

Abstract
Purpose:
Mounting evidence demonstrates that combining radiation therapy (RT) with immunotherapy can reduce tumor burden in a subset of patients. However, conventional systemic delivery of immunotherapeutics is often associated with significant adverse effects, which force treatment cessation. The aim of this study was to investigate a minimally invasive therapeutics delivery approach to improve clinical response while attenuating toxicity.
Methods and Materials:
We used a nanofluidic drug-eluting seed (NDES) for sustained intratumoral delivery of combinational antibodies CD40 and PDL1. To enhance immune and tumor response, we combined the NDES intratumoral platform with RT to treat the 4T1 murine model of advanced triple negative breast cancer. We compared the efficacy of NDES against intraperitoneal administration, which mimics conventional systemic treatment. Tumor growth was recorded, and local and systemic immune responses were assessed via imaging mass cytometry and flow cytometry. Livers and lungs were histologically analyzed for evaluation of toxicity and metastasis, respectively.
Results:
The combination of RT and sustained intratumoral immunotherapy delivery of CD40 and PDL1 via NDES (NDES CD40/PDL1) showed an increase in both local and systemic immune response. In combination with RT, NDES CD40/PDL1 achieved significant tumor burden reduction and liver inflammation mitigation compared with systemic treatment. Importantly, our treatment strategy boosted the abscopal effect toward attenuating lung metastatic burden.
Conclusions:
Overall, our study demonstrated superior efficacy of combination treatment with RT and sustained intratumoral immunotherapy via NDES, offering promise for improving therapeutic index and clinical response.
Introduction
Sweeping advances in radiation therapy (RT) and immunotherapy have led to the proliferation of clinical trials investigating the efficacy of combination therapy for cancer treatment.1,2 Given the mounting evidence supporting radiation as an immune stimulus,3–7 the Radiation Biology Task Force supports investigating the effectiveness of immunotherapy combined with RT.8 Although abscopal responses were observed with RT and immunotherapy,9–12 the limited scope of efficacy emphasizes the need to identify regimens fundamental to the success of combination therapy.3
Immunotherapy, when conventionally administered through systemic delivery, is associated with a high incidence of immune-related adverse events involving autoimmune and inflammatory toxicities.13,14 In this context, the combination of immunotherapy with RT raises concerns of incremental toxicities posing additional risks and clinical challenges. In view of this, intratumoral immunotherapy delivery represents a rational concept to locally modulate the tumor immune microenvironment (TIME) to transform nonresponsive tumors with minimal systemic toxicity.15–21 When delivered intratumorally, immunotherapeutics exploit existing immune infiltrates22 to direct an immunologic response to tumor, generating durable systemic immunity to activate an abscopal response.6,15 Although clinical findings have alluded to the potential benefit of intratumoral immunotherapy,19 adverse events occurred owing to the repeated injections needed to sustain local bioavailability.23 Moreover, reports of rapid tumor clearance and high serum exposure from bolus local injections24,25 motivate the need for technological interventions to achieve long-term controlled release intratumorally.
To improve the therapeutic index, we sought to harness the immune potentiating effect of RT with intratumoral immunotherapy. To this end, we leveraged our intratumoral drug-eluting fiducial marker, the nanofluidic drug-eluting seed (NDES),26,27 for local immunotherapy delivery. We examined the effect of local irradiation and NDES-mediated intratumoral immunotherapy delivery of immunomodulating antibodies, agonistic α-CD40 (CD40) and α-programmed death-ligand 1 (PDL1), on tumor growth and TIME. CD40 activation is critical for T cell priming and generating T cell immunity through dendritic cell (DC) activation and consequently “cold” to “hot” tumor conversion.28 In the tumor microenvironment (TME), PDL1 expression is critical for tumor cell immune evasion; its engagement with PD1 on T cells causes cellular dysfunction.29 As such, blocking PDL1 in the TME reactivates T cell–mediated antitumor immune response. Furthermore, we posit that intratumoral delivery via NDES could mitigate toxicities associated with CD40 and PDL1.30–33
Here, we demonstrated that sustained intratumoral delivery of CD40 and PDL1 in combination with local irradiation was superior to systemic administration in the 4T1 murine model of triple negative breast cancer (TNBC). With our intratumoral NDES treatment regimen, we observed significant amplification of tumor immune response and decreased tumor burden and lung metastasis. Furthermore, NDES-mediated intratumoral immunotherapeutics administration mitigated systemic exposure to drugs and had minimal liver toxicity. Although intraperitoneal (IP) administration of CD40 and PDL1 combined with local irradiation decreased tumor growth to a similar extent, this regimen necessitated repeated injections and was associated with systemic toxicity.
Methods and Materials
NDES fabrication
Nanochannel membranes were microfabricated following a protocol reported elsewhere.34 Membranes were affixed at the end of 3.5-mm-long stainless-steel reservoirs using implantable-grade thermal epoxy (EPO-TEK 354-T) and cured overnight at 60°C. The reservoir was loaded with lyophilized antibodies through the open extremity, capped with silicone adhesive (MED3–4213, Nusil), and dried at 37°C for 2 hours. To prevent drug leakage, ultraviolet (UV) epoxy was applied over the top of the silicone cap and UV cured for ~15 seconds. NDES weight was measured before and after drug loading to calculate total content. To prime the membrane for drug release, the assembled NDES was submerged for 30 minutes in 1 mL of 1X phosphate-buffered saline (PBS) solution under vacuum.
Antibodies
CD40 (FGK4.5; BE0016) and PDL1 (10F.9G2; BE0101) monoclonal antibodies were purchased from Bio X Cell. Stock solution of CD40 was concentrated using Amicon Ultracel-30k and labeled with fluorescent molecule Alex-aFluor 647 (AF647-CD40) via N-hydroxysuccinimide ester (A37573; Invitrogen). Stock solution of PDL1 was concentrated and combined with AF647-CD40 in a 1:1 ratio. The combination of antibodies (CD40/PDL1) was frozen at −80°C and lyophilized (LABCONCO Freezone 4.5; Hampton, NH) for 48 hours; trehalose dehydrate was added at 37% w/w for drug stabilization during lyophilization.
In vitro release analysis of CD40/PDL1 from the NDES
NDES was loaded with lyophilized antibodies (AF647-CD40+ PDL1) and immersed in 200 μL of 1X PBS sink solution within individual microcuvettes for UV/Vis spectroscopy measurements. Absorbance measurements at 280 nm and 650 nm were collected every 10 minutes for 14 days via a custom-built robotic carousel (Quantum Northwest, Inc) interfaced with a UV/Vis spectrophotometer (Cary 50; Agilent Technologies).35 The cumulative release of the combination of antibodies was determined through the absorbance at 280 nm, in accordance with the Lambert–Beer law. To demonstrate that CD40 mAb and PDL1 mAb released in a 1:1 ratio from the NDES, AF647-CD40 release was obtained from AF647 absorbance at 650 nm and PDL1 release was calculated by subtracting the concentration of CD40 from the total concentration of the 2 proteins.
Animals
Seven-week-old female BALB/c mice were obtained from Taconic Biosciences (Rensselaer, NY). Animals were housed at the Houston Methodist Research Institute (HMRI) Comparative Medicine under conditions outlined in the National Institutes of Health guide for Care and Use of Laboratory Animals and monitored daily. All animal studies were conducted in accordance with the protocol approved by the Institutional Animal Care and Use Committee.
