

Abstract
The life-sustaining air-blood interface of the respiratory system requires the exquisite integration of the epithelial lining with the mesenchymal capillary network, all supported by elastic smooth muscle and rigid cartilage keeping the expandable airways open. These intimate tissue interactions originate in the early embryo, where bidirectional paracrine signaling between the endoderm epithelium and adjacent mesoderm orchestrates lung and trachea development and controls the stereotypical branching morphogenesis. Although much attention has focused on how these interactions impact the differentiation of the respiratory epithelium, relatively less is known about the patterning and differentiation of the mesenchyme. Endothelial cells, smooth muscle cells, and chondrocytes together with other types of mesenchymal cells are essential components of a functional respiratory system, and malformation of these cells can lead to various congenital defects. In this review, we summarize the current understanding of mesenchymal development in the fetal trachea and lung, focusing on recent findings from animal models that have begun to shed light on the poorly understood respiratory mesenchyme lineages.
Keywords: mesenchyme, respiratory system, lung, trachea, development, tissue interaction
1. Overview of respiratory system development
There are several kinds of mesenchymal lineages in the adult lung: endothelial cells, smooth muscle cells (SMC), lipofibroblasts, myofibroblasts, macrophages, and some other less characterized fibroblasts. Embryonic development of the respiratory system begins in the first trimester, at about 28 days of gestation in humans and at embryonic day (E) 9.5 in the mouse, when the splanchnic mesoderm sends growth factor signals to the adjacent ventral foregut endoderm epithelium to specify a few hundred respiratory progenitors expressing the transcription factor Nkx2-1. Endothelial cells can be identified adjacent to the epithelium as early as respiratory specification [1], eventually constituting an indispensable component of the lung. Between E10.5 and E12.5 in the mouse, referred to as the embryonic stage of lung development, continued mesenchymal-epithelial interactions orchestrate the physical separation of the single foregut tube into a dorsal Sox2-positive esophagus and ventral Nkx2-1+ trachea, with bilateral primary lung buds evaginating into the surrounding splanchnic mesenchyme at the posterior end (Figure 1). During this morphogenesis, the foregut epithelium fuses in the middle and the lateral mesenchyme invades to completely surround the presumptive esophagus and trachea. The mesenchyme between the esophagus and trachea differentiates into the dorsal trachealis smooth muscle while the mesenchyme surrounding the ventral lateral aspect of the trachea develops into a series of cartilage rings separated by fibroelastic tissue [2] (Figure 2). Beginning in this embryonic stage, the primary lung buds elaborate branches that will form the major lobes of the lung.
Figure 1.

Timeline of lung development. At E9.5, the common foregut tube begins to express Nkx2.1 ventrally at the onset of respiratory specification. During the pseudoglandular stage, two lung buds arise from the trachea, now distinct from the esophagus, and begin to further develop the future lung lobes. By the end of the embryonic period, the tracheal mesenchyme has developed a distinct pattern of smooth muscle and cartilage, while the lung mesenchyme contains myofibroblasts, lipofibroblasts, smooth muscle, and vasculature.
Figure 2.
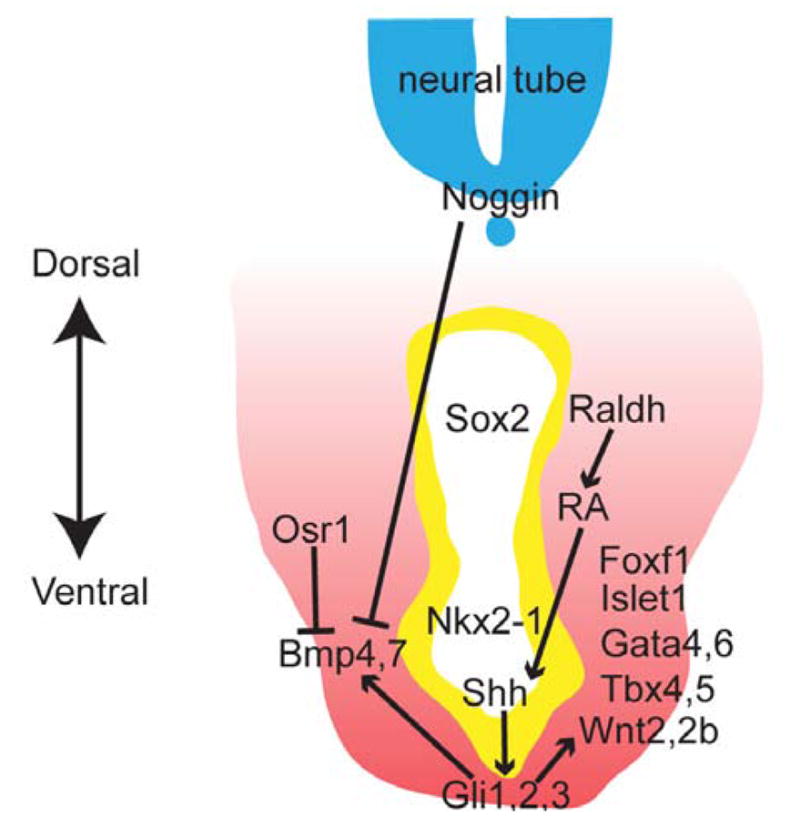
Epithelial-mesenchymal signals regulate respiratory specification. Raldh enzyme produces RA in the splanchnic mesenchyme surrounding the foregut. RA signals to the epithelium, promoting Shh ligand expression, which in turn functions through Gli effectors in the mesenchyme. HH activity is required for essential mesenchymal lung-inducing signals including Wnt2/2b and Bmp ligands. Those ventral Wnt and Bmp signals instruct ventral epithelium to initiate the respiratory program, marked by inducing Nkx2-1 while repressing Sox2 expression. RA signaling also coordinates patterning in the endoderm and mesoderm with appropriate competence to respond to inducing signals, including a range of mesenchymal identity genes such as Foxf1, Osr1, Gata4/6, and Tbx4/5. Additionally, dorsal signals such as Noggin antagonize ventral respiratory inducing signals, allowing for esophagus formation.
During the pseudoglandular stage (E12.5–E15.5), signaling interactions between the lung bud epithelium and the surrounding mesenchyme coordinate the stereotypical branching morphogenesis and proximal-distal (P-D) patterning of the growing fetal lung. By the end of the pseudoglandular period, the lung is highly arborized with distinct epithelial and mesenchymal cell types developing in different regions. The proximal branches will form the mainstem bronchi, with ciliated, basal, and secretory epithelial cells that are surrounded by alternating rings of cartilage and medial smooth muscle similar to the trachea. Airway smooth muscle cells (aSMC) first appear at E10.5 around bifurcating bronchi, and further extend in a cranial to rostral fashion to more distal airways, but are absent in the most terminal buds. In contrast, the distal lung tips, which will develop into the alveolar regions, are surrounded by a relatively thick mesenchyme that is still undifferentiated, with the exception of the vasculature plexus. During the canalicular and saccular stage [E16.5-postnatal day (P) 4], the distal small bronchioles undergo further branching and the surrounding mesenchyme thins to form alveolar sacs, where saccule epithelium is in close proximity to a distal vasculature network. The walls of such developing alveoli are called primary septae. The final phase of lung development is the alveolar stage (P4–P20), when further thinning of alveolar walls and the formation of secondary septae allow for efficient air exchange between alveolar epithelium and nearby capillaries [3]. Two major types of interstitial fibroblasts are identifiable in the maturing alveoli: lipofibroblasts and myofibroblasts, both of which are essential for alveolar development and play important roles in response to injury [4, 5].
The correct development of the lung mesenchymal lineages is crucial for respiratory physiology, and abnormalities during development can lead to respiratory diseases identified in patients. For example, alveolar capillary dysplasia with misalignment of pulmonary veins is a lethal neonatal lung disorder caused by mutations in the FOXF1 locus (OMIM 265380). This congenital disorder is characterized by decreased number of capillaries adjacent to alveolar epithelium, mislocation of pulmonary veins, and hypertrophy of pulmonary arteries [6]. Another example is malformation of trachea cartilage rings, which can lead to tracheal stenosis identified in human patients with breathing difficulties due to narrowed airways [7].
The pathophysiology of congenital respiratory diseases is beginning to be understood using animal model systems as well as human pluripotent stem cell-derived lung-like tissue. Historically, much of the research on respiratory system development and disease has focused on the epithelium, but recent research has begun to reveal important mechanisms in the mesenchymal lineages, which are the focus of this review. In particular, we will examine how a conserved collection of cell signaling pathways including Wnt, Bone Morphogenetic Proteins (BMP), Hedgehog (HH), retinoic acid (RA) and Fibroblast Growth Factor (FGF) are used reiteratively, with different functions at distinct times in fetal lung and trachea development. These repeated signals coordinate the integration of appropriate mesenchymal cells with the cognate epithelial tissue to generate the exquisite structure and physiological function of the respiratory system.
2. Early mesenchymal patterning and specification of the respiratory lineage
During early somite stages of embryogenesis (mouse E8.5–9.5), differential Wnt and BMP signals from the mesoderm pattern the foregut endoderm, with high ventral activity inducing a respiratory fate and low dorsal activity resulting in an esophageal fate (Figure 2). Expression of Wnt pathway ligands Wnt2 and Wnt2b (Wnt2/2b) in the ventral lateral splanchnic mesoderm activates the Wnt transcriptional effector β-catenin (β-cat) in the adjacent ventral foregut epithelium to induce respiratory fate as marked by expression of homeobox gene Nkx2-1 [8,9]. Bmp4 and Bmp7 are also expressed in the ventral splanchnic and cardiac mesoderm in cooperation with Wnt2/2b to promote a respiratory fate. Deletion of both type 1 BMP receptors (Bmpr1a;b) from the E9.5 foregut epithelium results in reduced Nkx2-1+ respiratory progenitors and ectopic expression of the dorsal foregut transcription factor Sox2 [10]. Epistasis experiments suggest that the main role of BMP is to suppress Sox2 expression in the ventral foregut endoderm and that this is required in order for Wnt/β-catenin to effectively induce Nkx2-1 [10]. Antagonists secreted from the dorsal mesoderm counteract the activity of ventral Wnt and BMP ligands in the presumptive esophageal region. Noggin expressed in the notochord protects the dorsal foregut from BMP signals and is necessary for proper tracheoesophageal morphology [11, 12]. Similarly, the secreted Wnt-antagonist Sfrp2, found in the dorsal-lateral mesoderm, appears to restrict Wnt activity to the ventral region of the foregut at E13.5 [13].
