

ABSTRACT
Cropping system diversity provides yield benefits that may result from shifts in the composition of root-associated bacterial and fungal communities, which either enhance nutrient availability or limit nutrient loss. We investigated whether temporal diversity of annual cropping systems (four versus two crops in rotation) influences the composition and metabolic activities of root-associated microbial communities in maize at a developmental stage when the peak rate of nitrogen uptake occurs. We monitored total (DNA-based) and potentially active (RNA-based) bacterial communities and total (DNA-based) fungal communities in the soil, rhizosphere, and endosphere. Cropping system diversity strongly influenced the composition of the soil microbial communities, which influenced the recruitment of the resident microbial communities and, in particular, the potentially active rhizosphere and endosphere bacterial communities. The diversified cropping system rhizosphere recruited a more diverse bacterial community (species richness), even though there was little difference in soil species richness between the two cropping systems. In contrast, fungal species richness was greater in the conventional rhizosphere, which was enriched in fungal pathogens; the diversified rhizosphere, however, was enriched in Glomeromycetes. While cropping system influenced endosphere community composition, greater correspondence between DNA- and RNA-based profiles suggests a higher representation of active bacterial populations. Cropping system diversity influenced the composition of ammonia oxidizers, which coincided with diminished potential nitrification activity and gross nitrate production rates, particularly in the rhizosphere. The results of our study suggest that diversified cropping systems shift the composition of the rhizosphere’s active bacterial and total fungal communities, resulting in tighter coupling between plants and microbial processes that influence nitrogen acquisition and retention.
IMPORTANCE Crops in simplified, low-diversity agroecosystems assimilate only a fraction of the inorganic nitrogen (N) fertilizer inputs. Much of this N fertilizer is lost to the environment as N oxides, which degrade water quality and contribute to climate change and loss of biodiversity. Ecologically inspired management may facilitate mutualistic interactions between plant roots and microbes to liberate nutrients when plants need them, while also decreasing nutrient loss and pathogen pressure. In this study, we investigate the effects of a conventional (2-year rotation, inorganic fertilization) and a diversified (4-year rotation, manure amendments) cropping system on the assembly of bacterial and fungal root-associated communities, at a maize developmental stage when nitrogen demand is beginning to increase. Our results indicate that agricultural management influences the recruitment of root-associated microbial communities and that diversified cropping systems have lower rates of nitrification (particularly in the rhizosphere), thereby reducing the potential for loss of nitrate from these systems.
KEYWORDS: agricultural management, nitrification, rhizosphere-inhabiting microbes
INTRODUCTION
Global maize (Zea mays L.) production (tons/year) exceeds all other crops (1). Over the last 50 years, increasingly available synthetic nitrogen (N) fertilizers and pesticides have allowed farmers to reduce or eliminate annual crop rotations, such that maize is grown continuously or in rotation with only one additional crop. However, crops in these simplified, low-diversity cropping systems assimilate only a fraction of the N from fertilizers (10 to 50%) (2) and may have lost mutualistic interactions with soil microbes that enhance crop N-use efficiency (3). Thus, there is increased attention toward developing economically viable and environmentally friendly agroecosystems that reduce the need for synthetic N fertilizer and herbicides by increasing crop rotation diversity and integrating livestock (4–6). Ecologically inspired management may facilitate mutualistic interactions between plant roots and microbes, increasing the depolymerization of soil organic matter (7–9) to liberate nutrients, decrease pathogen pressure (10), stimulate plant growth (11), and/or facilitate stress tolerance (12).
Plants provide a suitable environment for the soil microbiome by producing carbon (C)-rich rhizodeposits to stimulate microbial metabolic activity near the root (13–15). The root-associated microbiome shifts as the plant develops, likely a consequence of changes in rhizodeposition (14–17). For example, expression of N metabolism genes in the Arabidopsis thaliana rhizosphere microbiome changes with shifts in root exudation (16). Consequently, root exudates, which can diffuse up to 5 mm from the root surface, appear to shape the rhizosphere microbiome (15, 16, 18). Bacteria residing at the root surface are influenced by plant roots and the soil they interact with, and they may play a particularly important role in plant-microbiome-mediated nutrient uptake due to the advantage microbes have over plants for recently mobilized N (19). Moreover, rhizosphere microbial communities are further influenced by agricultural management (20–22), resulting in unique interactions between the effects of management on the microbial seed bank and the influence of plant rhizodeposition on microbiome assembly and function.
The influence of crop rotation diversity on soil and rhizosphere metabolic activity and microbiome assembly has been examined at the Marsden Long-Term Cropping System Experiment in the Midwest USA Corn-belt. The diversified system (4-year rotation with a reduction in tillage, pesticides, and inorganic fertilization) is equally productive as the conventional system (2-year rotation with inorganic fertilization) (4), and it has lower environmental N losses (23). It also supports higher microbial biomass, greater net production of bioavailable N from soil organic matter, and potentially mineralizable N (24–26). However, gross rates of ammonia production in the bulk soil (i.e., root-free) did not differ substantially between cropping systems, suggesting that plant-microbe interactions, rather than bulk plant-available soil N production, play an unspecified role in boosting yields in the diversified system (24). Following up on these observations, we showed that management influenced the assembly of prokaryotic and fungal communities near and on the root as the plant develops (27). Interestingly, bulk soil and rhizosphere prokaryotic communities in the conventional system were significantly different from each other only at select plant developmental stages (V11 and R2), suggesting there was a transient rhizosphere effect. This could be a consequence of differential exudation by roots, analogous to how soil type and plant development influence the rhizosphere effect (11, 28). Using arbuscular mycorrhizal fungi (AMF)-proficient and -deficient corn, we showed that AMF aid plants in nitrate uptake but have little influence on the growth of ammonia oxidizers, regardless of cropping system (29).
Here we explore the following questions about the effects of diversified cropping systems on root microbiomes. (i) Does diversification influence the plant roots’ selection of the rhizosphere and endosphere microbiome? (ii) Does diversification impact rhizosphere priming and does this, in turn, influence the metabolic properties of the root-associated microbiome? (iii) If so, does this shift in metabolic capabilities influence N-cycling processes? We hypothesize that rhizosphere priming (18, 30, 31) is likely shaped by agricultural management effects on the microbial seed bank, resulting in shifts in community structure and their metabolic potential, and that these differences are greatest during periods of high plant nutrient demand (e.g., N). As a consequence of greater coupling between roots and microbes, we further predict that nitrification is lower in the rhizosphere than in the bulk soil of diversified agricultural systems. To test this hypothesis, we assessed the prokaryotic and fungal communities of the maize root-associated microbiome (rhizosphere and endosphere) and soil (no living root influence) in rhizotrons filled with soil from the conventional and diversified cropping system plots of our previous work (29–32). Plants were grown to the V4/V5 developmental stage, i.e., when the rate of maize nutrient uptake becomes greatest (32). Moreover, we complemented our assessment of the resident prokaryotic community (DNA-based) with the potentially metabolically active (RNA-based) prokaryotic community (33); this would provide insight into whether management influences rhizosphere priming, also because bacteria in close physical proximity of a root (e.g., rhizoplane) exhibit a high degree of selectivity in terms of residents and potentially active populations (34).
RESULTS
Diversified cropping systems change the root architecture of maize.
Roots of plants grown in soil from the diversified system exhibited a finer, more ramified architecture than those grown in soil from the conventional system (Table 1). Moreover, root/shoot ratios indicated that diversified cropping systems led to an increased allocation of plant biomass to roots. As reported previously for this site (4), we also observed that soil from the diversified cropping system had substantially lower nitrate pools (see Table S1 in the supplemental material), which may have stimulated greater root development in these soils.
TABLE 1.
Maize root architectural features
| Root trait | Mean ± SE |
P valuea | |
|---|---|---|---|
| Conventional | Diversified | ||
| Total no. of roots | 102 ± 13 | 169 ± 17 | 0.01 |
| Length of primary root (cm) | 11.2 ± 0.7 | 14.9 ± 1.5 | 0.09 |
| Vol of primary root (cm3) | 1350 ± 188 | 736 ± 112 | 0.03 |
| Total surface area of roots (cm2) | 23.1 ± 2.4 | 29.4 ± 1.7 | 0.04 |
| Maximum depth of root system (cm) | 19.1 ± 1.8 | 26.4 ± 3.2 | 0.07 |
| Maximum horizontal width of roots (cm) | 7.5 ± 0.7 | 10.3 ± 1.1 | 0.06 |
| Root dry wt (g) | 0.25 ± 0.03 | 0.19 ± 0.02 | 0.08 |
| Root/shoot ratio (g/g) | 0.11 ± 0.01 | 0.18 ± 0.02 | 0.01 |
Separate one-way ANOVAs were performed on each root trait (n = 16 for each cropping system).