4T1 mouse mammary carcinoma model
4T1 mouse mammary tumor cell line (CRL-2539) was obtained from American Type Culture Collection and cultured according to their guidelines. 4T1 cells were routinely assessed for mycoplasma contamination. For inoculation, low passage cells were resuspended at 3 × 104 cells in 100 μL of 3:1 mixture of PBS and Matrigel. Cell mixture (100 μL) was injected at the fourth left mammary fat pad. Tumor volume growth was monitored thrice weekly via perpendicular tumor diameter measurements and calculated using the formula (mm3) 0.5 × (length) × (width)2.
4T1 mice treatment
Once tumors reached ~150 mm3 (day −17), mice were randomized to different groups before treatment (n = 7–8). Mice were anesthetized with inhaled isoflurane and IP dexmedetomidine (5 μg/g body weight) before RT treatment. RT was administered using the RS 2000 small animal irradiator (160 kV, 25 mA and mean beam as 2 Gy/min Rad Source; Brentwood, TN) at 8 Gy for 3 consecutive days36 before immunotherapy treatment (days −3, −2, and −1). A rigid exposed-flank shield (Precision X-ray Inc) and a flexible lead layer were used to shield the mice from irradiation, exposing only the tumor. After RT, atipamezole was administered to reverse dexmedetomidine anesthesia. RT-treated mice were given Baytril wafers and subcutaneous fluid daily for health maintenance.
IP treatment groups were administered with either PBS (control) or a combination of CD40 and PDL1 (100 μg each in 100 μL) on days 0, 2, 4, and 6. NDES was filled with either PBS (control) or ~800 μg lyophilized CD40 and PDL1 and primed for drug release as previously described. Intratumoral implantation occurred on day 0 via a minimally invasive trocar approach similar to brachytherapy seed insertion.26 Because sustained drug release occurs without requiring manipulation upon implantation, no further intervention was performed for the NDES group.
Response Evaluation Criteria in Solid Tumors
A modified Response Evaluation Criteria in Solid Tumors (mRECIST) assessment was performed based on the literature,37,38 with responses characterized by tumor growth inhibition. Tumor growth inhibition percentage was determined by calculating percent change of tumor volume at endpoint (Vt) versus treatment initiation day (V0): %ΔVt = 100% × (Vt − V0)/V0. The mRECIST criteria are defined as follows: complete response (mCR), −100% < %ΔVt < −40%; partial response (mPR), −40% < %ΔVt < −20%; stable disease (mSD), −20% < %ΔVt < 30%; progressive disease (mPD), %ΔVt > 30%. Mice that had not completed 14-day treatment were removed from the data set.
Progression-free analysis
A 4T1 survival study was conducted with an additional cohort of mice. Progression-free survival (PFS) was calculated from the last dose of treatment to the date of tumor doubling time or death from any cause. Tumor doubling time (DT) is the period when V2 = 2V1, which was calculated with the equation39 DT = ln2/ln (V2/V1)/(t2 − t1). Survival was analyzed by the Kaplan–Meier method using log-rank test.
Tumor-infiltrating immune cell isolation
To assess tumor immune cell infiltration, tumors and spleens were collected for flow cytometry analysis at the study endpoint. Tumors were minced into 1 to 3 mm sections and incubated for 1 hour at 37°C on a rocker in RPMI1640 with 1× collagenase/hyaluronidase (StemCell Technologies) and 20 U/mL of DNase I. After digestion, tumors were disaggregated into single cell suspensions via mechanical filtration through 70 μm cell strainers. Tumor-infiltrating leukocytes were enriched using Lymphoprep (StemCell Technologies). Tumor cells were collected after Lymphoprep separation. Spleens were dissociated into single-cell suspension by mechanical filtration through a 70 μm cell strainer with ACK Lysis buffer (118-156-101; Quality Biological) to lyse splenic red blood cells. Single cells were first blocked with antimouse CD16/CD32/Fc block (eBioscience) and stained using either myeloid or T cell panel (Table E1) for extracellular staining. For intra-cellular staining, cells were fixed with 1% formalin and membrane permeabilized before the addition of antibodies. Data were acquired on LSRII and Fortessa (BD Biosciences) flow cytometers and analyzed using FlowJo v10 software. Analysis was performed after exclusion of debris, doublets, and nonviable cells.
Imaging mass cytometry
Metal-labeled antibodies were prepared according to the Fluidigm protocol. Antibodies were obtained in carrier/protein-free buffer and then prepared using the MaxPar antibody conjugation kit (Fluidigm). After determining the percent yield by absorbance measurement at 280 nm, the metal-labeled antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience) for long-term storage at 4°C. Antibodies used in this study are listed in Table E1.
Tumor sections were baked at 60°C overnight, then dewaxed in xylene and rehydrated in a graded series of alcohol (ethanol absolute, ethanol:deionized water 90:10, 80:20, 70:30, 50:50, 0:100; 10 minutes each) for imaging mass cytometry. Heat-induced epitope retrieval was conducted in a water bath at 95°C in Tris-Tween20 buffer at pH 9 for 20 minutes. After immediate cooling for 20 minutes, the sections were blocked with 3% bovine serum albumin in tris-buffered saline (TBS) for 1 hour. For staining, the sections were incubated overnight at 4°C with an antibody master mix (Table E1). Samples were then washed 4 times with TBS/0.1% Tween20. For nuclear staining, the sections were stained with Cell-ID Intercalator (Fluidigm) for 5 minutes and washed twice with TBS/0.1% Tween20. Slides were air-dried and stored at 4°C for ablation.
The sections were ablated with Hyperion (Fluidigm) for data acquisition. Imaging mass cytometry data were segmented by ilastik and CellProfiler. Histology topography cytometry analysis toolbox (HistoCAT) and R scripts were used to quantify cell number, generate tSNE plots, and perform neighborhood analysis.40 For all samples, tumor and cellular densities were averaged across 2 images per tumor, with n = 3 per group.
Histologic analysis
Tumors were sectioned for histology before processing for flow cytometry. Tumors, livers, and lungs were fixed in 10% formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E) at the Research Pathology Core of HMRI.
Lung and liver histologic analysis
H&E images were acquired using an Olympus BX61 microscope (Center Valley, PA) mounted with a 10×/0.05 non-oil objective and 10× ocular attached to an Olympus DP72 digital camera. H&E sections were evaluated by a pathologist blinded to treatment groups. Livers were assigned pathologic numeric scores according to literature41 as follows: 0, normal liver, no lesions or hepatocellular damage noted; 1, rare portal and parenchymal infiltrates but no necrosis; 2, moderate parenchymal or portal infiltrates but no necrosis; 3, frequent and/or large portal or parenchymal infiltrates with occasional isolated islands of coagulative necrosis; and 4, extensive areas of inflammation with bridging coagulative necrosis. Area and count of spontaneous lung metastasis induced by orthotopic tumor inoculation in the mammary fat pad were analyzed via Image J.42
Serum enzyme-linked immunosorbent assay
Nunc Maxisorp plates (96-well; Thermo Fisher) were coated with 1.25 μg/mL antirat heavy chain antibody (#MCA278; Bio-Rad Laboratories, Hercules, CA) overnight at 4°C. Serum samples were added at a 1:10 dilution and standard curves were prepared using CD40. An antirat kappa/lambda light chain antibody conjugated to horse-radish peroxidase (#MCA1296P; Bio-Rad Laboratories) was used as the secondary antibody. One-Step UltraTMB (Thermo Fisher) was used as substrate. To stop the colorimetric reaction, ELISA Stop Solution (Invitrogen) was used. Enzyme-linked immunosorbent assay plates were analyzed in a UV-Vis plate reader (BioTek Synergy H4) for absorbance at 450 nm.
