

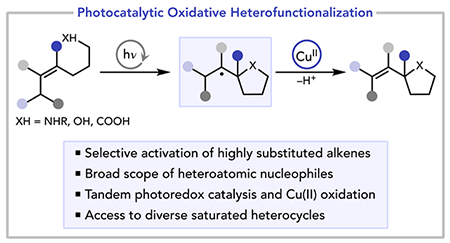
Abstract
Oxidative heterofunctionalization reactions are among the most attractive methods for the conversion of alkenes and heteroatomic nucleophiles into complex saturated heterocycles. However, the state-of-the-art transition metal-catalyzed methods to effect oxidative heterofunctionalizations are typically limited to unhindered olefins, and different nucleophilic partners generally require quite different reaction conditions. Herein, we show that Cu(II)-mediated radical–polar crossover allows for highly efficient and exceptionally mild photocatalytic oxidative heterofunctionalization reactions between bulky tri- and tetrasubstituted alkenes and a wide variety of nucleophilic partners. Moreover, we demonstrate that the broad scope of this transformation arises from photocatalytic alkene activation and thus complements existing transition metal-catalyzed methods for oxidative heterofunctionalization. More broadly, these results further demonstrate that Cu(II) salts are ideal terminal oxidants for photoredox applications and that the combination of photocatalytic substrate activation and Cu(II)-mediated radical oxidation can address long-standing challenges in catalytic oxidation chemistry.
Graphical Abstract
Oxidative alkene functionalizations1 are powerful methods for the rapid synthesis of saturated heterocycles commonly found in many important natural products, pharmaceuticals, and agrochemicals.2 These reactions are synthetically attractive for a number of reasons. First, they enable the direct oxidative coupling of alkenes with heteronucleophiles without the need to prepare preoxidized and often unstable heteroatom donors as internal oxidants.3–6 Moreover, and in contrast to redox-neutral alkene hydrofunctionalization reactions,7–9 oxidative alkene heterofunctionalizations conserve an olefin functionality that can be valuable for further synthetic elaborations. The state-of-the-art methods are Pd(II)-catalyzed processes that can proceed via either of two mechanistically distinct pathways (Scheme 1):10,11 syn-nucleopalladation involves migratory insertion of an alkene across a Pd–heteroatom bond,12 while anti-nucleopalladation involves attack of a heteroatomic nucleophile on a Pd(II)-coordinated alkene. The details of these mechanisms, however, engender several notable limitations. First, both mechanistic manifolds rely upon the coordination of the alkene to the Pd(II) center. Highly substituted alkenes that coordinate weakly, therefore, are poor reaction partners in all but a handful of highly specialized cases.13,14 Second, relatively subtle changes to the substrate structures and reaction conditions can switch the operative mechanistic pathway,15 requiring re-optimization of reaction conditions for heteroatom nucleophiles with different metal-binding propensities. Thus, the identification of conditions for the oxidative functionalization of sterically hindered alkenes with diverse heteronucleophiles remains an unsolved challenge that Pd(II) catalysis seems poorly positioned to address.
Scheme 1.
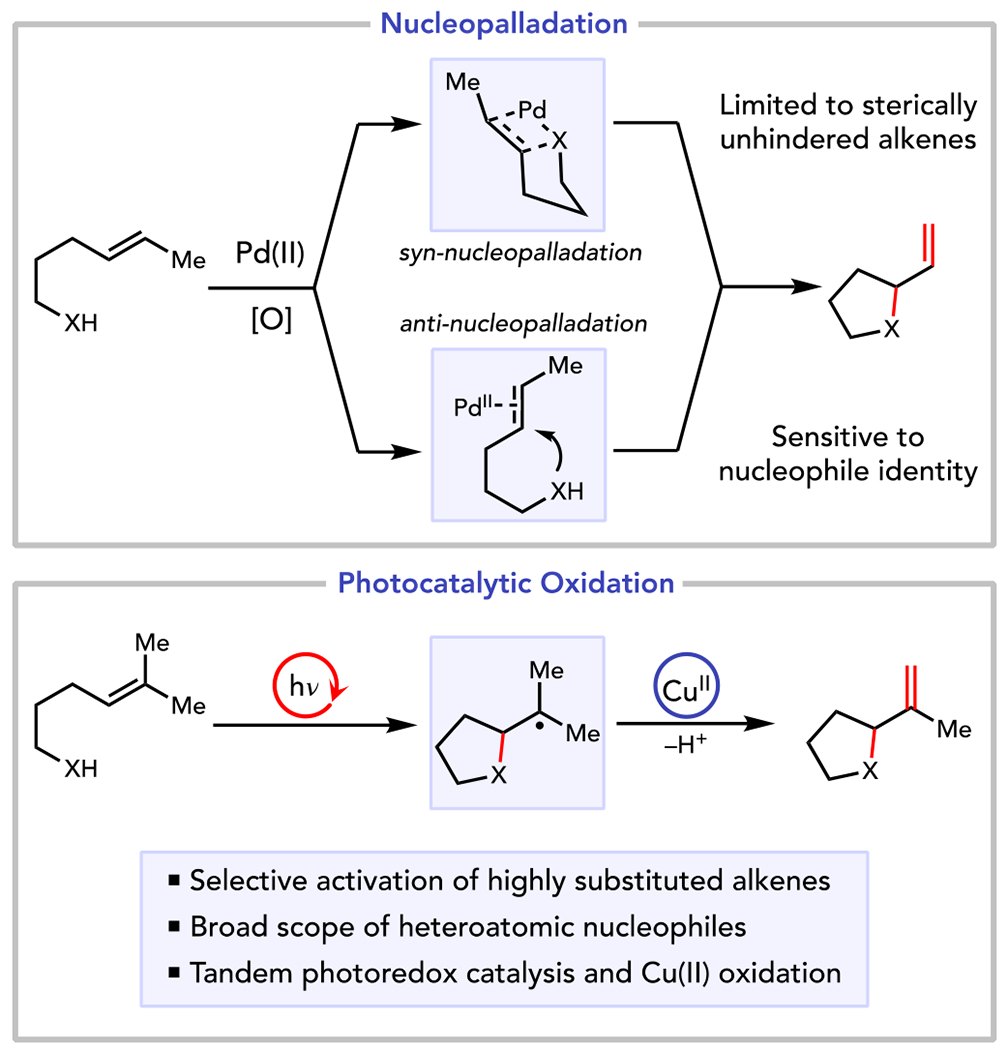
Oxidative Heterofunctionalization of Alkenes
