

Summary
Estrogen receptor α (ERα) is a hormone receptor and a key driver for over 70% of breast cancers that has been studied for decades as a transcription factor. Unexpectedly, we discover that ERα is a potent non-canonical RNA-binding protein. We show that ERα RNA-binding function is uncoupled from its activity to bind DNA and is critical for breast cancer progression. Employing genome-wide CLIP sequencing and a functional CRISPRi screen, we find that the ERα-associated mRNAs sustain cancer cell fitness and elicit cellular responses to stress. Mechanistically, ERα controls different steps of RNA metabolism. In particular, we demonstrate that ERα RNA-binding mediates the alternative splicing of XBP1 and translation of eIF4G2 and MCL1 mRNAs, which facilitates survival upon stress conditions and sustains tamoxifen resistance of cancer cells. Therefore, ERα is a multifaceted RNA-binding protein and this activity transforms our knowledge of post-transcriptional regulation underlying cancer development and drug response.
Keywords: ERα, RNA-binding protein, breast cancer, translation control, RNA splicing, integrated stress response, cell survival
Graphical Abstract

eTOC blurb
ERα, a transcription factor deregulated in breast cancer, can also reprogram gene expression at the post-transcriptional level by associating with RNAs to induce the production of stress response proteins and enhances breast cancer cell fitness.
Introduction
Breast cancer is one of the most common cancers in the world, with over 70% of breast cancers harboring the activation of the nuclear hormone receptor, estrogen receptor α (ERα) (Ali and Coombes, 2002). In the past few decades, a comprehensive picture of ERα function, predominantly at the level of transcriptional regulation in the nucleus, and its contribution to breast cancer progression has emerged. Inhibition of ERα with tamoxifen, an estrogen antagonist, is typically employed as a first-line therapeutic agent that blocks the activity of ERα in transcriptional regulation and prolongs patient survival (Howell et al., 2004; Shiau et al., 1998). However, a notable portion of patients eventually developed relapsed diseases and became insensitive to this antagonist (Metcalfe et al., 2018). In most cases of tamoxifen-resistant tumors, ERα remains active (Jeselsohn et al., 2015; Katzenellenbogen et al., 2018). Notably, ERα is a nucleocytoplasmic shuttling protein, rapidly translocating between the nucleus and cytoplasm (Lombardi et al., 2008). In this study, we have made a striking discovery that ERα is non-canonical RNA-binding protein (RBP) with a previously unknown function as a regulator of RNA metabolism.
Recent discoveries of non-canonical RBPs have expanded the RNA-binding repertoire to a broader category of proteins (Castello et al., 2012; Trendel et al., 2019). Their ability to bind RNA may either support their original functions or provide different functions such as in controlling post-transcriptional regulations. For example, the master transcription co-activator, CBP/p300 binds to a variety of enhancer RNAs that increase its histone acetylation activity and transcription control (Bose et al., 2017). Cyclin A2, a core cell cycle regulator is also an RBP that play a role in RNA metabolism, by binding and modulating the translation of the meiotic recombination 11 (Mre11) mRNA, which leads to a kinase-independent, RNA-binding-dependent role in repair replication errors (Kanakkanthara et al., 2016). All of these examples highlight the pleiotropic potential of key proteins in controlling distinct biological processes. Interestingly, a study in the ‘90s reported that nuclear receptors including ERα are colocalized and physically interacted with a non-coding RNA, which enhances the transcriptional potency of the nuclear receptors (Lanz et al., 1999). Yet, decades of research on ERα remains predominantly focused on its activity in binding DNA.
In this study, we surprisingly found that ERα is a non-canonical RBP and abolishing ERα RNA-binding activity does not affect its classical DNA-binding ability, yet renders growth defects of breast cancer cells both in vitro and in vivo. Employing unbiased genome-wide high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP) and functional genomic CRISPRi screens, we pinpointed unique networks of mRNAs crucial for cancer progression that are bound by ERα in the 3’ untranslated region (3’ UTR), notably including those involved in cellular response to stress during tumor development. Two well characterized adaptive responses, Integrated Stress Response (ISR) and the Unfolded Protein Response (UPR) (Costa-Mattioli and Walter, 2020; Hetz et al., 2015; Ron and Walter, 2007) sense various cellular stressors [e.g. nutrient deprivation, hypoxia, oxidative (Bi et al., 2005; Wouters et al., 2005), and proteotoxic stress (Hart et al., 2012; Nguyen et al., 2018)] and converge on post-transcriptional signaling pathways that rewire gene expression (Donnelly et al., 2013; Nguyen et al., 2018; Vattem and Wek, 2004). Here, we demonstrate that ERα controls the alternative splicing of XBP1 mRNA, a key component of the UPR pathway, as well as regulating the translation of stress response proteins such as eIF4G2 and MCL1 mRNAs. The ability of ERα to modulate these genes at the post-transcriptional level promotes cancer cell survival and sustains the tamoxifen resistance of cancer cells. Collectively, our studies show that ERα is a multifaceted RBP, which transforms our knowledge of post-transcriptional regulation underlying tumor cell survival and drug response.
Results
ERα is an RNA-binding protein associated with multiple oncogenic mRNAs
In order to understand whether and how ERα functions outside the nucleus in addition to its role in the signaling cascade (Acconcia et al., 2006; Cabodi et al., 2004; Kumar et al., 2007; Migliaccio et al., 1996; Simoncini et al., 2000; Song et al., 2004; Song et al., 2002; Song et al., 2005), we first performed ERα-immunoprecipitation followed by a quantitative mass spectrometry analysis to identify a possible ERα protein interactome in the cytoplasmic fraction of ERα-positive MCF7 breast cancer cells. Intriguingly, the most enriched protein category that interacts with ERα is proteins that bind RNA (Figure S1A and Table S1). These include key translation factors (e.g. eIF3C, eIF4E2, eIF4A1, and eEF1A1), a set of ribosomal proteins, proteins involved in mRNA translation regulation (e.g. LARP1 and YBX1), proteins involved in RNA splicing (e.g. RtcB), as well as those mediating RNA stability controls (e.g. ELAVL1, FMR1 and G3BP1/2).
To investigate whether ERα functions as an RBP, we employed Oligo(dT) beads to pull down polyA mRNAs and assessed the fraction of ERα bound to these mRNAs. These experiments show that a notable proportion of the cytoplasmic and soluble nuclear ERα is bound to polyA RNAs in MCF7 and T47D ERα-positive breast cancer cell lines (Figure 1A). ERα association with RNA is also evident in human breast tumor samples, which is significantly higher than that in normal adjacent tissues (Figure S1B). This is in contrast to other nuclear receptor superfamily members, including progesterone receptor (PR), androgen receptor (AR), retinoid X receptor α (RXRα) or peroxisome proliferator-activated receptor γ (PPARγ) that are not tightly associated with polyA RNA in MCF7 breast cancer cells. Interestingly however, the glucocorticoid receptor (GR) noticeably associates with RNAs (Figure S1C), which is in agreement with a previous report that GR is associated with some mRNAs (e.g. cytokines CCL2 and CCL7) (Ishmael et al., 2011).
Figure 1. ERα binds to RNA directly.
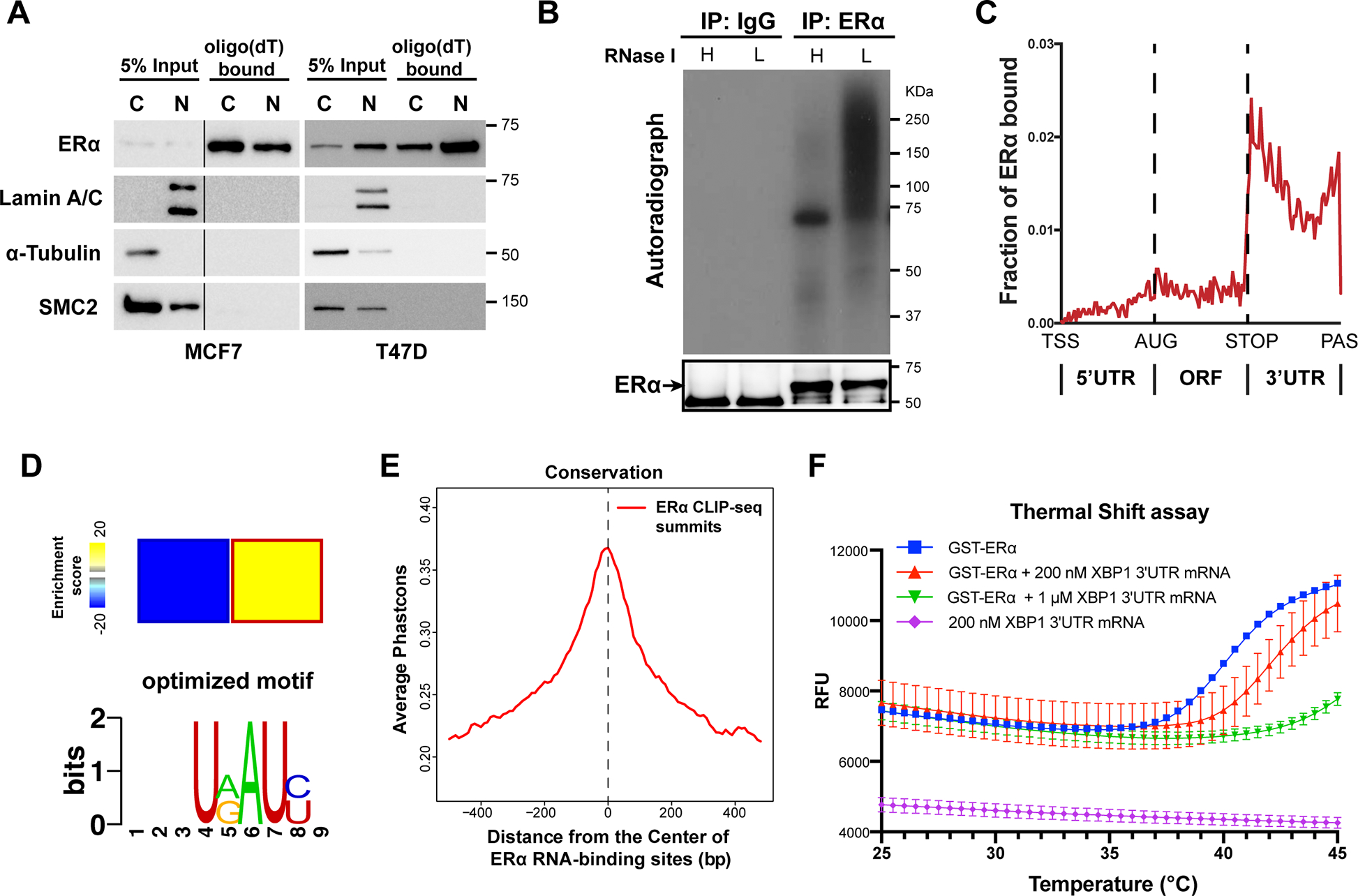
See also Figure S1.
(A) Representative western blots for ERα bound to Oligo(dT) beads in cytoplasmic (C) and nuclear (N) MCF7 and T47D human breast cancer cell lysates. Internal controls: Lamin A/C (N) and α-tubulin (C), and a DNA-binding protein SMC2 is used as a negative control for RNA-binding.
(B) Autoradiogram of 32P-labelled RNA crosslinked to endogenous ERα, showing ERα-RNA complexes shifting upwards from the size of ERα (66 KDa). RNA was partially digested using Low (L) or High (H) concentration of RNase.
(C) Distributions of ERα binding events on indicated mRNA regions. TSS: transcription starting sites; AUG: translation start codon; STOP: translation stop codon; PAS: Polyadenylation signal.
(D) Heatmap (upper panel) showing the enrichment of a sequence motif (lower pannel) in the 3’ UTRs of ERα-bound mRNAs.
(E) Phastcons conservation scores of ERα-bound RNA sequence compared with their adjacent regions. The window represents ±0.5 kb regions from the center of the ERα RNA-binding site.
(F) Thermal shift assay employing GST-ERα and in vitro transcribed 3’UTR fragment of XBP1 mRNA (unspliced isoform, 1–400 nt) at different concentrations. Protein melting temperature assessed by raw fluorescence (RFU) by real-time PCR.
N=3 biological replicates.
To further investigate whether ERα directly binds RNA, we used ultraviolet radiation crosslinking and co-immunoprecipitation [CLIP (Licatalosi et al., 2008; Zhang and Darnell, 2011)]. We observed that cross-linked ERα-RNA complexes migrated in a less defined band, reflecting cross-linkage with heterogeneous RNAs, which became defined at the size of the ERα protein upon high concentration of RNase treatment (Figure 1B). This result demonstrates that ERα is capable of binding RNA directly. To investigate the RNA targets of ERα across the genome, we next sequenced ERα CLIP libraries using HITS-CLIP (Figure S1D). ERα binds both exonic and intronic sites on RNAs, and within the transcript, ERα preferentially binds at 3’UTRs (Figure 1C, Figure S1E, F and Table S2). ERα-bound mRNAs are enriched for common gene ontology terms crucial for cancer progression, including cell-cell adhesion, response to endoplasmic reticulum stress (ER-stress), as well as negative regulation of the apoptotic processes (Figure S1G). Intriguingly, we identified a sequence motif that is enriched in ERα CLIP peaks over the background (Figure 1D), which is primarily detected in the 3’UTR regions of mRNAs. Importantly, this motif sequence is observed in many ERα-bound mRNAs and is conserved among mammals (Figure S1H). We also find that ERα RNA-binding sites are highly conserved compared to the immediate surrounding regions (Figure 1E), which is comparable with that of HuR, a classical RBP that binds the 3’UTR of mRNAs (Figure S1I). Therefore, the evolutionary conservation of the ERα RNA-binding site suggests a putative role in RNA regulation.
We further confirmed that ERα is an RBP by visualizing the direct interaction of ERα with RNA in vitro. We selected one of the top ERα-bound mRNAs, XBP1 (Figure S1E), and monitored the thermal stability of purified ERα protein with or without this mRNA (Figure 1F). In this experiment we observed that the melting temperature of ERα protein increased dramatically when incubated together with the XBP1 mRNA in a dose-dependent manner (Figure 1F), demonstrating that there is a direct association of ERα with this mRNA. Therefore, these data not only confirmed that ERα binds RNA directly but also suggest that ERα may bind to specific mRNAs important for cancer development.
ERα binds RNA utilizing an RNA-binding domain located in its hinge region
We next tested which domain of ERα is responsible for the association with mRNA. We first utilized RNABindRPlus (Terribilini et al., 2007) to predict the RNA-binding domain (RBD) of ERα. Intriguingly, one of the key predicted RBDs is located between amino acids 255 to 272 within the hinge domain of ERα after its DNA-binding domain (DBD) (Figure 2A). We determined that only the ERα construct containing the predicted RBD is directly associated with RNAs in vitro (Figure S2A). Moreover, this association is completely abolished when the predicted RBD is removed (Figure 2B), demonstrating that ERα interacts with RNA directly through this putative RBD.
Figure 2. ERα RNA-binding activity is a key determinant of its oncogenic role in breast cancer.
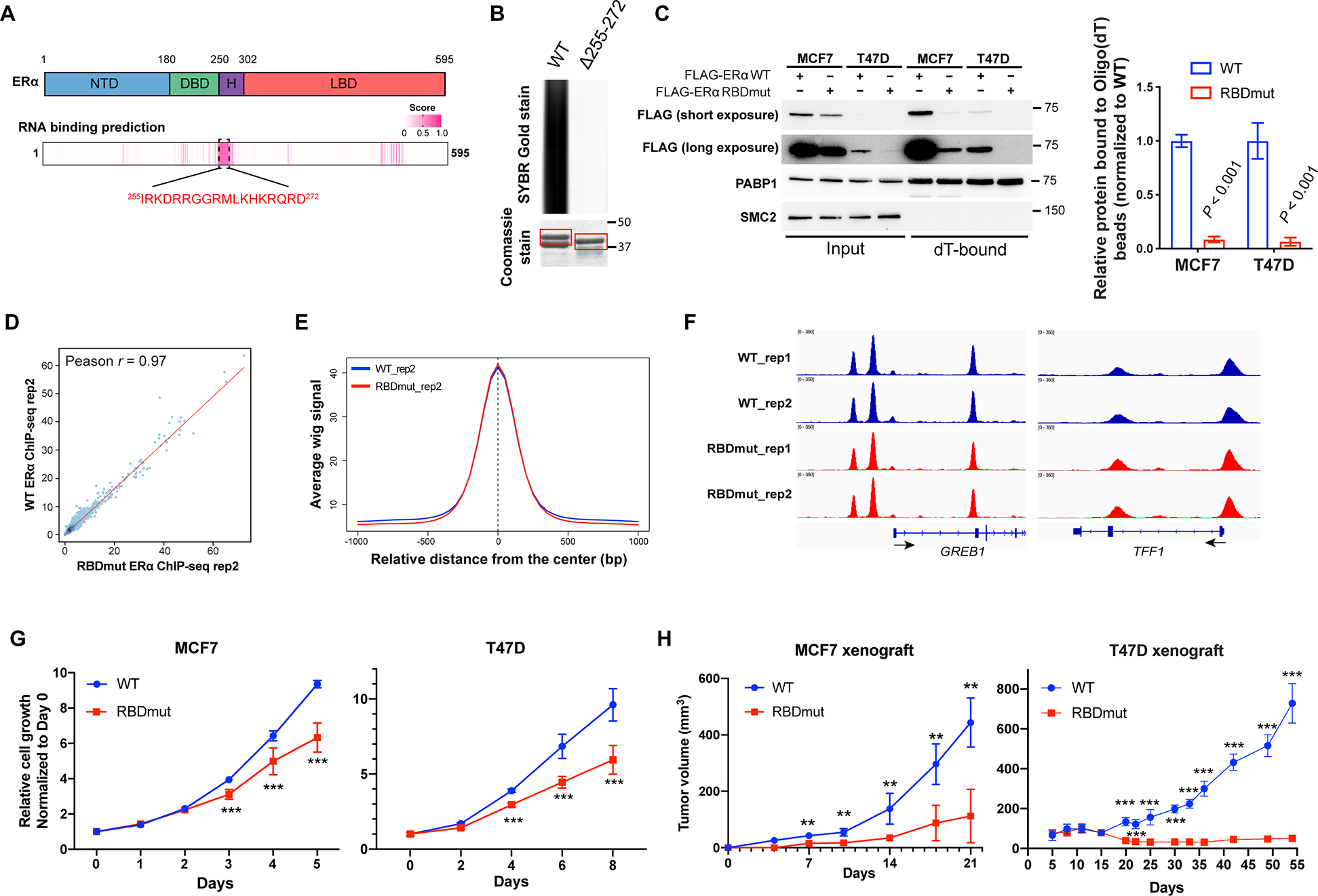
See also Figure S2.