Recent studies in mouse and Xenopus have revealed a conserved gene regulatory network that controls the localized expression of these critical lung-inducing Wnt and BMP ligands [14]. Shortly after gastrulation (~E8.0 in mice), the anterior lateral plate mesoderm (LPM) cells express the enzymes Raldh2 and Rdh10, which synthesize the RA signaling morphogen from vitamin A [15]. RA then acts in two different ways to pattern distinct gene expression domains in the LPM. First, RA acts within the mesoderm to promote expression of a cohort of transcription factors in the LPM that overlies the presumptive lung endoderm including Foxf1, Osr1, Gata4/5/6, and Tbx4/5, all of which have been implicated in cardiopulmonary development. At the same time, RA restricts the Islet1+ secondary heart field and Hand1+ intestinal LPM, which are anterior and posterior to the lung field, respectively [14]. Secondly, RA signals to the endoderm to promote expression of HH ligands. These epithelial HH ligands then signal back to the Foxf1/Osr1+ LPM to proteolytically activate the transcription factors Gli2 and Gli3, which are nuclear effectors of the HH pathway. This HH/Gli signaling is required to maintain the Foxf1+ mesenchyme and stimulate Wnt2/2b and Bmp4 expression. Xenopus or mouse embryos completely deficient for either RA or HH signaling fail to express Wnt2/2b and exhibit respiratory agenesis [14]. Thus the bidirectional RA-HH signaling coordinates the development of the lung-inducing mesenchyme with the responding respiratory epithelium. It is currently unknown however, which of the genes in this RA-HH-Wnt signaling cascade are direct or indirect transcriptional targets. Indeed, the gene regulatory networks controlling mesodermal patterning during respiratory specification are likely to be even more complex as evidence from Xenopus suggests that Osr1/2 negatively regulate Bmp4 expression and that too much BMP/Smad activity can actually inhibit lung-inducing Wnt signals [16].
As the RA-HH signaling cascade patterns different LPM domains, it coordinates development of the heart and lungs by promoting a multipotent cardiopulmonary progenitor (CPP) cell population at the intersection of the secondary heart field and the presumptive lung mesenchyme. At E8.5, the CPP cells are located at the posterior pole of the heart and in the splanchnic mesoderm adjacent to the ventral foregut, and are marked by the HH-dependent co-expression of Wnt2, Islet1, and Gli1 [14, 17]. Genetic lineage tracing and clonal analysis have shown that the CPP cells can generate the myocardium of the heart inflow tract, the pulmonary airway and vascular smooth muscle, as well as pericytes and proximal endothelium in the lung [17].
The importance of proper LPM patterning during respiratory specification is illustrated by the fact that mutations in some of the key genes involved result in severe congenital conditions. For example, an important target of the RA-HH signaling in the LPM of mice and Xenopus is Foxf1. As mentioned earlier, human mutations in the FOXF1 locus or the genomic sequences that control its expression result in alveolar capillary dysplasia with misalignment of pulmonary veins [18]. Moreover, as we will see in the following section, disruptions to the mesenchymal Wnt-BMP signals that pattern the foregut can result in congenital tracheo-esophageal birth defects, such as tracheo-esophageal fistula (TEF).
3. Mesodermal lineages in the trachea and mainstem bronchi
After patterning of the foregut into an Nkx2-1+ ventral domain and a Sox2+ dorsal domain, the single foregut tube separates into the trachea and esophagus between E10.5 and E11.5 in mice. During this morphogenesis, the lateral walls of the foregut epithelium fuse at the boundary between the Sox2 and Nkx2-1 domains, and Foxf1+ LPM invades between the presumptive trachea and esophagus as they separate into distinct tubes [19]. The precise cell behaviors that drive this morphogenesis are obscure, and remain an active area of research, but the end result is a layer of mesenchyme completely surrounding the trachea that, between E10.5–14.5, differentiates into either smooth muscle or cartilage. Smooth muscle precursors expressing Actin alpha 2 (Acta2) are initially found in the mesenchyme of the carina, the point at which the trachea bifurcates, and then in the dorsal trachea and along the medial aspect of the primary lung buds that will form the large airways [2]. These Acta2+ cells eventually differentiate into the dorsal trachealis smooth muscle and into the smooth muscle that lines the medial aspect of the main bronchi [2]. In contrast, the ventral lateral mesenchyme of the trachea and the peripheral sides of the future mainstem bronchi develop into continuous chondrogenic precursors expressing the transcription factor Sox9, which are subsequently differentiated and restricted into collagen type II (Col2a1)-expressing cartilage rings [2, 20–22]. By E14.5, the differentiation of the mesenchyme is largely complete with dorsal smooth muscle and a series of C-shaped cartilage rings separated by membranous mesenchymal tissue, the developmental origin of which remains unknown, surrounding the ventral trachea and lateral main bronchi (Figure 3) [21, 23].
Figure 3.

Tracheal mesenchyme patterning. Many signaling pathways intersect to regulate the characteristic muscle-cartilage pattern found in the trachea. Wnt and Shh signaling appear to be required to promote expression of Sox9, a common chondrocyte marker. FGF is thought to both promote Sox9 and regulate formation of distinct cartilage rings. Tbx4/5 mutants display reduced cartilage and disrupted smooth muscle.
3.1. Molecular mechanisms of trachea and esophagus separation
The same signaling pathways that control early foregut patterning and respiratory specification continue to be expressed in the developing foregut and regulate tracheo-esophageal separation. For example, disruptions in Wnt-BMP signaling along the dorsal-ventral (D–V) axis of the foregut can result in defective tracheo-esophageal separation. Mouse embryos with null mutations in the BMP-antagonist Noggin, or Barx1−/− mutants that have reduced mesodermal Sfrp2 and dorsally expanded Wnt signaling, both exhibit TEF, where the esophagus fails to separate properly from the trachea [12, 13]. Conversely, conditional deletion of Bmp4 in the foregut mesenchyme leads to tracheal agenesis despite successful specification of respiratory progenitors, possibly due to reduced tracheal cell proliferation, while deletion of both Noggin and BMP ligand Bmp7 or Bmp4 rescues the Noggin−/− TEF phenotype [12, 24, 25].
Whereas a total absence of all RA signaling results in a complete loss of respiratory fate [14], a partial loss of RA signaling or a disruption in specific RA pathway components after respiratory specification can result in tracheal defects. Mouse embryos lacking various individual nuclear hormone RA receptors (RARs) exhibit TEF as well as defects in formation of the tracheal cartilage rings [26, 27]. For example, loss of both RARα and RARβ results in failure of the trachea and esophagus to separate, where RARγ mutants exhibit fused and disrupted cartilage rings [26–28].
Similar to the RA pathway, mutation of individual HH ligands, or Gli transcription factors, reveals a continued role for epithelial HH signals in tracheal development [29]. Shh and/or Indian hedgehog (Ihh) ligands are continually expressed in the respiratory epithelium from E8.5 through the late fetal period [14, 30], where they are well known to promote the survival and proliferation of the Foxf1+ mesoderm along the entire gut tube, including the trachea and lungs [31]. Both Foxf1+/−mouse mutants and human patients carrying mutations in the Foxf1 locus display TEF, while Shh−/− mice exhibit a single foregut tube [18, 30–32]. Compound Gli2−/−;Gli3+/− mutant embryos do make respiratory tissue but exhibit TEF with esophageal atresia, additionally highlighting the role that HH signaling plays in foregut tube separation [32]. Similar to Shh mutants, Nkx2-1−/− embryos fail to undergo proper tracheo-esophageal morphogenesis and exhibit a single foregut tube connected to hypoplastic lungs. Interestingly, the mesenchyme surrounding the single Nkx2-1−/− foregut tube has fewer cartilaginous rings and posteriorly resembles esophageal smooth muscle [33], while Sox2−/− mutants exhibit esophageal agenesis with a single foregut tube of respiratory character and expanded smooth muscle at the expense of tracheal cartilage [34].
While these studies have revealed some of the signaling pathways and transcription factors regulating foregut D–V patterning and subsequent tracheo-esophageal separation, how these factors interact in a gene regulatory network to control the cell behaviors that drive this complex tissue morphogenesis is still unknown and an important area of study.
3.2. Epithelial signaling that regulates tracheal mesenchyme differentiation
The observation that mutations in epithelial transcription factors Nkx2-1 and Sox2 cause defects in tracheal cartilage differentiation indicates that the identity of the epithelium influences the surrounding mesenchyme. Here again Shh plays an important role as an epithelial signal regulating mesenchymal differentiation. Shh−/− embryos display a reduction in the prechondrogenic transcription factor Sox9 in the tracheal mesenchyme at E13.5 followed by a total absence of cartilaginous rings in later fetal development. Similarly Gli2−/− embryos exhibit reduced and malformed tracheal rings [23, 32, 35]. Interestingly, cartilage differentiation can be rescued in Shh−/− foregut explants in vitro through treatment with either Bmp4 or Noggin, as the BMP pathway has previously been established to play a significant role in chondrogenesis [23, 36]. This result suggests that Shh may regulate cartilage differentiation in part by modulating the appropriate levels of BMP within the mesoderm [23], as it does during earlier respiratory specification at E9.5 [16].
Recently Wnt signaling from the tracheal epithelium has also been implicated in regulating tracheal mesenchyme differentiation. Epithelial specific deletion of the Wntless (Wls) gene, which encodes an intracellular cargo trafficking protein that is essential for the secretion of Wnt ligands, results in an absence of tracheal cartilage and expansion of disorganized tracheal smooth muscle into the ventral region [20, 37]. Several candidate Wnt ligands are expressed in the mouse tracheal epithelium at the right time including Wnt7b, Wnt5a, Wnt4, and Wnt11 (the last two are also expressed in mesenchyme), all of which may redundantly regulate cartilage formation [20]. Wnt7b−/− embryos exhibit incomplete cartilage rings with some hypertrophic tracheal smooth muscle [38, 39], whereas Wnt5a−/− embryos have a short trachea with fewer cartilage rings [40]. Wnt4−/− mutants display smaller cartilage rings and failure of cartilage ring fusion, following a reduction in Sox9+ chondrogenic precursors [41].