Effects of cropping systems on bulk soil physicochemical properties. Download Table S1, PDF file, 0.08 MB (81.5KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Microbial community structure.
Cropping system and root proximity (i.e., whether samples originated from the bulk soil, rhizosphere, or endosphere) influenced prokaryotic and fungal community composition, with some significant interactions (Table 2). Prokaryotic and fungal communities separate clearly by proximity to the roots (horizontal spread), and less clearly by cropping system (vertical spread) (Fig. 1). The effect of the cropping system on prokaryotic communities was more clearly resolved at the RNA level than the DNA level, where soil, rhizosphere, and endosphere samples generally clustered separately by cropping system; similar trends were also observed in the separation by proximity to the roots (Fig. 1A and B). Likewise, the fungal community was more clearly resolved by root proximity, and to a lesser degree by cropping system (Fig. 1C).
TABLE 2.
Permutational multivariate analysis of variance of microbial β-diversity
| Factor | Prokaryotic |
Fungal |
||
|---|---|---|---|---|
| R 2 | P value | R 2 | P value | |
| Cropping system | 0.043 | <0.001 | 0.074 | <0.001 |
| Root proximitya | 0.327 | <0.001 | 0.326 | <0.001 |
| Cropping system × root proximitya | 0.024 | <0.001 | 0.054 | <0.001 |
| DNA vs RNA | 0.070 | <0.001 | ||
| Cropping system × DNA vs RNA | 0.006 | 0.070 | ||
| Root proximitya × DNA vs RNA | 0.035 | <0.001 | ||
| Cropping system × DNA vs RNA × root proximitya | 0.007 | 0.388 | ||
Root proximity tests whether there are differences between bulk soil, rhizosphere, and endosphere.
FIG 1.

Bray-Curtis dissimilarity distance principal-coordinate analysis (PCoA) plots of total bacterial resident (DNA-based) (A), metabolically active (RNA-based) (B), and total fungal (C) resident community profiles of maize grown in bulk soil from conventional (red) and diversified (blue) cropping systems. Symbols: ●, bulk soil; ■, rhizosphere; ▲, endosphere.
Microbial diversity.
The diversity of the microbial communities differed by proximity to the roots and by cropping system (Fig. 2 and Table S2). Root proximity tended to decrease prokaryotic species richness and to increase evenness. On the other hand, interpretation of whether diversified systems altered these two diversity indices was influenced by whether relying on DNA- or RNA-based analyses (Fig. 2 and Table 3), with RNA-based profiles indicating greater prokaryotic richness in diversified systems. Moreover, DNA-based prokaryotic species richness was also greater in the diversified system, with the exception that the bulk soil had higher richness in the DNA-based analyses. In contrast, the overall trend was the opposite for fungi, and the richness in the rhizosphere was significantly greater in the conventional system (Fig. 2). Evenness in the bulk soil differed between cropping systems for the RNA- but not DNA-based analyses, with a slight trend for increased evenness in the rhizosphere and endosphere of plants grown in the diversified cropping system. While the endosphere had a lower richness overall (both for prokaryotes and fungi), its community was more even, with little difference between DNA- and RNA-based analyses for either cropping system.
FIG 2.

Richness (observed and Chao1) and Simpson’s evenness diversity indices in bulk soil, rhizosphere, and endosphere. The prokaryote metabolically active community (red), prokaryote total resident community (black), and fungi total resident community (green) are shown. Asterisks indicate statistically significant differences between cropping systems based on a Fisher’s LSD (P < 0.05) post hoc test. While not visible, evenness in the soil prokaryotic community was greater in the diversified than the conventional cropping system. ▴, diversified; ●, conventional.
TABLE 3.
Contrasts comparing prokaryotic DNA- and RNA-based α-diversity indicesa
| Root proximity |
P value |
|||||
|---|---|---|---|---|---|---|
| Observed |
Chao1 |
Simpson |
||||
| Conv | Div | Conv | Div | Conv | Div | |
| Bulk soil | 0.008 | <0.001 | 0.008 | <0.001 | <0.001 | <0.001 |
| Rhizosphere | 0.050 | <0.001 | 0.431 | 0.012 | 0.186 | 0.803 |
| Endosphere | 0.001 | 0.183 | 0.198 | 0.981 | 0.951 | 0.405 |
Conv and Div refer to conventional and diversified cropping systems, respectively.
Analysis of variance of α-diversity indices. Download Table S2, PDF file, 0.1 MB (107.4KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential abundance of prokaryotic communities.
We performed linear discriminant effect size (LEfSe) analyses (35) at various taxonomic ranks to reveal how cropping system influenced prokaryotic communities (Fig. 3 and 4) and whether the total resident (DNA-based) and potentially metabolically active (RNA-based) community profiles differed (Fig. 5). Family level cladograms displaying significant enrichment of taxa (linear discriminant analysis [LDA], P < 0.05) are strikingly different, regardless of cropping system and DNA-/RNA-based profiles, most notably in the rhizosphere (Fig. 3 and 4). Cropping systems influenced the correspondence between DNA- and RNA-based profiles, particularly the bulk soil and rhizosphere communities (Fig. 5). While plant selection for taxa was generally similar, unique taxa were preferentially enriched in each cropping system.
FIG 3.

LDA effect size cladograms comparing the total resident (DNA-based) and metabolically active (RNA-based) prokaryotic community profiles. Separate analyses were performed on bulk soil, maize rhizosphere, and maize endosphere. Circles represent phylogenetic levels from kingdom to family from the center outwards. The colored nodes and shading denote significant differences in relative abundance between specific taxa in the conventional (red) or diversified (green) system; yellow node, no significant difference. 1, Alphaproteobacteria (1a, Sphingomonadaceae and Erythrobacteraceae; 1b, Burkholderiales; 1c, Rhizobiaceae; 1d, Bradyrhizobiaceae); 2, Betaproteobacteria; 3, Pseudomonadales; 4, Thaumarchaeota; 5, Actinobacteria; 6, Rubrobacteria; 7, Bacteroidetes; 8, Cyanobacteria; 9, Nitrospiraceae; 10, Nitrosomonadaceae; 11, Deltaproteobacteria; 12, Euryarchaeota; 13, Acidobacteria; 14, Elusimicrobia; 15, Planctomycetes; 16, Spirochaetae; 17, Verrucomicrobia; 18, Flavobacteria; 19, Ktedonobacteria; 20, Holophagae; 21, Chloroflexi.
FIG 4.

LDA effect size cladograms comparing the conventional and diversified cropping system prokaryotic community profiles. Separate analyses were performed on soil, maize rhizosphere, and maize endosphere. Colored nodes and shading denote significant differences in relative abundance between specific taxons in the total (red) or metabolically active (green) community; yellow node, no significant difference. 1, Proteobacteria (1a, Betaproteobacteria; 1b, Deltaproteobacteria; 1c, Gammaproteobacteria; 1d, Alphaproteobacteria); 2, Planctomycetes; 3, Nitrospirae; 4, Gemmatimonadetes; 5, Firmicutes; 6, Chloroflexi; 7, Bacteroidetes; 8, Solirubrobacterales; 9, Rubrobacteria; 10, Frankiales; 11, Acidimicrobiia; 12, Holophagae; 13, Thaumarchaeota; 14, Euryarchaeota; 15, Verrucomicrobia; 16, Fibrobacteres; 17, Cyanobacteria; 18, Chlorobi; 19, Chlamydiae; 20, Actinobacteria; 21, Sphingobacteriia; 22, Acidobacteria; 23, Cytophagia.
FIG 5.