Statistical analysis
Data were analyzed using GraphPad Prism 8.3.1. For parametric data sets, statistical significance was determined by unpaired t test for 2-tailed data. For multiple group comparisons, an analysis of variance (ANOVA) was performed. Unpaired 1-way, 2-way ANOVA and multiple comparisons were performed using Tukey corrections.
Results
Sustained and constant release of CD40 and PDL1 via NDES
Our group developed the NDES for sustained and constant intratumoral drug delivery without requiring repeated interventions (Fig. 1A). Smaller than a grain of rice, the NDES is intended for a one-time minimally invasive intratumoral trocar implantation,26,27 adopted from clinical fiducial marker and brachytherapy seed placement. Controlled release of immunotherapeutics is regulated via a microfabricated silicon nanofluidic membrane mounted on one end of the cylindrical device (Fig. 1A, B).26 Immunotherapeutics were released from the NDES via concentration-driven diffusion through the nanochannels (Fig. 1A, B).26,43,44 Nanoconfinement enhances electro-static and steric interactions between the drug molecules and nanochannel walls via saturated diffusive transport.45 Thus, sustained and constant delivery is achieved without pumping or actuation. In this study, we used NDES with nanochannels of 150 nm (Fig. 1B). To understand the release profile of the NDES, we performed an in vitro cumulative release assay of CD40 and PDL1. Over the course of 14 days, we observed sustained delivery profiles with cumulative release of 48.0 ± 11.9 μg and 57.8 ± 8.6 μg for CD40 and PDL1, respectively (Fig. 1C). In combination, NDES-mediated delivery of CD40 and PDL1 achieved a cumulative release of 105.8 ± 20.5 μg over 14 days, translating to an average of 7.6 ± 1.5 μg of CD40/PDL1 delivered per day (Fig. 1D).
Fig. 1.

Nanofluidic drug-eluting seed (NDES), nanofluidic membrane, and drug release. (A) Cross-sectional rendering of NDES containing antibody in powder form and picture of NDES. (B) Scanning electron microscopy (SEM) image of nanofluidic membrane. (C) In vitro cumulative release of CD40 and PDL1. (D) In vitro cumulative release of the combination of CD40 and PDL1.
Sustained intratumoral release of CD40/PDL1 via NDES in combination with RT reduces tumor burden
To evaluate the synergistic effects of RT and intratumoral immunotherapy delivered via NDES, we used the 4T1 orthotopic, syngeneic murine model of TNBC. 4T1, which metastasizes to the lung, is one of the few clinically relevant TNBC models that closely recapitulates human disease progression.46 Importantly, orthotopic inoculation in immunocompetent BALB/c mice allows assessment of immune response in TME.
In a pilot study, we investigated the efficacy of intratumoral CD40 via NDES (NDES CD40) in combination with RT. We have previously shown that intratumoral NDES CD40 significantly slowed tumor growth rate but did not achieve substantial burden reduction.26 Thus, we postulated that the addition of RT could improve the efficacy of intratumoral NDES CD40. As demonstrated in the literature,47 dose and fractionation of RT are crucial elements to improve local immune effect by inducing abscopal responses. Hypofractionated RT, delivered as 8 Gy × 3, in combination with immune checkpoint inhibitors, can induce abscopal responses in the 4T1 model.36,48 This effect is not recapitulated in single high dose radiation administration. Thus, we used hypofractionated RT, as 8 Gy × 3 consecutive days, in combination with CD40/PDL1 to induce an immune response and potentially generate an abscopal effect.
RT cohorts were pretreated with hypofractionated RT (8 Gy × 3) before NDES CD40 administration for 9 days. Flow cytometry analysis demonstrated significant increase in activated DCs (CD11c+CD86+ or CD11c+CD80+) in the tumors (Fig. E1A, B) and spleens (Fig. E1C, D), particularly with combination of RT and NDES CD40. Furthermore, combination treatment resulted in increased activation of splenic CD8 T cells, detected through CD69 (Fig. E1E) and Granzyme B (Fig. E1F) markers, as well as augmented proliferation intratumorally (Fig. E1G). Despite the amplified immune response with combination RT and CD40, the tumors maintained a slowed but steady growth rate (Fig. E1H).
Because RT increased tumor PD1/PDL1 expression and demonstrated synergistic efficacy with PDL1 blockade to promote antitumor immunity,49–51 we hypothesized that addition of PDL1 would enhance antitumor immune response. In this experimental arm, similar to the previous study, hypofractionated radiation pretreatment (8 Gy × 3) was initiated when the tumors reached 150 mm3, 17 days after 4T1 inoculation (Fig. 2A). Following this, mice were administered with CD40/PDL1 either via NDES (one-time intratumoral insertion) or conventional systemic administration through IP injection. IP injections were given for 4 doses, for a total dose equivalent to that loaded into NDES, on days 0, 2, 4, 6, and monitored for 14 days.
Fig. 2.

Tumor burden reduction with Rad and CD40/PDL1 treatment of 4T1 mice. (A) Treatment schematic depicting hypofractionated 8 Gy radiation therapy (RT) pretreatment for 3 consecutive days followed by CD40/PDL1 administration either via nanofluidic drug-eluting seed (NDES) or intraperitoneal (IP). IP CD40/PDL1 was administered at 100 μg each in 100 μL on days 0, 2, 4, and 6. NDES CD40/PDL1 intratumoral implantation occurred on day 0 in a one-time procedure. Mice were monitored for 14 days after CD40/PDL1 administration. (B) Tumor growth of control untreated (Ctrl UnTx), Ctrl IP phosphate-buffered saline (PBS), Ctrl NDES PBS, IP CD40/PDL1, NDES CD40/PDL1, Rad only, IP CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad mice (n = 7–8/group). Data are expressed as mean ± standard deviation. Significance was analyzed by 2-way analysis of variance (ANOVA). ***P ≤ .001, ****P ≤ .0001. (C) Final resected tumor weights (top) and images (bottom) of all groups at endpoint. Data are expressed as mean ± standard deviation. Significance was analyzed by 1-way ANOVA. *P ≤.05, ***P ≤ .001. (D) Tumor growth inhibition percentage (%ΔVt) of Rad only, IP CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad. Each bar represents an individual mouse. Complete response (mCR), −100% < %ΔVt < −40%; partial response (mPR), −40% < %ΔVt < −20%; stable disease (mSD), −20% < %ΔVt < 30%; progressive disease (mPD), %ΔVt > 30%. (E) Representative images of lung sections showing metastasis of each group. (F) Percentage of spontaneous lung metastasis area analyzed by Image J. Data are expressed as mean ± standard deviation whereas significance was analyzed by 1-way ANOVA (n = 7–8/group). **P ≤ .005. (G) Kaplan-Meier curves comparing progression-free survival of mice treated with Rad and with combination of IP or NDES-delivered CD40/PDL1 (log-rank test; n = 7–8/group; **P ≤ .005, ***P ≤ .001).
Compared with their respective controls, significant tumor control was achieved in both the NDES and IP CD40/PDL1 only groups, with an impaired but nevertheless upward trending growth rate (Fig. 2B). Tumor inhibition in radiation-only treatment was evident after day 8 and approximated baseline volume by study endpoint. The addition of CD40/PDL1, either via IP or NDES, to RT had a profound inhibitory effect on tumor growth; significant tumor burden reduction below baseline was achieved by day 11, indicative of treatment response (Fig. 2B). Final tumor weights confirmed radiation-induced tumor shrinkage across all radiation-treated groups. The addition of CD40/PDL1 through IP or NDES combined with RT further significantly reduced final tumor weights compared with their respective controls and radiation-only treatment (Fig. 2C).