We imagined that photoredox catalysis might offer a complementary strategy towards oxidative alkene heterofunctionalization that would be free of these long-standing limitations. Electron-rich tri- and tetrasubstituted alkenes that are generally poor substrates for Pd(II)-catalyzed oxidative heterofunctionalizations are easily activated by photoredox catalysts. Moreover, Nicewicz and others have shown that the resulting alkene radical cations are susceptible to highly regioselective nucleophilic attack, and because this step does not involve a discrete nucleophile–catalyst interaction, they are relatively agnostic towards the identity of the nucleophilic reaction partner.16–22 Thus, if an appropriate terminal oxidant could be identified to intercept the resulting organoradical intermediate and promote oxidative elimination, this process could constitute a new, alternative platform for oxidative alkene heterofunctionalization. Lei and coworkers recently demonstrated the feasibility of a similar approach in the oxidative coupling of olefins with alcohols or azoles using a dual photoredox/hydrogen evolution system.23 However, these reactions were limited to activated styrenic olefins and structurally simple nucleophiles, and thus the construction of complex saturated heterocyclic motifs has yet to be realized.
Inexpensive Cu(II) salts are ideal terminal oxidants for photoredox applications.24 Many electron-rich radical species are efficiently oxidized by Cu(II), which offers a means to divert the prototypical organoradical chemistry enabled by photoredox catalysis towards formally cationic reactivity via radical–polar crossover.25–27 We have leveraged this combination to design a photocatalytic protocol for the alkoxylation of benzylic C–H bonds28 and the photocatalytic oxyamination of electron-rich olefins.29 In both instances, oxidation of a photogenerated organoradical was followed by substitution with a heteroatomic nucleophile. To achieve oxidative heterofunctionalization, however, the radical mechanism would have to be terminated by an alternative oxidative elimination process. Seminal investigations by Kochi showed that thermally generated organoradicals could be diverted either towards oxidative substitution or oxidative elimination by tuning of reaction conditions,30–33 and more recent studies by Glorius and Tunge have demonstrated that the Cu(II)-mediated oxidative elimination pathway is operative for radicals produced by photochemical decarboxylation.34,35
We therefore propose a new mechanism for copper-enabled photoredox oxidative heterofunctionalization (Scheme 2). Photoexcitation of MesAcrPh+ affords a highly oxidizing excited state MesAcrPh+* (*E1/2red = + 2.12 V vs SCE).21 Rapid reductive quenching by alkene 1 would furnish radical cation 2 and MesAcrPh•. Radical cation 2 is a potent electrophile and should be readily trapped by a pendant heteroatomic nucleophile to generate carbon-centered radical 3. Radical oxidative addition of 3 by an appropriate Cu(II) oxidant would afford an organocopper(III) intermediate. Key to the successful realization of this strategy would be the optimization of the elimination step to favor contrathermodynamic elimination to 4. The alternate regiochemistry of oxidative elimination would afford an electron-rich enamine that would not survive the highly oxidizing conditions of this transformation. Finally, oxidation of MesAcrPh• by Cu(I) generated over the course of the reaction would close the photoredox catalytic cycle and regenerate ground-state MesAcrPh+.
Scheme 2.