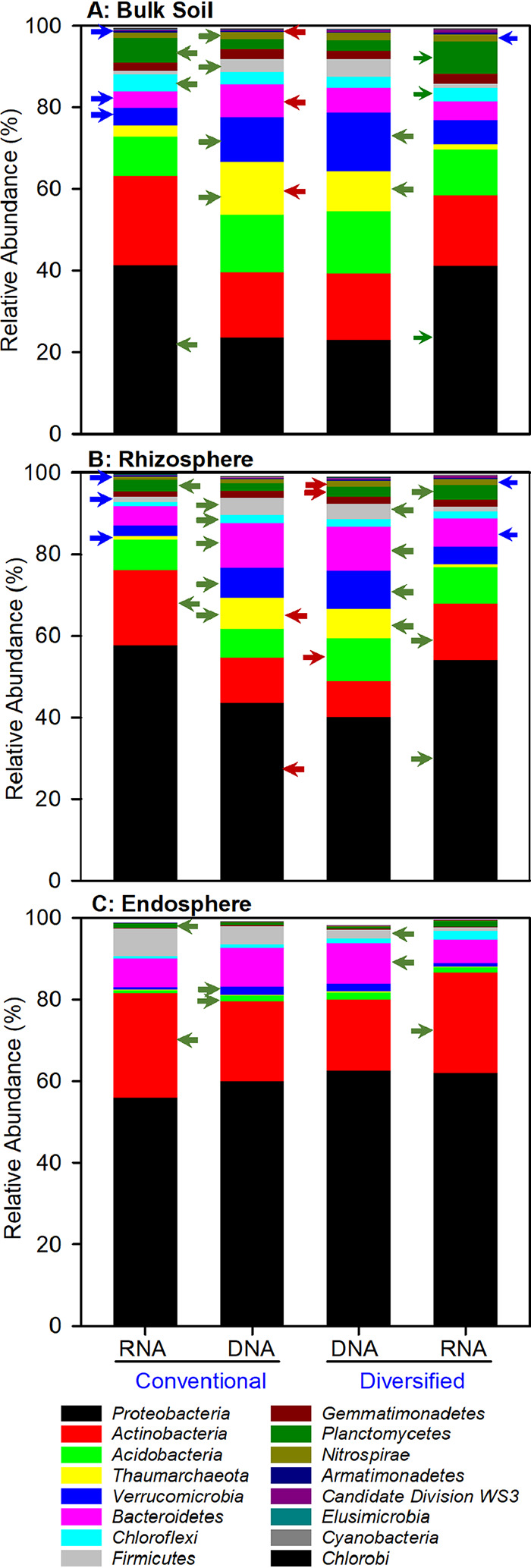
Phylum-level relative abundance of DNA- and RNA-based prokaryotic community profiles in conventional and diversified cropping systems. Red arrows identify statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundances between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different (LDA, P < 0.05) RNA-based relative abundances between cropping systems. Low-abundance phyla are not included.
Bulk soil.
The bulk soil of each cropping system harbored distinct DNA- and RNA-based community profiles (Fig. 3, 4, and 5). At the DNA level, there were more differentially abundant phyla and families in the conventional soil of which 59% (30 out of 51 families) were Proteobacteria and Actinobacteria families (Table 4 and Fig. 6A and D; see also Fig. S1A and S1D and Fig. S2A and S2D in the supplemental material). Twelve out of the 19 differentially abundant proteobacterial families were Alphaproteobacteria, primarily of the order Rhizobiales. In contrast, in the diversified system, there were fewer differentially abundant families, representing fewer phyla; Proteobacteria and Actinobacteria represented ∼40% of the differentially abundant taxa (Table 4), whereas no proteobacterial class dominated (Fig. 3, Fig. 6A and D, Fig. S1A and S1D, and Fig. S2A and S2D). The Actinobacteria and Acidobacteria contributed uniquely to differences between systems, with 8 differentially abundant Acidobacteria families in the diversified system compared to 11 actinobacterial families in the conventional system (Fig. S2A and S2D); many were of low abundance. While there were only a few differences in Planctomycetes and Verrucomicrobia (Fig. S3A and S3D), the conventional system had more differentially abundant Bacteroidetes (Fig. S4A), whereas the diversified system had more differentially abundant Firmicutes families (Fig. S4D).
TABLE 4.
Number of differentially abundant taxa at various taxonomic ranksa
| Communityb | Cropping system | Bulk soil |
Rhizosphere |
Endosphere |
|||
|---|---|---|---|---|---|---|---|
| Phylum/classc | Familyd | Phylum/class | Family | Phylum/class | Family | ||
| DNA | Conventional | 6 | 51 (13) | 2 | 41 (9) | 0 | 8 (3) |
| Diversified | 1 | 43 (10) | 7 | 72 (15) | 1 | 17 (7) | |
| RNA | Conventional | 0 | 30 (8) | 0 | 23 (5) | 0 | 9 (4) |
| Diversified | 3 | 41 (12) | 6 | 46 (13) | 1 | 14 (7) | |
| ITS | Conventional | 4 | 16 (4) | 5 | 22 (4) | 1 | 2 (1) |
| Diversified | 3 | 12 (4) | 3 | 7 (3) | 1 | 2 (1) | |
Based on individual LEfSe analyses comparing the conventional and diversified cropping systems.
DNA, RNA, and ITS refer to the total resident prokaryotic, potentially metabolically active prokaryotic, and total resident fungal communities, respectively.
Differentially abundant bacterial phyla or fungal classes.
Values in parentheses are the number of phyla represented by the differentially abundant families.
FIG 6.

DNA- and RNA-based family level relative abundance of Alphaproteobacteria (A to C) and Betaproteobacteria (D to F) in conventional and diversified cropping systems. Red, green, and blue arrows are as defined in the legend to Fig. 5. An asterisk over two adjacent bars indicates statistically significant differences between RNA- and DNA-based analyses. Low-abundance families are not visible.
DNA- and RNA-based family level relative abundance of Deltaproteobacteria (A to C) and Gammaproteobacteria (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance (<0.02%) Delta- and Gammaproteobacteria families are not visible. Download FIG S1, JPG file, 1.1 MB (1.1MB, jpg) .
{kind=link}
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA- and RNA-based family level relative abundance of Actinobacteria (A to C) and Acidobacteria (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance Actinobacteria (<0.08%) and Acidobacteria (<0.05% for soil and rhizosphere; <0.01% for endosphere) families are not visible. Download FIG S2, JPG file, 2.0 MB (2MB, jpg) .
{kind=link}
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA- and RNA-based family level relative abundance of Planctomycetes (A to C) and Verrucomicrobia (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance Planctomycetes (<0.05% for soil and rhizosphere; <0.005% endosphere) and Verrucomicrobia (<0.05% for soil and rhizosphere; <0.02% for endosphere) families are not visible. Download FIG S3, JPG file, 1.4 MB (1.4MB, jpg) .
{kind=link}
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA- and RNA-based family level relative abundance of Bacteroidetes (A to C) and Firmicutes (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance Bacteroidetes (<0.02%) and Firmicutes (<0.01%) families are not visible. Download FIG S4, JPG file, 1.4 MB (1.4MB, jpg) .
{kind=link}
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The RNA-based profiles revealed 33% more differentially abundant families representing 50% more phyla in the diversified bulk soil than in the conventional bulk soil (Table 4). The Proteobacteria, Actinobacteria, and Acidobacteria represent 51% of the differentially abundant taxa, the remainder of which are from nine other phyla (Table 4, Fig. 6A and D, Fig. S1A and S1D, and Fig. S2A and S2D). In the conventional bulk soil, these same three phyla comprise 73% of the differentially abundant families. Most of the differentially abundant Proteobacteria were Alphaproteobacteria (primarily Rhizobiales and Rhodospirillales), similar to what was observed with the DNA-based profiles (Fig. 6A). The increase in potentially metabolically active Betaproteobacteria was due primarily to the Comamonadaceae (Fig. 6D). There were few differences in the abundance of Deltaproteobacteria and Gammaproteobacteria (Fig. S1A and S1D), although Deltaproteobacteria were more abundant in the RNA-based profiles. The Actinobacteria were more potentially metabolically active in the conventional soils than in the diversified soils (Fig. S2A). As has been observed in various cropping systems (36, 37) and here in bulk soil and rhizosphere, the Planctomycetes were more abundant in the RNA- than DNA-based profiles (Fig. S3A and S3B), including the soil anammox clade OM190 (36). Few differences were found in Verrucomicrobia, Bacteroidetes, and Firmicutes (Fig. S3D and Fig. S4A and S4D).
Rhizosphere.