(A) Prediction of the RNA-binding domain (RBD) using the RNABindRPlus tool. NTD: N-terminal domain; DBD: DNA binding domain; H: Hinge domain; LBD: Ligand binding domain. Pink entries correspond to sequence with high prediction score of RNA-binding.
(B) SYBR Gold stain of Trizol-purified RNAs pulled down by GST-ERα protein [amino acids (aa) 144–314] with or without the putative RBD (aa 255–272). Coomassie stain of GST-purified ERα is shown.
(C) Left: Representative western blots for FLAG-ERα bound to Oligo(dT) beads in MCF7 and T47D cells with the RBD mutation of ERα (FLAG-ERα RBDmut, aa 259RRGG > 259AAAA), compared to those with wild-type (WT) ERα. PABP1 and SMC2 are used as positive and negative controls for RNA-binding respectively. Right: Quantifications are shown.
(D) The correlation of FLAG-ERα chromatin-binding signals using ChIP-seq wiggle files of MCF7 cells stably expressing WT or RBDmut ERα.
(E) The comparison of WT and RBDmut ERα enrichments around WT ERα ChIP-seq peaks using the SitePro tool (cistrome.org/ap).
(F) Integrative Genomics Viewer (IGV) views of WT and RBDmut ERα binding to GREB1 and TFF1 in MCF7 cells.
(G) Relative cell growth of MCF7 (left) and T47D (right) breast cancer cells with WT or RBDmut ERα measured by sulforhodamine B (SRB) assay.
(H) Tumor volumes of mouse xenografts implanted with MCF7 (left) and T47D (right) cells harboring WT or RBDmut ERα. N=5 mice per arm.
All values represent the mean ± SD. Two-sided t-test. Otherwise noted, N=3 biological replicates, ** P < 0.01, *** P < 0.001.
We created an RBD mutant of ERα by minimalizing the mutation to amino acid 259–262 (RRGG > AAAA), and generated stable ERα RNA-binding-deficient MCF7 and T47D breast cancer cell lines by replacing the endogenous ERα with FLAG-tagged wild-type (WT) and RBD-mutated (RBDmut) ERα (Figure S2B). In MCF7 and T47D cells, ERα RBDmut loses its RNA-binding ability (Figure 2C). As both WT and RBDmut ERα are able to localize to the nucleus (Figure S2C), we next asked whether the ERα RBD mutant may have an impact on the DNA-binding function of ERα. To test this, we first extracted the chromatin-bound fractions of MCF7 cell lysates, and confirmed that in both WT and RBDmut cell lines, ERα retains its DNA ability (Figure S2D). We further performed chromatin-immunoprecipitation followed by high-throughput sequencing (ChIP-seq), using FLAG antibody to immunoprecipitate both WT and RBDmut ERα and their associated DNAs. The ERα RBDmut possesses similar DNA-bindings with the WT one, with a strong Pearson correlation of the chromatin-bindings (r = 0.97, Figure 2D) and similar peak intensities surrounding the ERα binding events (Figure 2E, F, and Figure S2E). Furthermore, a volcano plot also shows very few differences in the DNA-binding events (Figure S2F), most of which are not located at annotated promoters or enhancers (Table S3), and are therefore unlikely to affect gene expression. As expected, the estrogen response element (ERE) is similarily most significantly enriched (Figure S2G). We further validated the ChIP-seq by ChIP-qPCR of two classical ERα transcriptional targets, GREB1 and TFF1. Our data confirmed that WT or RBDmut ERα binds to the promoters of these genes identically (Figure S2H).
Interestingly, within a total of 1193 mRNAs bound by ERα (including 3’UTR-, 5’UTR-, and CDS-bound mRNAs), 107 of them were overlapped with those ERα-bound on chromatin, including known estrogen regulated genes such as TFF1 and GREB1 (Figure S2I). This suggests that ERα may simultaneously interact with this subset of mRNAs both as a transcription factor and as a RNA-binding protein. However, the majority of mRNAs bound by ERα are not ERα-bound genes on the chromatin (Figure S2I). These results indicate that the ERα RNA-binding activity may regulate a different group of genes at the post-transcriptional level, indepently from its classical function as a transcription factor.
ERα RNA-binding activity contributes to breast cancer progression
We next investigated whether ERα RNA-binding is essential for cancer cell growth. Strikingly, mutating the RBD of ERα in both MCF7 and T47D breast cancer cells significantly impaired the proliferation of both ERα RBD mutated breast cancer cells (Figure 2G). We further extended our study in vivo by employing xenograft mouse models. Notably, abrogating the RNA-binding ability of ERα also suppressed tumor growth in vivo (Figure 2H). These data demonstrate that the RNA-binding activity of ERα contributes to breast cancer progression.
Functional genomic screen identifies essential ERα-bound transcripts important for breast cancer fitness
In order to understand how ERα-RNA association contributes to breast cancer progression at the molecular level, we employed a CRISPRi screen approach (Gilbert et al., 2014; Horlbeck et al., 2016) with a customized library to target the genes whose mRNAs were bound by ERα at their 3’ UTRs. The sgRNA library constructs (Table S4) were infected into MCF7 cells stably expressing the doxycycline-inducible, inactive form of Cas9 protein (dCas9), and the sgRNAs frequencies representing the growth phenotype were determined (Figure 3A, B and Figure S3A). With a cut-off of [|log10(P value) × average growth phenotype| > 1], this screen identified several transcripts important for the fitness of breast cancer cells and also narrowed down the list of ERα-bound transcripts to a set of functional gene networks (237 genes) whose silencing inhibits cell growth significantly (Figure 3B, and Table S5). We performed gene ontology analysis on the 237 transcripts essential for breast cancer growth. This revealed several significantly enriched biological function categories (P value < 0.01) that were grouped into major functional clusters (Figure 3C) associated with known key cellular hallmarks underlying breast cancer including cell cycle, response to estradiol stimulus, protein synthesis and cell motility. Notably enriched was a group of mRNAs involved in the adaptive response to stress (Figure 3C), a cellular process that is emerging as a key hallmark of tumor development and therapy response. Importantly, cancer cells are exposed to stressful environments in vivo such as nutrient deprivation, hypoxia, and oxidative stressors during tumor growth and metastasis formation.
Figure 3. Functional genomics identifying ERα-bound mRNAs essential for the fitness of breast cancer cells.
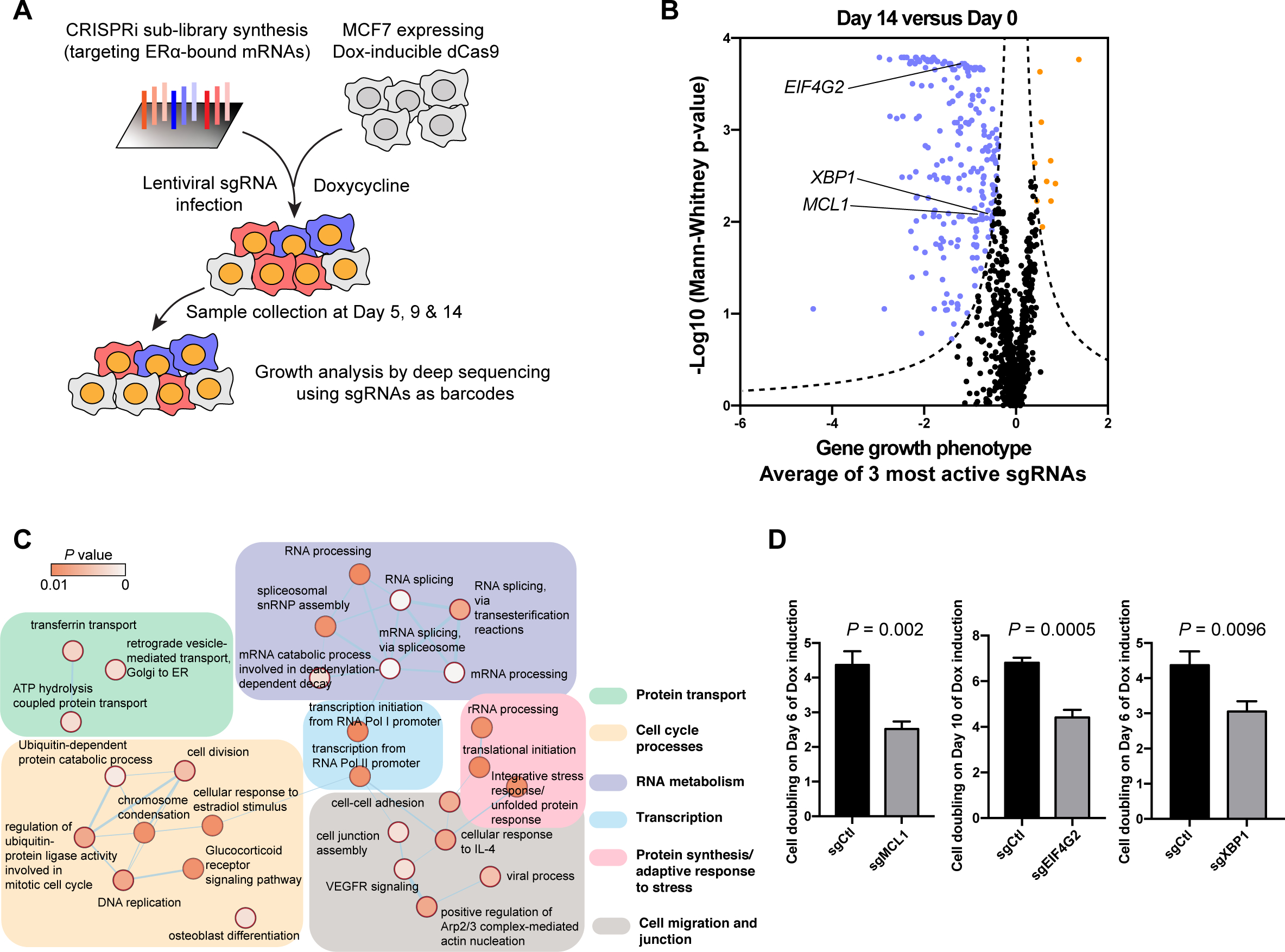
See also Figure S3.
(A) A schematic of the strategy for CRISPRi screen of ERα-bound mRNAs essential for the fitness of MCF7 breast cancer cells.
(B) Growth phenotype analysis of the CRISPRi screen on Day 14 compared with Day 0 shown as a volcano plot. The average growth phenotype for each gene with the 3 most active sgRNAs is shown in X-axis, and the significance of the growth phenotype is presented in Y-axis as log10(Mann-Whitney p-values) of all 5 sgRNAs. The Dash lines mark the threshold of significance in this study: |Growth phenotype × log10(Mann-Whitney p-values) | > 1. Genes that pass the threshold are shown, with genes that favor cell growth in blue, those inhibit cell growth in orange.
(C) Gene ontology of ERα-bound genes that favor cancer cell growth were analyzed by DAVID and presented utilizing Cytoscape software. For each ontology term the P values was calculated.
(D) Cell doublings on indicated days of cells infected with sgRNAs targeting MCL1 (sgMCL1), eIF4G2 (sgEIF4G2) and XBP1 (sgXBP1) compared to the control (sgCtl). N=3 biological replicates. Two-sided t-test. All values represent the mean + SD.
Therefore, in order to further understand how ERα as an RBP provides a survival advantage to cancer cells, we functionally investigated transcripts from our CRISPRi screen: the anti-apoptotic protein myeloid cell leukemia 1 (MCL1), eukaryotic translation initiation factor 4 gamma 2 (eIF4G2), and transcription factor X-box binding protein (XBP1), all of which are implicated in the cellular response to stress. MCL1 belongs to the BCL-2 anti-apoptotic protein family that prevents cancer cell death (Cory et al., 2016). eIF4G2 (also named DAP5, p97 and NAT1), is homologous to eIF4G1, a translation initiation factor. Importantly, eIF4G2 is involved in the translation of mRNAs crucial for cell growth and invasion (Liberman et al., 2015; Marash et al., 2008; Ramirez-Valle et al., 2008; Weingarten-Gabbay et al., 2014) in a cap-independent manner upon stress (de la Parra et al., 2018). XBP1 is a key transcription factor downstream of IRE1-mediated UPR, whose mRNA is unconventionally processed to generate a spliced form of XBP1 (XBP1s) (Walter and Ron, 2011) that has enhanced transcriptional activity. This activation of XBP1 promotes cell survival upon stress and thus is oncogenic in many cancer types, including breast cancer (Chen et al., 2014; Gomez et al., 2007). The CRISPRi screen revealed that silencing these genes resulted in noticeable cell growth inhibition as early as 5-day of dCas9 induction, which became more pronounced at later timepoints (Figure S3B). To validate their phenotype in regulating cell fitness, we performed a competition growth assay to determine the growth advantage/disadvantage of each gene. Consistent with the CRISPRi screen result, depletion of MCL1, eIF4G2 or XBP1 rendered a significant growth defect (Figure 3D), suggesting that these mRNAs bound by ERα are essential for breast cancer fitness.
ERα RNA-binding facilitates XBP1 splicing upon stress
ERα binding to selective ISR and UPR transcripts is suggestive of a previously unknown post-transcriptional program. Nevertheless how specific components of these response pathways such as XBP1, are regulated in cancer cells remain poorly understood. Using electrophoretic mobility shift assay (EMSA) with ERα and the 3’UTR of XBP1 mRNA, we observe a significant association of XBP1 3’UTR with ERα (Figure 4A and Figure S4A). Functionally, blocking the RNA-binding activity of ERα almost completely repressed the alternative splicing of XBP1 mRNA upon endoplasmic reticulum (ER)-stress in MCF7 cells (Figure 4B). A similar defect in XBP1 splicing was also observed in T47D cells with the ERα RBDmut (Figure S4B). The cleavage of XBP1 mRNA renders a shorter transcript, but also shifts its open reading frame (ORF), resulting in the production of a larger, but more potent transcription factor, XBP1s (Calfon et al., 2002; Yoshida et al., 2001). In this respect, the difference in the splicing of XBP1 mRNA between cells with WT or RBDmut ERα is also evident at the protein level (Figure 4C).
Figure 4. ERα RNA-binding facilitates XBP1 splicing upon stress.

See also Figure S4.
(A) IGV view of ERα HITS-CLIP showing ERα binding to XBP1 mRNA with indicated cross-link-induced mutation sites (CIMS) representing ERα-bound regions.
(B) Representative RT-PCR analysis of XBP1 splicing in MCF7 cells harboring WT or RBDmut ERα upon DMSO or tunicamycin treatment for 4 h. Upper bands indicate unspliced XBP1 mRNA and the lower indicate spliced XBP1 mRNA. β-actin mRNA (ACTB) is used as an internal control.
(C) Representative western blots for spliced and unspliced XBP1 protein (XBP1s and XBP1u), phosphor-eIF2α (p-eIF2α), eIF2α and GAPDH in MCF7 cells with WT and RBDmut ERα treated with DMSO (−) or tunicamycin (+) for 6 h.
(D) A cartoon demonstrating the processes of XBP1 splicing: cleaved by IRE1 and ligated by RtcB.
(E) Representative western blots for RtcB and ERα immunoprecipitated (IP) by ERα-specific and IgG (negative control) antibodies.
(F) IGV view of ERα (peaks and binding regions in blue) and RtcB [binding regions in red, PAR-CLIP (GSM936508) (Baltz et al., 2012)] association on the XBP1 mRNA.
(G) Left: Representative RT-PCR analysis of XBP1 splicing in MCF7 cells harboring WT and RBDmut ERα, with or without the RtcB silencing (sgRTCB) upon 4 h tunicamycin treatment. Right: Relative ratios of spliced XBP1 (XBP1s) compared to unspliced XBP1 (XBP1u) are quantified.