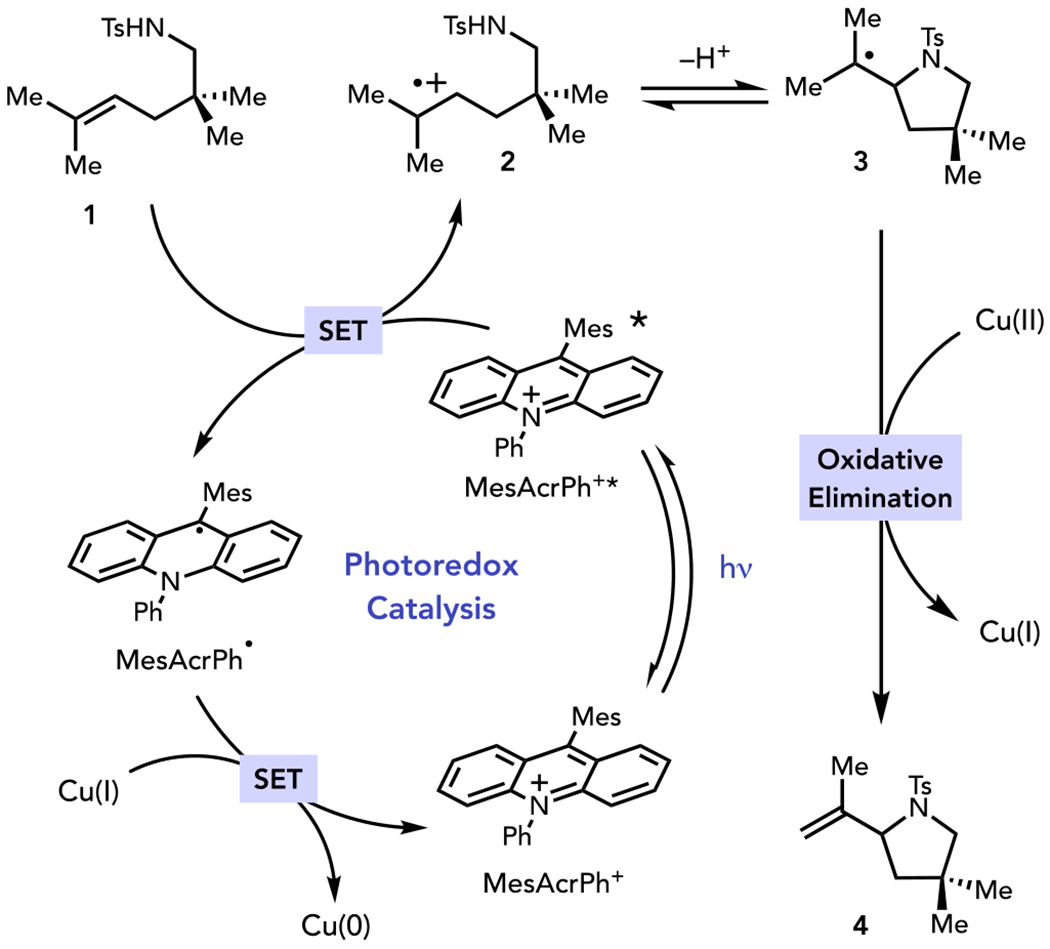
Proposed Reaction Design for Photocatalytic Oxidative Heterofunctionalization
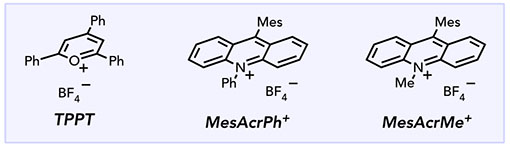
We began our investigations by examining the oxidative cyclization of trisubstituted alkene 1 upon irradiation in the presence of 2.5 mol% N-phenylacridinium tetrafluoroborate (MesAcrPh+), 1 equiv trifluoroacetic acid (TFA), and a variety of Cu(II) terminal oxidants (Table 1, entries 1–6). The formation of cyclized product 4 is observed in most cases, but as expected, the identity of the Cu(II) oxidant proved to be crucial. We hypothesize that sterically bulky carboxylate ligands improve the regioselectivity of alkene formation during Cu(II)-mediated oxidative elimination.36–37,38 Other highly oxidizing photocatalysts afford 4 in moderate yields (entries 7 and 8), but MesAcrPh+ proved optimal for this transformation. Empirically, we also observed that the addition of 1 equiv TFA improved the rate and reproducibility of oxidative cyclization (entry 9). This is most readily rationalized by the increased solubility of Cu(II) salts in organic media upon the addition of trifluoroacetic acid.39–41 Thus, optimized conditions were found to be: 2.5 mol% MesAcrPh+, 2 equiv copper(II) 2-ethylhexanoate (Cu(EH)2), 1 equiv 1, and 1 equiv TFA in 1,2-DCE with irradiation by two 15 W blue LED flood lamps (entry 6). Control reactions verified the photocatalytic nature of this process: omitting photocatalyst, Cu(II) oxidant, or light resulted in no formation of pyrrolidine 4 (entries 10–12).
Table 1.
Optimization of Oxidative Amination

| |||
|---|---|---|---|
|
| |||
| entry[a] | photocat. | oxidant | % yield[b] |
| 1 | MesAcrPh+ | CuBr2 | 0% |
| 2 | MesAcrPh+ | Cu(OTf)2 | 3% |
| 3 | MesAcrPh+ | Cu(OAC)2 | 48% |
| 4 | MesAcrPh+ | CU(TFA)2 | 57% |
| 5 | MesAcrPh+ | Cu(OPiv)2 | 57% |
| 6 | MesAcrPh+ | CU(EH)2 | 87% |
| 7 | MesAcrMe+ | CU(EH)2 | 52% |
| 8 | TPPT | Cu(EH)2 | 24% |
| 9[c] | MesAcrPh+ | Cu(EH)2 | 28% |
| 10 | none | CU(EH)2 | 0% |
| 11 | MesAcrPh+ | none | 0% |
| 12[d] | MesAcrPh+ | CU(EH)2 | 0% |
| |||
Reactions conducted using 1 (0.1 mmol), oxidant (2 equiv), TFA (1 equiv), and photocatalyst (2.5 mol%) in degassed 1,2-DCE and irradiated with a 15 W blue LED flood lamp for 16 h.
Yields were determined by 1H NMR analysis of the unpurified reaction mixtures using phenanthrene as an internal standard.
Reaction conducted in absence of 1 equiv TFA.
Reaction vessel was covered in aluminum foil.