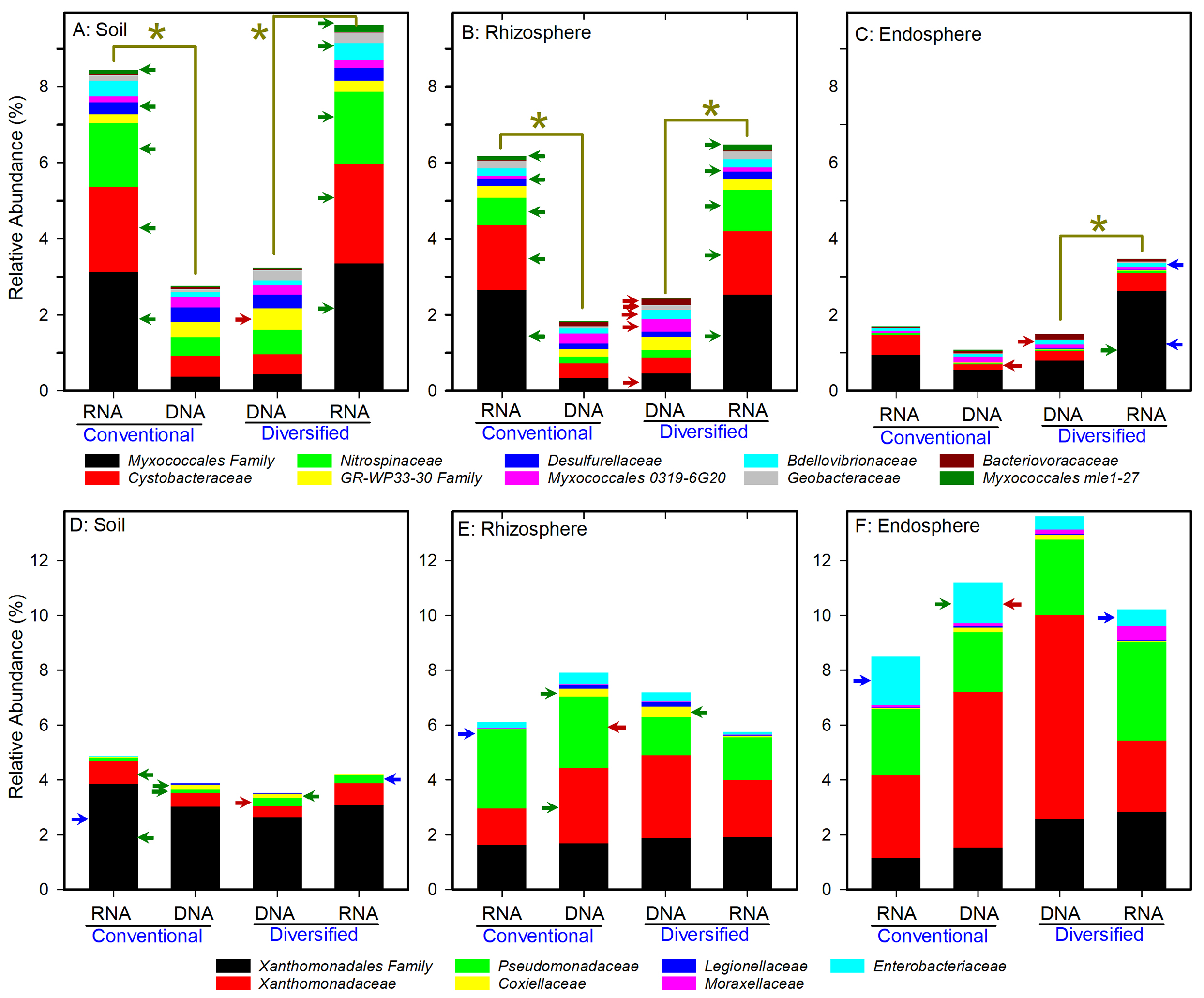
The rhizosphere of each cropping system harbored distinct DNA- and RNA-based community profiles (Fig. 3, 4, and 5), and the differences were particularly pronounced compared to bulk soil (Table 4). At the DNA level, Actinobacteria, Acidobacteria, and Proteobacteria comprised 50% and 76% of the differentially abundant families in the diversified and conventional rhizospheres, respectively. A key commonality with bulk soil was the preponderance of differentially abundant alphaproteobacterial families in the conventional (15 out of 18 Proteobacteria) compared to the diversified (8 out of 23 Proteobacteria) rhizospheres (Fig. 6B and D and Fig. S1B and S1D), including a >3-fold increase in Rhizobiaceae (Fig. 6B). Relative to bulk soil, the greatest enrichment in the rhizosphere were the Betaproteobacteria, and unlike in soil, several Betaproteobacteria families were more abundant in the RNA-based profiles in the diversified system (Fig. 6E). The Comamonadaceae and Oxalobacteraceae of the Burkholderiales were enriched in the diversified and conventional rhizospheres, respectively, but not the Burkholderiaceae (Fig. 6E). As in bulk soil, the Deltaproteobacteria were highly metabolically active in the rhizosphere, with some families exhibiting 2- to 5-fold differences between RNA- and DNA-based relative abundance (Fig. S1B), including the nitrite-oxidizing Nitrospinaceae. There were more Pseudomonadaceae in the conventional rhizosphere; in bulk soil, Pseudomonadaceae were more abundant in the diversified system (Fig. S1E).
Compared to bulk soil, there was an ∼33% decrease in Actinobacteria in the rhizosphere but unlike bulk soil, there were significant increases in RNA relative abundance, particularly in the conventional system (Fig. S2B). Similarly, both cropping systems were enriched in metabolically active Actinobacteria families. Furthermore, in both DNA- and RNA-based profiles, there were more differentially abundant Acidobacteria in the diversified system, many of which were in low abundance (Fig. S2E). Relative to bulk soil, there was a modest overall decrease in the abundance of Planctomycetes and Verrucomicrobia in the rhizosphere, and a modest overall increase in Bacteroidetes and Firmicutes, with low-abundance taxa distinguishing the two systems for Bacteroidetes (Fig. S3B and S3E and Fig. S4B and S4E).
Endosphere.
DNA- and RNA-based community profiles in the endosphere were more congruent than in the rhizosphere or bulk soil (Fig. 3, 4, and 5). This is likely a consequence of plant selection for bacteria capable of surviving intercellular spaces and lack of influence of relic DNA on community profiles. Yet, despite these similarities, there were several notable differences, with 28 families (9 phyla) being more abundant in the diversified, compared to 10 families (3 phyla) in the conventional system (Table 4), although many were low-abundance taxa (<0.1%).
Increased representation of Proteobacteria in the endosphere, relative to the rhizosphere, was cropping system specific (Fig. 6C and F and Fig. S1C and S1F). The Rhizobiaceae was particularly enriched in the conventional endosphere, similar to what was observed in the rhizosphere (Fig. 6C). Additionally, the Comamonadaceae and Oxalobacteraceae were enriched in the diversified and conventional endospheres, respectively (Fig. 6F). The Xanthomonadaceae were enriched in the endosphere relative to the rhizosphere (Fig. S1C and S1F). While the RNA-based relative abundance of Proteobacteria did not differ between the two cropping systems (Fig. 5C), there was a dramatic shift in community composition, with more Beta- and Deltaproteobacteria in the diversified and more Alphaproteobacteria (particularly the Sphingomonadaceae) in the conventional system (Fig. 3, Fig. 6C and F, and Fig. S1C and S1F).
The endosphere was enriched in Actinobacteria (Micromonosporaceae, Streptomycetaceae, and Pseudonocardiaceae) compared to the rhizosphere and soil, comprising a substantial proportion of the potentially active community, particularly in the conventional endosphere (Fig. 5 and Fig. S2C). The effect of cropping system on the dynamic selection process for growth in planta is also reflected in the differential enrichment of Verrucomicrobia and Bacteroidetes families (Fig. S3F and S4C). The conventional endosphere, however, was depleted in Acidobacteria (Fig. S2F) and Planctomycetes (Fig. S3C). Finally, the endosphere was enriched in Paenibacillaceae, likely reflecting their enrichment in the rhizosphere (Fig. S4F).
Differential abundance of fungal communities.
Conventional and diversified soil were each uniquely enriched in various fungal families (Fig. 7 and Fig. S5). In the diversified bulk soil, there were fewer differentially abundant families (Table 4), of which 83% were Ascomycota (primarily Sordariomycetes) and Basidomycota (primarily Tremellomycetes and Mortierellomycetes). There was a pronounced cropping system effect in the rhizosphere (Fig. 7), leading to three times more differentially abundant fungal families in the conventional rhizosphere than in the diversified rhizosphere (Table 4), primarily Ascomycota and Basidiomycota. The conventional rhizosphere and endosphere were enriched in numerous phytopathogenic fungal families (Botryosphaeriaceae, Glomerellaceae, Diaporthaceae, Pleosporaceae, and Ustilaginaceae). Furthermore, the Glomeromycetes were significantly more abundant in the diversified rhizosphere than in the conventional rhizosphere. There were few differences between conventional and diversified endospheres; the conventional endosphere was enriched in plant pathogens, while the diversified endosphere was enriched in Trichocomaceae and Microdochiaceae, which are known saprophytes and plant pathogens, respectively.
FIG 7.

LDA effect size cladograms comparing the total resident (DNA-based) fungal community profiles, categorized by cropping system. Separate analyses were performed on bulk soil, maize rhizosphere, and maize endosphere. Circles represent phylogenetic levels from kingdom to family from the center outwards as described in the legend to Fig. 3. The colored nodes and shading denote significant difference in relative abundance between specific taxa in the conventional (red) or diversified (green) system; yellow node, no significant difference. A, Eurotiomycetes; B, Agaricomycetes; C, Cystobasidiomycetes; D, Tremellomycetes; E, Lobulomycetes; F, Spizellomycetes; G, unidentified class 14173; H, Dothideomycetes; I, Sordariomycetes; J, Ustilaginomycetes; K, Glomeromycetes.