3.3. Signaling within the mesenchyme that regulates differentiation of the tracheal mesenchyme
One demonstrated mesenchymal signal critical for tracheal mesenchyme differentiation is Fgf10, which has multiple roles supporting lung development, as will be discussed in following sections [42, 43]. For example, one prominent role is that of inducing branching morphogenesis, as seen in Fgf10LacZ/−embryos, while Fgf10−/− embryos entirely lack lung tissue but maintain trachea [44, 45]. Evidence also suggests that there is a critical range of Fgf signaling that is required for tracheal mesenchymal differentiation. Starting around E14.5, Fgf10 becomes expressed in the ventral tracheal mesenchyme in between the cartilage rings where it appears to play a role in ring separation, although the detailed mechanism is largely unknown [43]. Consistent with this hypothesis, null mutations in Fgf10 or an epithelial splicing isoform of its receptor Fgfr2 exhibit a short trachea with disorganized cartilage rings [43, 46]. However, a mesenchymal Fgfr2 mutant containing an activating mutation found in the human Apert syndrome (OMIM 101200) demonstrates larger, fused tracheal rings, which can be rescued by a reduction in Fgf10 [47, 48]. The possibility that the FGF pathway may promote chondrogenesis is reinforced by the hyperplastic cartilage rings seen in mice overexpressing Fgf18, and promotion of tracheal cartilage in rats treated with exogenous FGF [21, 49, 50]. Additionally, treatment of respiratory tract explants beginning at E11.5 with pharmacological kinase inhibitors of FGFR and its downstream effector extracellular signal-regulated kinase (ERK) results in a significant reduction in Sox9 [21].
Other mesenchymal signals belonging to well-known signaling pathways play a role in tracheal mesenchymal differentiation. Mesenchyme-specific deletion of ERK1/2 kinases, known to play a role in cell proliferation, leads to a reduction in the number of cartilage rings [51]. Hypomorphic expression of Rspo2, which potentiates canonical Wnt signaling by binding to the Lrp6 receptor, leads to an absence of cartilage rings, further supporting the idea that β-catenin activation is required for tracheal cartilage ring formation [52]. Additionally, BMP signaling has been long established to play a role in chondrogenesis [36]. While these various signals are known to play some role in proper tracheal mesenchyme differentiation, their exact roles in this complex process remain undiscovered.
3.4. Transcription factors regulating tracheal mesenchyme differentiation
The signaling factors described above exert their effects largely by regulating the expression of key mesenchymal transcription factors such as Foxf1, Tbx4/5, and Sox9, which in turn execute the smooth muscle and cartilage differentiation programs. For example, mouse embryos lacking one allele of Foxf1, which appears to be regulated by Shh, Bmp4, and Fgf7, display hypoplastic and malformed tracheal cartilage [31]. The expression and regulation of these transcription factors are therefore critical for successful differentiation of the tracheal mesenchyme.
The essential role of Sox9, a transcription factor associated with cartilage development, in tracheal mesenchyme development is demonstrated by its conditional deletion in the mesenchymal cells, which leads to a reduction and disruption of cartilage ring formation [20]. Additionally, the opposing localization of ventral Sox9 and dorsal Acta2 leads to the possibility that the cartilage and smooth muscle lineages actively repress each other to promote proper localization. Interestingly, conditional respiratory lineage deletions of either Sox9 or serum response factor (Srf), a transcription factor that represents smooth muscle development, demonstrate proliferation but not expansion of the opposite tracheal mesenchymal cell type [2]. Conversely, in the mainstem bronchi, both proliferation and expansion of the opposite cell type are observed, suggesting a regional difference between the trachea and bronchi in regard to this cross-repression [2]. However, reduction of Sox9 in Col2a1+ cells does not expand Acta2 expression, leading to the possibility that the Sox9 and Acta2 expression domains in main bronchi are established before activation of Col2a1 at E10.5 [20, 22]. Distinct developmental mechanisms between the trachea and bronchi are further evidenced in Bmp4-null foreguts, which display bronchi but no trachea [24]. In addition, the refinement and maintenance of the Sox9 expression domain also remain mysterious. As mentioned earlier, the mechanism through which longitudinal stripes of Sox9 become laterally segmented into cartilage rings remains unknown. Additionally, the maintenance of Sox9 appears to depend on murine calcium channel Cav3.2, loss of which in tracheal chondrocytes leads to reduction of tracheal Sox9 without any changes in mesenchymal cell number or proliferation [53].
The T-box transcription factors Tbx4 and Tbx5 (referred to as Tbx4/5) are expressed throughout the mesenchyme of the respiratory region from E9.0 until E15.5 where their tracheal expression becomes limited to the mesenchyme in between cartilage rings [54]. Tbx4+/−;Tbx5−/− embryos contain fewer and malformed cartilage rings, but demonstrate relatively normal Col2a1 levels in the remaining cartilage rings accompanied by an expansion of mispatterned smooth muscle [54]. Additionally, while reduction of both Tbx4/5 and Fgf10 does not result in any tracheal phenotype distinct from those seen in Tbx4/5 mutants [54], the Fgfr2 knock-in model displays upregulation of Tbx4 and continuous Tbx5 expression that contrasts with its normal segmented pattern. This result suggests that Fgf signaling affects Tbx expression in certain contexts, some of which will be discussed below [47, 48]. While relatively less is known about the signals and transcription factors that regulate the tracheal smooth muscle development, mice carrying null mutations in Wls, Tbx4/5, or transmembrane protein Tmem16a, all display disrupted dorsal smooth muscle organization in addition to reduced cartilage [20, 54, 55].
These data suggest that there must be mechanisms that control the balance between cartilage and smooth muscle development in the trachea, which remain to be elucidated. Another important unresolved issue is how these different transcription factors are integrated into a gene regulatory network (GRN) to execute the differentiation program.
4. Fetal lung mesenchyme
4.1. Mesenchymal-epithelial signaling orchestrating proliferation, branching morphogenesis and proximal-distal patterning of the fetal lung
Between E9.5 and E10.5 in mice, a cascade of mesenchymal signaling including RA, Fgf10, Wnt, and Tgfβ2 promotes the initial outgrowth of the primary lung buds into the surrounding mesoderm at the posterior end of the forming trachea. RA seems to initiate this process. As discussed earlier, embryos completely lacking RA signaling such as Raldh2−/− mutants fail to specify the respiratory lineage and die in early gestation [56]. However, if Raldh2−/− fetuses are transiently supplemented with maternal RA between E7.5 and E8.5 to overcome early embryonic lethality, they do specify Nkx2-1+ progenitors but then fail to form lung buds similar to Fgf10−/− and Fgfr2b−/− mutants [56, 57]. On one hand, RA appears to repress the expression of the Wnt antagonist Dkk1 and thus generates a permissive territory where mesenchymal Wnt2/2b can maintain Nkx2-1 in the lung epithelium [58]. On the other hand, RA promotes Fgf10 expression in the mesenchyme by suppressing the activity of the TGFβ pathway [59]. RA may also indirectly maintain Fgf10 and Wnt2/2b levels by promoting the expression of mesenchymal transcription factors such as Tbx4, Tbx5, and Hoxa5. For example, depletion of potential RA targets Tbx4 and/or Tbx5 results in reduced Fgf10 and Wnt2b in chicken and mouse embryos [54, 60]. Together with previously described Fgf10 signals regulating Tbx4/5 expression during trachea development, it is possible that there might be a positive feedback between Fgf10 and Tbx4/5.
In the pseudoglandular and canalicular stages, several mesenchymal compartments can be identified in the lung: the mesothelial region, which is the outermost layer of the lung; the submesothelial region, which is immediately next to the mesothelial, with a relatively looser structure; and the subepithelial region, which is adjacent to the epithelium and demonstrates a more organized orientation. During this period of development, continued bidirectional mesenchymal-epithelial signaling coordinates growth, branching morphogenesis, and P–D patterning of the fetal lung. This is a well-studied system in mice where the mesenchyme at the tip of the growing lung buds is a dynamic signaling center that confers Sox9+ distal identity to the epithelium and promotes branching. As the distal tip grows away from the previous branch, the more proximal trunk segments are no longer influenced by signals from the distal signaling center, resulting in the adoption of a Sox2+ proximal identity by the trunk epithelium. Pulmonary mesenchyme also demonstrates P–D patterning. The mesenchymal cells surrounding the proximal airways differentiate into aSMC beginning at E11, whereas myofibroblasts and lipofibroblasts form in the distal region at later stages, facilitating alveologenesis. Additionally, the vasculature system continues to develop along with newly formed airways, with larger arteries forming along airways in contrast to more dispersedly distributed veins.
The distal tip signaling center is comprised of a series of reciprocal interactions mediated by Fgf10, Shh, Wnt2, and Bmp4 that form both positive and negative feedback loops (Figure 4A). Fgf10 binds to the receptor Fgfr2b in the adjacent epithelium, promoting branching morphogenesis and maintaining distal progenitor state. Fgf10 expression in the distal tip mesenchyme is negatively regulated by epithelial Shh, which is expressed throughout the epithelium, but is enriched in the distal tip. At the very tip of the branches, Shh activates the expression of HH interacting protein (Hhip), a negative feedback inhibitor that sequesters HH ligands, protecting Fgf10 expression at the very tip. This results in a dynamic expression of Fgf10, that is enriched at the tip, but becomes down regulated in the stalk, ensuring successful branching morphogenesis. Bmp4, expressed in the distal epithelium, also appears to have an inhibitory function on Fgf10-mediated branching initiation while maintaining distal tubule identity. Additionally, Wnt2/2b are expressed in the distal mesenchyme while Wnt7b is enriched in distal epithelium. Wnt2 and Wnt7b are suggested to promote branching and maintain distal epithelial identity. Consistently, loss of Wnt pathway effector β-catenin (β-cat) or overexpression of Wnt antagonist Dkk1 results in reduced branching and impaired epithelial differentiation, potentially through downregulating Fgfr2b and Bmp4 [61]. Finally, Tgfβ1 is enriched in the mesenchyme of the stalk region and is suggested to inhibit branching based on in vitro data [3].