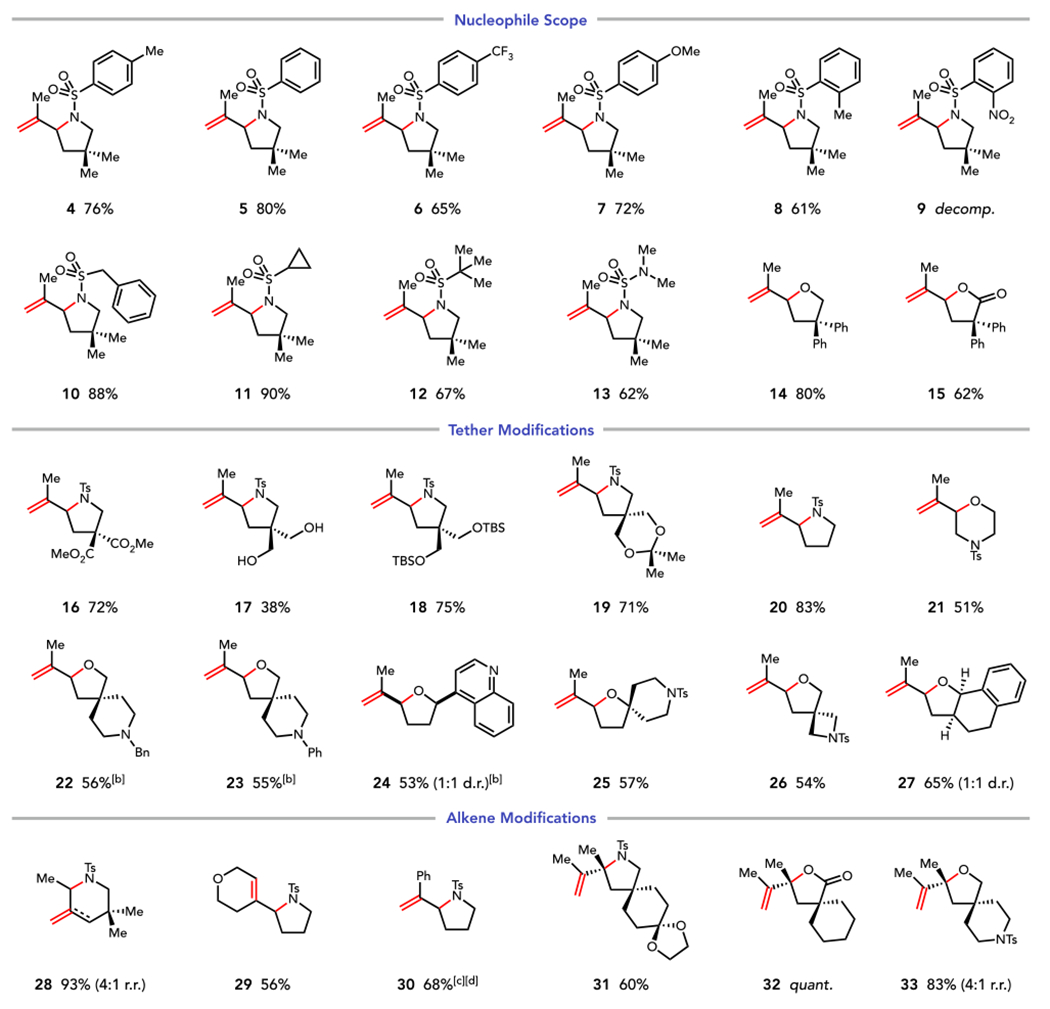
The degree of generality exhibited by this transformation is notable (Table 2). A wide variety of arylsulfonamides undergo efficient oxidative cyclization. Electron-rich and electron-deficient aryl sulfonamides react smoothly (4–8), including sterically hindered ortho-substituted sulfonamides (8). Unfortunately, 2-nosylsulfonamide was not tolerated (9), presumably due to the redox activity of the nitroarene. Alkyl sulfonamides are excellent reaction partners (10 and 11); these include a sterically hindered tert-butylsulfonamide, which gives 67% yield of 12. Sulfamoyl ureas (13), alcohols (14), and carboxylic acids (15) give high yields of cyclized product, offering efficient access to a diverse array of heterocyclic scaffolds. Importantly, no alteration of the catalyst system or reaction conditions is necessary in these cases despite the disparate nucleophiles examined.
Table 2.
Scope of Photocatalytic Oxidative Heterofunctionalization[a]
|
Reaction conditions: substrate (0.3 mmol, 1 equiv), MesAcrPh+ (2.5 mol%), Cu(EH)2 (2 equiv), TFA (1 equiv), and 1,2-DCE. irradiated for 8–96 h. Diastereomer and regioisomer ratios were determined by 1H NMR analysis of the unpurified reaction mixtures.
p-TsOH (2 equiv) instead of TFA.
p-TsOH (1 equiv) instead of TFA.
Reaction irradiated for 30 min.
Modifications to the length and identity of the tethering group are readily accommodated, and the functional group tolerance of this method is excellent. Common organic functional groups including esters (16), alcohols (17), silyl ethers (18), and acetals (19) are tolerated. These reactions exhibit excellent chemoselectivity as only cyclization of the sulfonamide is observed in the presence of nucleophilic free alcohols (17). While cyclizations affording five-membered rings are most efficient (20), six-membered rings are also readily accessible (21). Given the oxidizing nature of these conditions, we were pleased to find that tertiary amines (22) and anilines (23) are well tolerated. Lewis basic heterocycles that might coordinate to Cu(II) also do not interfere with the desired transformation (24); however, substrates containing Lewis basic functionalities react most efficiently with 2 equiv p-TsOH acid in place of TFA, presumably because the basic nitrogen is protonated under these conditions. These functionalities are also particularly noteworthy because they are common poisons for transition metal catalysts, further demonstrating the unique complementarity of this photocatalytic system to its Pd(II)-catalyzed counterparts. A sterically hindered tertiary nucleophile undergoes cyclization, delivering a densely functionalized spirocyclic tetrahydrofuran (25) in 57% yield. Azetidine 26 was prepared in 54% yield, showcasing the potential utility of this method in the synthesis of medicinally desirable structures.42 Bicyclic heterocycles could also be synthesized (27), demonstrating the application of this method in the construction of higher-order molecular architectures.