Class level relative abundance of fungal community profiles in conventional and diversified cropping systems. Arrows indicate statistically different (LDA, P < 0.05), DNA-based relative abundance of a class between the two cropping systems. Phyla below 1% were summed. Phyla are abbreviated as follows: Ascom, Ascomycota; Basid, Basidiomycota; Morti, Mortierellomycota; Chytr, Chytridiomycota; Glome, Glomeromycota. Download FIG S5, JPG file, 0.5 MB (563.1KB, jpg) .
{kind=link}
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ammonia oxidizer community structure and nitrification.
For both the DNA- and RNA-based profiles, the Nitrosomonadaceae relative abundance was highest in bulk soil, and they were enriched in the diversified rhizosphere (Fig. 3 and 6D). There were more Thaumarchaeota in the conventional bulk soil, but there were no differences in the rhizosphere (Fig. 8A and B). While the Nitrospiraceae were enriched in the diversified rhizosphere, there was also enrichment of other Nitrospirae in both the diversified bulk soil and rhizosphere (Fig. 8C and D). We observed a cropping system but not rhizosphere effect in the enrichment of specific operational taxonomic units (OTUs). We also measured ammonia-oxidizing bacteria (AOB) and archaea (AOA) amoA gene abundance by quantitative PCR (qPCR). While it was not surprising to detect higher AOB amoA gene abundance in the conventional bulk soil than in the diversified bulk soil, given differences in inorganic N fertilizer inputs (4), it was interesting to observe a similar pattern in the rhizosphere (Fig. 9A). Our results also suggest that there is an enrichment of Thaumarchaeota in the rhizosphere, independent of cropping system effect (Fig. 9B and C), and that there may be more Nitrosomonadaceae in the conventional soil than expected based on relative abundance (Fig. 6D and 9A).
FIG 8.

DNA- and RNA-based family level relative abundance of Thaumarchaeota and Nitrospirae in bulk soil and the rhizosphere of conventional and diversified cropping systems. (A to D) Thaumarchaeota (A and B); Nitrospirae (C and D). Red, green, and blue arrows are as defined in the legend to Fig. 5. An asterisk indicates statistically significant difference at the phylum level. Low-abundance families are not visible.
FIG 9.

Root and cropping system influences on ammonia oxidizer abundance and nitrification. (A to F) AOB amoA gene abundance (A); AOA amoA gene abundance (B); AOB/AOA ratio (C); potential nitrification activity (D); gross nitrate production (15NO3 pool dilution) (E); nitrate pool sizes (F). Values are means plus standard errors of the means (SEM) (error bars). Separate two-way ANOVAs were performed on each treatment and are summarized in Table S3 in the supplemental material. Bars with different letters above the bars indicate significant differences at P < 0.05 based on Tukey’s HSD test.
Analysis of variance of ammonia oxidizers and their potential metabolic activity. Download Table S3, PDF file, 0.09 MB (94.6KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bulk soil nitrification potentials, gross nitrate production rates, and nitrate pool sizes did not differ substantially between cropping systems (Fig. 9D to F) at the V4/V5 developmental stage. In contrast, rhizosphere soils exhibited a cropping system effect, although the trend of lower gross nitrate production rates in the diversified rhizosphere was not statistically significant (Table S3). Despite no apparent correlation between Thaumarchaeota, Nitrosomonadaceae, or Nitrospira relative abundance and potential nitrification rates, patterns in nitrate pool sizes and gross nitrate production rates were more similar to the AOB/AOA amoA gene abundance ratio (Fig. 9C to F), indicating the relationship between these two groups of organisms could explain the lower levels of nitrate detected in subsurface water in the diversified system (23).
DISCUSSION
We investigated the effects of conventional (2-year rotation, inorganic fertilization) and diversified (4-year rotation, manure amendments) cropping systems on the assembly of the prokaryotic and fungal root-associated communities at a maize developmental stage when N demand is poised to increase. The premise was that plant selection for a microbial community facilitating nutrient acquisition or retention would likely be in place before the onset of rapid nutrient uptake by the plant. We assessed whether cropping system influenced rhizosphere priming of the prokaryotic community by comparing the total resident (DNA-based) with the potentially metabolically active (RNA-based) community profiles, as well as its effects on gross rates and potential of nitrification. While our extraction protocols are considered nonquantitative, differences in taxon relative abundances do reflect differences in community composition. The root-soil compartments examined reflect the continuum from the soil to the plant, an important consideration when exploring the phytobiome (38). Our results show that due to the influence of agricultural management on the soil microbial community, the rhizosphere and endosphere microbial communities of diversified systems are distinct, as are the potential metabolic activities of rhizosphere communities.
DNA-based profiles reflect a range of metabolic states (dormant, growing) and relic (dead) while RNA analyses reflect the potentially metabolically active state (potential for protein synthesis) (33). The potential for protein synthesis likely reflects the ability of an organism to rapidly respond to changes in the environment (33, 39). Compositional changes in RNA profiles occur in response to nutrient inputs, including root exudates (40–42), and active communities have been shown to correlate better with CO2 production following a rain event (43) than total community composition. Prokaryotic species richness was higher in the RNA- than DNA-based analyses, suggesting that RNA-based profiling may provide greater differentiation of how environmental factors influence community composition. The greater congruence between the DNA- and RNA-based profiles within and on the root may reflect a reduction in the potential influence of relic DNA on community profiles (44, 45) or the greater stimulation of metabolic activities by nutrients provided by the root (34, 40). This is consistent with a prior report of greater correspondence between DNA- and RNA-based profiles in the rhizoplane of maize and other plant species (34). Convergence of community indices (i.e., relative abundance and species richness/evenness) in the endosphere likely reflects the strong selective environment of living within plants (46–48). Thus, management influences the soil microbial seed bank from which the plant enriches specific populations via rhizodeposits in the rhizosphere, providing a niche-specific community from which endophytes are selected. Collectively, diversified systems promote greater prokaryotic species richness in the maize root-associated microbiome, possibly due to the effects of different crop species, manure amendments, and/or reduced tillage on microbial community structure (34, 47, 49, 50). This is consistent with reports showing that long-term chemical-only fertilization induces diversity decline of soil bacteria (51). In contrast, species richness of fungal rhizosphere communities was greater in conventional systems, suggesting management can influence fungal and prokaryotic species diversity differently.
Reduced N availability in the diversified system likely influences maize root architecture and, in turn, the rhizosphere microbial community structure. Fine roots are known to drive microbial community dynamics, as well as nutrient cycling and water acquisition (52, 53). Enhanced root formation may increase rhizodeposits, and with a larger, more ramified root system exploring a greater soil volume, more C is likely being provided to a larger proportion of soil microbes in the diversified system. This could partially explain the greater prokaryotic richness as well as differences in potential metabolic activity in the diversified rhizosphere. Consistent with this premise is the greater relative abundance of AMF (Glomeromycetes) in the diversified rhizosphere, although not within the root where arbuscules form. Whether there is greater AMF biomass in the rhizosphere, where fungal hyphae acquire nutrients, needs to be determined. These observations suggest that plants in diversified systems increase soil resource acquisition not only via roots, but also possibly via AMF that could tighten N coupling between plants and microbial processes (53), perhaps contributing to yield benefits in these systems. Future work should explore this further by better assessing AMF abundance and composition with AMF-specific primer sets as well as by measuring AMF hyphal biomass in the rhizosphere soil compared to bulk soil (29).
There were more prokaryotic families, representing a greater number of phyla, that were more abundant in the diversified rhizosphere than in the conventional rhizosphere, which likely influences differences in the assembly of the endosphere community. Enrichment of Acidobacteria, Bacteroidetes, and Verrucomicrobia families in the diversified rhizosphere compared to the conventional rhizophere is consistent with previous reports describing maize rhizoplane communities at this same experimental site (27). These organisms are known for the breakdown of complex organic materials, such as plant polysaccharides (54–57). In contrast, Proteobacteria and Actinobacteria were selectively enriched in the conventional rhizosphere, which likely influenced the preferential enrichment of the Proteobacteria in the endosphere. This is consistent with the notion that the conventional rhizosphere community is more reliant on simple C substrates (56). Considered together, this suggests that conventional systems are less suited to complex C decomposition and, by extension, ammonia production, which is consistent with previous findings about the organic matter dynamics at this site (4, 24, 26, 58).