Figure 4.

Mesenchymal-epithelial crosstalk directing branching and mesenchymal differentiation into vasculature and smooth muscle cells. A) Several signals direct branching and proximal-distal patterning in lung endoderm. Submesothelial Fgf10 signals to the nearby distal epithelium, inducing new branching formation. Shh is expressed in the epithelium and is enriched in the distal tip. Shh inhibits Fgf10 expression, while at the distal tip, high Shh signaling activates Hhip expression, which in turn sequesters Shh ligand, thus protecting Fgf10 expression at the distal tip. Bmp4 appears to have autocrine inhibitory effects on budding. Wnt signals, activated by mesenchymal Wnt2/2b and epithelial Wnt7b, cooperate with Fgf10 to promote branching morphogenesis. B) Vegf, which is gradually restricted to the distal epithelium, promotes vasculature development, partly through supporting RA and Hgf signals in endothelial cells. The Wnt pathway appears to have multiple functions supporting vasculature formation. Epithelial Wnt ligands are critical during this process; Wnt activity in the epithelium is indicated to promote Vegf signaling, while Wnt signaling in endothelial cells is required for endothelial cell maturation. Other important endothelial autonomous transcription factors include Foxm1, Prx1 and Sox17. C) Wnt2 and Wnt7b, through effector β-cat, promote smooth muscle differentiation from multipotent mesenchymal cells, while Hox5 appears to be upstream of Wnt signaling. Fgf10 appears downstream of Wnt2, marks SMC progenitors, and is also required for SMC development, potentially through upregulation of epithelial Bmp4. Fgf9, from epithelial and mesothelial cells, signals through mesenchymal Fgfr1/2 and prevents SMC differentiation distally. Additionally, Shh is required for both airway and vascular SMC development.
This same signaling network that controls branching morphogenesis also coordinates the growth of both the mesenchymal and epithelial compartments to ultimately allow for the generation of sufficient lung volume. Lineage labeling of single Tbx4+ mesenchymal progenitors suggests that these progenitors appear to be separated into either right or left lobes from as early as E9 [62]. During the pseudoglandular stage, Fgf9 is expressed in the epithelium and mesothelium, and acts to maintain Shh, which in turn promotes mesenchymal proliferation and cell survival [63]. Fgf9 also signals to Fgfr1/2c in the mesenchyme and promotes Wnt2 expression, which in turn acts through β-cat to promote mesenchymal proliferation through Cyclin D1 [64]. Another epithelial signal is Wnt7b. In addition to regulating branching morphogenesis as mentioned earlier, Wnt7b coordinates epithelial and mesenchymal proliferation by activating canonical Wnt signaling in adjacent mesenchyme and supporting Bmp4 expression in the epithelial tip. Interestingly, though Wnt7b-null mutants display hypoplastic lungs, general patterning and differentiation is not affected [38]. For more details on the complex molecular mechanisms controlling lung branching morphogenesis, we refer readers to these excellent reviews [3, 65].
One outstanding question is how the locations of initial domain branches, which set up the overall structure of the lung, are determined. The position of a new branch during later branching is suggested to be an intrinsic property of the distal signaling center and the feedback inhibition. During early lobe initiation, the location of five lobes appears rather stereotypical and genetically wired but the mechanisms that regulate this process remain mysterious. Another interesting question is which signals are responsible for regulating the size of the developing lung tissue, since inadequate proliferation can lead to congenital defects such as lung hypoplasia. During epithelial branching and P–D identity establishment, mesenchymal cells also undergo differentiation and patterning in different regions of the lung.
4.2. Genetic lineage tracing of lung mesenchyme
Genetic cell labeling experiments have begun to shed light on the relationship between various mesenchymal lineages, their ontogeny, and how they are formed. The ontogeny of mesenchymal cells has long been an intriguing topic for lung researchers. Injection of a retroviral library into the airway of cultured E11.5 lungs demonstrates clonality of cells localized initially near the hilum that then migrate down the airways to the subpleural regions, mostly giving rise to peribronchial SMCs [66]. With the development of various genetic lineage tracing tools such as Cre and CreER together with reporters, progenitors expressing such recombinases driven by certain markers will recombine the stop codon in front of the reporters; thus cells can be permanently labeled with expression of certain markers such as lacZ or various fluorescent reporters. In the case of creER, the recombinase is only activated in the presence of tamoxifen which can be administered at specific time points, thus achieving both tissue and timing specificity. Lineages of pulmonary mesenchymal progenitors will be discussed below and summarized in table 1.
Table 1.
Lineage tracing of pulmonary progenitors. Various progenitors labeled at different stages using genetic tools and the differentiated lineages they give rise to as lung develops.
| Progenitor | Time of label | Expression when labeled | vSMC | aSMC | Endo | Peri | Myof | Lipo |
|---|---|---|---|---|---|---|---|---|
| Wnt2, Islet1, Gli1 | E8–E9 | Cardiopulmonary progenitor | ✓ | ✓ | (proximal) | ✓ | ||
| Gli1 | E10–E11 | Scattered in mesenchyme | ✓ | ✓ | ✓ | |||
| Tbx4 | E6≫E11 | All mesenchymal cells | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Tbx4 | >E11 | Majority of mesenchyme | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Tbx4 | >E15 | Majority of mesenchyme | ✓ | ✓ | ✓ | ✓ | ||
| Fgf10 | E11.5 | Submesothelial cells | ✓ | ✓ | ✓ | |||
| Fgf10 | P2–P14 | Mesenchyme subpopulation | ✓ | |||||
| WT1 | E10 | Mesothelial cells | ✓ | ✓ | ✓ | ✓ | ||
| Notch N1IP | E13.5 | Subpopulation of mesothelial and mesenchymal cells | ✓ | ✓ | ||||
| Pdgfrα | >E14 | Mesenchyme surrounding proximal airway to scattered in distal mesenchyme | ✓ | ✓ |
Endo, endothelial cells; peri, pericytes; myof, myofibroblast; lipo, lipofibroblasts.
The earliest multipotent cardiopulmonary progenitor cells are marked by the co-expression of Gli1, Wnt2, and Islet1. As mentioned earlier, lineage tracing of these cells starting at E8–9 shows that they contribute to the heart, pulmonary vasculature and some lung mesenchyme [17]. However, Gli1-expressing cells restrict their potential as lung develops. When they are labeled at E10 and E11, they give rise only to mesothelial cells, parabronchial and perivascular SMC at E12 as well as secondary alveolar crest myoblasts at P14 [67]. As previously mentioned, Tbx4 is expressed in the splanchnic mesoderm surrounding the presumptive lung as early as E9.25 [54]. Using reverse tetracycline transactivator (rtTA) together with TetO-Cre and transgenic reporter mice, Cre is expressed in rtTA-positive cells when given Doxycycline (Dox). A Tbx4-rtTA system, where rtTA is driven under a Tbx4 enhancer, targets almost all mesenchymal cells surrounding lung at E10.5, with Dox induction from E6.5 to E10.5. Tbx4 expression appears to overlap with Gli1, Wnt2 and Islet1, but a direct lineage comparison has not yet been done. By providing Dox at various stages, the dynamics of this system is also identified. While vascular SMC and endothelial cells can only be targeted when given Dox prior to E15.5 and E11.5, respectively, other cell types, including aSMC, myofibroblasts, lipofibroblasts, and pericytes, can be labeled during the entire developmental period [68].
As early as E10, mesothelial cells are identified with expression of Wilm’s tumor suppressor gene Wt1. Using both Wt1-cre or creERT lineage tracing and ex vivo dye labeling, mesothelium is shown to undergo epithelial to mesenchymal transition (EMT) and migrate into the bulk region of the lung. This population of cells contributes to lung mesenchyme, differentiating into vascular endothelial and smooth muscle cells, bronchial smooth muscle, tracheal cartilage, pericytes, and platelet-derived growth factor receptor (Pdgfr) β-expressing cells along pulmonary blood vessels. The entry of mesothelial cells into lung mesenchyme occurs until E17.5, and appears to be dependent on HH signaling [69]. Cells derived from the Wt1 lineage persist postnatally. Interestingly, fetal but not adult Wt1-traced cells increase proliferation during fibrosis [70]. The Wt1 mutant shows lung lobe fusion, reduced pleural cavities and diaphragmatic hernia [71].
In addition to the previously mentioned function of Fgf10 during branching morphogenesis, lineage tracing shows that two distinct waves of Fgf10+ progenitors give rise to distinct descendants. The first wave comprises Fgf10-expressing cells in the submesothelial region during the early pseudoglandular stage and contributes to parabronchial and vascular smooth muscle cells, as well as lipofibroblasts at later stages. The second wave of Fgf10+ progenitors during the late pseudoglandular stage develops into lipofibroblasts and some other uncharacterized mesenchymal cells in the lung [72]. Furthermore, Fgf10 signals through Fgfr1b/2b are also required for lipofibroblast formation [73].
Canonical Notch signaling appears to regulate arterial SMC recruitment and Clara cell selection. Cells that have experienced Notch activation are labeled using a knock-in mouse line with Notch1 Intramembrane Proteolysis (N1IP)-cre, where cre activity is induced when the receptor Notch1 is proteolysed. These cells are first identified at E13.5 in a subpopulation of mesothelial and mesenchymal cells. Later, the vascular plexus, arterial endothelium, and vascular SMC (vSMC) are labeled, which is also dependent on adequate Notch response [74]. Additionally, Notch promotes EMT in mesothelial cells, and Notch deficiency during this process can be rescued with the activation of a general EMT inducer Tgfβ signal [74].