(H) Representative western blots for ERα hotspot mutants (E380Q, L536H, Y537S and D538G) and WT ERα immunoprecipitated (IP) by an antibody specifically recognizing RtcB.
(I) Relative cell growth of MCF7 cells treated with IRE1 inhibitor (IRE1i, STF-083010) for 72 h compared to DMSO. Cells were cultured in normal cell culture conditions.
(J) Tumor volumes of mice implanted with MCF7 cells in the mammary fat pad of mice dosed with vehicle or 30 mg/kg of IRE1 inhibitor (IRE1i) STF-083010 once per week (N=7 mice per arm).
(K) Representative RT-PCR analysis of XBP1 splicing in tamoxifen-resistant (TamR-1) and its parental tamoxifen-sensitive MCF7 cells without tunicamycin treatment.
(L) Relative cell apoptosis as shown by relative Annexin V+ populations of TamR-1 cells treated with IRE1 inhibitor for 24 h compared to DMSO in stressful cell culture conditions: in ultra-low attachment dishes (Suspension) or serum free media (Serum deprivation).
N=3 biological replicates. Two-sided t-test. All values represent the mean + SD. Otherwise noted, * P < 0.05, ** P < 0.01, *** P < 0.001.
Upon ER-stress, UPR signaling and particularly the inositol-requiring enzyme 1 (IRE1) endonuclease is activated and mediates the cleavage of XBP1 mRNA (Yoshida et al., 2001), which is subsequently ligated by an RNA 2’,3’-cyclic phosphate and 5’-OH ligase RtcB (Lu et al., 2014) (Figure 4D). Interestingly, ERα physically interacts with RtcB (Figure 4E), likely through its ligand binding domain (LBD) (Figure S4C). The ERα-bound regions on XBP1 mRNA are very close to where RtcB binds (Figure 4F). Interestingly, abrogating ERα RNA-binding activity or silencing RtcB resulted in notable defects in XBP1 splicing upon stress; the ERα RBD mutation together with RtcB silencing however, did not further suppress XBP1 splicing (Figure 4G), suggesting that ERα controls XBP1 alternative splicing through RtcB.
Studies show that approximately 20% of patients that are ERα-positive and received endocrine therapies gain hotspot mutations in the ERα LBD (Jeselsohn et al., 2015) which account for acquired endocrine resistance. Interestingly, we observed a dramatic increase in the physical interaction of RtcB with ERα harboring these clinical relevant mutations (particularly L536H) compared to WT ERα (Figure 4H), revealing that the gain-of-function mutants of ERα might also possess a previously unknown function, such as facilitating XBP1 splicing through RtcB. This further supports that the post-transcriptional role of ERα may be clinical relevant for the endocrine therapy response in human breast cancers.
To extend the functional significance of the XBP1 splicing pathway (Figure 4D) in breast cancer, we employed a pharmacological approach by inhibiting the cleavage of XBP1 mRNA through an IRE1 inhibitor, STF-083010, which specifically blocks its endonuclease but not the kinase activity (Papandreou et al., 2011). Inhibiting the activity of IRE1 dramatically inhibited ERα-positive MCF7 cancer cell growth (Figure 4I). Furthermore, we monitored tumor growth in the mammary environment of NSG mice, with or without STF-083010. Dosing the mice with STF-083010 notably blocked tumor formation (Figure 4J), suggesting that targeting this pathway can be beneficial for treating breast cancer. Previous reports have suggested that overexpression of XBP1s in ERα+ breast cancer cells may lead to estrogen-independent growth and reduced sensitivity to antiestrogens such as tamoxifen (Gomez et al., 2007; Ming et al., 2015), which predicts a poorer survival of ERα+ breast cancer patients receiving endocrine therapies (Davies et al., 2008). Importantly, a tamoxifen-resistant breast cancer cell line generated from long-term tamoxifen treatment in MCF7 cells, TamR-1, has naturally increased XBP1 splicing events compared to its parental MCF7 cells (Figure 4K). The naturally augmented XBP1 splicing may promote survival and adaptation to stress of cancer cells. To mimic common stressors tumor cells encountered in vivo, for example stressors upon tumor dissemination and nutrient shortage, we in vitro cultured TamR-1 cells in ultra-low attachment plates or the serum-depleted media, respectively. Under both stress conditions, IRE1 inhibition significantly increased the apoptosis of TamR-1 cells upon tamoxifen treatment (Figure 4L), indicating that blocking XBP1 splicing can efficiently reverse tamoxifen resistance. This pro-apoptotic effect was largely inhibited upon ERα silencing (Figure S4D), revealing that the sensitivity of cells to the IRE1 inhibitor is, at least in part, dependent on ERα. Together, our data reveal a previously unknown post-transcriptional function of ERα in XBP1 splicing, which possesses both a clinical relevance and the potential for targeted therapies in breast cancer.
ERα modulates the translation of eIF4G2 and MCL1 mRNAs
Besides mRNA splicing, binding to mRNA by RBPs may also contribute to other post-transcriptional regulations such as mRNA translation and degradation. Interestingly, blocking the RNA-binding ability of ERα or silencing ERα dramatically reduces the protein abundances of eIF4G2 and MCL1 in both MCF7 and T47D cells (Figure 5A, B and Figure S5A), without effects on their mRNA expression (Figure 5C and Figure S5B). Moreover, blocking proteasome-mediated protein degradation with a specific proteasome inhibitor MG-132 did not fully rescue the reduction in eIF4G2 and MCL1 proteins induced by ERα RNA-binding deficiency (Figure S5C). To ascertain whether MCL1 and eIF4G2 are regulated by ERα at the translation level, we examined their distributions in polysomes (translationally active ribosome fractions) on sucrose gradient fractionation. While the mutation of the ERα RBD did not result in noticeable alterations in global protein synthesis (Figure 5D), it affects the translation of specific mRNAs. Specifically, we observed an accumulation of MCL1 and eIF4G2 mRNAs in less translationally active polysome (light polysomes) or transaltionally inactive fractions (Figure 5E), but not the control α-tubulin mRNA (Figure S5D). Interestingly, ERα does not bind to other anti-apoptotic BCL-2 family mRNAs (BCL2 and BCL2L1) (Figure S5E), suggesting that ERα controls mRNA translation in a transcript-specific manner.
Figure 5. ERα RNA-binding increases the translation of eIF4G2 and MCL1 mRNA, and its targeting abrogates cell survival and reverses tamoxifen resistance.
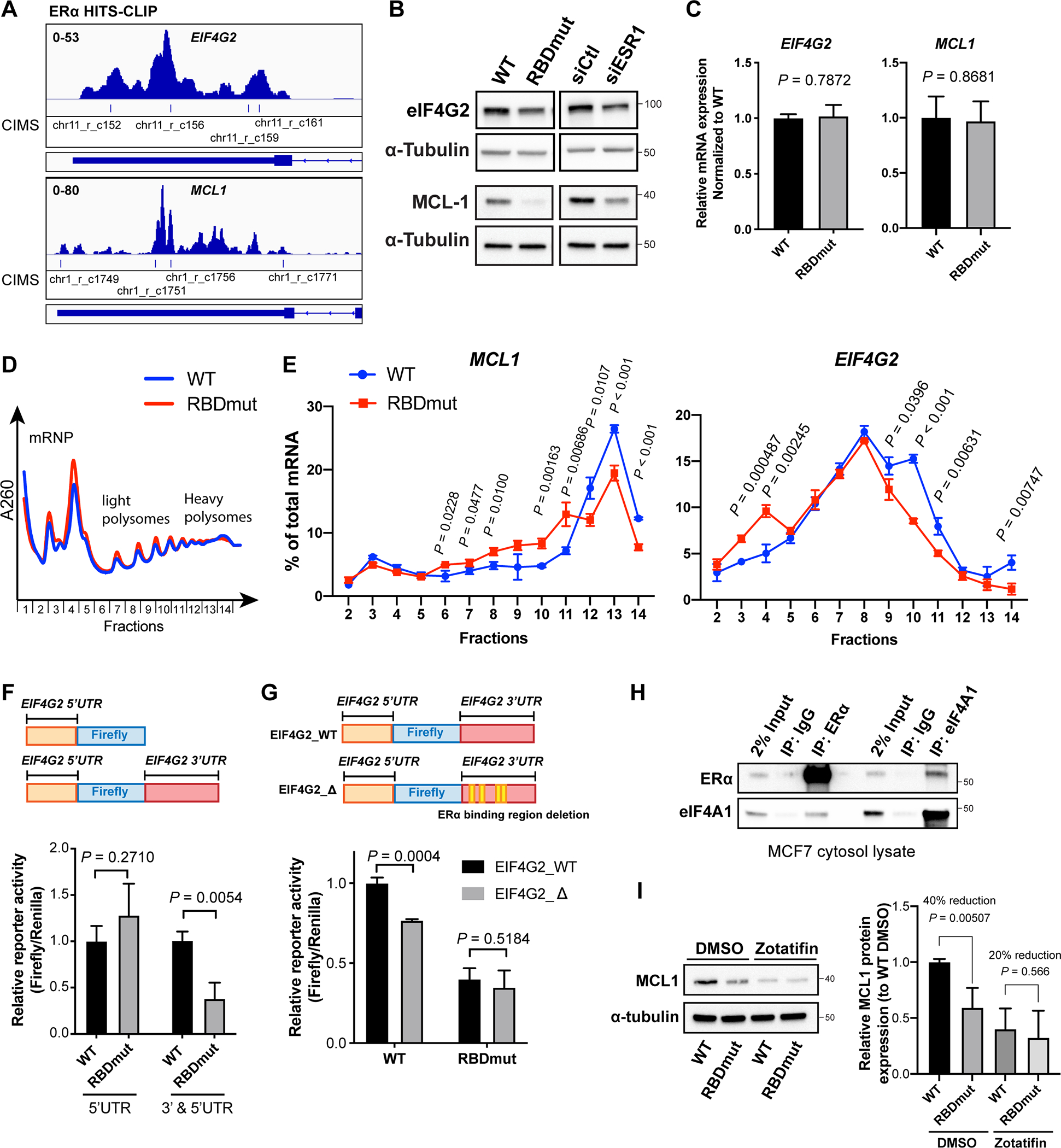
See also Figure S5.
(A) IGV views of ERα HITS-CLIP analysis showing ERα binding to eIF4G2 and MCL1 mRNAs with indicated CIMS representing ERα-bound regions.
(B) Representative western blots for eIF4G2, MCL1 and α-tubulin in MCF7 cells with WT or RBDmut ERα, or with and without ERα silencing (siESR1) for 72 h.
(C) Relative mRNA expressions of eIF4G2 and MCL1 in MCF7 cells with WT or RBDmut ERα measured by qPCR.
(D) Representative polysome traces of MCF7 cells with WT or RBDmut ERα. Fractions 2–5: mRNA ribonucleoprotein (mRNP)/monosome; Fractions 6–10: light polysome; Fractions 11–14: heavy polysome.
(E) Percentages of MCL1 (left) and EIF4G2 (right) mRNAs distributed in each fraction against total mRNA of them are shown.
(F) Relative reporter activities of eIF4G2 (5’UTR)-luciferase and eIF4G2 (3’ and 5’UTR)-luciferase in MCF7 cells with WT or RBDmut ERα.
(G) Relative reporter activities of eIF4G2 (3’ and 5’UTR)-luciferase with the deletion of ERα-binding sequences (EIF4G2_Δ) compared to WT (EIF4G2_WT) in MCF7 cells with WT or RBDmut ERα.
(H) Representative western blots for eIF4A1 and ERα immunoprecipitated (IP) by ERα, eIF4A1 and IgG antibodies.
(I) Left: representative western blots for MCL1 and α-tubulin in MCF7 cells with WT or RBDmut ERα treated with eIF4A inhibitor Zotafitin for 6 h; Right: quantifications of relative MCL1 protein abundances normalized to α-tubulin from independent experiments.
N=3 biological replicates. Two-sided t-test. All values represent the mean + SD.
Translation of mRNAs are frequently regulated particularly by their 5’UTRs and 3’UTRs (Mazumder et al., 2003; Wells et al., 1998). We generated two constructs, where one only has the full-length eIF4G2 5’UTR cloned upstream the firefly reporter, the other has both its 5’UTR and 3’UTR incorporated upstream and downstream the firefly respectively (Figure 5F). The reporter activity (Firefly/Renilla) of these constructs in both ERα WT and RBDmut MCF7 cells were normalized to their luciferase mRNA levels. Comparing cells harboring WT and RBDmut ERα, we observed that while there is no significant difference in the activity of a reporter containing only the 5’UTR of eIF4G2, there is a dramatic reduction in the 3’UTR-containing reporter activity when ERα cannot bind RNA (Figure 5F). We next asked whether altering the ERα-bound sequences of eIF4G2 may also block its translation. To this end, deleting the ERα-bound sequences in eIF4G2 3’UTR (EIF4G2_Δ) rendered a notable inhibition in the reporter activity in cells with WT ERα, which is less evident in those with ERα RBDmut (Figure 5G), suggesting that blocking either the RNA-binding ability of ERα or the ability of the eIF4G2 transcript to bind ERα is sufficient to repress the translation of eIF4G2 mediated by ERα.
Intriguingly, from our ERα-IP mass spectrometry data we observed that ERα may interact with eIF4A1, an RNA helicase of the translation initiation complex and importantly one of the key translation regulators of MCL1 (Robert et al., 2014). To determine whether the regulation of MCL1 translation by ERα is through eIF4A1, we first validated the interaction between ERα and eIF4A1 (Figure 5H). Furthermore, treatment with a clinical eIF4A-specific inhibitor, zotatifin, decreases MCL1 protein significantly, whereas this reduction is less evident in cells with ERα RBDmut cells, suggesting that the regulation of MCL1 by ERα is at least in part through eIF4A1 (Figure 5I).
Together, our data reveal that ERα is a multifaceted RNA-binding protein involved in a variety of RNA post-transcriptional regulation processes including translation control and RNA splicing.
ERα post-transcriptional targets are overexpressed in ERα+ breast cancer
Given that ERα controls the post-transcriptional regulations of eIF4G2, MCL1 and XBP1, we next asked whether these proteins are overexpressed in human breast cancers. We first obtained human breast tumors together with their normal adjacent tissues from 14 patients diagnosed with ERα+ breast cancer. We observed a significant increase in eIF4G2, MCL1 and XBP1s protein abundance in tumors compared with the normal adjacent tissues (Figure 6A and Figure S6A). We further performed immunohistochemistry (IHC) staining of these proteins on breast cancer tissue microarrays (TMA), which contain in total 65 ERα+ invasive breast carcinomas cases and 8 normal adjacent tissue biopsies. In accordance with our western blot data, we observed significantly augmented expression of these proteins in tumor compared to normal tissues (Figure 6B and S6B). Importantly, we have also analyzed the correlation between protein and mRNA expression data of eIF4G2, MCL1, and GREB1 (an ERα-transcriptional target as a control) in breast cancer patient tumor samples from a published database (Krug et al., 2020). We observed a notable correlation between GREB1 protein and mRNA expression (Figure 6C, Pearson r = 0.7586), consistent with the fact that GREB1 is often regulated at the transcription level. In comparison, there is a lack of correlation between protein and mRNA expression of eIF4G2 and MCL1 (Figure 6C, Pearson r = 0.3137 and 0.2087 respectively). The lack of RNA and protein correlation suggests that in human breast cancer, post-transcriptional means of regulation, notably translation mediated by ERα, can be a key determinant of gene expression changes.
Figure 6. ERα post-transcriptional stress response targets are overexpressed in ERα+ breast cancer at the protein level, which can be targetable.