Trisubstituted alkenes are excellent substrates in this reaction, and in all cases, we observe exclusive anti-Markovnikov selectivity in the initial bond-forming event.43–45 Thus, cyclization can proceed in endo fashion, consistent with generation of a more stable radical intermediate upon nucleophilic trapping (28). In this experiment, the oxidative elimination gives a modest preference for the exocylic terminal olefin (4:1), in line with the regioselectivites observed by Glorius for decarboxylative olefination. Additionally, oxidative elimination affording endocyclic olefins can be conducted without issue (29). Styrenic olefins afford 1,1-disubstituted styrenes (30) as the sole products. Importantly, and in contrast to many Pd(II)-catalyzed oxidative amination methods, no alkene isomerization is observed.46–47,48,49 Short reaction times were crucial to obtain good yields of cyclized product, however, as 30 undergoes slow oxidative decomposition under the reaction conditions. To our delight, tetrasubstituted alkenes, typically among the most challenging substrates for Pd(II)-catalyzed heterofunctionalization methods, react smoothly, and, consistent with the absence of a discrete catalyst–nucleophile interaction, the identity of the heteroatomic nucleophile has little effect on the efficiency of cyclization. Sulfonamides (31), carboxylic acids (32), and alcohols (33) all afford good to excellent yields of the desired heterocycles.
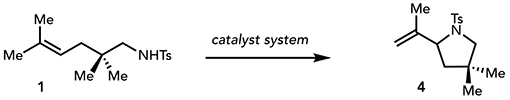
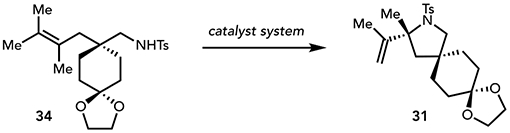
Importantly, the scope of this new oxidative heterofunctionalization reaction provides a synthetic capacity that state-of-the-art Pd(II)-catalyzed methods do not. To demonstrate this complementarity, we subjected trisubstituted alkene 1 and tetrasubstituted alkene 34 to several known, highly active Pd(II)-based catalyst systems for oxidative amination (Tables 3a and 3b).50–51,52,53 These systems uniformly afforded unsatisfactory yields of oxidative cyclization with significant decomposition of the starting alkenes. The highest yields were obtained with the catalyst system reported by Stahl (Pd(OAc)2/pyridine)52 but only after extended reaction times at elevated temperatures (13% and 22% yield of 4 and 31, respectively). In contrast, our photocatalytic protocol effects rapid and efficient oxidative cyclization of both 1 and 34 to their corresponding heterocycles.
Table 3.
Survey of Oxidative Amination Methods
| A Oxidative Amination of Trisubstituted Alkene 1 | |||
|---|---|---|---|
| |||
|
| |||
| entry | catalyst system | % yield | % RSM |
| 1a | MesAcrPh+/hν, Cu(EH)2 | 87 | 0 |
| 2b | Pd(OAc)2/DMSO, O2 | trace | 70 |
| 3c | Pd(OAc)2/pyridine, O2 | 13 | 59 |
| 4d | Pd(TFA)2/(−)-sparteine, O2 | trace | 69 |
| B Oxidative Amination of Tetrasubstituted Alkene 34 | |||
| |||
|
| |||
| entry | catalyst system | % yield | % RSM |
|
| |||
| 1a | MesAcrPh+/hν, Cu(EH)2 | 56 | 31 |
| 2b | Pd(OAc)2/DMSO, O2 | 4 | 66 |
| 3c | Pd(OAc)2/pyridine, O2 | 22 | 60 |
| 4d | Pd(TFA)2/(−)-sparteine, O2 | 7 | 78 |
Reaction conducted using MesAcrPh+ (2.5 mol%), Cu(EH)2 (2 equiv), TFA (1 equiv), and 1,2-DCE. irradiated for 16-18 h.
Reaction conducted using Pd(OAc)2 (5 mol%), NaOAc (2 equiv), O2, DMSO, rt, 72 h.
Reaction conducted using Pd(OAc)2 (5 mol%), pyridine (10 mol%), O2, toluene (0.1 M), 80 °C, 24 h.
Reaction conducted using Pd(TFA)2 (10 mol%), (−)-sparteine (40 mol%), DIPEA (2 equiv), MS 3Å, O2, toluene (0.1 M), 80 °C, 26 h.
The origin of this complementary reactivity can readily be rationalized upon examination of each reaction component by cyclic voltammetry (Figure 1). Trisubstituted alkenes are oxidized at significantly less positive potentials than sulfonamides, alcohols, or carboxylic acids in 1,2-DCE. Moreover, the oxidation of functionalized alkene 1 occurs at significantly less positive potentials than either alkenes or sulfonamides alone. As observed by Moeller in studies of the reactions of heteronucleophiles with electrochemically generated alkene radical cations,54,55 this observation would be consistent with a relatively slow alkene oxidation followed by a rapid cyclization event. Thus, the activation of the polysubsituted alkene moiety by photochemical oxidation rather than by transition metal coordination is responsible for the complementary reactivity profile we observe.56
Figure 1.