Pdgfrα is required for secondary septum formation and development of lipofibroblasts during postnatal lung development [75]. Pdgfrα+ cells identified using a green fluorescent protein (GFP) knock-in reporter during the pseudoglandular stage are associated mostly with proximal airways, but can be found in the distal mesenchyme by the canalicular stage. These cells eventually co-express either myofibroblast markers at the tip of the secondary septae or lipofibroblast markers in the primary septae during alveolarization [76]. To lineage trace those cells, both a Pdgfrα:cre line as well as a tamoxifen-inducible Pdgfrα:creERT were used. Constitutive cre suggests this population contributes to bronchial SMC, myofibroblasts and lipofibroblasts. Furthermore, the inducible line reveals that the commitment to myofibroblasts and lipofibroblasts occurs before secondary septation [76]. In addition, Pdgfrβ lineage tracing reveals that all pulmonary artery wall smooth muscle cells derive from this population [77].
Besides differentiation of mesenchymal lineages that originate from the splanchnic mesoderm around the foregut, the lung also acquires additional lineages from the hematopoietic system. Two major populations of macrophages reside in the adult lung. Alveolar macrophages locate at the lumen of alveoli and are exposed to the environment. They phagocytose foreign particles and also catabolize surfactants. Interstitial macrophages reside in the interalveolar space together with other mesenchymal cells important for tissue remodeling and antigen presentation. During development, three waves of macrophage formation are recognized. First, macrophages marked by the cell surface glycoprotein F4/80 are diffusely distributed in lung starting from E10.5, with lineage tracing suggesting their origin in the yolk sac. About two days later, a second wave of macrophages, marked by galactose-specific lectin Mac2 and potentially derived from fetal liver, intermingle with the first population in the lung interstitium. Within the first week of postnatal life, the Mac2+ macrophages enter the alveolar space, constituting alveolar macrophages, while the F4/80+ macrophages are restricted to submesothelial and perivascular regions. In the meantime, bone marrow-derived circulating macrophages populate the lung mesenchyme, forming the definitive interstitial macrophages [78].
Though the various lineage tracings mentioned above have uncovered several important mesenchymal progenitor populations, the comprehensive mesenchymal lineage ontogeny map is still lacking. The relative relationships between different progenitor populations or how different lineages arise from the same progenitor populations are not well understood. Despite that, there is increasing understanding about the development of several major mesenchymal lineages, such as vasculature, smooth muscle, myofibroblasts and lipofibroblasts. We will focus here on the vasculature and smooth muscle cells, which differentiate in the prenatal period. For a discussion on myofibroblasts and lipofibroblasts, which differentiate postnatally, please refer to other recent reviews [79, 80].
4.3. Pulmonary vasculature
In the lung, the pulmonary arterial system brings deoxygenated blood from the right heart to the alveolar capillary surface, which is then returned to the left heart via the pulmonary venous system while the lymphatic system collects the interstitial fluid. While the arterial system develops largely alongside the airway structures, the venous and lymphatic systems have their own unique pattern. As mentioned earlier, the pulmonary endothelial plexus can be identified in association with lung buds as early as E9.5. Moreover, the vasculature connection between heart and lung is dependent on HH signaling in the previously mentioned CPP [17]. Emerging pulmonary vasculature lacks fate specification [81]. By E10.5, pulmonary arteries can be identified with rudimentary lumens, connecting the aortic sac with the developing lung region [82]. By E15.5, more organized vascular networks with distinguishable arterial and venous identities are present to support circulation. As the lung develops, the pulmonary vascular system continues to expand and mature in the form of a tree-like structure in a proximal to distal manner. The supporting structures of the vessels also vary from proximal to distal; for example, smooth muscle layers are present proximally whereas the distal plexus is mainly surrounded by only pericytes. The alveolar capillary system expands extensively during the saccular and alveolar stages in close proximity to the alveolar epithelial cells. By day 34 of human gestation, continuous flow can be observed connecting the aortic sac, the pulmonary artery, the capillary plexus, the pulmonary veins, and the left atrium [83].
It is still not entirely clear whether pulmonary vasculature is derived through angiogenesis, sprouting from pre-existing vessels, or vasculogenesis, de novo differentiation into endothelial cells, or a combination of both processes. Analysis of Flk1-lacZ endothelial reporter mice suggests that much of the pulmonary vasculature is mostly derived through vasculogenesis [82]. Another model suggests a distal angiogenesis mechanism, where pulmonary endothelial cells are always in connection with the whole body circulatory system and expand to form new networks as the lungs develop [84]. A third model proposes that the central vessels form through angiogenesis while the vascular plexus in the distal fetal lung forms through vasculogenesis, and that those two systems of vessels then somehow connect to form the pulmonary circulatory system [85, 86]. The formation of pulmonary veins that do not track with the distal airway is less understood than that of arteries, but immunostaining of different stages of human embryos suggests that they form through vasculogenesis during the pseudoglandular stage and then via angiogenesis at later stages [83]. A recent paper identified two enhancers of the Foxf1 locus, one that promotes Foxf1 expression in proximal vessels and another in distal vasculature, reinforcing the regional differences of blood vessel development [87]. Temporally controlled lineage tracing, such as examination of a pulse labeling of all vasculature at early stages, should provide more definitive information on this issue. Nonetheless, several signal pathways have been identified as critical regulators of pulmonary vasculogenesis as will be discussed below (Figure 4).
The vascular endothelial growth factor (Vegf) is essential for all endothelial development in embryos. The Vegf receptor Flk1 mutant mouse embryos die before E9.5, with blood vessels completely absent [88]. During lung development, Vegf is initially produced in both epithelial cells and mesenchymal cells, and later is more restricted to distal epithelium and then most likely only in type II cells, a specialized distal cell type that produces surfactant protein-C (SP-C). Both Vegf receptors Flt1 and Flk1 are expressed in the lung mesenchyme [89]. Inhibition of Vegf signaling using soluble receptors in renal capsule grafts leads to compromised vasculature formation, demonstrating its necessity in supporting vasculature development [90]. Conditional loss of Vegf in type II cells results in decreased capillary vessels, partially through RA signaling in endothelial cells [89]. Vegf signaling from the epithelium also induces the expression of hepatocyte growth factor (Hgf) in endothelial cells that acts in an autocrine manner to promote vascular development [91]. However, excessive Vegf can also be detrimental to vascular development. Overexpression of various Vegf isoforms can disrupt distal vasculature networks, leading to atypical evaginations of small vessels into proximal large airways [92] or the promotion of lymphatic vasculature over blood vessels [93]. Recent evidence suggests that pulmonary Vegf signaling is also regulated by microRNAs, as anti-angiogenic miR122 and pro-angiogenic miR130a, expressed in both epithelium and mesenchyme, might inhibit and promote mesenchymal Vegfr2 expression, respectively [94].
Several previously described signals that are important for fetal lung growth are also apparent upstream regulators of Vegf signaling, such as the FGF and Wnt pathway (Figure 4). Fgf9, which is expressed in both epithelial and mesothelial cells, is required for lung mesenchymal Vegfa expression [95]. The Nemo-like kinase (Nlk) is an upstream negative regulator of Vegf. By phosphorylating the Wnt pathway effector lymphoid enhancing factor (Lef), Nlk inhibits the potential direct role of Lef in activating Vegf expression in epithelial cells [96]. Moreover, Wnt7b−/− mutants display vasculature defects and smooth muscle leakage, resulting in pulmonary hemorrhage [39]. In addition, epithelial-specific deletion of Wls, which is required for Wnt secretion, not only results in trachea cartilage malformation as mentioned earlier, but also leads to reduced pulmonary vasculature [37, 97]. Consistent with this, β-cat is required in Flk1+ angioblasts for their differentiation into more mature Pecam+ endothelial cells [98]. Conversely, mesenchymal-specific deletion of adenomatosis polyposis coli (APC), a negative regulator of the Wnt/β-cat pathway, leads to gradual loss of vasculature beginning at E13.5 [99], suggesting that too much Wnt activity compromises endothelial cell survival.
A number of transcription factors are known to be critical for pulmonary endothelial cells. For example, the previously mentioned Foxf1 promotes vascular development, potentially through directly activating the transcription of important endothelial genes including Tie2, Pecam1, Flt1, and Flk1 [100]. Another Fox family member, Foxm1, is also required for sufficient blood vessel formation and mesenchyme proliferation. Mutants of Foxm1 display arteriolar smooth muscle hypertrophy but inadequate peripheral vascular plexus formation. One direct target of Foxm1 in the developing lungs is the extracellular matrix protein Laminin a4, indicating an important role of extracellular matrix in supporting vascular development [101]. Another endothelial autonomous factor is the Paired related homeobox gene 1 (Prx1), which promotes endothelial differentiation from mesenchyme in vitro. Lack of Prx1 in vivo also leads to pulmonary vascular defects [102]. Lastly, loss of Sox17 results in dilated arteries and veins as well as sparse peripheral vasculature in the lung [103].
4.4. Vascular and airway smooth muscle
Based on histological analysis in humans, intrapulmonary artery SMC develop and mature from hilum to periphery, similar to mouse development [83]. Elegant lineage tracing experiments suggest that in mice the pulmonary artery smooth muscle is derived from Tbx4+ lung mesenchyme rather than from endothelial or mesothelial tissue. Pulmonary artery walls are apparently constructed radially, with the inner smooth muscle layer being recruited by the endothelial cells from surrounding Pdgfrβ-expressing mesenchyme cells. The SMC first align longitudinally, then downregulate Pdgfrβ and reorient circumferentially between E11.5 and E14.5. A similar program occurs in the second layer of surrounding mesenchyme. However, the third layer of mesenchyme arrests at the Pdgfrβ expression stage, fails to express SMC and remains longitudinal, and eventually constitutes the adventitial layer of the arterial wall. Interestingly, radial mitosis is observed only in the innermost smooth muscle layer to potentially facilitate the second layer of SMC formation. This process appears to be dependent on radial gradient signals such as Pdgf, which is expressed in endothelial cells and whose receptors Pdgfrα/β are expressed in the surrounding mesenchyme in a complementary fashion. However, the lack of phenotype in germline Pdgfrβ mutants alone and the lack of even the first layer of SMC in Pdgfrα (f/−)/β (−/−);Tbx4cre mutants indicate that other signals may additionally regulate the arterial SMC program [77]. Proper arterial SMC development is critical, as their malformation is involved in pulmonary hypertension [104]. Though less understood, human venous SMC appear to derive from mesenchyme directly, as evidenced by staining on different stages of human tissue [83].