In response to rhizodeposits, microbial growth in the rhizosphere increases, and this growth may stimulate microbial demand for N, leading to direct competition with the plant for N. This microbial demand for N could be met with increased N mineralization (ammonium production), decreased nitrification, or both (59–61). Prior work at the site used to collect soils for this study concluded that N mineralization likely did not contribute to the yield benefit of diversified cropping systems (24, 58). Here we show that nitrification (potential activity, gross rates) is suppressed more in the diversified rhizosphere than in the conventional rhizosphere at the V4/V5 developmental stage. This is consistent with another report indicating that potential nitrification activity is lower in the maize rhizosphere at an early reproductive stage (62) and with lower nitrate levels detected in soil water in the diversified system at this study site (23). Lower nitrification in the rhizosphere reflects a complex shift in nitrifier community structure, with proportionally more AOA relative to AOB in the rhizosphere than bulk soil, in a cropping system-specific manner. The trend of lower nitrification in the diversified rhizosphere is likely due to a combination of a shift toward a nitrifying community that is possibly more adept at subsisting on lower levels of ammonium, smaller populations of microbes that perform nitrification, and/or lower metabolic activity of nitrifying bacteria. While our data support a more efficient coupling of N supply with plant and microbial demand in the diversified rhizosphere, more in-depth exploration into how management and the rhizosphere influence N cycling activities is essential for confirming or refuting our conclusions.
In summary, we show that in a diversified cropping system at a stage at which the plant is poised for rapid nutrient uptake, the plant selected a more diverse prokaryotic community, with altered N cycling populations and their metabolism that may contribute to yield benefits and reduced N losses. While there is convergence in community structure during plant selection for bacterial endophytes, that structure is influenced by the rhizosphere community structure. In the diversified system, maize increased soil resource acquisition (more ramified root system, increased AMF relative abundance), which will alter the microbial habitat (sites for colonization, rhizodeposition patterns). One possible consequence of the altered habitat is the greater competition for ammonium during decomposition processes which could decrease nitrification. It is tempting to conclude that one mechanism by which the diversified system at the Marsden experimental site is able to support high productivity with reduced inorganic fertilization is more efficiently coupled C and N cycles. Yet this alone may not explain the yield benefit entirely, since plant health may be better in the diversified system due to decreased fungal pathogen pressure, as well as selection of a plant-associated microbiome which stimulates plant growth directly.
MATERIALS AND METHODS
The following is a summarized version of Materials and Methods used in this study; the detailed version is available as Text S1 in the supplemental material.
Detailed Materials and Methods. Download Text S1, PDF file, 0.2 MB (201.6KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Experimental site description, soil collection, and soil analyses.
Soil was collected from Iowa State University’s Marsden Long-term Cropping System Experiment, USA, in June 2014. Soils and management practices at the site have previously been described (4). We sampled from a conventional system (2-year rotation of maize and soybean) and a diversified system (4-year rotation of maize, soybean, oat/alfalfa, and alfalfa). There were four replicated blocks for each management system.
Five samples were aseptically collected in each corn plot; samples from the same plot were pooled and sieved; packing of rhizotrons occurred within 48 h of soil collection. Soil physicochemical properties are described in Table S1 in the supplemental material.
Rhizotrons, growth conditions, and sampling protocols.
For each cropping system, two rhizotrons were filled with soil from each block to the average bulk density of the upper soil layer at the sampling site. Two surface-sterilized, pregerminated maize seedlings were planted in each rhizotron. The growth conditions were monitored (65% water-holding capacity, 24°C, 16-h/8-h light/dark photoperiod) until the plants reached vegetative stage V4/V5.
Bulk soil samples were aseptically collected (∼1 g; >1 cm from a root), and the root system of a plant was then extracted. The rhizosphere was obtained by vortexing and sonicating the roots in phosphate buffer (27). After pooling and filtering the washates/sonicates, the filtrate was centrifuged. The pellet obtained was resuspended in sterile water, centrifuged, and decanted to obtain a rhizosphere sample. Prior to obtaining endosphere samples, soil-free roots were sonicated in sterile water and scanned for automatic root image analysis (63). They were then freeze-dried, weighed, and pulverized for endosphere sampling. All samples were kept at −80°C.
DNA and RNA extractions.
DNA and RNA were extracted from all samples with commercially available kits. After DNase treating the RNA extracts, first- and second-strand cDNA synthesis was performed. The DNA concentrations were determined by spectrophotometry, while the quality of the cDNA was assessed with a 2100 Bioanalyzer. All samples were stored at −80°C.
Amplicon sequencing and processing.
For the 16S rRNA V4 region, amplicon library preparation and sequencing using the 515/806R primers (64) were performed by Argonne National Laboratory, USA. For the fungal internal transcribed spacer (ITS) region, DNA was sequenced at the University of Minnesota Genome Center using the ITS1/ITS2 primers. Amplicons of 16S rRNA and ITS were paired end sequenced (250 bp) on Illumina MiSeq instruments in separate runs. Three replicates of bacterial or fungal mock community DNA (65) were included with the respective libraries.
Following the removal of alien sequences (e.g., adapters, mitochondria), quality filtering (Q25), sample sorting, OTU clustering, and the filtering out of chimeras/singletons (66), taxonomy was assigned to the 16S rRNA sequences (67). They were then aligned (68), and a phylogenetic tree was produced in QIIME. A total of 5,453,802 reads were generated, with a median sequence length of 253 bp.
Similarly, an OTU table was generated for fungal ITS sequences after removal of alien sequences, quality filtering (Q25), phiX DNA filtering, sequence dereplication, and generation of a parametric error model (69). Chimeras were then removed, and taxonomy was assigned (70).
Data analysis.
Distance matrices of 16S rRNA OTUs were created in QIIME using weighted and unweighted UniFrac distances (71), and Bray-Curtis dissimilarity distances, also used to examine ITS OTUs. All analyses were performed on nonrarefied data (72). Further analyses were carried out using PHYLOSEQ (73) and LEfSe (35). Based on the analysis of the mock bacterial communities (Table S4), family level resolution was used for identifying differentially abundant taxa, and OTU level resolution was used to quantify the total number of differentially abundant taxa.
Mock community identification at the genus level. Download Table S4, PDF file, 0.1 MB (108.4KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Assessment of ammonia oxidizer abundance.
Assessment of AOA and AOB abundances was performed by targeting the amoA gene of DNA samples by qPCR (based on SYBR green fluorescence); the primer sets for AOB and AOA were amoA-1F/2R (74) and Arch-amoAF/R (75), respectively.
Nitrification potential and gross nitrate production.
Separate rhizotrons, with and without maize plants, were prepared for determining the nitrification potential and gross nitrate production rates. Nitrification potential was determined over a 24-h period as described previously (76). For determining gross NO3 production rates, a 15KNO3 solution was first homogenously distributed in the soil of each rhizotron, samples were then collected after 15-min, 3-h, and 24-h incubation and immediately subjected to a KCl extraction. 15N/14N ratio determination occurred at Utah State University, as described previously (60, 77). The amount of 15NO3 to apply was determined by measuring inorganic N pool sizes in five rhizotrons (with and without plants). Subsamples of bulk and rhizosphere soils were collected for measuring soil physicochemical properties.
Statistical analyses.
Amplicon data analyzed with permutational multivariate analyses of variance (PERMANOVAs) using distance matrices (78), and elemental and root traits analyzed with analyses of variance (ANOVAs) (in R, on transformed data [79–81]). ANOVAs of qPCR data, nitrification potentials, nitrate pool sizes, and gross nitrate production rates performed using JMP13 (on transformed data; post hoc test, Tukey’s honestly significant difference test [HSD]). Richness indices tested by ANOVAs (post hoc test, Fisher’s least significant difference [LSD]), and Simpson index examined by Kruskal-Wallis tests, in JMP13.
Data availability.
All sequences were deposited into the NCBI Sequence Read Archive (PRJNA686799 and PRJNA685216).
ACKNOWLEDGMENTS
We thank Albert Lea Seed House for providing the corn seeds used in this study; Nigel Lee for the root trait measurements; Garrett Smith, Anna Olsen, and Cassandra Wattenburger for technical assistance; and Amanda Heiderscheit with generation of plots.
We declare that we have no conflicts of interests.
This research was supported by the USDA AFRI grant program (grant 2014-67019-21628) and by the Iowa Agriculture and Home Economics Experiment Station. This material is based upon work supported by Larry Halverson while serving at the U.S. National Science Foundation (NSF). Any opinion, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the U.S. NSF.
G.B., M.J.C., K.S.H., and L.J.H. designed research. G.B., C.L., C.C., and N.K.M. performed research. G.B., C.C., C.L., N.K.M., and L.J.H. analyzed data. G.B., C.L., M.J.C., K.S.H., and L.J.H. wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Kirsten S. Hofmockel, Email: kirsten.hofmockel@pnnl.gov.
Larry J. Halverson, Email: larryh@iastate.edu.
Nick Bouskill, Lawrence Berkeley National Laboratory.