Furthermore, we adapted the mRECIST to determine tumor growth inhibition percentage by comparing percent change of tumor volume before and after treatment. The combination of RT with immunotherapy (CD40/PDL1) induced mPR and mSD (Fig. 2D). In contrast, we observed mPD in the radiation-only cohort with few cases of mSD (Fig. 2D), indicative of the inadequacy of monotherapy. Thus, the combination of radiation and immunotherapy imparts tumor control efficacy that is superior to standalone treatment.
NDES CD40/PDL1 in combination with RT demonstrated an abscopal effect via reduction of spontaneous lung metastasis and improved PFS
Mounting evidence suggests the abscopal effect, induced by the combination of immunotherapy and RT, improves patient survival and reduces metastatic burden.52,53 We found that local treatment via NDES CD40/PDL1, with or without RT, had significantly lower spontaneous lung metastasis compared with radiation-only treatment (Fig. 2E, F). To further evaluate the effects of this combination therapy, we conducted a survival study comparing Ctrl UnTx, Rad only, IP CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad. PFS analysis demonstrated statistically significant prolonged survival in Rad only, IP CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad compared with Ctrl UnTx (Fig. 2G). Specifically, local therapy via NDES CD40/PDL1 + Rad significantly enhanced survival compared with RT only (P < .01) or Ctrl UnTx (P < .0001) (Fig. 2G). IP CD40/PDL1 + Rad showed prolonged survival compared with Ctrl UnTx (P < .0001); however, no significant improvement was shown compared with Rad only (P > .99). This suggests the addition of localized immunotherapy enhances the duration of tumor control compared with systemic immunotherapy.
Treatment-associated toxicity and adverse effects demonstrated in IP CD40/PDL1-treated groups
To monitor for signs of treatment-induced toxicity, we monitored mice weight (Fig. 3A, B) and rectal temperature (Fig. E2A, B). Mice in IP CD40/PDL1 cohorts with and without RT demonstrated notable weight loss as early as day 4 (Fig. 3A,B) and did not completely recover compared with matched controls by study endpoint. Significant weight loss in the IP CD40/PDL1 + Rad persisted until day 11 (Fig. 3B). In addition, all mice receiving IP treatment showed wide variability and fluctuations in rectal temperature (Fig. E2A, B). This decrease occurred primarily in IP CD40/PDL1 + Rad post-IP injections, on days 1, 5, and 7, as well as after treatment completion on day 11, in comparison to Ctrl IP (Fig. E2A, B). Although the temperature varied across treatment groups, no hypo- or hyperthermia was observed. In comparison, NDES-treated mice with and without RT exhibited a healthy weight gain and range throughout the study, compared with matched controls (Fig. 3A, B). Variation in rectal temperature in NDES CD40/PDL1 + Rad was observed on day 11 compared with Ctrl NDES and Rad only. However, this was likely secondary to the global effect of radiation because NDES CD40/PDL1 did not exhibit weight loss (Fig. 3A, B).
Fig. 3.

Toxicity evaluation for body weight, liver inflammation, and lung metastasis. (A) Body weights of control (Ctrl) intraperitoneal (IP) phosphate-buffered saline (PBS), Ctrl nanofluidic drug-eluting seed (NDES) PBS, IP CD40/PDL1, and NDES CD40/PDL1 during treatment. Significance annotation of NDES CD40/PDL1 versus IP CD40/PDL1 indicated in the graph. Significance was analyzed by 2-way analysis of variance (ANOVA) (n = 7–8/group). *P ≤ .05. Detailed significance annotation is shown in Table E2. (B) Body weights of Ctrl UnTx, Rad only, IP CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad, during treatment. Significance annotation of Ctrl UnTx versus IP CD40/PDL1 Rad + indicated in the graph. Significance was analyzed by 2-way ANOVA (n = 7–8/group), **P ≤ .01, ****P ≤ .0001. Detailed significance annotation is shown in Table E2. (C) Liver inflammation scoring by pathologist. (D) Representative figures of liver inflammation of each group.
To assess treatment-associated adverse effects, liver inflammation was assessed via histologic analysis.26 All RT groups demonstrated a clear benefit in reducing inflammation compared with control (Fig. 3C, D). Conversely, IP CD40/PDL1 treatment profoundly worsened pathologic liver inflammation compared with NDES CD40/PDL1 groups (Fig. 3C, D). Serum from all groups was analyzed via ELISA for CD40 as a measure of systemic drug exposure. Only IP CD40/PDL1 + Rad had detectable CD40 in the serum at study endpoint, indicating evidence of systemic exposure. All other groups were below the lower limit of quantification. Thus, the CD40/PDL1 released by NDES remained local and did not enter systemic circulation (Fig. E3). Taken together, these results demonstrate the safety profile of NDES treatment in comparison to systemic administration.
NDES and IP CD40/PDL1 + Rad affected immune cell populations locally and systemically
To characterize treatment-induced immune response, we performed flow cytometry to identify immune cell populations in tumors and spleens as a measure of local and systemic responses, respectively.
With IP or NDES CD40/PDL1 + Rad treatment, we observed a significant increase in intratumoral infiltrated CD8 T cells, which are fundamental for cancer elimination (Fig. 4A). On the contrary, CD8 T cells were markedly decreased in the spleen (Fig. 4B), possibly attributable to the lymphopenic effects of radiation.54 Despite this, the splenic CD8 T cells in the NDES and IP CD40/PDL1 + Rad cohorts had a significant increase in Granzyme B expression, a signature of cytotoxic T cell activation (Fig. 4C). We used Granzyme B+ CD8+ T cells as a surrogate measure of systemic immune response induction. We observed that local treatment with NDES induced an increase in splenic Granzyme B+ CD8+ T cells; however, future studies are needed to demonstrate tumor-specific reactivity. Additionally, we observed augmented intratumoral proliferating CD4 T cells in both IP CD40/PDL1 + Rad and NDES CD40/PDL1 + Rad groups (Fig. 4D).
Fig. 4.

Flow cytometry analysis showing immune cell alteration locally (tumor) and systemically (spleen). CD8 T cells in the (A) tumor and (B) spleen. (C) Granzyme B (GrzB) expressing CD8 T cells in the spleen. (D) Ki67+CD4+ T cells in the tumor. (E) CD206+F4/80+CD11b+ M2-like macrophage in tumor. (F) PDL1-expressing 4T1 cancer cells (n = 7–8/group). Data are plotted as mean ± standard deviation. Significance was analyzed by unpaired t test. *P ≤ .05, **P ≤ .005, ***P ≤ .0005, and ****P ≤ .0001.
Macrophage response within TME is both location and treatment specific. Dependent on phenotype, macrophages can promote (M1) or suppress (M2) adaptive immunity. The polarized suppressive M2 macrophages (CD206+F4/80+CD11b+) were markedly decreased intratumorally across Rad only, IP CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad compared with control (Fig. 4E). Systemically, we did not detect a significant difference in M2 macrophages between NDES and IP CD40/PDL1 (Fig. E4A). In addition to M2 macrophages, myeloid-derived suppressive cells (MDSCs) are anti-inflammatory regulatory cells. If increased, MDSCs (Gr-1+CD11b+) potentiate a suppressive environment, leading to possible downstream inhibitory effects to immunotherapy. With respect to untreated control, a pronounced decrease in MDSCs was detected in the TME upon treatment, although not statistically significant in the NDES CD40/PDL1 + Rad cohort (Fig. E4B). Systemically, MDSCs were reduced in the IP CD40/PDL1 + Rad compared with control (Fig. E4C), indicating a possible systemic effect stemming from systemic treatment. In response to immunotherapy treatment, PDL1-expressing 4T1 tumor cells were considerably diminished in NDES and IP CD40/PDL1 + Rad treated groups (Fig. 4F). This effect was independent of radiation treatment.