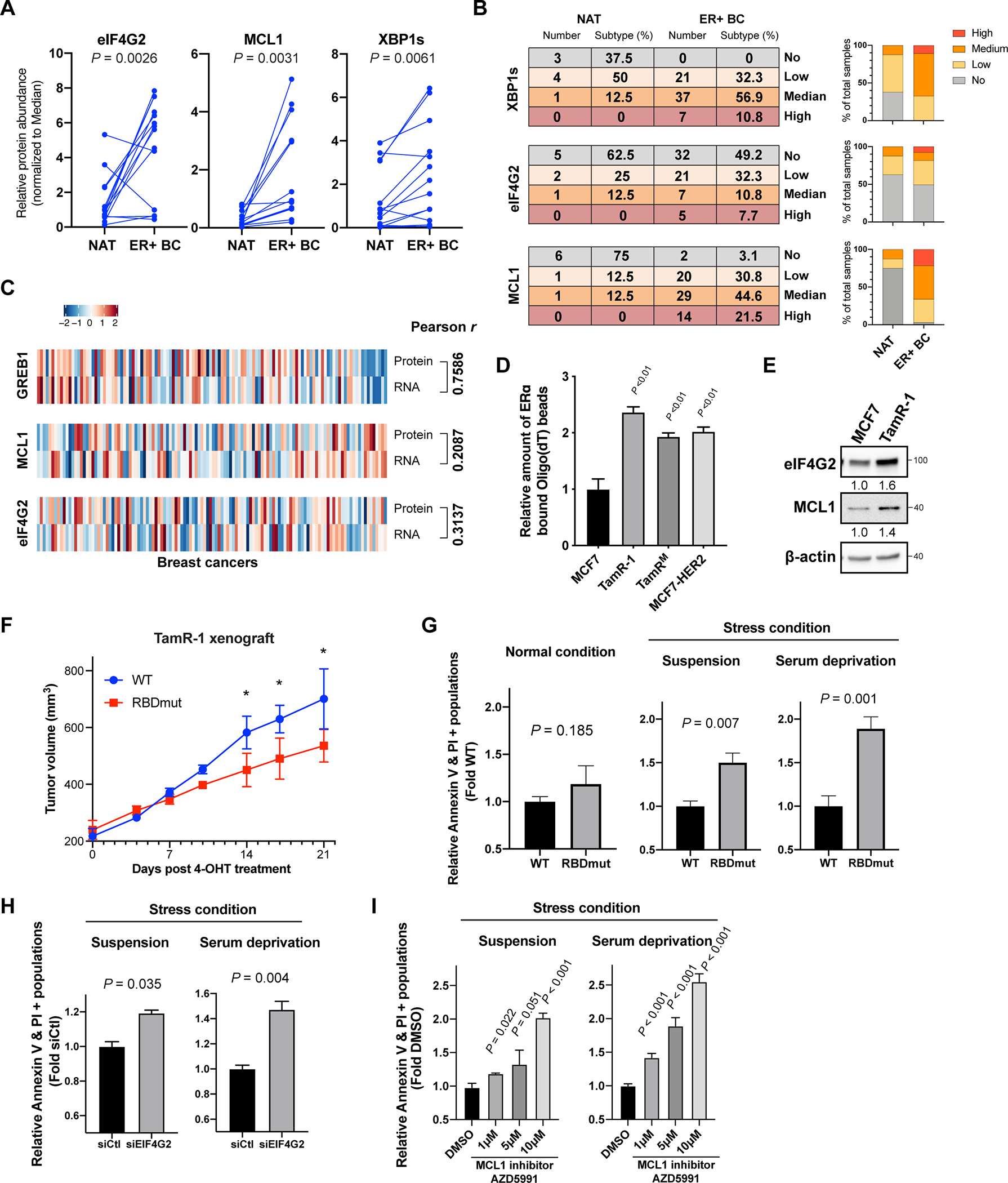
See also Figure S6.
(A) Quantifications of eIF4G2, MCL1 and XBP1s protein expressions in 14 freshly collected ERα+ breast cancer tumors (ER+ BC) and their paired normal adjacent tissues (NAT). Two-sided paired t-test.
(B) ER+ BC and NAT from tissue microarrays (TMAs) were stained and assessed for the protein expressions of XBP1s, eIF4G2 and MCL1. No: no expression, Low: low expression, Median: median expression, and High: high expression. Counts for normal tissues or tumors expressing indicated proteins at each level were quantified on the right.
(C) Multi-omic landscape of eIF4G2, MCL1 and GREB1 comparing their protein and mRNA expressions are shown. The heatmap are generated with the CPTAC-BRCA2020 data viewer. Pearson correlation test.
(D) Relative amount of ERα pulled-down by Oligo(dT) beads in tamoxifen-resistant cells (TamR-1, TamRM, and MCF7-HER2) compared to parental MCF7 cells.
(E) Western blot analysis of eIF4G2, MCL1 and β-actin in TamR-1 comparing to MCF7 cells.
(F) Tumor volumes of mouse xenografts implanted with TamR-1 cells harboring WT or RBDmut ERα, dosed with 20mg/kg of 4-hydroxytamoxifen (4-OHT) intraperitoneally (i.p.) every 2 days for 21 days. N=5 mice per arm. * P < 0.05.
(G) Relative fold differences of ERα RBDmut TamR-1 cells undergo apoptosis (Annexin V+, PI+) comparing to the WT cells, under normal or the stress conditions-suspension and serum deprivation, where WT and RBDmut TamR-1 cells were cultured for 24h in ultra-low attachment dishes, or in serum-free media respectively.
(H) Relative fold differences of TamR-1 cells undergo apoptosis (Annexin V+, PI+) with 72 h eIF4G2 silencing (siEIF4G2) to siCtl under indicated stress conditions for 24 h.
(I) Relative fold differences of TamR-1 cells undergo apoptosis (Annexin V+, PI+) upon 24 h MCL1 inhibitor AZD5991 treatment, under indicated stress conditions.
N=3 biological replicates. Two-sided t-test. All values represent the mean + SD.
Targeting ERα post-transcriptional signaling abrogates cell survival and reverses tamoxifen resistance
Cancer cells are usually exposed to stressful environments when forming tumors in vivo, which includes not only the challenging tumor growth conditions such as hypoxia and nutrient shortage, but also the stress induced by anti-cancer therapies (Andruska et al., 2015; Costa-Mattioli and Walter, 2020; Urra et al., 2016). As ERα-target mRNAs particularly XBP1, MCL1 and EIF4G2 are all overexpressed in human breast cancer, and essential for cells to overcome cellular stressors and survive (Fritsch et al., 2007; Lee et al., 2003; Marash et al., 2008; Romero-Ramirez et al., 2004), we hypothesized that ERα RNA-binding may also be crucial for cancer cells to survive and overcome unfavorable growth conditions and anti-cancer therapies. Pharmacological targeting the ERα transcription signaling by endocrine therapies, such as the estrogen receptor modulator 4-hydroxy-tamoxifen (4-OHT) shows high efficacy in the majority of ERα-positive diseases, however a notable portion of patients eventually developed drug resistance (Ali and Coombes, 2002). Interestingly, ERα remains functionally important in the resistance disease, and multiple mechanisms responsible for endocrine resistance have been proposed (Hanker et al., 2020; Jeselsohn et al., 2015; Musgrove and Sutherland, 2009; Osborne and Schiff, 2011). Importantly, ERα is a nucleocytoplasmic shuttling protein whose cytoplasmic proportion increases upon long-term tamoxifen treatment (Fan et al., 2007), suggesting that the non-genomic function of ERα in the cytoplasm may also play an important role in the resistance. Intriguingly, treating breast cancer cells with either tamoxifen or the selective estrogen receptor degrader (SERD) fulvestrant, induced cellular stress, such as endoplasmic reticulum stress (ER-stress) represented by the increased phosphorylation of eIF2α (Figure S6C), which if not overcome will lead to cell death. Thus, handling stress by ERα RNA-binding may be crucial for cancer cells to survive and develop resistance. In this regard, the RNA-bound proportions of ERα approximately doubled in tamoxifen-resistant MCF7 cells (TamR-1 and TamRM) generated from long-term tamoxifen treatment (Lykkesfeldt et al., 1994; Raha et al., 2015), as well as MCF7 cells stably overexpressing human epidermal growth factor receptor 2 (HER2), one of the phenotypes for tamoxifen resistance in human breast cancer (Shou et al., 2004) (Figure 6D). These cells are all insensitive to tamoxifen, resistant to tamoxifen-induced G1 arrest and apoptosis. Notably, similar to that of XBP1 splicing (Figure 4I), TamR-1 cells exhibit increased eIF4G2 and MCL1 protein abundances (Figure 6E) without significant changes in their transcript levels compared to tamoxifen-sensitive MCF7 cells (Figure S6D). These results indicate that ERα RNA-binding and its post-transcriptional targets may have a functional role in the tamoxifen response of breast cancer.
We next implanted TamR-1 cells harboring RBDmut and WT ERα into NSG mice, and observed a tumor growth inhibition upon tamoxifen administration of TamR-1 cells when the RBD of ERα is mutated (Figure 6F). To investigate the mechanism underlying tamoxifen re-sensitization, we cultured the ERα WT and RBDmut TamR-1 cells and detected a significant but mild G1 arrest when ERα RBD was mutated (Figure S6E). We further examined ERα WT and RBDmut TamR-1 cells under stress conditions including culturing in ultra-low attachment plates or serum depleted media together with tamoxifen treatment. Intriguingly, TamR-1 cells without ERα RNA-binding activity are more sensitive to these stressors together with tamoxifen treatment, leading to a notable increase in apoptosis, which is not observed under normal conditions (Figure 6G). These results indicate that ERα-RNA association may be particularly important for cancer cells to overcome cellular stress.
We next sought to functionally characterize the role of ERα targets in facilitating breast cancer survival and during endocrine therapy. Similar to the phenotype of TamR-1 cells with ERα RBDmut, under normal cell culture conditions, silencing eIF4G2 using siRNA (siEIF4G2) together with tamoxifen treatment only resulted in increased G1 cell cycle arrest (Figure S6F), whereas no significant cell apoptosis was observed (Figure S6G). Importantly, providing eIF4G2 silenced cells with cell stressors dramatically increased the percentages of cells undergo apoptosis (Figure 6H). This phenomenon was also observed when MCL1 is pharmacologically targeted with a specific inhibitor, where the effect of MCL1 inhibition on cell apoptosis were more evident under conditions such as nutrient deprivation or suspension, under tamoxifen treatment (Figure 6I and Figure S6H). Together, these data strongly suggest that ERα-mediated post-transcriptional regulation plays an essential role for cancer cell adaptation to stressful environments and maintenance of cell survival.
Discussion
The role of ERα in RNA metabolism underlying breast cancer
ERα is known as a master transcription factor controlling the expression of key genes involved in breast cancer progression. On the other hand, our work shows the importance of ERα post-transcriptional regulation as another key determinant of its oncogenic potential. Emerging evidence has revealed that alterations in the expression and function of RBPs can serve as key oncogenic events and may amplify the effects of cancer drivers in promoting tumor progression (Pereira et al., 2017). Although recent Encyclopedia of DNA Elements (ENCODE) project has mapped the RNA-interactome of hundreds of human canonical RBPs using enhanced CLIP (eCLIP) assays (Van Nostrand et al., 2020), it remains poorly understood how non-canonical RBPs associate with RNAs, as well as how these non-canonical functions are related to cancer and disease. It is therefore intriguing to discover that the RNA-binding activity of ERα is uncoupled from its classical chromatin-binding function, which provides an ideal platform to study at which step of tumor progression the function of a non-canonical RBP becomes specifically crucial. Importantly, the RNA-binding function can also apply to other nuclear receptors such as GR, which may also participate in the cancer development (Ishmael et al., 2011). Employing unbiased genome-wide CLIP-seq and functional genomics screen, we have uncovered an oncogenic program comprised of hundreds of mRNAs bound by ERα, particularly a group of mRNAs involved in the adaptive response to stress that may represent a unique role of the post-transcriptional regulation function of ERα in cancer. Cancer cells are constantly exposed to multiple stressors during tumor growth and metastasis formation including external signals such as hypoxia, nutrient shortage, immune attack, as well as the delivery of anticancer drugs. Endocrine therapy results in a rapid activation of stress signaling (Andruska et al., 2015; Cook et al., 2014) such as ER-stress. Overcoming cellular stress is therefore critical for cancer cell survival, which may also serve as a potential drug target (Chen and Cubillos-Ruiz, 2021; Gaillard et al., 2015; Hayes et al., 2020; Jin and Saatcioglu, 2020). In this respect, the translational control or alternative splicing of stress response mRNAs by ERα may offer a fast way of controlling protein production, and may be particularly important for breast cancer cells to adapt to various stressors in the tumor microenvironment. Thus, this functional preference of the non-canonical activity of ERα in adaptive response to stress may differ from that of its classical transcriptional role (e.g. in stimulating cell proliferation), which may provide extra advantages for the fitness of breast cancer cells.
Targeting ERα-mediated post-transcription networks in breast cancer
Historically, breast cancer research has focused mainly on genomic and transcriptional alterations underlying tumor development. However, post-transcriptional regulation is known to be a key determinant for final protein production required for the adaptation of cells to altered tumor miroenvironments (Liu et al., 2016; Xu and Ruggero, 2020). In addition, the post-transcriptional of gene regulation is not typically detected by genomics and RNA-seq studies, therefore limiting the predictive power of mRNA-based prognostic biomarkers, especially when the production of key pro-tumor proteins are not associated with changes in mRNA abundance [e.g. eIF4G2 and MCL1 in this study (Figure 6C)]. In this respect, our results are vital in delineating ERα non-genomic functional partners (e.g. RtcB) as well as downstream post-transcriptional effectors (e.g. XBP1s, eIF4G2, and MCL1) that may also serve as relevant biomarkers, and offer an innovative line of therapies targeting selective vulnerabilities for advanced breast cancer. Post-transcriptional regulation, especially translation control, may be hijacked by cancer cells as a means to rapidly escape anti-cancer therapy (Herviou et al., 2020; Rapino et al., 2018). To this end, targeting the translation control processes that ERα mediates, through inhibiting the functional partners of ERα in regulating mRNA translation, may offer a distinctive therapeutic window in treating breast cancers, especially those resistant to endocrine therapies (Baselga et al., 2012; Geter et al., 2017). More specifically, ERα+ breast cancer cells may be sensitive to therapies that target the stress response for cancer cell survival such as the ISR. Indeed, we show that targeting one node of the ISR, the IRE1-XBP1 signaling, through an IRE1 inhibitor is already sufficient to inhibit tumor progression in vivo, and promote cell death of tamoxifen resistant cells, revealing a “Achilles’ Heel” of ERα+ breast cancer. In this respect, our discovery that a non-genomic function of ERα regulates post-transcriptional gene expression of mRNAs implicated in stress response may provide a unique opportunity for researchers to revisit the role and mechanism of this nuclear receptor in broader physiological events and human diseases.
Limitations of the Study
Although our study demonstrated that the RNA-binding function of ERα triggers post-transcriptional regulation of specific mRNAs for cancer cells to overcome stress, several outstanding issues remain. Given that ERα is a key transcription factor in breast cancer, and certain genes (e.g. XBP1) are regulated by ERα at both the transcriptional and post-transcriptional levels, future work is needed to investigate how the RNA-binding and the classical DNA-binding function of ERα are coordinated. For example, does the agonist and antagonist of this nuclear receptor for DNA-binding (e.g. E2, Diethylstilbestrol, Tamoxifen, Fulvestrant, etc.) alter its RNA-binding activity? In addition, emerging evidence shows limited correlations between protein and mRNA expressions in cells, where the post-transcriptional means of protein production come to the forefront, particularly during cancer progressions, Although we show that the post-transcriptional targets of ERα (e.g. eIF4G2, XBP1 and MCL1) may be crucial for cancer cells to survive stress, Kaplan-Meier analyses should be done in the future to determine patient prognosis and clinical outcomes, especially for those relapse and become insensitive to therapies.
STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ERα, rabbit (for CLIP) | Santa Cruz | Cat# sc-542; RRID:AB_631470 |
| Anti-ERα, rabbit (for IP & mass spec) | Cell Signaling Technology | Cat# 8644S; RRID:AB_2617128 |
| Anti-FLAG (M2), mouse | Sigma | Cat# F1804; RRID:AB_262044 |
| Anti-IgG, rabbit | Cell Signaling Technology | Cat# 2729S; RRID:AB_1031062 |
| Anti-Lamin A/C, mouse | Santa Cruz | Cat# sc-7292; RRID:AB_627875 |
| Anti-α-tubulin, mouse | Abcam | Cat# ab18251; RRID:AB_2210057 |
| Anti-SMC2, rabbit | Cell Signaling Technology | Cat# 5329S; RRID:AB_10693789 |
| Anti-PABP1, rabbit | Cell Signaling Technology | Cat# 4992S; RRID:AB_10693595 |
| Anti-eIF4A1, mouse (for IP and western) | MyBioSource | Cat# MBS8504039 |
| Anti-eIF4G2, mouse (for western and IHC) | Proteintech | Cat# 67428-1-Ig; RRID:AB_2882667 |
| Anti-RtcB, rabbit | Proteintech | Cat# 19809-1-AP; RRID:AB_10695047 |
| Anti-MCL1, rabbit (for western and IHC) | Santa Cruz | Cat# sc-819; RRID:AB_2144105 |
| Anti-β-actin, mouse | Sigma | Cat# A5316; RRID:AB_476743 |
| Anti-Histone H3, | Cell Signaling Technology | Cat# 14269S; RRID:AB_2756816 |
| Anti-XBP1s, rabbit (for western and IHC) | Cell Signaling Technology | Cat# 12782S; RRID:AB_2687943 |
| Bacterial and virus strains | ||
| MAX Efficiency DH5α Competent Cells | Life Technologies | Cat# 18258012 |
| One Shot, BL21(DE3), E. coli | Life Technologies | Cat# C601003 |
| Biological samples | ||
| Human breast cancer TMAs | US Biomax | https://www.biomax.us/tissue-arrays/Breast/BC081116d |
| Chemicals, peptides, and recombinant proteins | ||
| Oligo(dT)-cellulose beads | Sigma | Cat# O3131 |
| TRIzol | Life Technologies | Cat# 15596026 |
| Glycoblue | Life Technologies | Cat# AM9515 |
| IPTG | Thermo Fisher Scientific | Cat# 15529-019 |
| BSA | Sigma | Cat# A7030 |
| TURBO DNA-free Kit | Thermo Fisher Scientific | Cat# AM1907 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail tablet | Roche | Cat# 11836170001 |
| Protein A Dynabeads | Thermo Fisher Scientific | Cat# 10008D |
| DNase RQ1 (1unit/ul) | Promega | Cat# M6101 |
| RNasin plus (40unit/uL) | Promega | Cat# N2615 |
| RNase A | Affymetrix | Cat# 70194Z |
| Calf alkaline phosphatase (10 unit/ul) | NEB | Cat# M0290 |
| T4 Polynucleotide kinase (10 unit/ul) | NEB | Cat# M0201 |
| T4 RNA ligase (10 unit/ul) | Thermo Fisher Scientific | Cat# EL0021 |
| Chloroform:isoamyl alcohol 49:1 | Sigma | Cat# 25668 |
| Acid phenol | Sigma | Cat# P4682 |
| Proteinase K (lyophilized) | Roche | Cat# 03115879001 |
| 3M NaOAc pH 5.5 | Thermo Fisher Scientific | Cat# AM9740 |
| Superscript III reverse transcriptase | Thermo Fisher Scientific | Cat# 18080093 |
| Accuprime pfx supermix | Thermo Fisher Scientific | Cat# 12344-040 |
| IGEPAL CA-630 | Sigma | Cat# I8896 |
| PhosSTOP™ | Sigma | Cat# 4906837001 |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| QIAprep Spin Miniprep Kit (250) | Qiagen | Cat# 27106 |
| RNA Clean & Concentrator™ Kits | Zymo Research | Cat# R1016 |
| ATP, [γ-32P]-6000Ci/mmol 10mCi/ml EasyTide | Perkinelmer | Cat# BLU502Z250UC |
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28706 |
| MinElute Reaction Cleanup Kit | Qiagen | Cat# 28204 |
| QuantiFluor dsDNA Sample Kit | Promega | Cat# E2671 |
| High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| NEBNext Ultra II DNA Library Prep with Sample Purification Beads | NEB | Cat# E7103S |
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 1)–24 rxns | NEB | Cat# E7335S |
| Deposited data | ||
| Sequencing data | This Study | GEO: GSE173631 |
| Mass spectrometry data | This Study | ProteomeXchange: PXD025018 |
| Experimental models: Cell lines | ||
| MCF7 | ATCC | Cat# HTB-22; RRID:CVCL_0031 |
| T47D | ATCC | Cat# HTB-133; RRID:CVCL_0553 |
| TamR-1 | Sigma | Cat# SCC101; RRID:CVCL_M436 |
| TamRM | (Raha et al., 2015) | N/A |
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| Software and algorithms | ||
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/ Salzberg, 2012) index.shtml |
| Prism 8 | GraphPad | https://graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij |
| CPTAC-BRCA2020 data viewer | (Krug et al., 2020) | http://prot-shiny-vm.broadinstitute.org:3838/CPTAC-BRCA2020 |
| Cistrome | (Liu et al., 2011) | http://cistrome.org/ap/ |
| phastCons | (Siepel et al., 2005) | http://compgen.cshl.edu/phast/phastCons-HOWTO.html |
| RStudio | RStudio | Rstudio.com |
| IGV | (Robinson et al., 2011) | http://software.broadinstitute.org/software/igv/download |
| MACS2 | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| ChAsE | (Younesy et al., 2016) | https://chase.cs.univie.ac.at |
| Flow Jo | Flow Jo | https://www.flowjo.com/ |
| Cutadapt v.1 | https://cutadapt.readthedocs.io/en/stable/ | |
| BWA v.0.7 | (Li and Durbin, 2009) | https://sourceforge.net/projects/bio-bwa/ |
| The CLIP tool kit (CTK) | (Shah et al., 2017) | https://zhanglab.c2b2.columbia.edu/index.php/CTK_Documentation |
| Kmplot | (Gyorffy et al., 2010) | https://kmplot.com/analysis/index.php?p=service&cancer=breast |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Davide Ruggero (davide.ruggero@ucsf.edu).
Materials availability
Proprietary material is available upon request from the authors.
Data and code availability
All sequencing data have been deposited in the GEO database under the accession number GEO: GSE173631. Proteomics data have been deposited in the ProteomeXchange: PXD025018. No software was generated for this project.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
All cells were cultured in a 37 °C 5% CO2 humidified incubator. 293T cells were cultured in DMEM medium supplemented with 10% FBS, GlutaMAX (1x) (Gibco), and Penicillin-Streptomycin (100 U/mL) (Gibco). The MCF7 and T47D cancer cell lines were cultured in RPMI-1640 medium supplemented with 10% FBS, GlutaMAX (1x) (Gibco), and Penicillin-Streptomycin (100 U/mL) (Gibco). TamRM and TamR-1 cells were cultured in DMEM Ham’s F12 medium containing 10% FBS, GlutaMAX (1x) (Gibco), Penicillin-Streptomycin (100 U/mL) (Gibco) and 1 μM of 4-Hydroxytamoxifen (Sigma). All cell lines were confirmed to be mycoplasma-free by the MycoAlert™ Mycoplasma Detection Kits (Lonza).
Xenograft and orthotopic mouse models
Eight-week-old NOD scid gamma (NSG) female mice (Bred by UCSF mouse facility) were used for breast xenograft experiments. 60-day slow-release 17β-Estradiol pellets (0.72mg, Innovative Research of America) were first implanted two days before tumor injection. To prepare cancer cells for injection, we briefly trypsinized adherent cancer cells, quenched them with 10% FBS RPMI media, and resuspended them in 1X PBS. Cells were pelleted again and mixed with Matrigel matrix (Corning) on ice. The Matrigel cell suspension was transferred into a 1 mL syringe and remained on ice until the time of implantation. 200 μl of cells (4 × 106 for MCF7, 8 × 106 for T47D and TamR-1) were then injected subcutaneously and tumor volumes were measured twice per week. For orthotopic model with STF-083010 treatment, 5 × 106 MCF7 cells were injected into the mammary fat pad of the NSG mice. When tumors reach 150 mm3, mice were randomly separated into two groups, and dosed with 30 mg/kg of STF-083010 once per week. Tumor volumes were measured every two weeks. For orthotopic model with Zotatifin treatment, 1mg/kg of Zotatifin were dosed every 2–3 days intraperitoneally.
Human samples
Human female breast cancer tumor and normal adjacent tissue samples were collected at the Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University. Patients are aged from 28 to 76. All analyses of human data were carried out in compliance with the relevant ethical regulations.
METHOD DETAILS
Generating ERα RBDmut stable cell lines.
For generating ERα WT and RBDmut stable cell lines, 3 × FLAG-tagged ERα coding sequence with or without the mutations (259RRGG > 259AAAA) was first cloned into the BamHI and ECoRI sites of the pWPXLd plasmid (Addgene plasmid #12258). The constructs were than packaged using the lentiviral packaging system by using PolyFect (Qiagen) and Opti-MEM (Invitrogen) to transfect the constructs with packaging plasmids into 293T cells. Virus was harvested 48 hours post-transfection and passed through a 0.45 mm filter. Transduced cells were selected by treatment with 2 mg/mL puromycin for 2–3 days. The endogenous ERα was then removed using Cas9-guide RNA ribonucleoprotein (RNP) complexes targeting the introns of ESR1 gene. sgRNAs targeting human ESR1 were designed using the Zhang Lab design tool (crispr.mit.edu). Chemically modified synthetic sgRNAs were purchased from Synthego (Menlo Park, CA, USA) and Cas9-NLS purified protein was from the QB3 MacroLab (UC Berkeley, CA, USA). Cas9 RNP was prepared immediately prior to nucleofection by incubating Cas9 protein with sgRNA at 1:1.3 molar ratio in 20 mM HEPES (pH 7.5), 150 mM KCl, 1 mM MgCl2, 10% glycerol and 1 mM TCEP at 37°C for 10 min. Cells were dissociated using trypsin, pelleted by centrifugation, and washed once with D-PBS. Nucleofection of human MCF7 cell line was performed using Amaxa Cell Line Nucleofector Kit V (Lonza, Allendale, NJ, USA) and program P-020 on an Amaxa Nucleofector II system, T47D cells was performed with X-005, and TamR-1 cells with E-014 programs. Each nucleofection reaction consisted of ~5×105 cells in 50 μL of nucleofection reagent mixed with two distinct 10 μl RNP mixtures containing two sgRNAs (to allow specific deletion of first exon ESR1 coding sequence). 4 days after nucleofection, cells were cultured into 96-well plates with 1 cell per well for single clone selection. The expression of ERα RBDmut and the removal of endogenous ERα were confirmed by sequencing and western blot.
For sample (e.g. western blot lysate) collection, MCF7 and T47D cells harboring WT or RBDmut ERα were plate on cell culture dishes with a 20% confluency, and maintained in the dishes for 48 h, when cell reaches approximately 90% confluency. Cells were washed twice with ice-cold PBS, scrapped off the dish in PBS, and palleted for further processes. For Zotafitin treatment, WT or RBDmut cells were counted, plated equal amount on cell culture dishes, and maintained in the dishes for 40–48 h until cell reaches 80–90% confluency and then replaced with fresh media containing Zotafitin.
sgRNA and siRNA-mediated knockdown
For stable knockdown using CRISPR sgRNAs, sgRNAs were first cloned into pLG15 vector (Gilbert lab) using Blp1 and BstX1 sites. The constructs were than packaged using the lentiviral packaging system by using PolyFect (Qiagen) and Opti-MEM (Invitrogen) to transfect the constructs with packaging plasmids into 293T cells. Virus was harvested 48 hours post-transfection and passed through a 0.45 mm filter. Transduced cells were selected by treatment with 2 mg/mL puromycin for 2–3 days.
For transient knockdown of target genes, siRNAs (Qiagen) were used: siESR1: Hs_ESR1_8 FlexiTube siRNA; siEIF4G2: FlexiTube GeneSolution GS1982 for EIF4G2. 20nM (final concentration) of each siRNA was transfected into 1×105 cancer cells using Hiperfect (Qiagen) per the manufacturer’s protocol. Cells were harvested 48–72 hours post-transfection. Knockdown of target genes was assessed by qRT-PCR as described below.
RNA Isolation and quantitative RT-PCR
RNA was isolated using TRIzol (Invitrogen) purification with PureLink RNA Mini Kit (Thermo Fisher, 12183018), or by RNeasy Mini Kit (Qiagen). RNAs were converted into cDNAs using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). cDNA samples were diluted 1:10 and 1 μl of template was used in a PowerUP SYBR Green master mix reaction run on an Applied Biosystems QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher). For measuring XBP1 splicing, RNAs were purified using RNeasy Mini Kit (Qiagen), converted into cDNAs using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). cDNA samples were diluted 1:10, and amplified using primers F: 5’-TTACGAGAGAAAACTCATGGC-3’. R: 5’-GGGTCCAAGTTGTCCAGAATGC-3’ with RT-PCR. PCR products were run on 2% TAE gels.
Cancer cell proliferation
2,000 cells per well of MCF7 and T47D with and without the RBD mutation of ERα were plated in 96-well plates. Cells were fixed every 24 hours by gentle addition of 100 ml of 40% trichloroacetic acid (TCA) (w/v) (final concentration of 10% TCA) per well. Plates were incubated for 1 hour at 4°C, washed five times with distilled water, and stained with 100 ml of sulforhodamine B (SRB) solution 0.4% (w/v) in 1% acetic acid for 1 hour at room temperature. Unbound dye was removed by washing plates five times with 1% acetic acid. Bound stain was solubilized by the addition of 100 ml of 10 mM tris base. Absorbance was determined on an automated plate reader (96-well microtiter) at 492 nm. In addition, CellTiter-Glo Luminescent Cell Viability Assay (Promega, WI, USA) was performed following manufacturer’s instructions with luminescence measurements made using a Glomax 96-well plate luminometer (Promega). Proliferation data were generated by first normalizing luminescence intensity in each well to the wells with ERα WT cells, and normalized luminescence data was plotted (± SD) from at least three independent experiments. Unpaired t test was used to test for significant variations.
Co-immunoprecipitation and TMT-labelled quantitative mass spectrometry
MCF7 cells were collected by scraping, then centrifuged to pellet. The pellets were then resuspended in hypotonic buffer (20 mM Tris-HCl pH 7.4, 10 mM KCl, 5 mM MgCl2 and 1X protease inhibitors), incubated on ice for 15 minutes, supplied with 0.5% IGEPAL CA-630, homogenized for 10 strokes and spun for 10 minutes at 2000 × g at 4°C. The supernatant was collected (cytosolic fraction). Immunoprecipitation of ERα was carried out using anti-ERα and rabbit IgG antibodies with dynabeads (Invitrogen), all according to the manufacturer’s protocol. The cytosolic lysates were then incubated with antibody-conjugated beads with end-over-end rotation at 4°C overnight. The beads were then washed three times with ice-cold hypotonic buffer containing 0.5% IGEPAL CA-630, and then three times with ice-cold PBS. Proteins were eluted from beads with 1X LDS loading dye and separated on TGX Stain-free protein gels (Bio-Rad), allowing the bromophenol blue marker to reach 1 cm inside the gel. Gel was stained using ProtoBlue Safe Colloidal Coomassie Blue G-250 stain. The upper portion of the lanes containing the proteins was excised and digested in-gel with trypsin as described previously (Rosenfeld et al., 1992). The extracted digests were vacuum-evaporated and dried samples were labeled according TMT 6-plex kit instructions (ThermoFisher Scientific), with minor modifications. Shortly, peptides were resuspended in 4 μl of 50 mM triethyl ammonium bicarbonate buffer. TMT reagents were resuspended in 41 μl acetonitrile per vial, and 20 μl of this solution were added to the individual samples (Era IP and mock control) to be labelled. After incubating for 1 h at 22°C, reactions were quenched by adding 4 μl 5% hydroxylamine and incubated for additional 15 min. After that, the labelling reactions were combined, partially evaporated to close to 5 μl, diluted in 100 μl 0.1% formic and desalted using a ZipTip C18 (Millipore) as indicated by the manufacturer. Peptides were eluted in 2×7 μl aliquots of 50% MeCN 0.1% formic acid, dried and resuspended in 5 μl 0.1% formic acid for mass spectrometry analysis on a QExactive Plus mass spectrometer (Thermo Scientific) connected to a NanoAcquity™ Ultra Performance UPLC system (Waters). A 15-cm EasySpray C18 column (Thermo Scientific) was used to resolve peptides (60-min 2–30% B gradient with 0.1% formic acid in water as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B, at a flow rate of 300 nl/min). MS was operated in positive mode in data-dependent mode to automatically switch between MS and MS/MS. MS spectra were acquired between 350 and 1500 m/z with a resolution of 70000. For each MS spectrum, the top 10 ions with a charge state of 2+ or higher were selected with an isolation window of 1 m/z. Precursor ions were fragmented by HCD using stepped relative collision energies of 25, 35 and 40 in order to ensure efficient generation of sequence ions as well as TMT reporter ions. MS/MS spectra were acquired in centroid mode with resolution 17500 from m/z=100. A dynamic exclusion window was applied which prevented the same m/z from being selected for 10s after its acquisition. Peak lists were generated using PAVA in-house software (Guan et al., 2011). All generated peak lists were searched against the human subset of the SwissProt database, using Protein Prospector (2015.12.1 release), using Protein Prospector (Clauser et al., 1999) with the following parameters: Enzyme specificity was set as Trypsin, and up to 2 missed cleavages per peptide were allowed. Carbamidomethylation of cysteine residues, and TMT labeling of lysine residues and N-terminus of the protein were allowed as fixed modifications. N-acetylation of the N-terminus of the protein, loss of protein N-terminal methionine, pyroglutamate formation from of peptide N-terminal glutamines, oxidation of methionine were allowed as variable modifications. Mass tolerance was 10 ppm in MS and 30 ppm in MS/MS. The false positive rate was estimated by searching the data using a concatenated database which contains the original SwissProt database, as well as a version of each original entry where the sequence has been randomized. A 1% FDR was permitted at the protein and peptide level. For quantitation only unique peptides were considered; peptides common to several proteins were not used for quantitative analysis. Relative quantization of peptide abundance was performed via calculation of the intensity of reporter ions corresponding to the different TMT labels, present in MS/MS spectra. Intensities were determined by Protein Prospector. Summed intensity on each TMT channel for all identified spectra were used to normalize individual intensity values. Relative abundances were calculated as ratios of the intensities in ERα IP channel vs the channel corresponding to mock (IgG) pulldown. For total protein relative levels, peptide ratios were aggregated to the protein levels using median values of the log2 ratios.
Co-immunoprecipitation of RtcB
Co-immunoprecipitation of ERα and RtcB was performed using lysate prepared from MCF7 cells. Cells were washed with ice-cold 1X PBS, collected by scraping, then centrifuged to pellet. Cell pellet was resuspended in hypotonic buffer (20 mM Tris-HCl pH 7.4, 10 mM KCl, 5 mM MgCl2 and 1X protease inhibitors), incubated on ice for 15 minutes, supplied with 0.5% IGEPAL CA-630, homogenized for 10 strokes and spun for 10 minutes at 2000 × g at 4°C. The supernatant was collected (cytosolic fraction). ERα- or IgG-conjugated dynabeads were added to lysate and incubated for overnight at 4°C with end-over-end rotation. Beads were then washed three times with ice-cold hypotonic buffer containing 0.5% IGEPAL CA-630. Proteins were eluted by resuspending beads in loading buffer (1x NuPAGE LDS loading buffer, 50mM DTT) and incubating for 5 minutes at 95°C. The presence of RtcB in input and immunoprecipitated fractions was assessed by western blot as described below.