REFERENCES
- 1.Ranum P, Peña-Rosas JP, Garcia-Casal MN. 2014. Global maize production, utilization, and consumption. Ann N Y Acad Sci 1312:105–112. doi: 10.1111/nyas.12396. [DOI] [PubMed] [Google Scholar]
- 2.Meena VS, Meena SK, Verma JP, Kumar A, Aeron A, Mishra PK, Bisht JK, Pattanayak A, Naveed M, Dotaniya ML. 2017. Plant beneficial rhizospheric microorganism (PBRM) strategies to improve nutrients use efficiency: a review. Ecol Eng 107:8–32. doi: 10.1016/j.ecoleng.2017.06.058. [DOI] [Google Scholar]
- 3.Sawers RJH, Gutjahr C, Paszkowski U. 2008. Cereal mycorrhiza: an ancient symbiosis in modern agriculture. Trends Plant Sci 13:93–97. doi: 10.1016/j.tplants.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Davis AS, Hill JD, Chase CA, Johanns AM, Liebman M. 2012. Increasing cropping system diversity balances productivity, profitability and environmental health. PLoS One 7:e47149. doi: 10.1371/journal.pone.0047149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liebman M, Gibson LR, Sundberg DN, Heggenstaller AH, Westerman PR, Chase CA, Hartzler RG, Menalled FD, Davis AS, Dixon PM. 2008. Agronomic and economic performance characteristics of conventional and low-external-input cropping systems in the central corn belt. Agron J 100:600–610. doi: 10.2134/agronj2007.0222. [DOI] [Google Scholar]
- 6.Foley JA, Ramankutty N, Brauman KA, Cassidy ES, Gerber JS, Johnston M, Mueller ND, O’Connell C, Ray DK, West PC, Balzer C, Bennett EM, Carpenter SR, Hill J, Monfreda C, Polasky S, Rockström J, Sheehan J, Siebert S, Tilman D, Zaks DPM. 2011. Solutions for a cultivated planet. Nature 478:337–342. doi: 10.1038/nature10452. [DOI] [PubMed] [Google Scholar]
- 7.van der Heijden MGA, Bardgett RD, van Straalen NM. 2008. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11:296–310. doi: 10.1111/j.1461-0248.2007.01139.x. [DOI] [PubMed] [Google Scholar]
- 8.Schimel JP, Bennett J. 2004. Nitrogen mineralization: challenges of a changing paradigm. Ecology 85:591–602. doi: 10.1890/03-8002. [DOI] [Google Scholar]
- 9.Miki T, Ushio M, Fukui S, Kondoh M. 2010. Functional diversity of microbial decomposers facilitates plant coexistence in a plant–microbe–soil feedback model. Proc Natl Acad Sci USA 107:14251–14256. doi: 10.1073/pnas.0914281107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Innerebner G, Knief C, Vorholt JA. 2011. Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl Environ Microbiol 77:3202–3210. doi: 10.1128/AEM.00133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumann G, Bott S, Ohler M, Mock H-P, Lippmann R, Grosch R, Smalla K. 2014. Root exudation, and root development of lettuce (Lactuca sativa L. cv. Tizian) as affected by different soils. Front Microbiol 5:2. doi: 10.3389/fmicb.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez RJ, Henson J, Van Volkenburgh E, Hoy M, Wright L, Beckwith F, Kim YO, Redman RS. 2008. Stress tolerance in plants via habitat-adapted symbiosis. ISME J 2:404–416. doi: 10.1038/ismej.2007.106. [DOI] [PubMed] [Google Scholar]
- 13.Singh BK, Millard P, Whiteley AS, Murrell JC. 2004. Unravelling rhizosphere–microbial interactions: opportunities and limitations. Trends Microbiol 12:386–393. doi: 10.1016/j.tim.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Bais H, Weir T, Perry L, Gilroy S, Vivanco J. 2006. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol 57:233–266. doi: 10.1146/annurev.arplant.57.032905.105159. [DOI] [PubMed] [Google Scholar]
- 15.Zhalnina K, Louie K, Hao Z, Mansoori N, da Rocha U, Shi S, Cho H, Karaoz U, Loque D, Bowen B, Firestone M, Northen T, Brodie E. 2018. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3:470–480. doi: 10.1038/s41564-018-0129-3. [DOI] [PubMed] [Google Scholar]
- 16.Chaparro JM, Badri DV, Vivanco JM. 2014. Rhizosphere microbiome assemblage is affected by plant development. ISME J 8:790–803. doi: 10.1038/ismej.2013.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badri D, Vivanco J. 2009. Regulation and function of root exudates. Plant Cell Environ 32:666–681. doi: 10.1111/j.1365-3040.2008.01926.x. [DOI] [PubMed] [Google Scholar]
- 18.Chaparro JM, Badri DV, Bakker MG, Sugiyama A, Manter DK, Vivanco JM. 2013. Root exudation of phytochemicals in Arabidopsis follows specific patterns that are developmentally programmed and correlate with soil microbial functions. PLoS One 8:e55731. doi: 10.1371/journal.pone.0055731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuzyakov Y, Xu X. 2013. Competition between roots and microorganisms for nitrogen: mechanisms and ecological relevance. New Phytol 198:656–669. doi: 10.1111/nph.12235. [DOI] [PubMed] [Google Scholar]
- 20.Zhu S, Vivanco JM, Manter DK. 2016. Nitrogen fertilizer rate affects root exudation, the rhizosphere microbiome and nitrogen-use-efficiency of maize. Appl Soil Ecol 107:324–333. doi: 10.1016/j.apsoil.2016.07.009. [DOI] [Google Scholar]
- 21.Hargreaves SK, Williams RJ, Hofmockel KS. 2015. Environmental filtering of microbial communities in agricultural soil shifts with crop growth. PLoS One 10:e0134345. doi: 10.1371/journal.pone.0134345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartmann M, Frey B, Mayer J, Mader P, Widmer F. 2015. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J 9:1177–1194. doi: 10.1038/ismej.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomer MD, Liebman M. 2014. Nutrients in soil water under three rotational cropping systems, Iowa, USA. Agric Ecosyst Environ 186:105–114. doi: 10.1016/j.agee.2014.01.025. [DOI] [Google Scholar]
- 24.Osterholz W, Liebman M, Castellano M. 2018. Can soil nitrogen dynamics explain the yield benefit of crop diversification? Field Crops Res 219:33–42. doi: 10.1016/j.fcr.2018.01.026. [DOI] [Google Scholar]
- 25.Osterholz WR, Rinot O, Shaviv A, Linker R, Liebman M, Sanford G, Strock J, Castellano MJ. 2017. Predicting gross nitrogen mineralization and potentially mineralizable nitrogen using soil organic matter properties. Soil Sci Soc Am J 81:1115–1126. doi: 10.2136/sssaj2017.02.0055. [DOI] [Google Scholar]
- 26.King AE, Hofmockel KS. 2017. Diversified cropping systems support greater microbial cycling and retention of carbon and nitrogen. Agric Ecosyst Environ 240:66–76. doi: 10.1016/j.agee.2017.01.040. [DOI] [Google Scholar]
- 27.Wattenburger CJ, Halverson LJ, Hofmockel KS. 2019. Agricultural management affects root-associated microbiome recruitment over maize development. Phytobiomes J 3:260–272. doi: 10.1094/PBIOMES-03-19-0016-R. [DOI] [Google Scholar]
- 28.Schreiter S, Ding G-C, Heuer H, Neumann G, Sandmann M, Grosch R, Kropf S, Smalla K. 2014. Effect of the soil type on the microbiome in the rhizosphere of field-grown lettuce. Front Microbiol 5:144. doi: 10.3389/fmicb.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wattenburger CJ, Gutknecht J, Zhang Q, Brutnell TP, Hofmockel KH, Halverson LJ. 2020. The rhizosphere and cropping system, but not arbuscular mycorrhizae, affect ammonia oxidizing archaea and bacteria abundances in two agricultural soils. Appl Soil Ecol 151:103540. doi: 10.1016/j.apsoil.2020.103540. [DOI] [Google Scholar]
- 30.Mendes R, Garbeva P, Raaijmakers JM. 2013. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol Rev 37:634–663. doi: 10.1111/1574-6976.12028. [DOI] [PubMed] [Google Scholar]
- 31.Berendsen RL, Pieterse CMJ, Bakker P. 2012. The rhizosphere microbiome and plant health. Trends Plant Sci 17:478–486. doi: 10.1016/j.tplants.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Abendroth LJ, Elmore RW, Boyer MJ, Marlay SK (ed). 2011. Corn growth and development, PMR 1009. Iowa State University Extension, Ames, IA. [Google Scholar]
- 33.Blazewicz SJ, Barnard RL, Daly RA, Firestone MK. 2013. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J 7:2061–2068. doi: 10.1038/ismej.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ofek M, Voronov-Goldman M, Hadar Y, Minz D. 2014. Host signature effect on plant root-associated microbiomes revealed through analyses of resident vs. active communities. Environ Microbiol 16:2157–2167. doi: 10.1111/1462-2920.12228. [DOI] [PubMed] [Google Scholar]