NDES CD40/PDL1 + Rad treatment augmented CD8+ T cells and reduced cancer cells and endothelial cells within the TIME
Imaging mass cytometry enables multiplexed assessment of abundance and protein expression localization, revealing spatial distribution of immune cells across treatments. Ctrl UnTx, Ctrl NDES PBS, Rad only, IP CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad were stained for immune cell spatial distribution (Fig. 5A).
Fig. 5.
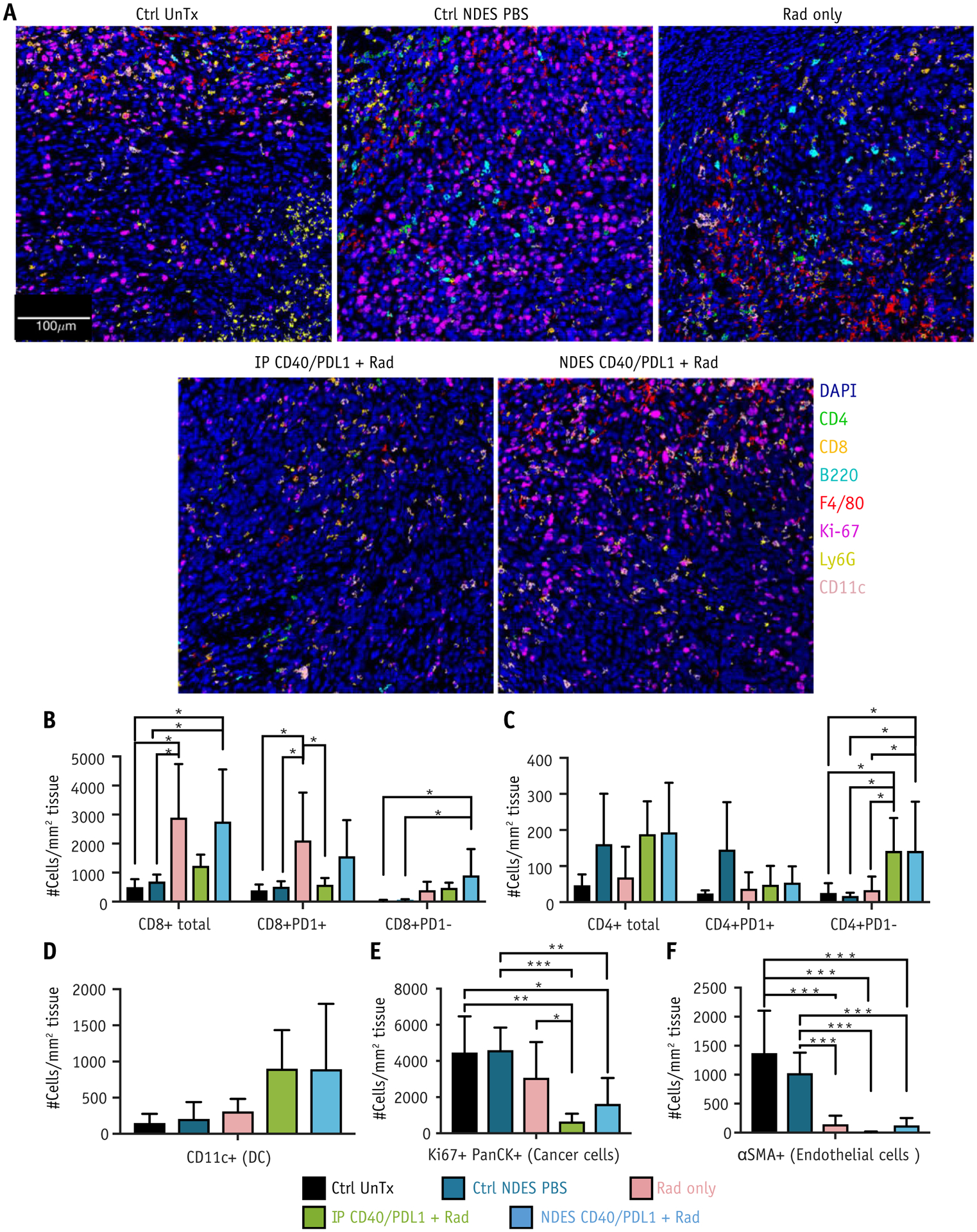
Imaging mass cytometry analysis of tumor immune microenvironment. (A) Representative overlaid images with 7 markers (B220, CD11c, F4/80, Ly6G, CD4, CD8, Ki67) of different treatment groups. (B-F) Number of cells were identified per mm2 area in stained tissue from Ctrl UnTx, Ctrl nanofluidic drug-eluting seed (NDES) phosphate-buffered saline (PBS), Rad only, intraperitoneal (IP) CD40/PDL1 + Rad, and NDES CD40/PDL1 + Rad. (B) Total CD8+, CD8+PD1+, and CD8+PD1− T cells; (C) total CD4+, CD4+PD1+, and CD4+PD1− T cells; (D) CD11c+ (DC); (E) Ki67+PanCK+ (cancer cells); (F) αSMA+ (endothelial cells). For all samples, tumor and cellular densities were averaged across 2 images per tumor with n = 3 per group. Significance was analyzed by unpaired t test, *P ≤ .05, **P ≤ .01, ***P ≤ .005.
To identify proximate cell-cell interactions and spatial features of immune cells within each treatment group, HistoCAT analysis was performed.40 Twenty-one cell populations based on marker expression were clustered via PhenoGraph (Fig. E5) and displayed on tSNE plots (Fig. E6) for each condition. These clusters identified key cell populations of interest within the TIME. Most notably, a significant increase in total CD8 T cells was observed in Rad only and NDES CD40/PDL1 + Rad compared with Ctrl UnTx and Ctrl NDES PBS (Fig. 5B). Exhausted CD8 T cells (CD8+PD1+) were augmented in Rad only compared with Ctrl UnTx, Ctrl NDES PBS, and IP CD40/PDL1 + Rad (Fig. 5B). Concurrently, a significant increase in effector CD8 T cells (CD8+PD1−) was seen in NDES CD40/PDL1 compared with Ctrl UnTx and Ctrl NDES PBS (Fig. 5B). Likewise, effector CD4 T cells (CD4+PD1−) showed significant presence in both NDES and IP CD40/PDL1 + Rad compared with Ctrl UnTx, Ctrl NDES PBS, and Rad only (Fig. 5C). There was no significant difference in exhausted CD4 T cells (CD4+PD1+) amongst the groups (Figure 5C). Additionally, an increase of intratumoral DCs was observed in both IP CD40/PDL1+Rad (P=0.07) and NDES CD40/PDL1+Rad (P=0.07), relative to Ctrl UnTx, Ctrl NDES PBS and Rad only (Figure 5D). Moreover, NDES CD40/PDL1+Rad augmented both CD8 and effector CD4 T cell infiltration in TIME compared to IP CD40/PDL1+Rad.
NDES CD40/PDL1 + Rad and IP CD40/PDL1 + Rad cohorts had pronounced reduction in cancer cells (Ki67+PanCK+) compared with Ctrl UnTx, Ctrl NDES PBS, and Rad only (Fig. 5E), suggesting effective tumor cell elimination. Compared with nonirradiated cohorts, radiation treatment resulted in decreased intratumoral angiogenesis, as observed via endothelial cell (αSMA+) reduction (Fig. 5F).