Airway SMC first appear at the carina at E10.5 in mouse. As SMC develop, neuronal precursors can be identified in lung associated with SMC at E11. Neural tissue continues to grow as the lung develops and mostly covers the proximal SMC, with some extensions into the mesenchyme [105]. Though aSMC also develop and mature in a proximal to distal fashion similar to the vascular SMC, the unique progenitor pool appears distant. In contrast to the apparent local condensations that form vascular SMC, clonal lineage tracing shows that aSMC progenitors reside in the mesenchyme ahead of the developing branches and, dependent on Wnt1, migrate along the bronchi to differentiate into SMC lineages. This migration process is also evidenced by live imaging of labeled mesenchyme grafts in lung culture. Interestingly, the SMC associated with domain branching appear not to be derived from proliferation of existing SMC, but instead from mesenchyme surrounding a stalk region that can be induced to form SMC only by another domain branching, with Wnt1 being the priming signal [62]. Moreover, a recent study suggests that Ezh2, a Polycomb repressive complex 2 component, in mesothelial cells prevents their differentiation into ectopic SMC at the lung periphery [106]. The contractility of aSMC might contribute to the airway branching through either mechanical stretching of epithelium or regulating growth factor release.
Wnt signaling has been shown to be critical for SMC development (Figure 4), in addition to previously discussed functions for lung specification and branching. During lung development, the Wnt receptors Fzd1/4/7 are expressed in the lung mesenchyme, whereas the Wnt co-receptor Lrp6 is expressed in the muscle component of large blood vessels [107]. Wnt2 initiates the SMC program in Pdgfrα/β-expressing multipotent mesenchyme and also induces Fgf10 in primitive lung mesenchyme, which promotes proliferation and maturation of aSMC (Figure 4) [108]. The number of aSMC is further reduced without both Wnt2 and Wnt7b, which act in an apparently cooperative fashion in the mesenchyme, dependent on Pdgf signals [109]. Deletion of β-cat from the pulmonary SMC results in reduced number of aSMC and vSMC as well as leakage between SMC, potentially due to insufficient proliferation [97]. In contrast, too much Wnt activity caused by mesenchymal deletion of the negative Wnt regulator APC leads to malformation of smooth muscle cells around both the airway and blood vessels [99]. Wnt signaling regulates smooth muscle precursor development in the mouse lung in part by supporting the expression of extracellular matrix protein tenascin C, which appears to be necessary and sufficient for Pdgfrα/β expression in explants [110]. Several factors have been suggested to regulate the Wnt pathway. Besides previously mentioned Wnt upstream regulators Tbx4/5 [54], Hox5a/b/c also supports Wnt2/2b expression and SM development [111]. Finally, miR-142 supports Wnt potentially through direct binding to APC mRNA, thus sustaining mesenchymal progenitor proliferation and preventing premature differentiation into smooth muscle [112].
Fgf10+ mesenchymal cells are shown to migrate along the airway and differentiate into parabronchial SMC. Moreover, Fgf10 is also required for such differentiation, potentially via supporting epithelial Bmp4 expression [45]. However, hyper-activation of the FGF pathway in the mesenchyme also inhibits SMC commitment as suggested in the Fgfr2c+/Δ mice, where deletion of one copy of the Fgfr2 exon 9 results in co-expression of both splice isoforms Fgfr2b and Fgfr2c in mesenchyme, leading to hyper-responsiveness to a broader range of FGF ligands [113]. While Fgf10 mostly signals through the IIIb isoform of Fgfr expressed in the epithelium, Fgf9, expressed in the epithelium and the mesothelium, signals through the IIIc isoform of Fgfr in the mesenchyme and appears to inhibit airway smooth muscle differentiation in mouse lung. Either mesenchymal conditional deletion of Fgfr1/2 or loss of Fgf9 leads to ectopic airway SM differentiation in the distal airway, with normal proximal-distal airway patterning [114]. The specificity of Fgf9 and Fgf10 responses is likely due to their differential binding capacity to different receptors [115].
Two other signals important in pulmonary smooth muscle development are Shh and Tgfβ. Shh signaling is required for both airway and vascular SM [35, 114], while Shh-mediated bronchial SMC formation appears to be independent of the effector Gli3 [116]. On the other hand, mesodermal TGFβ receptor Alk5 is required for both vascular and aSMC development, through supporting Pdgfrα+ precursors and inhibiting the alternative lipofibroblast fate [117]. A recent paper suggests that epithelial Notch activity is required to support distal airway SM development, which is dependent on epithelial integrity [118].
Given the tremendous advancement of our understanding of pulmonary vasculature and smooth muscle development, an exciting future direction will be to further explore how different signals interact with each other, and how multipotent mesenchymal progenitors respond to different signals and differentiate into unique lineages in particular patterns. Not only is this a fascinating developmental biology question, but also it will provide guidance to understand and treat various pathological diseases associated with pulmonary mesenchymal malformation.
5. Future directions
The last decade has revealed many of the mechanisms regulating development of the respiratory mesenchyme and how its development is closely coordinated with the better-studied epithelium. One interesting area of research focuses on using directed tissue engineering to solve congenital respiratory problems such as tracheal stenosis. Another direction for further investigation is how mesenchymal patterning is initially established during early specification and lobe formation. For example, it is currently unknown how Wnt, BMP, and other signals are induced only on the ventral side and how FGF10 establishes lobe formation at the correct locations. In addition, much more work is needed to systematically determine the origin and lineage relationship between all of the mesenchymal cell types in the lung, particularly fibroblasts. In addition to careful lineage tracing, single cell sequencing should also prove to be useful to identify multi-lineage progenitors as well as cellular signaling crosstalks that generate the exquisite structure-function of the respiratory system.
Acknowledgments
We thank Anne-Karina Perl, John Shannon, Debora Sinner, Malcolm Fisher, and Scott Rankin for comments on the manuscript. Respiratory system research in the Zorn lab is funded by NIH grant HL114898 to AMZ. TN was supported by National Institute of General Medical Sciences T32 GM063483-14.
Footnotes
CONFLICT OF INTEREST STATEMENT
There are no conflicts of interest.
References
- 1.Peng T, Morrisey EE. Pulm Circ. 2013;3:176–178. doi: 10.4103/2045-8932.109954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hines EA, Jones MK, Verheyden JM, Harvey JF, Sun X. Proc Natl Acad Sci USA. 2013;110:19444–19449. doi: 10.1073/pnas.1313223110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swarr DT, Morrisey EE. Annu Rev Cell Dev Biol. 2015;31:553–573. doi: 10.1146/annurev-cellbio-100814-125249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim N, Vu TH. Birth Defects Res C Embryo Today. 2006;78:80–89. doi: 10.1002/bdrc.20062. [DOI] [PubMed] [Google Scholar]
- 5.McGowan SE, McCoy DM. Am J Physiol Lung Cell Mol Physiol. 2014;307:L618–631. doi: 10.1152/ajplung.00144.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, Godiwala N, Yatsenko SA, Sebastian J, Madan-Khetarpal S, Surti U, Abellar RG, Bateman DA, Wilson AL, Markham MH, Slamon J, Santos-Simarro F, Palomares M, Nevado J, Lapunzina P, Chung BH, Wong WL, Chu YW, Mok GT, Kerem E, Reiter J, Ambalavanan N, Anderson SA, Kelly DR, Shieh J, Rosenthal TC, Scheible K, Steiner L, Iqbal MA, McKinnon ML, Hamilton SJ, Schlade-Bartusiak K, English D, Hendson G, Roeder ER, DeNapoli TS, Littlejohn RO, Wolff DJ, Wagner CL, Yeung A, Francis D, Fiorino EK, Edelman M, Fox J, Hayes DA, Janssens S, De Baere E, Menten B, Loccufier A, Vanwalleghem L, Moerman P, Sznajer Y, Lay AS, Kussmann JL, Chawla J, Payton DJ, Phillips GE, Brosens E, Tibboel D, de Klein A, Maystadt I, Fisher R, Sebire N, Male A, Chopra M, Pinner J, Malcolm G, Peters G, Arbuckle S, Lees M, Mead Z, Quarrell O, Sayers R, Owens M, Shaw-Smith C, Lioy J, McKay E, de Leeuw N, Feenstra I, Spruijt L, Elmslie F, Thiruchelvam T, Bacino CA, Langston C, Lupski JR, Sen P, Popek E, Stankiewicz P. Hum Genet. 2016;135:569–586. doi: 10.1007/s00439-016-1655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hewitt RJ, Butler CR, Maughan EF, Elliott MJ. Semin Pediatr Surg. 2016;25:144–149. doi: 10.1053/j.sempedsurg.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, Yamaguchi TP, Morrisey EE. Dev Cell. 2009;17:290–298. doi: 10.1016/j.devcel.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris-Johnson KS, Domyan ET, Vezina CM, Sun X. Proc Natl Acad Sci USA. 2009;106:16287–16292. doi: 10.1073/pnas.0902274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domyan ET, Ferretti E, Throckmorton K, Mishina Y, Nicolis SK, Sun X. Development. 2011;138:971–981. doi: 10.1242/dev.053694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fausett SR, Brunet LJ, Klingensmith J. Dev Biol. 2014;391:111–124. doi: 10.1016/j.ydbio.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Litingtung Y, Ten Dijke P, Chiang C. Dev Dyn. 2007;236:746–754. doi: 10.1002/dvdy.21075. [DOI] [PubMed] [Google Scholar]