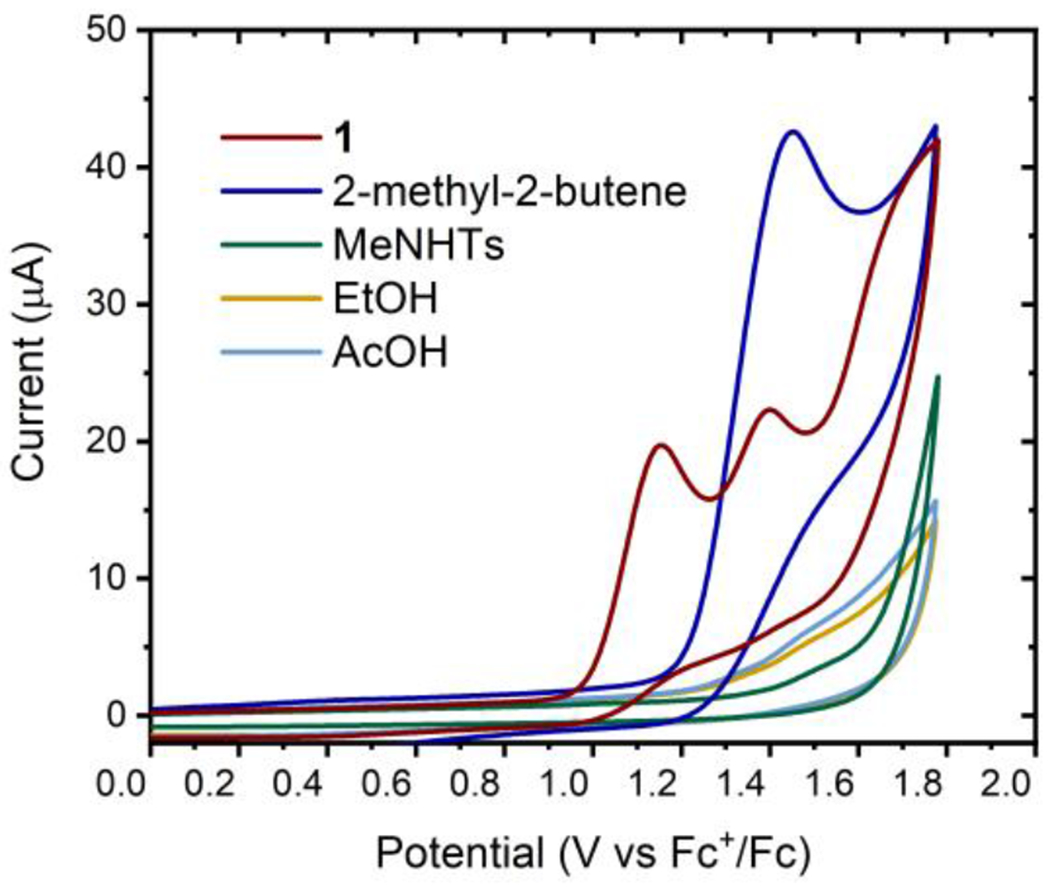
Cyclic Voltammetry Experiments
In summary, we have shown that a photocatalytic system consisting of a highly oxidizing photocatalyst and a Cu(II) terminal oxidant enables the oxidative heterofunctionalization of highly substituted alkenes with a diverse range of nucleophilic partners. This reaction addresses several long-standing synthetic limitations of state-of-the-art oxidative alkene heterofunctionalizations. The mechanism of photoactivation involving the generation of an alkene radical cation intermediate obviates the need for discrete metal–alkene or metal–nucleophile interactions, and the scope of this photocatalytic method thus complements those of existing Pd(II)-catalyzed reactions. More generally, this reaction adds to the growing evidence that Cu(II) salts are ideal terminal oxidants for photoredox oxidation reactions. While we have previously shown that a similar system enables oxidative substitution reactions of photochemically generated organoradical intermediates, here we show that their reactivity can be diverted towards oxidative elimination reactions. Together, these results suggest a powerful and potentially general strategy for the development of a wide range of novel oxidative functionalization reactions that exploit the versatility of photoredox activation.
Supplementary Material
ACKNOWLEDGMENT
Funding for this project was provided by the NIH (GM095666). G.L. is grateful to 3M for a 3M Science & Technology Fellowship. NMR and MS facilities at UW–Madison are funded by the NIH (1S10 OD020022-1) and a generous gift from the Paul J. and Margaret M. Bender Fund.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures; characterization data; spectra for all new compounds (PDF)
REFERENCES
- 1.For a review, see:; Nakamura I; Yamamoto Y Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis. Chem. Rev 2004, 104, 2127–2198. [DOI] [PubMed] [Google Scholar]
- 2.Heterocycles in Natural Product Synthesis; Majumdar KC, Chattopadhyay SK, Eds.; Wiley-VCH Verlag & Co. KGaA: Weinheim. 2011. [Google Scholar]
- 3.Stahl SS Palladium Oxidase Catalysis: Selective Oxidation of Organic Chemicals via Direct Dioxygen-Coupled Catalytic Turnover. Angew. Chem., Int. Ed 2004, 43, 3400–3420. [DOI] [PubMed] [Google Scholar]
- 4.Stahl SS Palladium-Catalyzed Oxidation of Organic Chemicals with O2. Science 2005, 309, 1824–1826. [DOI] [PubMed] [Google Scholar]
- 5.Kitamura M; Narasaka K Synthesis of Aza-Heterocycles from Oximes by Amino-Heck Reaction. Chem. Rec 2002, 2, 268–277. [DOI] [PubMed] [Google Scholar]
- 6.Race NJ; Hazelden IR; Faulkner A; Bower JF Recent developments in the use of aza-Heck cyclizations for the synthesis of chiral N-heterocycles. Chem. Sci 2017, 8, 5248–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- 8.Crossley SWM; Obradors C; Martinez RM; Shenvi RA Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Margrey KA; Nicewicz DA A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalization Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res 2016, 49, 1997–2006. [DOI] [PubMed] [Google Scholar]
- 10.McDonald RI; Liu G; Stahl SS Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kočovský P; Bäckvall J-E The syn/anti-Dichotomy in the Palladium-Catalyzed Addition of Nucleophiles to Alkenes. Chem. Eur. J 2015, 21, 36–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanley PS; Hartwig JF Migratory Insertion of Alkenes into Metal–Oxygen and Metal–Nitrogen Bonds. Angew. Chem., Int. Ed 2013, 52, 8510–8525. [DOI] [PubMed] [Google Scholar]
- 13.Yang G; Shen C; Zhang W An Asymmetric Aerobic Aza-Wacker-Type Cyclization: Synthesis of Isoindolinones Bearing Tetrasubstituted Carbon Stereocenters. Angew. Chem., Int. Ed 2012, 51, 9141–9145. [DOI] [PubMed] [Google Scholar]
- 14.Kou X; Shao Q; Ye C; Yang G; Zhang W Asymmetric Aza-Wacker-Type Cyclization of N-Ts Hydrazine-Tethered Tetrasubstituted Olefins: Synthesis of Pyrazolines Bearing One Quaternary or Two Vicinal Stereocenters. J. Am. Chem. Soc 2018, 140, 7587–7597. [DOI] [PubMed] [Google Scholar]
- 15.Trend RM; Ramtohul YK; Stoltz BM Oxidative Cyclizations in a Nonpolar Solvent Using Molecular Oxygen and Studies on the Stereochemistry of Oxypalladation. J. Am. Chem. Soc 2005, 127, 17778–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gassman PG; Bottoroff KJ Anti-Markovnikov Addition of Nucleophiles to a Non-conjugated Olefin via Single Electron Transfer Photochemistry. Tetrahedron Lett. 1987, 28, 5449–5452. [Google Scholar]
- 17.Mizuno K; Nakanishi I; Ichinose N; Otsuji Y Structural Dependence on Photoaddition of Methanol to Arylalkenes. Solvent and Additive Effects on Photoinduced Electron Transfer Reaction. Chem. Lett 1989, 1095–1098. [Google Scholar]
- 18.Hamilton DS; Nicewicz DA Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols. J. Am. Chem. Soc 2012, 134, 18577–18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen TM; Nicewicz DA Anti-Markovnikov Hydroamination of Alkenes Catalyzed by an Organic Photoredox System. 2013, 135, 9588–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perkowski AJ; Nicewicz DA Direct Catalytic Anti-Markovnikov Addition of Carboxylic Acids to Alkenes. J. Am. Chem. Soc 2013, 135, 10334–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilger DJ; Grandjean JM; Lammert T; Nicewicz DA The Direct Anti-Markovnikov Addition of Mineral Acids to Styrenes. Nature Chem. 2014, 6, 720–72. [DOI] [PubMed] [Google Scholar]
- 22.Wang H; Man Y; Xiang Y; Wang K; Li N; Tang B Regioselective Intramolecular Markovnikov and anti-Markovnikov hydrofunctionalization of alkenes via photoredox catalysis. Chem. Commun 2019, 55, 11426–11429. [DOI] [PubMed] [Google Scholar]
- 23.Yi H; Niu L; Song C; Li Y; Dou B; Singh AK; Lei A Photocatalytic Dehydrogenative Cross-Coupling of Alkenes with Alcohols or Azoles without External Oxidant. Angew. Chem., Int. Ed 2017, 56, 1120–1124. [DOI] [PubMed] [Google Scholar]
- 24.Reed NL; Yoon TP Oxidase reactions in photoredox catalysis Chem. Soc. Rev 2021, 50, 2954–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lampard C; Murphy JA; Lewis N Tetrathiafulvalene as a catalyst for radical-polar crossover reactions. J. Chem. Soc., Chem. Commun 1993, 295–297. [Google Scholar]
- 26.Murphy JA; Rasheed F; Roome SJ; Lewis N Termination of radical-polar crossover reactions by intramolecular nucleophiles. Chem. Commun 1996, 737–738. [Google Scholar]
- 27.Murphy JA; Rasheed F; Gastaldi S; Ravishanker T; Lewis N Synthesis of functionalized indolines by radical-polar crossover reactions. J. Chem. Soc., Perkin Trans. 1, 1997, 1549–1558. [Google Scholar]
- 28.Lee BJ; DeGlopper KS; Yoon TP Site-Selective Alkoxylation of Benzylic C–H Bonds by Photoredox Catalysis. Angew. Chem., Int. Ed 2019, 59, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed NL; Herman MI; Miltchev VP; Yoon TP Photocatalytic Oxyamination of Alkenes: Copper(II) Salts as Terminal Oxidants in Photoredox Catalysis. Org. Lett 2018, 20, 7345–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kochi JK Oxidation of Allylic Radicals by Electron Transfer: Effect of Complex Copper Salts. J. Am. Chem. Soc 1962, 84, 3271–3277. [Google Scholar]
- 31.Kochi JK The Decomposition of Peroxides Catalyzed by Copper Compounds and the Oxidation of Alkyl Radicals by Cupric Salts. J. Am. Chem. Soc 1963, 85, 1958–1968. [Google Scholar]
- 32.Kochi JK; Bemis A Carbonium Ions from Alkyl Radicals by Electron Transfer. J. Am. Chem. Soc 1968, 90, 4038–4051. [Google Scholar]
- 33.Kochi JK; Bemis A; Jenkins CL Mechanism of Electron Transfer Oxidation of Alkyl Radicals by Copper(II) Complexes. J. Am. Chem. Soc 1968, 90, 4616–4625. [Google Scholar]
- 34.Tlahuext-Aca A; Candish L; Garza-Sanchez RA; Glorius F Decarboxylative Olefination of Activated Aliphatic Acids Enabled by Dual Organophotoredox/Copper Catalysis. ACS Catal. 2018, 8, 1715–1719.. [Google Scholar]
- 35.Cartwright KC; Lang SB; Tunge JA Photoinduced Kochi Decarboxylative Elimination for the Synthesis of Enamides and Enecarbamates from N-Acyl Amino Acids. J. Org. Chem 2019, 84, 2933–2940. [DOI] [PubMed] [Google Scholar]
- 36.Faulkner A; Race NJ; Scott JS; Bower JF Copper catalyzed Heck-like cyclizations of oxime esters. Chem. Sci 2014, 5, 2416–2421. [Google Scholar]