Oligo(dT) pull down of RNA-associated proteins
20 mg/mL Oligo(dT)-cellulose beads (Sigma, O3131, or comparable brands with >40 A260 units per gram polyA RNA binding capacity) were swelled in wash buffer [20 mM Tris-HCl pH 7.4, 250 mM NaCl, 10 mM KCl, 5 mM MgCl2 RNasin (promega)] overnight at 4°C with end-over-end rotation. Cell lysates from MCF7, T47D and TamR cells were washed twice with ice-cold PBS and scrape into 1 mL ice-cold lysis buffer [20 mM Tris-HCl pH 7.4, 250 mM NaCl, 10 mM KCl, 5 mM MgCl2, 0.1% Triton-X, RNasin (promega) and 1X protease inhibitors]. Samples were incubated on ice for 10 min and lysed with ten strokes of a chilled dounce homogenizer. Lysates were spun down at 14,000 xg at 4°C, and supernatant were collected. Protein concentrations were determined using Braford Assay (Bio-Rad), and lysate containing equal amount of proteins were added to the swelled Oligo(dT) beads and samples were end-over-end rotated for 2 h at 4°C. Samples were spun down at 2,500 xg for 2 min, and beads were subsequently washed 5x with wash buffer, and samples were eluted from the beads with 2x Laemmili loading buffer with 10 mM DTT, boiled at 95°C for 10 min, with votexting every 1 min. Samples were analyzed by western blot.
Western blotting
Cell lysates were prepared by lysing cells in ice-cold RIPA buffer (25mM Tris-HCl pH 7.6, 0.15M NaCl, 1% IGEPAL CA-630, 1% sodium deoxycholate, 0.1% SDS) containing 1X protease inhibitors (Roche). Lysate was cleared by centrifugation at 14,000 × g for 15 min at 4°C. Samples were boiled in 1X LDS loading buffer (Invitrogen) and 100mM DTT. Proteins were separated by SDS-PAGE using 4%–12% Bis-Tris NuPAGE gels, transferred to 0.2 μm Nitrocellulose Membrane (Thermo Scientific). Membranes were blocked with 5% nonfat milk and probed using target-specific antibodies in 2.5% BSA in PBST. Bound antibodies were detected using horseradish peroxidase–conjugated secondary antibodies and ECL Western Blotting Substrate (Thermo Scientific), according to the manufacturer’s instructions.
ERα HITS-CLIP
HITS-CLIP was performed as previously described. Biological replicates of MCF7 cells were crosslinked with 400 mJ/cm2 254nm UV. Crosslinked cells were then lysed on ice in low salt buffer (1X PBS, 0.1% SDS, 0.5% sodium deoxycholate, 0.5% IGEPAL CA-630) supplemented with Recombinant RNasin Ribonuclease Inhibitor (Promega) and 1X protease inhibitors (Roche). Lysate was then treated with DNase I (Promega) at 37°C for 5 minutes. Lysate was then treated with RNase A (low dilution: 1:1500; high dilution: 1:30) (Thermo Scientific) and incubated at 37°C for 5 minutes. Lysate was clarified by spinning at 20,000 × g at 4°C for 20 minutes. The clarified lysate was transferred to protein A dynabeads (Invitrogen) conjugated to anti-ERα antibody (Santa Cruz sc-542) and rotated end-over-end at 4°C for 3 hours. The beads were washed twice with low salt buffer, high salt buffer (5X PBS, 0.1% SDS, 0.5% sodium deoxycholate, 0.5% IGEPAL CA-630), and PNK buffer (50mM Tris pH 7.4, 10mM MgCl2, 0.5% IGEPAL CA-630), respectively. The immunoprecipitated protein-RNA complexes were first dephosphorylated on-bead with CIP (NEB), washed once with PNK buffer, once with PNK + EGTA buffer (50mM Tris pH 7.4, 20mM EGTA, 0.5% IGEPAL CA-630), lastly twice with PNK buffer. T4 RNA ligase (Thermo Scientific) was used to ligate a 5’ RNA linker [32P-labeled RL3: 5’(-OH)-GUGUCAGUCACUUCCAGCGG-3’-(3InvdT)] to the samples on-bead overnight at 16°C, followed by 1X low salt buffer, 1X high salt buffer, and 2X PNK buffer washings. The samples were then phosphorylated on-bead using PNK (NEB), followed by 2X PNK washes, and then eluted by 70°C heating for 10 minutes in 1X NuPAGE LDS loading buffer at 1,000rpm. The eluates were separated on a 4%–12% Bis-Tris NuPAGE gel (Invitrogen), transferred to Whatman BA85 nitrocellulose (Sigma), and exposed to film (24–72 h) to determine the migration of the RNA-protein complexes, and the relevant region was cut from the membrane for library preparation and cut into small pieces on a whatman paper and transferred to Eppendorf tubes. The RNA from the membrane was then isolated by digesting with 200 μL of proteinase K solution [4mg/mL proteinase K (Invitrogen), 100mM Tris pH 7.5, 50mM NaCl, 10mM EDTA] and incubating at 37°C for 20 minutes at 1,000rpm. 200 μL PK-urea solution (100mM Tris pH 7.5, 50mM NaCl, 10mM EDTA, 7M urea) were subsequently added and samples were incubated at 37°C for another 20 minutes at 1,000rpm. Lastly, 400 μL acid phenol (Sigma) and 130 mL chloroform (Sigma) were added and samples were continuously incubated at 37°C for 20 minutes at 1,000rpm. Tubes were vortexed and spun, and RNA was precipitated from the aqueous layer. The RNA pellet was washed and ligated to the RL5D linker [5’(-OH)-AGGGAGGACGAUGCGGr(N)r(N)r(N)r(N)G-3’(-OH)] using T4 RNA ligase and incubating overnight at 16°C. The ligation reaction was then treated with DNase I (Promega), extracted with acid phenol chloroform, and the aqueous layer was precipitated. The RNA was purified and cDNA was synthesized using Superscript III reverse transcriptase (Invitrogen) with customized DP3 primer (5’-CCGCTGGAAGTGACTGACAC-3’), using 50°C for 45 minutes, 55°C for 15 minutes, and 90°C for 5 minutes program. The first round of PCR was then carried out using DP5 (5’-AGGGAGGACGATGCGG-3’) and DP3 primers with Accuprime Pfx Supermix (Invitrogen) using the following cycle conditions: 1. 95°C 2min, 2. 95°C 20 s, 3. 58°C 30 s, 4. 68°C 20 s, 5. 68°C 5min, with Steps 2–4 repeated 24 times. The PCR products were gel purified using 10% Urea-TBE PAGE with Qiagen Gel extraction kit. A second PCR step was then performed to attach Illumina flowcell adaptor sequences using DSFP5 (5’-AATGATACGGCGACCACCGACTATGGATACTTAGTCAGGGAGGACGATGCGG-3’) and DSFP3 (5’-CAAGCAGAAGACGGCATACGACCGCTGGAAGTGACTGACAC-3’) primers, with the following cycle conditions: 1. 95°C 2min, 2. 95°C 20 s, 3. 58°C 30 s, 4. 68°C 40 s, 5. 68°C 5min, with Steps 2–4 repeated 6 times. Lastly, the PCR products were gel purified using a 2% metaphor agarose (Lonza) gel. The resulting libraries were sequenced using SSP1 primer (5’-CTATGGATACTTAGTCAGGGAGGACGATGCGG-3’) as the custom sequencing primer.
CLIP-seq processing and analysis
To identify ERα-bound sites from the ERα CLIP-seq data, first the adaptor sequences were removed and quality trimming was performed Cutadapt. Reads were then mapped to the human genome (build hg19) using BWA (v.0.7.) with the default parameters. The ERα binding sites were then identified using the CTK package. For our analysis, the CLIP-derived ERα binding sites from all of the samples were combined to create the list of ERα binding sites.
For the conservation analysis, the PhastCons (Siepel et al., 2005) scores of CLIP-derived ERα binding sites were calculated and plotted using the Conservation Plot tool of Cistrome (http://cistrome.org/ap/).
Thermal shift assay
The XBP1 3’UTR DNA was synthesized by IDT, and PCR amplified with a 5’ primer containing a T7 promoter sequence. The XBP1 3’UTR mRNA fragment (1–400 nt) were synthesized using MEGAscript T7 in vitro transcription kit (Thermo Fisher) followed by Turbo DNase digestion to remove residue DNA. Purified GST-ERα protein was resuspended in RNase-free water to required concentrations, and 5 μL of the proteins were added into each well of a 96-well qPCR plate. XBP1 3’UTR mRNAs were diluted using RNase-free water, and 5 μL of them or RNase-free water (control) were added to each well containing different concentrations of GST-ERα protein. 2.5 μL of 50X SYPRO Orange Fluorescent Dye (Thermo Fisher) were added to each well to make a final concentration of 5X. Protein melt curve was measured using CFX RT-PCR detection systems (Bio-Rad).
Sucrose gradient fractionation
MCF7 cells harboring WT or RBDmut ERα were counted, plated equal number of cells on 15-cm cell culture dish at the concentration of approximately 20–25%, maintained for 48 h until cells became 90% confluent. Cell were then treated with 0.1 mg/ml Cycloheximide for 5 min, prior to lysing in 300 μl of lysis buffer (20 mM Tris pH 7.5, 200 mM NaCl, 15 mM MgCl2, 1 mM DTT, 1% Triton X-100, 0.1 mg/ml cycloheximide, 200 U/ml RNasin (Promega)). Nuclei and membrane debris were then removed by centrifuging at 10,000 g, 5 min. The lysate was loaded onto a sucrose gradient (10–50% sucrose(w/v) made with Gradient Master™ (BIOCOMP), 20 mM Tris pH7.5, 100 mM NaCl, 15 mM MgCl2) and centrifuged in a SW41Ti rotor (Beckman) for 2.5 h at 38,000 rpm at 4°C. Fractions were collected by density gradient fractionation system Piston Gradient Fractionator ™ (BIOCOMP).
Reporter assay
The 5’UTR and 3’UTR of eIF4G2 mRNA were cloned into the pGL3-promoter vector, before and after the Firefly reporter coding sequence, respectively. For deleting the ERα-binding sites in eIF4G2 3’UTR, gBlock DNA of eIF4G2 3’UTR with ERα-binding sites deleted were ordered from IDT. Details of the ERα-bound eIF4G2 3’UTR regions are uploaded to GEO, which can be accessed with the accession code: GSM5272770. MCF7 cells were transfected with 200 ng of pGL3-eIF4G2 constructs and 20 ng of pRL (Renilla luciferase) plasmid using Amaxa Cell Line Nucleofector Kit V (Lonza, Allendale, NJ, USA) and program P-020 on an Amaxa Nucleofector II system. Cells were collected 48 h post-transfection and half of the cells were assayed using Dual luciferase kit (Promega), the other half were proceeded for TRIzol (Invitrogen) purification of RNA. Purified RNAs were treated twice with Turbo DNase to remove Firefly and Renilla plasmids, and reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The Firefly luciferase activity was normalized to Renilla activity, and further normalized to Firefly and Renilla luciferase RNA levels quantified by RT-qPCR.
Cloning of pooled CRISPRi library sgRNAs targeting ERα-bound mRNAs
To generate customized CRISPRi sub-library targeting ERα-bound mRNAs, sgRNA oligo pool was first generated with Twist Bioscience (Table S4), and was used as a template for PCR amplifications using Phusion High-Fidelity DNA Polymerase (Thermo Fisher), and the following cycle condition: 1. 98°C 30 s, 2. 98°C 15 s, 3. 56°C 15 s, 4. 72°C 15 s, 5. 72°C 10 min, with Steps 2–4 repeated 15 times. PCR products were purified using Qiagen’s MinElute Reaction Cleanup Kit. The expected PCR product size is 84bp, which can be determined by TBE gel. 5 μg of pLG1 library vector (Gilbert Lab) and 1 μg of PCR products were digested using FastDigest BstXI and BlpI (1 h for vector, 4 h for PCR products) at 37°C, Digested pLG1 vector was gel purified using Gel extraction kit (Qiagen). Digested PCR products (inserts) were run on 10% acrylamide gel, crushed using 18.5-gauge needle and by spinning down through a 0.5 mL nonstick tube with a hole to 1.5 mL tube. 200 μL water was added to the gel pieces and samples were incubated for 1 h at 70°C, followed by spinning down for 3 min at 20,000 xg through Costar Spin-X columns. Elutes free of gel debris were proceeded to isopropanol precipitation. Purified vector and inserts (1:1 ratio) were ligated using T4 ligase at 16°C for 16 h, followed by purifications with ethanol precipitation. Transformations were performed using electroporation with Mega X cells (1 μL of ligation with 20 μL of cells), followed by recovery at 37°C for 1 h, and culture in 100 mL LB overnight at 37°C. Plasmids were purified using Qiagen Midiprep kit.
CRISPRi screen
For CRISPRi screen, MCF7 cells were first stably overexpressed doxycycline-inducible dCas9, and cultured in RPMI-1640 medium with Tet-free serum. MCF7-dCas9 cells were then infected with CRISPRi sub-library plasmids followed by 48 h puromycin selection. The efficient infection of the library can be determined by flow cytometry. After the selection, cells were treated with doxycycline for 4 days to induce the dCas9 expression, and the Day 0 timepoint was collected snap frozen at −80°C, and cell numbers were determined by flow cytometry. Cells were continuously cultured in the RPMI-1640 medium with Tet-free serum containing doxycycline for required days, and for each timepoints cell numbers were counted by flow cytometry. Cells at each timepoints were lysed and genomic DNAs (gDNAs) were harvested using NucleoSpin Blood XL kit following manufactural instructions. Purified gDNAs were digested with SbfI-HF (NEB) at the concentration of 400 U per 1 mg of gDNA, followed by purifications. gDNAs from each timepoints and replicates were amplified using Q5 PCR MasterMix (NEB) with the following cycle conditions: 1. 98°C 30 s, 2. 98°C 10 s, 3. 65°C 75 s, 4. 65°C 5 min, with Steps 2–3 repeated 22 times. PCR products were purified and sequenced.
ChIP-qPCR and sequencing
MCF7 cells with ERα WT and RBDmut were grown in complete RPMI-1640 media to 80–90% confluency. The media were removed and replaced with media containing 1% formaldehyde and crosslinked for 8 min at 37°C, followed by quenching with Glycine at a final concentration of 0.2 M. The cells were washed with ice-cold PBS twice and lysed in 10 mL of LB1 buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% IGEPAL CA-630, and 0.25% Triton X-100) for 10 min at 4°C with end-over-end rotation. Cells were pelleted, resuspended in 10 mL of LB2 buffer (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA), and mixed at 4°C for 5 min with end-over-end rotation. Cells were then pelleted and resuspended in 300 mL of LB3 buffer (10 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-deoxycholate, and 0.5% N-lauroylsarcosine) and sonicated in Diagenode Bioruptor. Triton X-100 was added to the lysate at a final concentration of 1%, and lysate was centrifuged for 10 min at 20,000 xg. The supernatant was then incubated with 100 μL of protein A dynabeads (Invitrogen) prebound with FLAG M2 (Sigma) antibody, and immunoprecipitation (IP) was conducted overnight at 4°C with end-over-end rotation.
Beads were washed 6 times in RIPA buffer (50 mM HEPES pH 7.6, 1 mM EDTA, 0.7% Na deoxycholate, 1% IGEPAL CA-630 and 0.5 M LiCl), followed by 1-time wash with TE buffer. 100 μL of elution buffer (% SDS, 0.1M NaHCO3) were added to the beads, and incubated overnight at 65°C to reverse crosslink. 170 μL of elution buffer was added to 30 μL input samples. Samples were then added with 200 μL of TE, treated with RNase A for 1 h at 37°C, followed by Proteinase K for 2 h at 55°C. Samples were purified with Phenol: Chloroform: Isoamyl alcohol and ethanol precipitations. DNA pallets were washed once with 75% ice-cold ethanol and resuspended in 50 μL of 10mM Tris HCl pH 8.0. Samples can be assessed by qPCR (ChIP-qPCR) or processed for sequencing library preparation (ChIP-seq) following the manufactural instructions of NEBNext Ultra II DNA library Prep Kit (NEB).
ChIP-seq analyses
Sequences generated by the Illumina Genome Analyzer were processed by the Illumina analysis pipeline and aligned to the Genome Reference Consortium Human Build 37 (GRCh37), using Bowtie2 (Langmead and Salzberg, 2012). Enriched regions of the genome were determined using MACS2 peak caller (Feng et al., 2012) by comparing the FLAG-ERα ChIP samples with the input controls. Further analyses were performed with Cistrome (Liu et al., 2011).
For differential binding analysis, the peaks called from each sample were merged using samtools and the unique records were stored as a union bed file. The aligned reads were then intersected with the union of the peaks. The number of intersections for each peak were counted and a counts matrix was generated. The count matrix was then analysed with two-way comparison using DESeq2 (Love et al., 2014) for statistical testing comparing RBDmut with WT samples, normalized to their input controls. Promoters or an enhancers annotations were performed with GenomicRanges (Lawrence et al., 2013) and annotatr package (Cavalcante and Sartor, 2017).