- 13.Woo J, Miletich I, Kim BM, Sharpe PT, Shivdasani RA. PLoS One. 2011;6:e22493. doi: 10.1371/journal.pone.0022493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rankin SA, Han L, McCracken KW, Kenny AP, Anglin CT, Grigg EA, Crawford CM, Wells JM, Shannon JM, Zorn AM. Cell Rep. 2016;16(1):66–78. doi: 10.1016/j.celrep.2016.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham TJ, Duester G. Nat Rev Mol Cell Biol. 2015;16:110–123. doi: 10.1038/nrm3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rankin SA, Gallas AL, Neto A, Gomez-Skarmeta JL, Zorn AM. Development. 2012;139:3010–3020. doi: 10.1242/dev.078220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng T, Tian Y, Boogerd CJ, Lu MM, Kadzik RS, Stewart KM, Evans SM, Morrisey EE. Nature. 2013;500:589–592. doi: 10.1038/nature12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, Bolivar J, Bauer M, Zackai EH, McDonald-McGinn D, Nowaczyk MM, Murray M, Hustead V, Mascotti K, Schultz R, Hallam L, McRae D, Nicholson AG, Newbury R, Durham-O’Donnell J, Knight G, Kini U, Shaikh TH, Martin V, Tyreman M, Simonic I, Willatt L, Paterson J, Mehta S, Rajan D, Fitzgerald T, Gribble S, Prigmore E, Patel A, Shaffer LG, Carter NP, Cheung SW, Langston C, Shaw-Smith C. Am J Hum Genet. 2009;84:780–791. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Que J. Wiley Interdisciplinary Reviews Dev Biol. 2015;4:419–430. doi: 10.1002/wdev.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snowball J, Ambalavanan M, Whitsett J, Sinner D. Dev Biol. 2015;405:56–70. doi: 10.1016/j.ydbio.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elluru RG, Thompson F, Reece A. The Laryngoscope. 2009;119:1153–1165. doi: 10.1002/lary.20157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elluru RG, Whitsett JA. Arch Otolaryngol Head Neck Surg. 2004;130:732–736. doi: 10.1001/archotol.130.6.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park J, Zhang JJ, Moro A, Kushida M, Wegner M, Kim PC. Dev Dyn. 2010;239:514–526. doi: 10.1002/dvdy.22192. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Gordon J, Manley NR, Litingtung Y, Chiang C. Dev Biol. 2008;322:145–155. doi: 10.1016/j.ydbio.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Que J, Choi M, Ziel JW, Klingensmith J, Hogan BL. Differentiation. 2006;74:422–437. doi: 10.1111/j.1432-0436.2006.00096.x. [DOI] [PubMed] [Google Scholar]
- 26.Lohnes D, Kastner P, Dierich A, Mark M, LeMeur M, Chambon P. Cell. 1993;73:643–658. doi: 10.1016/0092-8674(93)90246-m. [DOI] [PubMed] [Google Scholar]
- 27.Luo J, Sucov HM, Bader JA, Evans RM, Giguere V. Mech Dev. 1996;55:33–44. doi: 10.1016/0925-4773(95)00488-2. [DOI] [PubMed] [Google Scholar]
- 28.Dolle P, Ruberte E, Leroy P, Morriss-Kay G, Chambon P. Development. 1990;110:1133–1151. doi: 10.1242/dev.110.4.1133. [DOI] [PubMed] [Google Scholar]
- 29.Kugler MC, Joyner AL, Loomis CA, Munger JS. Am J Respir Cell Mol Biol. 2015;52:1–13. doi: 10.1165/rcmb.2014-0132TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Litingtung Y, Lei L, Westphal H, Chiang C. Nat Genet. 1998;20:58–61. doi: 10.1038/1717. [DOI] [PubMed] [Google Scholar]
- 31.Mahlapuu M, Enerback S, Carlsson P. Development. 2001;128:2397–2406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- 32.Motoyama J, Liu J, Mo R, Ding Q, Post M, Hui CC. Nat Genet. 1998;20:54–57. doi: 10.1038/1711. [DOI] [PubMed] [Google Scholar]
- 33.Minoo P, Su G, Drum H, Bringas P, Kimura S. Dev Biol. 1999;209:60–71. doi: 10.1006/dbio.1999.9234. [DOI] [PubMed] [Google Scholar]
- 34.Que J, Luo X, Schwartz RJ, Hogan BL. Development. 2009;136:1899–1907. doi: 10.1242/dev.034629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller LA, Wert SE, Clark JC, Xu Y, Perl AK, Whitsett JA. Dev Dyn. 2004;231:57–71. doi: 10.1002/dvdy.20105. [DOI] [PubMed] [Google Scholar]
- 36.Zou H, Wieser R, Massague J, Niswander L. Genes Dev. 1997;11:2191–2203. doi: 10.1101/gad.11.17.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cornett B, Snowball J, Varisco BM, Lang R, Whitsett J, Sinner D. Dev Biol. 2013;379:38–52. doi: 10.1016/j.ydbio.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajagopal J, Carroll TJ, Guseh JS, Bores SA, Blank LJ, Anderson WJ, Yu J, Zhou Q, McMahon AP, Melton DA. Development. 2008;135:1625–1634. doi: 10.1242/dev.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shu W, Jiang YQ, Lu MM, Morrisey EE. Development. 2002;129:4831–4842. doi: 10.1242/dev.129.20.4831. [DOI] [PubMed] [Google Scholar]
- 40.Li C, Xiao J, Hormi K, Borok Z, Minoo P. Dev Biol. 2002;248:68–81. doi: 10.1006/dbio.2002.0729. [DOI] [PubMed] [Google Scholar]
- 41.Caprioli A, Villasenor A, Wylie LA, Braitsch C, Marty-Santos L, Barry D, Karner CM, Fu S, Meadows SM, Carroll TJ, Cleaver O. Dev Biol. 2015;406:222–234. doi: 10.1016/j.ydbio.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, Hogan BL. Development. 2007;134:2521–2531. doi: 10.1242/dev.003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sala FG, Del Moral PM, Tiozzo C, Alam DA, Warburton D, Grikscheit T, Veltmaat JM, Bellusci S. Development. 2011;138:273–282. doi: 10.1242/dev.051680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. Nat Genet. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- 45.Mailleux AA, Kelly R, Veltmaat JM, De Langhe SP, Zaffran S, Thiery JP, Bellusci S. Development. 2005;132:2157–2166. doi: 10.1242/dev.01795. [DOI] [PubMed] [Google Scholar]
- 46.Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS. Genes Dev. 1998;12:3156–3161. doi: 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Xiao R, Yang F, Karim BO, Iacovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, Yu K, Ornitz DM, Elisseeff J, Huso DL, Jabs EW. Development. 2005;132:3537–3548. doi: 10.1242/dev.01914. [DOI] [PubMed] [Google Scholar]
- 48.Tiozzo C, De Langhe S, Carraro G, Alam DA, Nagy A, Wigfall C, Hajihosseini MK, Warburton D, Minoo P, Bellusci S. Pediatr Res. 2009;66:386–390. doi: 10.1203/PDR.0b013e3181b45580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishimaru T, Komura M, Sugiyama M, Komura H, Arai M, Fujishiro J, Uotani C, Miyakawa K, Kakihara T, Hoshi K, Takato T, Tabata Y, Komuro H, Iwanaka T. J Pediatr Surg. 2015;50:255–259. doi: 10.1016/j.jpedsurg.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 50.Komura M, Komura H, Konishi K, Ishimaru T, Hoshi K, Takato T, Tabata Y, Iwanaka T. J Pediatr Surg. 2014;49:296–300. doi: 10.1016/j.jpedsurg.2013.11.040. [DOI] [PubMed] [Google Scholar]
- 51.Boucherat O, Nadeau V, Berube-Simard FA, Charron J, Jeannotte L. Development. 2015;142:3801. doi: 10.1242/dev.131821. [DOI] [PubMed] [Google Scholar]
- 52.Bell SM, Schreiner CM, Wert SE, Mucenski ML, Scott WJ, Whitsett JA. Development. 2008;135:1049–1058. doi: 10.1242/dev.013359. [DOI] [PubMed] [Google Scholar]
- 53.Lin SS, Tzeng BH, Lee KR, Smith RJ, Campbell KP, Chen CC. Proc Natl Acad Sci USA. 2014;111:E1990–1998. doi: 10.1073/pnas.1323112111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arora R, Metzger RJ, Papaioannou VE. PLoS Genet. 2012;8:e1002866. doi: 10.1371/journal.pgen.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rock JR, Futtner CR, Harfe BD. Dev Biol. 2008;321:141–149. doi: 10.1016/j.ydbio.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Dolle P, Cardoso WV, Niederreither K. Dev Biol. 2006;297:433–445. doi: 10.1016/j.ydbio.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 57.Desai TJ, Chen F, Lu J, Qian J, Niederreither K, Dolle P, Chambon P, Cardoso WV. Dev Biol. 2006;291:12–24. doi: 10.1016/j.ydbio.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 58.Chen F, Cao Y, Qian J, Shao F, Niederreither K, Cardoso WV. J Clin Invest. 2010;120:2040–2048. doi: 10.1172/JCI40253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen F, Desai TJ, Qian J, Niederreither K, Lu J, Cardoso WV. Development. 2007;134:2969–2979. doi: 10.1242/dev.006221. [DOI] [PubMed] [Google Scholar]
- 60.Sakiyama J, Yamagishi A, Kuroiwa A. Development. 2003;130:1225–1234. doi: 10.1242/dev.00345. [DOI] [PubMed] [Google Scholar]
- 61.Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM, Piccolo S, Birchmeier W, Whitsett JA, Millar SE, Morrisey EE. Dev Biol. 2005;283:226–239. doi: 10.1016/j.ydbio.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 62.Kumar ME, Bogard PE, Espinoza FH, Menke DB, Kingsley DM, Krasnow MA. Science. 2014;346:1258810. doi: 10.1126/science.1258810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White AC, Xu J, Yin Y, Smith C, Schmid G, Ornitz DM. Development. 2006;133:1507–1517. doi: 10.1242/dev.02313. [DOI] [PubMed] [Google Scholar]