- 37.Wu X; Riedel J; Dong VM Transforming Olefins into γ,δ-Unsaturated Nitriles through Copper Catalysis. Angew. Chem., Int. Ed 2017, 56, 11589–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C; Liu R-H; Tian M-Q; Hu X-H; Loh T-P Regioselective Copper-Catalyzed Oxidative Coupling of α-Alkylated Styrenes with Tertiary Alkyl Radicals. Org. Lett 2018, 20, 4032–4035. [DOI] [PubMed] [Google Scholar]
- 39.Antilla JC; Buchwald SL Copper-Catalyzed Coupling of Arylboronic Acids and Amines. Org. Lett 2001, 3, 2077–2079. [DOI] [PubMed] [Google Scholar]
- 40.Yoo W-J; Tsukamoto T; Kobayashi S Visible-Light-Mediated Chan–Lam Coupling Reactions of Aryl Boronic Acids and Aniline Derivatives. Angew. Chem., Int. Ed 2015, 54, 6587–6590. [DOI] [PubMed] [Google Scholar]
- 41.Brewer AC; Hoffman PC; Martinelli JR; Kobierski ME; Mullane N; Robbins D Development and Scale-Up of a Continuous Aerobic Oxidative Chan–Lam Coupling. Org. Process Res. Dev 2019, 23, 1484–1498. [Google Scholar]
- 42.Marson CM New and unusual scaffolds in medicinal chemistry. Chem. Soc. Rev 2011, 40, 5514–5533. [DOI] [PubMed] [Google Scholar]
- 43.Neunteufel RA; Arnold DR Radical Ions in Photochemistry. I. 1,1-Diphenylethylene Cation Radical. J. Am. Chem. Soc 1973, 95, 4080–4081. [Google Scholar]
- 44.Arnold DR; Du X; Chen J The Effect of Meta- or Para-Cyano Substitution on the Reactivity of the Radical Cations of Arylalkenes and Alkanes. Radical Ions in Photochemistry, Part 34. Can. J. Chem 1995, 73, 307–318. [Google Scholar]
- 45.Arnold DR; Chan MS; McManus KA Photochemical Nucleophile-Olefin Combination, Aromatic Substitution (photo-NOCAS) Reaction, Part 12. Factors Controlling the Regiochemistry of the Reaction with Alcohol as the Nucleophile. Can. J. Chem 1996, 74, 2143–2166. [Google Scholar]
- 46.Kotov V; Scarborough CC; Stahl SS Palladium-Catalyzed Aerobic Oxidative Amination of Alkenes: Development of Intra- and Intermolecular Aza-Wacker Reactions. Inorg. Chem 2007, 46, 1910–1923. [DOI] [PubMed] [Google Scholar]
- 47.Kohler DG; Gockel SN; Kennemur JL; Waller PJ; Hull KL Palladium-catalyzed anti-Markovnikov selective oxidative amination. Nature Chem. 2018, 10, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen JR; Bahamonde A; Furukawa Y; Sigman MS Enantioselective N-Alkylation of Indoles via an Intermolecular Aza-Wacker-Type Reaction. J. Am. Chem. Soc 2019, 141, 8670–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bahamonde A; Al Rifaie B; Martin-Heras V; Allen JR; Sigman MS Enantioselective Markovnikov Addition of Carbamates to Allylic Alcohols for the Construction of α-Secondary and α-Tertiary Amines. J. Am. Chem. Soc 2019, 141, 8708–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu G; Stahl SS Two-Faced Reactivity of Alkenes: cis-versus trans-Aminopalladation in Aerobic Pd-Catalyzed Intramolecular Aza-Wacker Reactions. J. Am. Chem. Soc 2007, 129, 6328–6335. [DOI] [PubMed] [Google Scholar]
- 51.Larock RC; Hightower T,R; Hasvold LA; Peterson KP Palladium(II)-Catalyzed Cyclization of Olefinic Tosylamides. J. Org. Chem 1996, 61, 3584–3585. [DOI] [PubMed] [Google Scholar]
- 52.Fix SR; Brice JL; Stahl SS Efficient Intramolecular Oxidative Amination of Olefins through Direct Dioxygen-Coupled Palladium Catalysis. Angew. Chem. Int. Ed 2002, 41, 164–166. [DOI] [PubMed] [Google Scholar]
- 53.Yip K-T; Yang M; Law K-L; Zhu N-Y; Yang D Pd(II)-Catalyzed Enantioselective Oxidative Tandem Cyclization Reactions. Synthesis of Indolines through C–N and C–C Bond Formation. J. Am. Chem. Soc 2006, 128, 3130–3131. [DOI] [PubMed] [Google Scholar]
- 54.Moeller K; Tinao LV Intramolecular Anodic Olefin Coupling Reactions: The Use of Bis Enol Ether Substrates. J. Am. Chem. Soc 1992, 114, 1033–1041. [Google Scholar]
- 55.Xu H-C; Moeller KD Intramolecular Anodic Olefin Coupling Reactions: Use of the Reaction Rate to Control Substrate/Product Selectivity. Angew. Chem. Int. Ed 2010, 49, 8004–8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.When a 1,2-disubstituted alkene was subjected to our photocatalytic conditions only unreacted starting material was observed (see supporting information). This result is inconsistent with an alternate pathway involving initial oxidation of the heteroatomic nucleophile by the excited state photocatalyst.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.