Immunohistochemistry (IHC) staining of breast cancer tissue microarray (TMA)
Tissue microarray slides (US Biomax, BC081116d, patient information including pathology grade, TNM, clinical stage, and Her2/ER/PR/Ki67 IHC results can be found on www.biomax.us/tissue-arrays/Breast/BC081116d) were deparaffinized by baking slides at 60 °C for 10 min and incubated in xylene for 20 min, twice. The slides were rehydrated in series of ethanol solutions (100% ethanol, 95% ethanol, and 70% ethanol) and distilled water. Antigen retrieval was performed by boiling slides for 20 min in a citrate-based antigen unmasking solution (Vector labs, H3300). After cooling down to room temperature, endogenous peroxidase activities were quenched by 1% H2O2 in distilled water for 20 min. After three washes in TT buffer (500 mM NaCl, 10 mM Trizma, and 0.05% Tween-20), slides were blocked in 2% normal goat serum and 4% BSA in TT buffer for 1 h. Next, tissue slides were incubated with various primary antibodies at 4 °C overnight. After three washes in TT buffer, biotinylated secondary antibody was added onto these slides for 1 h, following by 1 h treatment of the Vectastain Elite ABC reagents (Vector Labs, PK-6100). After three TT washes, the slides were processed with DAB peroxidase substrate kit (Vector Labs, SK-4100), and hematoxylin solutions for immunohistochemistry staining, dehydrated in a standard ethanol/xylenes series, and mounted in 75% v/v Permount (Fischer Scientific, SP15–500) in xylenes.
Ethical compliance
All experiments involving live vertebrates performed at UCSF were done in compliance with ethical regulations approved by the UCSF IACUC committee. Protocols for human sample collection and analysis were approved by Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, and all analyses of human data were carried out in compliance with the relevant ethical regulations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Differences in gene expression as measured by qRT-PCR (Figures 5B, S4C and S4G), reporter activity normalized to qRT-PCR results (Figures 5C, 5D), significance differences between samples reported for each timepoint of the cell proliferation assays (Figures 2A, 2B, 3D and 4F), and significance reported for protein and cDNA abundance quantifications from western blots or agarose gels were calculated with unpaired t test (Figures 1E, 4E, 5E, S4E). Significance reported for xenograft tumor injections (Figures 2A, 2B, and 5G), unpaired t test was used to calculate the volume differences at each timepoints. Graphs were generated using Graphpad Prism software. Significance reported for scatterplots between HITS-CLIP replicates were calculated with Pearson’s Correlation Coefficient Assay (Figure S1B). Significant differences between samples on cell cycle accumulation and cell apoptosis were determined by flow cytometry and calculated based on unpaired t test (Figures 4H, 5H, 5I, 5J, S5I, S5J, S5K). For all figures: otherwise noted, *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
(A) Gene ontology for the mass spectrometry analysis of ERα-interacting proteins in the cytoplasmic lysate of MCF7 breast cancer cells.
(B) Representative western blots for ERα bound to Oligo(dT) beads in cytoplasmic and soluble nuclear lysate of an ER+ human breast tumor with its paired normal adjacent tissue.
(C) Representative western blots for GR, PR, AR, RXRα and PPARγ bound to Oligo(dT) beads in cytoplasmic and soluble nuclear MCF7 lysate.
(D) Associations between ERα HITS-CLIP replicates shown as correlations calculated on peak levels. Every point represents a peak, and x and y axes show the counts in that peak for the given replicates. Correlation scores are calculated with Pearson r.
(E) Integrative Genomics Viewer (IGV) view of the three replicates of ERα HITS-CLIP analysis showing ERα binding to XBP1, MCL1 and eIF4G2 mRNAs.
(F) Pie charts presenting the distribution of ERα-RNA interactions by genome region.
(G) Gene ontology of ERα-bound genes were analyzed by DAVID and presented utilizing Cytoscape tool. For each ontology term the P values was calculated.
(H) The presence of ERα-bound motif in the 3’UTR of XBP1, MCL1 and eIF4G2 mRNAs in indicated mammal species.
(I) Phastcons conservation scores of HuR-bound RNA regions compared with their adjacent regions. The window represents ±0.5 kb regions from the center of the HuR RNA-binding events.
(A) Left: SYBR Gold stain of Trizol purified RNAs pulled down by GST-purified ERα proteins on the top, and Coomassie stain of GST-purified ERα on the bottom. Right: Quantifications of relative amounts of RNA bound to each GST-purified ERα proteins are shown.
(B) A schematic of the strategy for generating MCF7 cells with WT and RBDmut ERα.
(C) Fluorescent microscope image showing the subcellular localization of GFP-ERα (WT and RBDmut), DAPI was used to stain the nucleus.
(D) Representative western blots for FLAG-ERα from chromatin-bound lysate fractions of MCF7 cells with WT and RBDmut ERα. Histone H3 is used as an internal control. N=3 independent experiments.
(E) Heat map showing binding peak intensity of ERα chromatin binding events in MCF7 cells with WT and RBDmut ERα. The window represents ±2.5 kb regions from the center of the binding events.
(F) Volcano plot showing the difference between ERα-bound chromatin locus on overlapped binding sites in MCF7 cells with WT and RBDmut ERα. Y-axis represents −log10 adjust P values (FDR), and X-axis represents log2 fold change (FC). Total 23193 WT and RBDmut ERα DNA-binding events are shown, 238 hits down in RBDmut are marked with blue, and 23 hits up in RBDmut are marked with orange.
(G) Estrogen responsive element (ERE) motif enriched within ERα-binding sites from FLAG ChIP-seq analysis with MCF7 cells with WT and RBDmut ERα.
(H) FLAG ChIP-qPCR in MCF7 cells with WT and RBDmut ERα on GREB1 and TFF1 promoter, a random genomic region is used as a negative control. IgG ChIP-qPCR is used as a negative control for IP. N=3 biological replicates. Two-sided t-test. All values represent the mean + SD.
(I) Venn diagram showing the overlap of ERα chromatin-bound and RNA-bound genes.
(A) Growth phenotype analysis of the CRISPRi screen on Day 9 and Day 5 compared with Day 0 shown as a volcano plot. The average growth phenotype for each gene with the 3 most active sgRNAs is shown in X-axis, and the significance of the growth phenotype is presented in Y-axis as log10(Mann-Whitney p-values). The Dash lines mark the threshold of significance in this study: |Growth phenotype × log10(Mann-Whitney p-values) | > 1. Genes that pass the threshold are shown with genes that favor the cell growths in blue, while those that inhibit cell growth are in orange.
(B) The average growth phenotype for XBP1, EIF4G2 and MCL1 genes with the 3 most active sgRNAs on Day 5, 9 and 14 are shown. Adjusted P values (Padj) of all 5 sgRNAs were shown.
(A) RNA electrophoretic mobility shift assay (EMSA) of the XBP1 3’UTR mRNA using GST-tagged ERα. Lower arrow: free XBP1 mRNA, upper arrow: XBP1 mRNA bound by ERα.
(B) Representative images for RT-PCR analysis of XBP1 splicing in T47D cells harboring WT and RBD-mutated ERα respectively, upon DMSO or tunicamycin treatment for 4 h. The upper band indicates unspliced XBP1 mRNA and the lower band indicates spliced XBP1 mRNA. β-actin mRNA (ACTB) is used as an internal control.
(C) Representative western blots for RtcB pulled down by GST-purified ERα protein constructs, with Coomassie stain of ERα protein constructs at the bottom.
(D) Relative cell apoptosis as shown by Annexin V+ populations of TamR-1 cells treated with 25 μM of IRE1 inhibitor (IRE1i) for 24 h compared with DMSO, with and without ERα silencing (siESR1).
All experiments are performed with N=3 biological replicates.
(A) Representative western blots for eIF4G2, MCL1 and α-tubulin in T47D cells with and without the RBDmut of ERα.
(B) Relative mRNA expressions of eIF4G2 and MCL1 measured by quantitative PCR (qPCR) analysis in T47D cells with and without the RBDmut of ERα.
(C) Representative western blots for eIF4G2, MCL1 and α-tubulin in MCF7 cells with and without the RBDmut of ERα, treated with proteasome inhibitor MG132 for 4 h.
(D) qPCR with reverse transcription (RT-qPCR) analysis of α-tubulin (TUBA) mRNA levels in fractions, percentages of α-tubulin mRNA distributed in each fraction against total α-tubulin mRNA are shown.
(E) IGV views of ERα HITS-CLIP analysis showing lack of ERα bindings on BCL2 and BCL2L1 mRNAs.
All values represent the mean + SD. Two-sided t test.
(A) Representative western blots for eIF4G2, MCL1, XBP1s and α-tubulin are shown in 14 freshly collected ERα+ breast cancer tumors (T) and their paired normal adjacent tissues (N).
(B) Representative immunohistochemistry (IHC) staining of XBP1s, eIF4G2 and MCL1 on ER+ breast cancer tissue microarray. The protein expressions were evaluated and scored. No: no expression, Low: low expression, Median: median expression and High: high expression.
(C) Representative western blots for eIF2α phosphorylation (p-eIF2α), eIF2α and α-tubulin in MCF7 cells treated with 4-Hydroxytamoxifen (4-OHT) and Fulvestrant for 24 or 72 hours.
(D) Relative mRNA expressions of eIF4G2 and MCL1 measured by qPCR analysis in MCF7 and TamR-1 cells. </p/> (E) Relative populations of cells in G1 cell cycle phase in TamR-1 cells with and without the RBDmut of ERα under normal cell culture conditions.
(F) Relative populations of cells in G1 cell cycle phase in TamR-1 cells with or without eIF4G2 silencing (siEIF4G2) under normal cell culture conditions.
(G) Relative fold differences of eIF4G2-silenced (siEIF4G2) TamR-1 cells undergo apoptosis (Annexin V+, PI+) comparing to the control (siCtl) cells, under normal cell culture conditions. (H) Relative fold differences of TamR-1 cells undergo apoptosis (Annexin V+, PI+) when treated with MCL1 inhibitor AZD5991 for 24 h comparing to DMSO under normal cell culture conditions.
All experiments are performed with n=3 biological replicates. Two-sided t-test. All values represent the mean + SD.
ERα interacting partners in the cytoplasm were detected by quantitative mass spectrometry using TMT labeling, related to Figure S1A. Unique peptide numbers for each gene are shown along with coverage in percentage. The enrichment for proteins interacting with ERα over IgG were calculated by log2FC (ERα/IgG).
Chromatin regions and matched gene names for ERα-associated RNAs identified by HITS-CLIP are shown, related to Figure 1 and Figure S1. For each binding event, binding intensities (k) are calculated, and k > 10 are used as a cut-off for positive bindings.
ERα-associated chromatin regions (regions with a light blue background are significantly altered between WT and RBDmut), log2 fold change of the binding intensities comparing RBDmut to WT, P values, False discovery rate (FDR), and the annotation of the chromatin regions are shown. Enhancer: enhancer regions; promoter: promoter regions; #N/A: no annotation.
sgRNA target sequences, with their predicted/empirical scores, off-target stringency and oligo sequences ordered from Twist Bioscience for each ERα-bound mRNAs are shown, related to Figure 3A. 5 sgRNAs for each of the1090 mRNAs bound by ERα with binding intensities k > 10 (from Supplementary Table 1) and 117 non-targeting control sgRNAs were employed.
Normalized average growth phenotypes and Mann-Whitney p-values for each ERα-bound mRNAs from two biological replicates of the CRISPRi analysis at Day 14, Day 9 and Day 5 time points are shown respectively, related to Figure 3B. Negative growth phenotype values together with Mann-Whitney p-values at a cut-off of [|log10(P value) × average growth phenotype| > 1] are used to indicate essential mRNAs for the fitness of MCF7 breast cancer cells.
Highlights.
ERα, a transcription factor critical for breast cancer, is an RNA-binding protein.
ERα regulates post-transcriptional expression of stress response genes.
ERα RNA binding activity is important for translational control and XBP1 splicing.
ERα RNA binding activity is critical for tumor growth and therapeutic response.
Acknowledgments
We would like to thank members of the Ruggero Laboratory, especially M. McMahon and X. Pang as well as Dr. Schneider for discussion of the project. M. Barna for reading and critical discussion on the manuscript. We thank L. Gilbert (UCSF) for providing reagents and technical supports on CRISPRi cloning and screen processes; L. Fish (UCSF) for providing technical support on HITS-CLIP experiments; S. Thomas and P.N. Munster (UCSF) for providing the TamRM cell line; B. Yang, Q. He, M. Ying and Z. Zhou (Zhejiang University) for providing reagents and supports; H. Wang, K. Z. Guiley, and V. Stojkovic (UCSF) for technique assistance; G. Giamas (University of Sussex) and J. Stebbing (Imperial College London) for providing GST-ERα constructs; eFFECTOR for providing Zotatifin; Mass Spectrometry was provided by the Mass Spectrometry Resource at UCSF (A.L. Burlingame, Director) supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF) and NIH P41GM103481 and 1S10OD016229; H.G. Nguyen was supported by Department of Defense and Prostate Cancer Foundation Young Investigator and Challenge grants; H. Goodarzi has received research support from the National Institutes of Health (NIH) R01CA24098; Y. Xu was supported by the Damon Runyon Postdoctoral Fellowship (DRG 2288–16) and the start-up Grant at Zhejiang University; D. Ruggero has received research support from the NIH (grants R01CA140456 and R35CA242986) and the American Cancer Society (grant American Cancer Society Research Professor Award).
Footnotes
Declaration of Interests
D.R. is a shareholder of eFFECTOR Therapeutics, Inc., and a member of its scientific advisory board.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Acconcia F, Manavathi B, Mascarenhas J, Talukder AH, Mills G, and Kumar R (2006). An inherent role of integrin-linked kinase-estrogen receptor alpha interaction in cell migration. Cancer Res 66, 11030–11038. [DOI] [PubMed] [Google Scholar]
- Ali S, and Coombes RC (2002). Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer 2, 101–112. [DOI] [PubMed] [Google Scholar]
- Andruska ND, Zheng X, Yang X, Mao C, Cherian MM, Mahapatra L, Helferich WG, and Shapiro DJ (2015). Estrogen receptor alpha inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc Natl Acad Sci U S A 112, 4737–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46, 674–690. [DOI] [PubMed] [Google Scholar]
- Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. (2012). Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. (2005). ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 24, 3470–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, and Berger SL (2017). RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168, 135–149 e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodi S, Moro L, Baj G, Smeriglio M, Di Stefano P, Gippone S, Surico N, Silengo L, Turco E, Tarone G, et al. (2004). p130Cas interacts with estrogen receptor alpha and modulates non-genomic estrogen signaling in breast cancer cells. J Cell Sci 117, 1603–1611. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, and Ron D (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406. [DOI] [PubMed] [Google Scholar]
- Cavalcante RG, and Sartor MA (2017). annotatr: genomic regions in context. Bioinformatics 33, 2381–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, and Cubillos-Ruiz JR (2021). Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer 21, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et al. (2014). XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 508, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauser KR, Baker P, and Burlingame AL (1999). Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 71, 2871–2882. [DOI] [PubMed] [Google Scholar]
- Cook KL, Clarke PA, Parmar J, Hu R, Schwartz-Roberts JL, Abu-Asab M, Warri A, Baumann WT, and Clarke R (2014). Knockdown of estrogen receptor-alpha induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. Faseb J 28, 3891–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S, Roberts AW, Colman PM, and Adams JM (2016). Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends Cancer 2, 443–460. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MP, Barraclough DL, Stewart C, Joyce KA, Eccles RM, Barraclough R, Rudland PS, and Sibson DR (2008). Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int J Cancer 123, 85–88. [DOI] [PubMed] [Google Scholar]
- de la Parra C, Ernlund A, Alard A, Ruggles K, Ueberheide B, and Schneider RJ (2018). A widespread alternate form of cap-dependent mRNA translation initiation. Nat Commun 9, 3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly N, Gorman AM, Gupta S, and Samali A (2013). The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci 70, 3493–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, Wang J, Santen RJ, and Yue W (2007). Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res 67, 1352–1360. [DOI] [PubMed] [Google Scholar]
- Feng JX, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nature Protocols 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch RM, Schneider G, Saur D, Scheibel M, and Schmid RM (2007). Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J Biol Chem 282, 22551–22562. [DOI] [PubMed] [Google Scholar]
- Gaillard H, Garcia-Muse T, and Aguilera A (2015). Replication stress and cancer. Nat Rev Cancer 15, 276–289. [DOI] [PubMed] [Google Scholar]
- Geter PA, Ernlund AW, Bakogianni S, Alard A, Arju R, Giashuddin S, Gadi A, Bromberg J, and Schneider RJ (2017). Hyperactive mTOR and MNK1 phosphorylation of eIF4E confer tamoxifen resistance and estrogen independence through selective mRNA translation reprogramming. Gene Dev 31, 2235–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez BP, Riggins RB, Shajahan AN, Klimach U, Wang A, Crawford AC, Zhu Y, Zwart A, Wang M, and Clarke R (2007). Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J 21, 4013–4027. [DOI] [PubMed] [Google Scholar]
- Guan S, Price JC, Prusiner SB, Ghaemmaghami S, and Burlingame AL (2011). A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Mol Cell Proteomics 10, M111 010728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li QY, and Szallasi Z (2010). An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Tr 123, 725–731. [DOI] [PubMed] [Google Scholar]
- Hanker AB, Sudhan DR, and Arteaga CL (2020). Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 37, 496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al. (2012). ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 122, 4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT, and Tew KD (2020). Oxidative Stress in Cancer. Cancer Cell 38, 167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herviou P, Le Bras M, Dumas L, Hieblot C, Gilhodes J, Cioci G, Hugnot JP, Ameadan A, Guillonneau F, Dassi E, et al. (2020). hnRNP H/F drive RNA G-quadruplex-mediated translation linked to genomic instability and therapy resistance in glioblastoma. Nat Commun 11, 2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, and Oakes SA (2015). Proteostasis control by the unfolded protein response. Nat Cell Biol 17, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell SJ, Johnston SR, and Howell A (2004). The use of selective estrogen receptor modulators and selective estrogen receptor down-regulators in breast cancer. Best Pract Res Clin Endocrinol Metab 18, 47–66. [DOI] [PubMed] [Google Scholar]
- Ishmael FT, Fang X, Houser KR, Pearce K, Abdelmohsen K, Zhan M, Gorospe M, and Stellato C (2011). The human glucocorticoid receptor as an RNA-binding protein: global analysis of glucocorticoid receptor-associated transcripts and identification of a target RNA motif. J Immunol 186, 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R, Buchwalter G, De Angelis C, Brown M, and Schiff R (2015). ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 12, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, and Saatcioglu F (2020). Targeting the Unfolded Protein Response in Hormone-Regulated Cancers. Trends Cancer 6, 160–171. [DOI] [PubMed] [Google Scholar]
- Kanakkanthara A, Jeganathan KB, Limzerwala JF, Baker DJ, Hamada M, Nam HJ, van Deursen WH, Hamada N, Naylor RM, Becker NA, et al. (2016). Cyclin A2 is an RNA binding protein that controls Mre11 mRNA translation. Science 353, 1549–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL, and Chandarlapaty S (2018). Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat Rev Cancer 18, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LC, et al. (2020). Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 183, 1436–1456 e1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wu Q, Chambliss KL, Yuhanna IS, Mumby SM, Mineo C, Tall GG, and Shaul PW (2007). Direct interactions with G alpha i and G betagamma mediate nongenomic signaling by estrogen receptor alpha. Mol Endocrinol 21, 1370–1380. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, and O’Malley BW (1999). A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97, 17–27. [DOI] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, and Glimcher LH (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23, 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman N, Gandin V, Svitkin YV, David M, Virgili G, Jaramillo M, Holcik M, Nagar B, Kimchi A, and Sonenberg N (2015). DAP5 associates with eIF2beta and eIF4AI to promote Internal Ribosome Entry Site driven translation. Nucleic Acids Res 43, 3764–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang XN, et al. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–U422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. (2011). Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, and Aebersold R (2016). On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550. [DOI] [PubMed] [Google Scholar]
- Lombardi M, Castoria G, Migliaccio A, Barone MV, Di Stasio R, Ciociola A, Bottero D, Yamaguchi H, Appella E, and Auricchio F (2008). Hormone-dependent nuclear export of estradiol receptor and DNA synthesis in breast cancer cells. J Cell Biol 182, 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Liang FX, and Wang X (2014). A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell 55, 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykkesfeldt AE, Madsen MW, and Briand P (1994). Altered expression of estrogen-regulated genes in a tamoxifen-resistant and ICI 164,384 and ICI 182,780 sensitive human breast cancer cell line, MCF-7/TAMR-1. Cancer Res 54, 1587–1595. [PubMed] [Google Scholar]
- Marash L, Liberman N, Henis-Korenblit S, Sivan G, Reem E, Elroy-Stein O, and Kimchi A (2008). DAP5 promotes cap-independent translation of Bcl-2 and CDK1 to facilitate cell survival during mitosis. Mol Cell 30, 447–459. [DOI] [PubMed] [Google Scholar]
- Mazumder B, Seshadri V, and Fox PL (2003). Translational control by the 3’-UTR: the ends specify the means. Trends Biochem Sci 28, 91–98. [DOI] [PubMed] [Google Scholar]
- Metcalfe C, Friedman LS, and Hager JH (2018). Hormone-Targeted Therapy and Resistance. Annu Rev Canc Biol 2, 291–312. [Google Scholar]
- Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, and Auricchio F (1996). Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J 15, 1292–1300. [PMC free article] [PubMed] [Google Scholar]
- Ming J, Ruan S, Wang M, Ye D, Fan N, Meng Q, Tian B, and Huang T (2015). A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget 6, 40692–40703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove EA, and Sutherland RL (2009). Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 9, 631–643. [DOI] [PubMed] [Google Scholar]
- Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, Hsieh AC, Cunningham JT, Truillet C, Tameire F, et al. (2018). Development of a stress response therapy targeting aggressive prostate cancer. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CK, and Schiff R (2011). Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, et al. (2011). Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117, 1311–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira B, Billaud M, and Almeida R (2017). RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 3, 506–528. [DOI] [PubMed] [Google Scholar]
- Raha P, Thomas S, Thurn KT, Park J, and Munster PN (2015). Combined histone deacetylase inhibition and tamoxifen induces apoptosis in tamoxifen-resistant breast cancer models, by reversing Bcl-2 overexpression. Breast Cancer Res 17, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Valle F, Braunstein S, Zavadil J, Formenti SC, and Schneider RJ (2008). eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol 181, 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, Sin O, Shostak K, Schmitz S, Piepers J, et al. (2018). Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609. [DOI] [PubMed] [Google Scholar]
- Robert F, Roman W, Bramoulle A, Fellmann C, Roulston A, Shustik C, Porco JA Jr., Shore GC, Sebag M, and Pelletier J (2014). Translation initiation factor eIF4F modifies the dexamethasone response in multiple myeloma. Proc Natl Acad Sci U S A 111, 13421–13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Ramirez L, Cao HB, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, et al. (2004). XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Research 64, 5943–5947. [DOI] [PubMed] [Google Scholar]
- Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rosenfeld J, Capdevielle J, Guillemot JC, and Ferrara P (1992). In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal Biochem 203, 173–179. [DOI] [PubMed] [Google Scholar]
- Shah A, Qian Y, Weyn-Vanhentenryck SM, and Zhang C (2017). CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, and Greene GL (1998). The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95, 927–937. [DOI] [PubMed] [Google Scholar]
- Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, and Schiff R (2004). Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst 96, 926–935. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, and Liao JK (2000). Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, and Santen RJ (2004). The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc Natl Acad Sci U S A 101, 2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song RX, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, and Santen RJ (2002). Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol Endocrinol 16, 116–127. [DOI] [PubMed] [Google Scholar]
- Song RX, Zhang Z, and Santen RJ (2005). Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab 16, 347–353. [DOI] [PubMed] [Google Scholar]
- Terribilini M, Sander JD, Lee JH, Zaback P, Jernigan RL, Honavar V, and Dobbs D (2007). RNABindR: a server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res 35, W578–W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendel J, Schwarzl T, Horos R, Prakash A, Bateman A, Hentze MW, and Krijgsveld J (2019). The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 176, 391–403 e319. [DOI] [PubMed] [Google Scholar]
- Urra H, Dufey E, Avril T, Chevet E, and Hetz C (2016). Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2, 252–262. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen JY, Cody NAL, Dominguez D, et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem KM, and Wek RC (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101, 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Weingarten-Gabbay S, Khan D, Liberman N, Yoffe Y, Bialik S, Das S, Oren M, and Kimchi A (2014). The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 33, 611–618. [DOI] [PubMed] [Google Scholar]
- Wells SE, Hillner PE, Vale RD, and Sachs AB (1998). Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2, 135–140. [DOI] [PubMed] [Google Scholar]
- Wouters BG, van den Beucken T, Magagnin MG, Koritzinsky M, Fels D, and Koumenis C (2005). Control of the hypoxic response through regulation of mRNA translation. Semin Cell Dev Biol 16, 487–501. [DOI] [PubMed] [Google Scholar]
- Xu YC, and Ruggero D (2020). The Role of Translation Control in Tumorigenesis and Its Therapeutic Implications. Annu Rev Canc Biol 4, 437–457. [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, and Mori K (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. [DOI] [PubMed] [Google Scholar]
- Younesy H, Nielsen CB, Lorincz MC, Jones SJ, Karimi MM, and Moller T (2016). ChAsE: chromatin analysis and exploration tool. Bioinformatics 32, 3324–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, and Darnell RB (2011). Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Gene ontology for the mass spectrometry analysis of ERα-interacting proteins in the cytoplasmic lysate of MCF7 breast cancer cells.
(B) Representative western blots for ERα bound to Oligo(dT) beads in cytoplasmic and soluble nuclear lysate of an ER+ human breast tumor with its paired normal adjacent tissue.
(C) Representative western blots for GR, PR, AR, RXRα and PPARγ bound to Oligo(dT) beads in cytoplasmic and soluble nuclear MCF7 lysate.
(D) Associations between ERα HITS-CLIP replicates shown as correlations calculated on peak levels. Every point represents a peak, and x and y axes show the counts in that peak for the given replicates. Correlation scores are calculated with Pearson r.
(E) Integrative Genomics Viewer (IGV) view of the three replicates of ERα HITS-CLIP analysis showing ERα binding to XBP1, MCL1 and eIF4G2 mRNAs.
(F) Pie charts presenting the distribution of ERα-RNA interactions by genome region.
(G) Gene ontology of ERα-bound genes were analyzed by DAVID and presented utilizing Cytoscape tool. For each ontology term the P values was calculated.
(H) The presence of ERα-bound motif in the 3’UTR of XBP1, MCL1 and eIF4G2 mRNAs in indicated mammal species.
(I) Phastcons conservation scores of HuR-bound RNA regions compared with their adjacent regions. The window represents ±0.5 kb regions from the center of the HuR RNA-binding events.
(A) Left: SYBR Gold stain of Trizol purified RNAs pulled down by GST-purified ERα proteins on the top, and Coomassie stain of GST-purified ERα on the bottom. Right: Quantifications of relative amounts of RNA bound to each GST-purified ERα proteins are shown.
(B) A schematic of the strategy for generating MCF7 cells with WT and RBDmut ERα.
(C) Fluorescent microscope image showing the subcellular localization of GFP-ERα (WT and RBDmut), DAPI was used to stain the nucleus.
(D) Representative western blots for FLAG-ERα from chromatin-bound lysate fractions of MCF7 cells with WT and RBDmut ERα. Histone H3 is used as an internal control. N=3 independent experiments.
(E) Heat map showing binding peak intensity of ERα chromatin binding events in MCF7 cells with WT and RBDmut ERα. The window represents ±2.5 kb regions from the center of the binding events.
(F) Volcano plot showing the difference between ERα-bound chromatin locus on overlapped binding sites in MCF7 cells with WT and RBDmut ERα. Y-axis represents −log10 adjust P values (FDR), and X-axis represents log2 fold change (FC). Total 23193 WT and RBDmut ERα DNA-binding events are shown, 238 hits down in RBDmut are marked with blue, and 23 hits up in RBDmut are marked with orange.
(G) Estrogen responsive element (ERE) motif enriched within ERα-binding sites from FLAG ChIP-seq analysis with MCF7 cells with WT and RBDmut ERα.
(H) FLAG ChIP-qPCR in MCF7 cells with WT and RBDmut ERα on GREB1 and TFF1 promoter, a random genomic region is used as a negative control. IgG ChIP-qPCR is used as a negative control for IP. N=3 biological replicates. Two-sided t-test. All values represent the mean + SD.
(I) Venn diagram showing the overlap of ERα chromatin-bound and RNA-bound genes.
(A) Growth phenotype analysis of the CRISPRi screen on Day 9 and Day 5 compared with Day 0 shown as a volcano plot. The average growth phenotype for each gene with the 3 most active sgRNAs is shown in X-axis, and the significance of the growth phenotype is presented in Y-axis as log10(Mann-Whitney p-values). The Dash lines mark the threshold of significance in this study: |Growth phenotype × log10(Mann-Whitney p-values) | > 1. Genes that pass the threshold are shown with genes that favor the cell growths in blue, while those that inhibit cell growth are in orange.
(B) The average growth phenotype for XBP1, EIF4G2 and MCL1 genes with the 3 most active sgRNAs on Day 5, 9 and 14 are shown. Adjusted P values (Padj) of all 5 sgRNAs were shown.
(A) RNA electrophoretic mobility shift assay (EMSA) of the XBP1 3’UTR mRNA using GST-tagged ERα. Lower arrow: free XBP1 mRNA, upper arrow: XBP1 mRNA bound by ERα.
(B) Representative images for RT-PCR analysis of XBP1 splicing in T47D cells harboring WT and RBD-mutated ERα respectively, upon DMSO or tunicamycin treatment for 4 h. The upper band indicates unspliced XBP1 mRNA and the lower band indicates spliced XBP1 mRNA. β-actin mRNA (ACTB) is used as an internal control.
(C) Representative western blots for RtcB pulled down by GST-purified ERα protein constructs, with Coomassie stain of ERα protein constructs at the bottom.
(D) Relative cell apoptosis as shown by Annexin V+ populations of TamR-1 cells treated with 25 μM of IRE1 inhibitor (IRE1i) for 24 h compared with DMSO, with and without ERα silencing (siESR1).
All experiments are performed with N=3 biological replicates.
(A) Representative western blots for eIF4G2, MCL1 and α-tubulin in T47D cells with and without the RBDmut of ERα.
(B) Relative mRNA expressions of eIF4G2 and MCL1 measured by quantitative PCR (qPCR) analysis in T47D cells with and without the RBDmut of ERα.
(C) Representative western blots for eIF4G2, MCL1 and α-tubulin in MCF7 cells with and without the RBDmut of ERα, treated with proteasome inhibitor MG132 for 4 h.
(D) qPCR with reverse transcription (RT-qPCR) analysis of α-tubulin (TUBA) mRNA levels in fractions, percentages of α-tubulin mRNA distributed in each fraction against total α-tubulin mRNA are shown.
(E) IGV views of ERα HITS-CLIP analysis showing lack of ERα bindings on BCL2 and BCL2L1 mRNAs.
All values represent the mean + SD. Two-sided t test.
(A) Representative western blots for eIF4G2, MCL1, XBP1s and α-tubulin are shown in 14 freshly collected ERα+ breast cancer tumors (T) and their paired normal adjacent tissues (N).
(B) Representative immunohistochemistry (IHC) staining of XBP1s, eIF4G2 and MCL1 on ER+ breast cancer tissue microarray. The protein expressions were evaluated and scored. No: no expression, Low: low expression, Median: median expression and High: high expression.
(C) Representative western blots for eIF2α phosphorylation (p-eIF2α), eIF2α and α-tubulin in MCF7 cells treated with 4-Hydroxytamoxifen (4-OHT) and Fulvestrant for 24 or 72 hours.
(D) Relative mRNA expressions of eIF4G2 and MCL1 measured by qPCR analysis in MCF7 and TamR-1 cells. </p/> (E) Relative populations of cells in G1 cell cycle phase in TamR-1 cells with and without the RBDmut of ERα under normal cell culture conditions.
(F) Relative populations of cells in G1 cell cycle phase in TamR-1 cells with or without eIF4G2 silencing (siEIF4G2) under normal cell culture conditions.
(G) Relative fold differences of eIF4G2-silenced (siEIF4G2) TamR-1 cells undergo apoptosis (Annexin V+, PI+) comparing to the control (siCtl) cells, under normal cell culture conditions. (H) Relative fold differences of TamR-1 cells undergo apoptosis (Annexin V+, PI+) when treated with MCL1 inhibitor AZD5991 for 24 h comparing to DMSO under normal cell culture conditions.
All experiments are performed with n=3 biological replicates. Two-sided t-test. All values represent the mean + SD.
ERα interacting partners in the cytoplasm were detected by quantitative mass spectrometry using TMT labeling, related to Figure S1A. Unique peptide numbers for each gene are shown along with coverage in percentage. The enrichment for proteins interacting with ERα over IgG were calculated by log2FC (ERα/IgG).
Chromatin regions and matched gene names for ERα-associated RNAs identified by HITS-CLIP are shown, related to Figure 1 and Figure S1. For each binding event, binding intensities (k) are calculated, and k > 10 are used as a cut-off for positive bindings.
ERα-associated chromatin regions (regions with a light blue background are significantly altered between WT and RBDmut), log2 fold change of the binding intensities comparing RBDmut to WT, P values, False discovery rate (FDR), and the annotation of the chromatin regions are shown. Enhancer: enhancer regions; promoter: promoter regions; #N/A: no annotation.
sgRNA target sequences, with their predicted/empirical scores, off-target stringency and oligo sequences ordered from Twist Bioscience for each ERα-bound mRNAs are shown, related to Figure 3A. 5 sgRNAs for each of the1090 mRNAs bound by ERα with binding intensities k > 10 (from Supplementary Table 1) and 117 non-targeting control sgRNAs were employed.
Normalized average growth phenotypes and Mann-Whitney p-values for each ERα-bound mRNAs from two biological replicates of the CRISPRi analysis at Day 14, Day 9 and Day 5 time points are shown respectively, related to Figure 3B. Negative growth phenotype values together with Mann-Whitney p-values at a cut-off of [|log10(P value) × average growth phenotype| > 1] are used to indicate essential mRNAs for the fitness of MCF7 breast cancer cells.
Data Availability Statement
All sequencing data have been deposited in the GEO database under the accession number GEO: GSE173631. Proteomics data have been deposited in the ProteomeXchange: PXD025018. No software was generated for this project.