Concurrently, IP CD40/PDL1 + Rad and NDES CD40/PDL1 + Rad exhibited significantly more inflammatory myeloid cells (CD11b+ and CD11b+Ly6G+) compared with Ctrl UnTx, Ctrl NDES PBS, and Rad only (Fig. E7). However, an excessive number of myeloid cells could induce an adverse immunosuppressive effect and inadvertently promote tumor development.55 Thus, further investigation of myeloid cells within TIME of NDES and IP CD40/PDL1 + Rad is required to fully elucidate their roles. Overall, spatial intratumoral immune cell distribution analysis suggests that NDES and IP CD40/PDL1 + Rad treatment promoted DC infiltration and effector CD8 and CD4 T cell mobilization to the TIME, leading to a decrease in cancer cells and consequently tumor regression.
Neighborhood analysis revealed DC-CD8 T cell interaction in NDES CD40/PDL1 + Rad
Neighborhood analysis was performed to examine the prevalence of cell-cell interaction and cell microenvironment (Fig. 6 and Fig. E7). The neighboring interaction between each pair of phenotypes was visualized as a heatmap, with rows representing cell phenotype neighborhood and columns as either enrichment or depletion within other neighborhoods.40 DCs (CD11c+) and cytotoxic T cell interaction was enriched (black solid box; row 12, column 3) in NDES and IP CD40/PDL1 + Rad compared with Ctrl, Ctrl NDES PBS, and Rad only (Fig. 6 and Fig. E8). Concurrently, a higher interaction of exhausted T cells and DCs was found in IP CD40/PDL1 + Rad compared with NDES CD40/PDL1 + Rad (black box with dashed lines; row 12, column 1). Furthermore, cancer cells (Ki67+PanCK+pSTAT1+) showed enrichment of cytotoxic T cells in NDES CD40/PDL1 + Rad. This interaction was inversed in Rad only and neutral in IP Cd40/PDL1 + Rad, Ctrl UnTx, and Ctrl NDES PBS (green solid circle; row 14, column 3) (Fig. 6 and Fig. E8). Immune suppressive cell types, including M2 macrophages (CD163+F4/80+) and neutrophils (CD11b+Ly6G+), were distinct from proliferative cancer cells (Ki67+PanCK+) in NDES CD40/PDL1 + Rad and IP CD40/PDL1 + Rad (purple solid box; rows 14 and 15, columns 8 and 10, respectively), relative to Ctrl UnTx, Ctrl NDES PBS, and Rad only groups. Notably, the M1-like macrophage (F4/80+) showed higher interaction with cancer cells (Ki67+PanCK+pSTAT1+) in NDES CD40/PDL1 + Rad but demonstrated lower interaction in IP CD40/PDL1 + Rad (purple solid box; row 14, column 9) (Fig. 6 and Fig. E8). This suggests that CD40/PDL1 delivered by either IP or NDES augmented the interaction of CD8 T cells and DCs in local immune response and avoided inherent immunosuppressive cells in TIME.
Fig. 6.
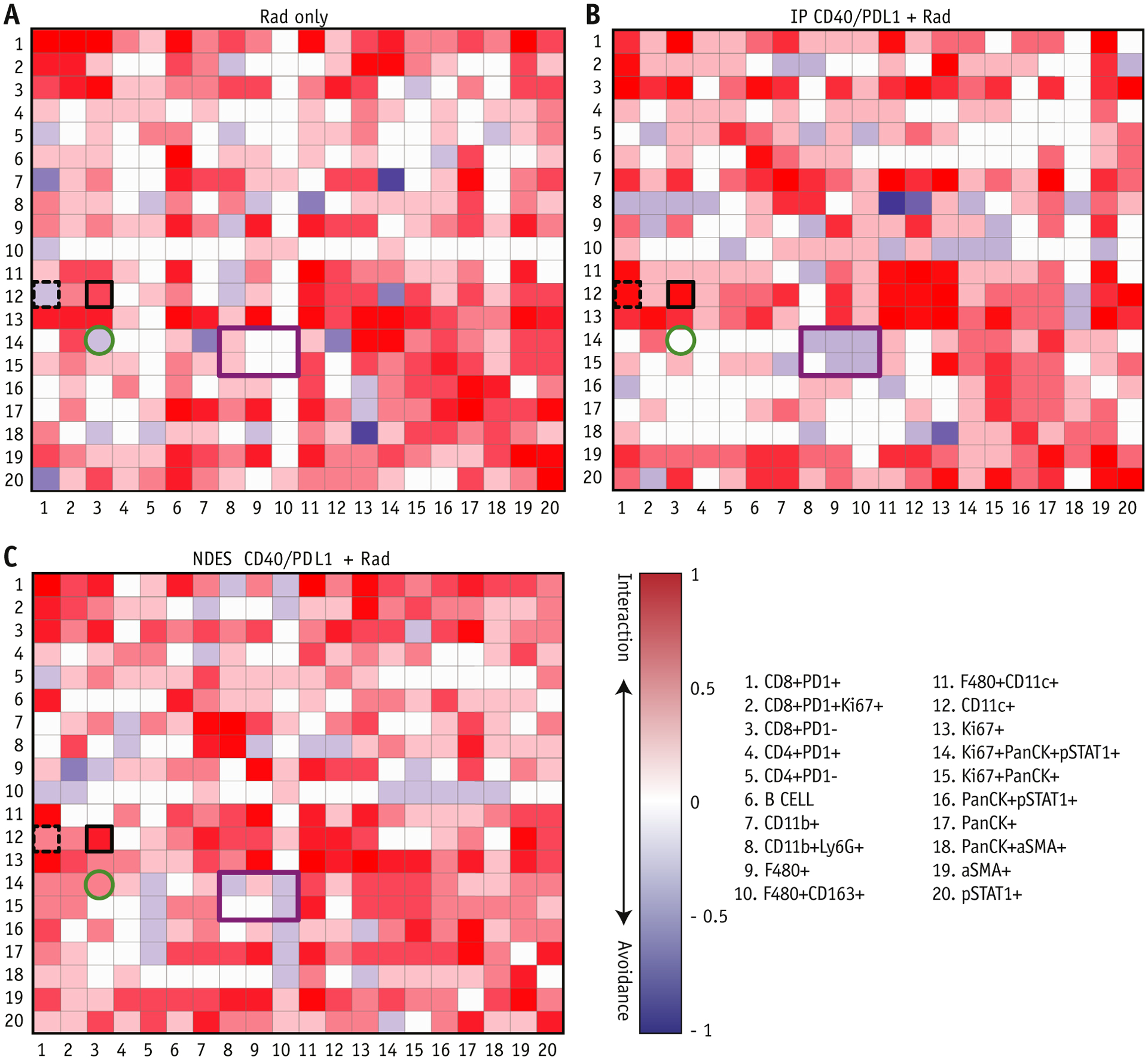
Neighborhood analysis of Rad only, intraperitoneal (IP) CD40/PDL1 + Rad, and nanofluidic drug-eluting seed (NDES) CD40/PDL1 + Rad groups. Cellular interactions present in (A) Rad only, (B) IP CD40/PDL1 + Rad, and (C) NDES CD40/PDL1 + Rad from imaging mass cytometry are represented as a heatmap. The cell type in the row is significantly neighbored (pink) or avoided (blue) by the cell type in the column. Highlighted squares indicate examples of directional interaction: phenotype #12 significantly surrounds cell type #3 (black box). IP CD40/PDL1 + Rad and NDES CD40/PDL1 + Rad DCs (CD11c +) (row 12) showed stronger interaction with CD8+ effector T cells (CD8+PD1−) (column 3) compared with Rad only; however, the DCs in IP CD40/PDL1 + Rad also showed an increased interaction with PD1+CD8+ T cells (column 1; black box with dashed lines) compared with NDES CD40/PDL1 + Rad. The cancer cells (row 14) in NDES CD40/PDL1 + Rad showed an increased interaction with CD8+ effector cells (column 3) compared with other groups. Also, cancer cells (row 14 and 15) avoided interaction with the possibly negative effect of CD11b+Ly6G+ and M2-like macrophage (F4/80+CD163+) (column 8 and 10) in NDES and IP CD40/PDL1 + Rad. However, an increased interaction of cancer cells and macrophages (column 9) was seen in NDES CD40/PDL1 + Rad (purple box).