- 35.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buckley DH, Huangyutitham V, Nelson TA, Rumberger A, Thies JE. 2006. Diversity of Planctomycetes in soil in relation to soil history and environmental heterogeneity. Appl Environ Microbiol 72:4522–4531. doi: 10.1128/AEM.00149-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buckley DH, Schmidt TM. 2003. Diversity and dynamics of microbial communities in soils from agro-ecosystems. Environ Microbiol 5:441–452. doi: 10.1046/j.1462-2920.2003.00404.x. [DOI] [PubMed] [Google Scholar]
- 38.Richter-Heitmann T, Eickhorst T, Knauth S, Friedrich MW, Schmidt H. 2016. Evaluation of strategies to separate root-associated microbial communities: a crucial choice in rhizobiome research. Front Microbiol 7:773. doi: 10.3389/fmicb.2016.00773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emerson JB, Adams RI, Román CMB, Brooks B, Coil DA, Dahlhausen K, Ganz HH, Hartmann EM, Hsu T, Justice NB, Paulino-Lima IG, Luongo JC, Lymperopoulou DS, Gomez-Silvan C, Rothschild-Mancinelli B, Balk M, Huttenhower C, Nocker A, Vaishampayan P, Rothschild LJ. 2017. Schrödinger’s microbes: tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 5:86. doi: 10.1186/s40168-017-0285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vieira S, Sikorski J, Dietz S, Herz K, Schrumpf M, Bruelheide H, Scheel D, Friedrich MW, Overmann J. 2020. Drivers of the composition of active rhizosphere bacterial communities in temperate grasslands. ISME J 14:463–475. doi: 10.1038/s41396-019-0543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foesel BU, Nägele V, Naether A, Wüst PK, Weinert J, Bonkowski M, Lohaus G, Polle A, Alt F, Oelmann Y, Fischer M, Friedrich MW, Overmann J. 2014. Determinants of Acidobacteria activity inferred from the relative abundances of 16S rRNA transcripts in German grassland and forest soils. Environ Microbiol 16:658–675. doi: 10.1111/1462-2920.12162. [DOI] [PubMed] [Google Scholar]
- 42.Frenk S, Dag A, Yermiyahu U, Zipori I, Hadar Y, Minz D. 2015. Seasonal effect and anthropogenic impact on the composition of the active bacterial community in Mediterranean orchard soil. FEMS Microbiol Ecol 91:fiv096. doi: 10.1093/femsec/fiv096. [DOI] [PubMed] [Google Scholar]
- 43.Placella SA, Brodie EL, Firestone MK. 2012. Rainfall-induced carbon dioxide pulses result from sequential resuscitation of phylogenetically clustered microbial groups. Proc Natl Acad Sci USA 109:10931–10936. doi: 10.1073/pnas.1204306109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lennon JT, Muscarella ME, Placella SA, Lehmkuhl BK. 2018. How, when, and where relic DNA affects microbial diversity. mBio 9:e00637-18. doi: 10.1128/mBio.00637-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carini P, Marsden PJ, Leff JW, Morgan EE, Strickland MS, Fierer N. 2016. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol 2:16242. doi: 10.1038/nmicrobiol.2016.242. [DOI] [PubMed] [Google Scholar]
- 46.Lebeis S, Paredes S, Lundberg D, Breakfield N, Gehring J, McDonald M, Malfatti S, del Rio T, Jones C, Tringe S, Dangl J. 2015. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349:860–864. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- 47.Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, del Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL. 2012. Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu H, Carvalhais LC, Crawford M, Singh E, Dennis PG, Pieterse CMJ, Schenk PM. 2017. Inner plant values: diversity, colonization and benefits from endophytic bacteria. Front Microbiol 8:2552. doi: 10.3389/fmicb.2017.02552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaparro J, Sheflin A, Manter D, Vivanco J. 2012. Manipulating the soil microbiome to increase soil health and plant fertility. Biol Fertil Soils 48:489–499. doi: 10.1007/s00374-012-0691-4. [DOI] [Google Scholar]
- 50.Tkacz A, Cheema J, Chandra G, Grant A, Poole PS. 2015. Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. ISME J 9:2349–2359. doi: 10.1038/ismej.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Q, Ling N, Chen H, Duan Y, Wang S, Shen Q, Vandenkoornhuyse P. 2020. Long-term chemical-only fertilization induces a diversity decline and deep selection on the soil bacteria. mSystems 5:e00337-20. doi: 10.1128/mSystems.00337-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bardgett RD, Mommer L, De Vries FT. 2014. Going underground: root traits as drivers of ecosystem processes. Trends Ecol Evol 29:692–699. doi: 10.1016/j.tree.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Moreau D, Bardgett RD, Finlay RD, Jones DL, Philippot L. 2019. A plant perspective on nitrogen cycling in the rhizosphere. Funct Ecol 33:540–552. doi: 10.1111/1365-2435.13303. [DOI] [Google Scholar]
- 54.Fierer N, Ladau J, Clemente JC, Leff JW, Owens SM, Pollard KS, Knight R, Gilbert JA, McCulley RL. 2013. Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342:621–624. doi: 10.1126/science.1243768. [DOI] [PubMed] [Google Scholar]
- 55.Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, Robeson M, Edwards RA, Felts B, Rayhawk S, Knight R, Rohwer F, Jackson RB. 2007. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol 73:7059–7066. doi: 10.1128/AEM.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pepe-Ranney C, Campbell AN, Koechli CN, Berthrong S, Buckley DH. 2016. Unearthing the ecology of soil microorganisms using a high resolution DNA-SIP approach to explore cellulose and xylose metabolism in soil. Front Microbiol 7:703. doi: 10.3389/fmicb.2016.00703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, Xie G, Haft DH, Sait M, Badger J, Barabote RD, Bradley B, Brettin TS, Brinkac LM, Bruce D, Creasy T, Daugherty SC, Davidsen TM, DeBoy RT, Detter JC, Dodson RJ, Durkin AS, Ganapathy A, Gwinn-Giglio M, Han CS, Khouri H, Kiss H, Kothari SP, Madupu R, Nelson K, Nelson WC, Paulsen I, Penn K, Ren Q, Rosovitz MJ, Selengut JD, Shrivastava S, Sullivan SA, Tapia R, Thompson LS, Watkins KL, Yang Q, Yu C, Zafar N, Zhou L, Kuske CR. 2009. Three genomes from the phylum Acidobacteria provide insight into their lifestyles in soils. Appl Environ Microbiol 75:2046–2056. doi: 10.1128/AEM.02294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Osterholz W, Rinot O, Liebman M, Castellano M. 2017. Can mineralization of soil organic nitrogen meet maize nitrogen demand? Plant Soil 415:73–84. doi: 10.1007/s11104-016-3137-1. [DOI] [Google Scholar]
- 59.Zhu B, Gutknecht J, Herman D, Keck D, Firestone M, Cheng W. 2014. Rhizosphere priming effects on soil carbon and nitrogen mineralization. Soil Biol Biochem 76:183–192. doi: 10.1016/j.soilbio.2014.04.033. [DOI] [Google Scholar]
- 60.Herman DJ, Johnson KK, Jaeger CH, III, Schwartz E, Firestone MK. 2006. Root influence on nitrogen mineralization and nitrification in Avena barbata rhizosphere soil. Soil Sci Soc Am J 70:1504–1511. doi: 10.2136/sssaj2005.0113. [DOI] [Google Scholar]
- 61.Frank DA, Groffman PM. 2009. Plant rhizospheric N processes: what we don’t know and why we should care. Ecology 90:1512–1519. doi: 10.1890/08-0789.1. [DOI] [PubMed] [Google Scholar]
- 62.Ai C, Liang G, Sun J, Wang X, He P, Zhou W. 2013. Different roles of rhizosphere effect and long-term fertilization in the activity and community structure of ammonia oxidizers in a calcareous fluvo-aquic soil. Soil Biol Biochem 57:30–42. doi: 10.1016/j.soilbio.2012.08.003. [DOI] [Google Scholar]
- 63.Pace J, Lee N, Naik HS, Ganapathysubramanian B, Lubberstedt T. 2014. Analysis of maize (Zea mays L.) seedling roots with the high-throughput image analysis tool ARIA (Automatic Root Image Analysis). PLoS One 9:e108255. doi: 10.1371/journal.pone.0108255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bakker MG. 2018. A fungal mock community control for amplicon sequencing experiments. Mol Ecol Resour 18:541–556. doi: 10.1111/1755-0998.12760. [DOI] [PubMed] [Google Scholar]
- 66.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17:10–12. doi: 10.14806/ej.17.1.200.. [DOI] [Google Scholar]
- 67.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caporaso JG, Bittinger K, Bushman FD, Desantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nilsson RH, Larsson KH, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, Kennedy P, Picard K, Glöckner FO, Tedersoo L, Saar I, Kõljalg U, