the phylogenetic tree was constructed by the maximum-likelihood method using MEGA. Bootstrap values less than 75 are not shown. The phytoglobins of nonleguminous plants are highlighted in green. The red star indicates the putative duplication event generating symbiotic hemoglobins in the Papilionoids. Download FIG S3, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Basic information on bacterial expression analysis (versus wild type [WT]) within G. soja WSD nodules. Download Table S5, XLSX file, 3.0 MB (3MB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Overview of genome information for Sinorhizobium strains used in this work. Download Table S6, XLSX file, 0.01 MB (10.8KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Core genes used in chronogram construction. Download Table S7, XLSX file, 0.01 MB (10.2KB, xlsx) .
Copyright © 2021 Cui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Raw sequence data from our RNA-seq analyses can be accessed via NCBI Sequence Read Archive (PRJNA652809). Complete genomes of five wild-type strains used in this study have been deposited in the GenBank database (BioProject no. PRJNA353922: SF8, SS1, and SJ4; BioProject no. PRJNA285929: SF4 and SF2).