Discussion
Our results are of fundamental clinical significance because TNBC is associated with high metastatic burden, and current therapeutic strategies remain dismal. Of relevance, atezolizumab, a PDL1 inhibitor, represents the first Food and Drug Administration–approved immunotherapy for triple negative breast cancer.56 Thus, we posit that combination of immunotherapy with RT could impart a superior clinical response.
The abscopal effect is historically considered a clinically rare achievement, with isolated cases observed with RT.6 Abscopal regression is dependent on generating local immune responses that translate systemically to target distant lesions. As such, rationally, the combination of immunotherapy with RT is an emerging strategy to boost the abscopal response. For example, in patients with follicular lymphoma, low-dose radiation combined with an intratumorally delivered Toll-like receptor-9 agonist, SD-101, demonstrated high efficacy in inducing abscopal response.57 Furthermore, combining RT with CD4058 or CTLA44 has been shown to enhance the abscopal effects in lung cancer and melanoma, respectively. In line with this, we showed that RT in combination with intratumorally delivered CD40 and PDL1 effectively induced an abscopal effect, resulting in substantial attenuation of spontaneous lung metastasis and prolonged survival in the 4T1 model of TNBC.
Minimizing treatment-associated adverse effects without compromising therapeutic efficacy is a clinically elusive goal thus far, particularly for immunotherapy, where toxicities are treatment limiting or sometimes fatal. Here, another key aspect of our work is that sustained local treatment via the NDES effectively reduced tumor burden to a similar extent as systemic treatment without inducing overt toxicity. In this study, we used readily detectable physiological parameters of body weight and temperature, as well as liver inflammation at study endpoint, as surrogate markers of toxicity. However, we do not preclude the possibility of subclinical toxicity, which may have been present but undetected. Immune-related adverse events or treatment-related toxicities are difficult to demonstrate in murine models and present a longstanding challenge for clinical translatability. In addition, observable adverse effects may not have manifested in this preclinical model owing to the brief experimental timeline in comparison with a human clinical course of disease.59
We demonstrated indirect evidence of induced systemic response as an effect of local treatment through increased splenic Granzyme B+ CD8+ T cells. Although we did not pursue experiments to show tumor-specific immune reactivity, a stark reduction in spontaneous lung metastasis was observed in the NDES treatment groups. These systemic effects were substantial and indicative of an abscopal response because our treatment was completely localized. However, further studies are needed to more definitively demonstrate tumor-specific immune reactivity. These studies include in vivo investigations involving either bilateral tumors with treatment of 1 tumor only or tumor rechallenge experiments. Thus, we do not preclude the possibility that NDES treatment enhances local control by RT, preventing the tumor from metastasizing.
Leveraging our sustained intratumoral delivery platform, we obviate the need for repeated interventions conventional to that of immunotherapy administration. Furthermore, by confining the drug locally, a smaller overall dosage is needed, equivalent to that of microdosing. Envisioned to be implanted intratumorally as a neoadjuvant therapy before tumor resection, the NDES represents a technological advancement that could transform the treatment regimen of locally advanced and metastatic solid tumors. Using existing clinical trocar methods for brachytherapy seed insertion, we anticipate rapid translation to treat solid tumors beyond that of TNBC.
Conclusions
When combined with hypofractionated RT, sustained intratumoral delivery of CD40 and PDL1 via NDES effectively modulated the TIME, with the recruitment of DCs and CD8 T cells. These immunologic responses demonstrate “cold” to “hot” tumor transformation, which translated to tumor burden reduction and abscopal regression of metastasis. Overall, our treatment strategy offers promise for enhancing therapeutic index and transforming the landscape of solid tumor therapy.
Supplementary Material
Acknowledgments—
We thank Dr Jianhua (James) Gu from the electron microscopy core; Dr Andreana L. Rivera, Yulan Ren, and Sandra Steptoe from the research pathology core of HMRI; Dr David L. Haviland and Nicole Vaughn from the Flow Cytometry Core of HMRI; and ImmunoMonitoring Core at the Immunotherapy Center and Cancer Center, HMRI.
Funding support from the Nancy Owens Breast Cancer Foundation (A.G.), Golfers Against Cancer (A.G.), and the development of the nanochannel membrane was funded by NIH-NIGMS R01GM127558 (A.G.). A.G. and his research group received support through the Frank J. and Jean Raymond Centennial Chair Endowment. This study was partially supported by Emily Hermann endowed chair fund to S.-H.C., and the following grants to SHC (NIH grant nos. R01CA127483, R01CA204191, R01CA208703, Houston Methodist Cancer Center 2019 High Impact in Cancer Research Grant Award) and DoD grant no. BC191397P1 (S.-H.C. & A.G.).
Disclosures:
X.L. reports a patent null pending. S.D. reports grants from Nanobiotix, grants and other from Lytix Biopharma, other from EMD Serono, and other from Mersana Therapeutics, outside the submitted work. A.G. reports a U.S. patent application no. 62/161,986 pending.
Footnotes
Supplementary material for this article can be found at https://doi.org/10.1016/j.ijrobp.2020.07.2326.
References
- 1.Kang J, Demaria S, Formenti S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer 2016;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keung EZ, Ukponmwan EU, Cogdill AP, Wargo JA. The rationale and emerging use of neoadjuvant immune checkpoint blockade for solid malignancies. Ann Surg Oncol 2018;25:1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marciscano AE, Walker JM, McGee HM, et al. Incorporating radiation oncology into immunotherapy: Proceedings from the ASTRO-SITC-NCI immunotherapy workshop. J Immunother Cancer 2018;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demaria S, Coleman CN, Formenti SC. Radiotherapy: Changing the game in immunotherapy. Trends Cancer 2016;2:286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer 2018;18:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol 2015;25:11–17. [DOI] [PubMed] [Google Scholar]
- 8.Wallner PE, Anscher MS, Barker CA, et al. Current status and recommendations for the future of research, teaching, and testing in the biological sciences of radiation oncology: Report of the American Society for Radiation Oncology Cancer Biology/Radiation Biology Task Force, executive summary. Int J Radiat Oncol Biol Phys 2014;88:11–17. [DOI] [PubMed] [Google Scholar]
- 9.Hiniker SM, Chen DS, Reddy S, et al. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol 2012;5:404–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012; 366:925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stamell EF, Wolchok JD, Gnjatic S, Lee NY, Brownell I. The abscopal effect associated with a systemic anti-melanoma immune response. Int J Radiat Oncol Biol Phys 2013;85:293–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Formenti SC, Rudqvist N-P, Golden E, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med 2018;24: 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoushtari AN, Friedman CF, Navid-Azarbaijani P, et al. Measuring toxic effects and time to treatment failure for nivolumab plus ipilimumab in melanoma. JAMA Oncol 2018;4:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson JA, Schneider BJ, Brahmer J, et al. Management of immunotherapy-related toxicities, version 1.2019. J Natl Compr Canc Netw 2019;17:255–289. [DOI] [PubMed] [Google Scholar]
- 15.Aznar MA, Tinari N, Rullan AJ, Sanchez-Paulete AR, Rodriguez-Ruiz ME, Melero I. Intratumoral delivery of immunotherapy-act locally, think globally. J Immunol 2017;198:31–39. [DOI] [PubMed] [Google Scholar]
- 16.Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: Using the tumor as the remedy. Ann Oncol 2017;28:xii33–xii43. [DOI] [PubMed] [Google Scholar]
- 17.van Herpen CM, De Mulder PH. Locoregional immunotherapy in cancer patients: Review of clinical studies. Ann Oncol 2000;11:1229–1239. [DOI] [PubMed] [Google Scholar]
- 18.Weide B, Derhovanessian E, Pflugfelder A, et al. High response rate after intratumoral treatment with interleukin-2: Results from a phase 2 study in 51 patients with metastasized melanoma. Cancer 2010;116: 4139–4146. [DOI] [PubMed] [Google Scholar]
- 19.Ray A, Williams MA, Meek SM, et al. A phase I study of intratumoral ipilimumab and interleukin-2 in patients with advanced melanoma. Oncotarget 2016;7:64390–64399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danielli R, Patuzzo R, Di Giacomo AM, et al. Intralesional administration of L19-IL2/L19-TNF in stage III or stage IVM1a melanoma patients: Results of a phase II study. Cancer Immunol Immunother 2015;64:999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sagiv-Barfi I, Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013;39:782–795. [DOI] [PubMed] [Google Scholar]
- 23.Irenaeus SMM, Nielsen D, Ellmark P, et al. First-in-human study with intratumoral administration of a CD40 agonistic antibody, ADC-1013, in advanced solid malignancies. Int J Cancer 2019;145:1189–1199. [DOI] [PubMed] [Google Scholar]
- 24.Milling L, Zhang Y, Irvine DJ. Delivering safer immunotherapies for cancer. Adv Drug Deliv Rev 2017;114:79–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwong B, Liu H, Irvine DJ. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials 2011;32:5134–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chua CYX, Jain P, Susnjar A, et al. Nanofluidic drug-eluting seed for sustained intratumoral immunotherapy in triple negative breast cancer. J Control Release 2018;285:23–24. [DOI] [PubMed] [Google Scholar]
- 27.Hood RL, Bruno G, Jain P, et al. Nanochannel implants for minimally-invasive insertion and intratumoral delivery. J Biomed Nanotechnol 2016;12:1907–1915. [DOI] [PubMed] [Google Scholar]
- 28.Vonderheide RH. The immune revolution: A case for priming, not checkpoint. Cancer Cell 2018;33:563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front Pharmacol 2017;8:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knorr DA, Dahan R, Ravetch JV. Toxicity of an Fc-engineered anti-CD40 antibody is abrogated by intratumoral injection and results in durable antitumor immunity. Proc Natl Acad Sci USA 2018;115:11048–11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res 2013;19:1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pillai RN, Behera M, Owonikoko TK, et al. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer: A systematic analysis of the literature. Cancer 2018;124:271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018;379: 2108–2121. [DOI] [PubMed] [Google Scholar]
- 34.Di Trani N, Silvestri A, Sizov A, et al. Electrostatically gated nanofluidic membrane for ultra-low power controlled drug delivery. Lab Chip 2020;20:1562–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geninatti T, Small E, Grattoni A. Robotic UV-Vis apparatus for long-term characterization of drug release from nanochannels. Meas Sci Technol 2014;25. 10.1088/0957-0233/25/2/027003. [DOI] [Google Scholar]
- 36.Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 2009; 15:5379–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao H, Korn JM, Ferretti S, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature Med 2015;21:1318–1325. [DOI] [PubMed] [Google Scholar]
- 38.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92:205–216. [DOI] [PubMed] [Google Scholar]
- 39.Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res 2007;67:3970–3975. [DOI] [PubMed] [Google Scholar]
- 40.Schapiro D, Jackson HW, Raghurman S, et al. histoCAT: Analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat Meth 2017;14:873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahonen CL, Wasiuk A, Fuse S, et al. Enhanced efficacy and reduced toxicity of multifactorial adjuvants compared with unitary adjuvants as cancer vaccines. Blood 2008;111:3116–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruno G, Di Trani N, Hood RL, et al. Unexpected behaviors in molecular transport through size-controlled nanochannels down to the ultra-nanoscale. Nat Commun 2018;9:1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrati S, Fine D, You J, et al. Leveraging nanochannels for universal, zero-order drug delivery in vivo. J Control Release 2013;172:1011–1019. [DOI] [PubMed] [Google Scholar]
- 45.Di Trani N, Pimpinelli A, Grattoni A. Finite-size charged species diffusion and pH change in nanochannels. ACS Appl Mater Interfaces 2020;12:12246–12255. [DOI] [PubMed] [Google Scholar]
- 46.DuPre SA, Redelman D, Hunter KW Jr. The mouse mammary carcinoma 4T1: Characterization of the cellular landscape of primary tumours and metastatic tumour foci. Int J Exp Pathol 2007;88:351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanpouille-Box C, Formenti SC, Demaria S. Toward precision radiotherapy for use with immune checkpoint blockers. Clin Cancer Res 2018;24:259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanpouille-Box C, Alard A, Aryankalayil MJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dovedi SJ, Illidge TM. The antitumor immune response generated by fractionated radiation therapy may be limited by tumor cell adaptive resistance and can be circumvented by PD-L1 blockade. Oncoimmunology 2015;4:e1016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong J, Le TQ, Massarelli E, Hendifar AE, Tuli R. Radiation therapy and PD-1/PD-L1 blockade: The clinical development of an evolving anticancer combination. J Immunother Cancer 2018;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deng L, Liang H, Burnette B, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124:687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dagoglu N, Karaman S, Caglar HB, Oral EN. Abscopal effect of radiotherapy in the immunotherapy era: Systematic review of reported cases. Cureus 2019;11:e4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi F, Wang X, Teng F, Kong L, Yu J. Abscopal effect of metastatic pancreatic cancer after local radiotherapy and granulocyte-macrophage colony-stimulating factor therapy. Cancer Biol Ther 2017;18:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Byun HK, Kim N, Yoon HI, et al. Clinical predictors of radiation-induced lymphopenia in patients receiving chemoradiation for glioblastoma: Clinical usefulness of intensity-modulated radiotherapy in the immuno-oncology era. Radiat Oncol 2019;14:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szebeni GJ, Vizler C, Nagy LI, Kitajka K, Puskas LG. Pro-tumoral inflammatory myeloid cells as emerging therapeutic targets. Int J Mol Sci 2016;17:1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atezolizumab combo approved for PD-L1-positive TNBC. Cancer Discov 2019;9:OF2. [DOI] [PubMed] [Google Scholar]
- 57.Frank MJ, Reagan PM, Bartlett NL, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov 2018;8:1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moeendarbari S, Tekade R, Mulgaonkar A, et al. Theranostic nano-seeds for efficacious internal radiation therapy of unresectable solid tumors. Scientific Reports 2016;6:20614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J, Blake SJ, Smyth MJ, Teng MW. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin Transl Immunology 2014;3:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.