- 64.Yin Y, White AC, Huh SH, Hilton MJ, Kanazawa H, Long F, Ornitz DM. Dev Biol. 2008;319:426–436. doi: 10.1016/j.ydbio.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ornitz DM, Yin Y. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008318. pii: a008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shan L, Subramaniam M, Emanuel RL, Degan S, Johnston P, Tefft D, Warburton D, Sunday ME. Dev Dyn. 2008;237:750–757. doi: 10.1002/dvdy.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li C, Li M, Li S, Xing Y, Yang CY, Li A, Borok Z, De Langhe S, Minoo P. Stem Cells. 2015;33:999–1012. doi: 10.1002/stem.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W, Menke DB, Jiang M, Chen H, Warburton D, Turcatel G, Lu CH, Xu W, Luo Y, Shi W. BMC Bio. 2013;11:111. doi: 10.1186/1741-7007-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dixit R, Ai X, Fine A. Development. 2013;140:4398–4406. doi: 10.1242/dev.098079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.von Gise A, Stevens SM, Honor LB, Oh JH, Gao C, Zhou B, Pu WT. Am J Respir Cell Mol Biol. 2016;54:222–230. doi: 10.1165/rcmb.2014-0461OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cano E, Carmona R, Munoz-Chapuli R. Am J Physiol Lung Cell Mol Physiol. 2013;305:L322–332. doi: 10.1152/ajplung.00424.2012. [DOI] [PubMed] [Google Scholar]
- 72.El Agha E, Herold S, Al Alam D, Quantius J, MacKenzie B, Carraro G, Moiseenko A, Chao CM, Minoo P, Seeger W, Bellusci S. Development. 2014;141:296–306. doi: 10.1242/dev.099747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al Alam D, El Agha E, Sakurai R, Kheirollahi V, Moiseenko A, Danopoulos S, Shrestha A, Schmoldt C, Quantius J, Herold S, Chao CM, Tiozzo C, De Langhe S, Plikus MV, Thornton M, Grubbs B, Minoo P, Rehan VK, Bellusci S. Development. 2015;142:4139–4150. doi: 10.1242/dev.109173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morimoto M, Liu Z, Cheng HT, Winters N, Bader D, Kopan R. J Cell Sci. 2010;123:213–224. doi: 10.1242/jcs.058669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Green J, Endale M, Auer H, Perl AT. Am J Respir Cell Mol Biol. 2016;54:532–545. doi: 10.1165/rcmb.2015-0095OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ntokou A, Klein F, Dontireddy D, Becker S, Bellusci S, Richardson WD, Szibor M, Braun T, Morty RE, Seeger W, Voswinckel R, Ahlbrecht K. Am J Physiol Lung Cell Mol Physiol. 2015;309:L942–958. doi: 10.1152/ajplung.00272.2014. [DOI] [PubMed] [Google Scholar]
- 77.Greif DM, Kumar M, Lighthouse JK, Hum J, An A, Ding L, Red-Horse K, Espinoza FH, Olson L, Offermanns S, Krasnow MA. Dev Cell. 2012;23:482–493. doi: 10.1016/j.devcel.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tan SY, Krasnow MA. Development. 2016;143:1318–1327. doi: 10.1242/dev.129122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choi CW. Korean J Pediatr. 2010;53:979–984. doi: 10.3345/kjp.2010.53.12.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chao CM, El Agha E, Tiozzo C, Minoo P, Bellusci S. Front Med (Lausanne) 2015;2:27. doi: 10.3389/fmed.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwarz MA, Caldwell L, Cafasso D, Zheng H. Am J Physiol Lung Cell Mol Physiol. 2009;296:L71–81. doi: 10.1152/ajplung.90452.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schachtner SK, Wang Y, Scott Baldwin H. Am J Respir Cell Mol Biol. 2000;22:157–165. doi: 10.1165/ajrcmb.22.2.3766. [DOI] [PubMed] [Google Scholar]
- 83.Hall SM, Hislop AA, Haworth SG. Am J Respir Cell Mol Biol. 2002;26:333–340. doi: 10.1165/ajrcmb.26.3.4698. [DOI] [PubMed] [Google Scholar]
- 84.Parera MC, van Dooren M, van Kempen M, de Krijger R, Grosveld F, Tibboel D, Rottier R. Am J Physiol Lung Cell Mol Physiol. 2005;288:L141–149. doi: 10.1152/ajplung.00148.2004. [DOI] [PubMed] [Google Scholar]
- 85.deMello DE, Reid LM. Pediatr Dev Path. 2000;3:439–449. doi: 10.1007/s100240010090. [DOI] [PubMed] [Google Scholar]
- 86.deMello DE, Sawyer D, Galvin N, Reid LM. Am J Respir Cell Mol Biol. 1997;16:568–581. doi: 10.1165/ajrcmb.16.5.9160839. [DOI] [PubMed] [Google Scholar]
- 87.Seo H, Kim J, Park GH, Kim Y, Cho SW. Histochem Cell Biol. 2016;146:289–300. doi: 10.1007/s00418-016-1445-4. [DOI] [PubMed] [Google Scholar]
- 88.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 89.Yun EJ, Lorizio W, Seedorf G, Abman SH, Vu TH. Am J Physiol Lung Cell Mol Physiol. 2016;310:L287–298. doi: 10.1152/ajplung.00229.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao L, Wang K, Ferrara N, Vu TH. Mech Dev. 2005;122:877–886. doi: 10.1016/j.mod.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 91.Seedorf GJ, Metoxen AJ, Rock R, Markham NE, Ryan SL, Vu TH, Abman SH. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1098–110. doi: 10.1152/ajplung.00423.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Akeson AL, Greenberg JM, Cameron JE, Thompson FY, Brooks SK, Wiginton D, Whitsett JA. Dev Biol. 2003;264:443–455. doi: 10.1016/j.ydbio.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 93.Mallory BP, Mead TJ, Wiginton DA, Kulkarni RM, Greenberg JM, Akeson AL. Microvasc Res. 2006;72:62–73. doi: 10.1016/j.mvr.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 94.Mujahid S, Nielsen HC, Volpe MV. PLoS One. 2013;8:e55911. doi: 10.1371/journal.pone.0055911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.White AC, Lavine KJ, Ornitz DM. Development. 2007;134:3743–3752. doi: 10.1242/dev.004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ke H, Masoumi KC, Ahlqvist K, Seckl MJ, Rydell-Tormanen K, Massoumi R. Sci Rep. 2016;6:23987. doi: 10.1038/srep23987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang M, Ku WY, Fu J, Offermanns S, Hsu W, Que J. Development. 2013;140:3589–3594. doi: 10.1242/dev.095471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.De Langhe SP, Carraro G, Tefft D, Li C, Xu X, Chai Y, Minoo P, Hajihosseini MK, Drouin J, Kaartinen V, Bellusci S. PLoS One. 2008;3:e1516. doi: 10.1371/journal.pone.0001516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Luo Y, El Agha E, Turcatel G, Chen H, Chiu J, Warburton D, Bellusci S, Qian BP, Menke DB, Shi W. BMC Biol. 2015;13:42. doi: 10.1186/s12915-015-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ren X, Ustiyan V, Pradhan A, Cai Y, Havrilak JA, Bolte CS, Shannon JM, Kalin TV, Kalinichenko VV. Circ Res. 2014;115:709–720. doi: 10.1161/CIRCRESAHA.115.304382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. J Biol Chem. 2005;280:22278–22286. doi: 10.1074/jbc.M500936200. [DOI] [PubMed] [Google Scholar]
- 102.Ihida-Stansbury K, McKean DM, Gebb SA, Martin JF, Stevens T, Nemenoff R, Akeson A, Vaughn J, Jones PL. Circ Res. 2004;94:1507–1514. doi: 10.1161/01.RES.0000130656.72424.20. [DOI] [PubMed] [Google Scholar]
- 103.Lange AW, Haitchi HM, LeCras TD, Sridharan A, Xu Y, Wert SE, James J, Udell N, Thurner PJ, Whitsett JA. Dev Biol. 2014;387:109–120. doi: 10.1016/j.ydbio.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hall SM, Hislop AA, Pierce CM, Haworth SG. Am J Respir Cell Mol Biol. 2000;23:194–203. doi: 10.1165/ajrcmb.23.2.3975. [DOI] [PubMed] [Google Scholar]
- 105.Tollet J, Everett AW, Sparrow MP. Dev Dyn. 2001;221:48–60. doi: 10.1002/dvdy.1124. [DOI] [PubMed] [Google Scholar]
- 106.Snitow M, Lu M, Cheng L, Zhou S, Morrisey EE. Development. 2016;143:3733–3741. doi: 10.1242/dev.134932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Z, Shu W, Lu MM, Morrisey EE. Mol Cell Biol. 2005;25:5022–5030. doi: 10.1128/MCB.25.12.5022-5030.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goss AM, Tian Y, Cheng L, Yang J, Zhou D, Cohen ED, Morrisey EE. Dev Biol. 2011;356:541–552. doi: 10.1016/j.ydbio.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miller MF, Cohen ED, Baggs JE, Lu MM, Hogenesch JB, Morrisey EE. Proc Natl Acad Sci USA. 2012;109:15348–15353. doi: 10.1073/pnas.1201583109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cohen ED, Ihida-Stansbury K, Lu MM, Panettieri RA, Jones PL, Morrisey EE. J Clin Invest. 2009;119:2538–2549. doi: 10.1172/JCI38079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hrycaj SM, Dye BR, Baker NC, Larsen BM, Burke AC, Spence JR, Wellik DM. Cell Rep. 2015;12:903–912. doi: 10.1016/j.celrep.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Carraro G, Shrestha A, Rostkovius J, Contreras A, Chao CM, El Agha E, Mackenzie B, Dilai S, Guidolin D, Taketo MM, Gunther A, Kumar ME, Seeger W, De Langhe S, Barreto G, Bellusci S. Development. 2014;141:1272–1281. doi: 10.1242/dev.105908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Langhe SP, Carraro G, Warburton D, Hajihosseini MK, Bellusci S. Dev Biol. 2006;299:52–62. doi: 10.1016/j.ydbio.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 114.Yi L, Domyan ET, Lewandoski M, Sun X. Dev Dyn. 2009;238:123–137. doi: 10.1002/dvdy.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li Y, Zhang H, Choi SC, Litingtung Y, Chiang C. Dev Biol. 2004;270:214–231. doi: 10.1016/j.ydbio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 116.Li A, Ma S, Smith SM, Lee MK, Fischer A, Borok Z, Bellusci S, Li C, Minoo P. BMC Biol. 2016;14:19. doi: 10.1186/s12915-016-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tsao PN, Matsuoka C, Wei SC, Sato A, Sato S, Hasegawa K, Chen HK, Ling TY, Mori M, Cardoso WV, Morimoto M. Proc Natl Acad Sci USA. 2016;113:8242–8247. doi: 10.1073/pnas.1511236113. [DOI] [PMC free article] [PubMed] [Google Scholar]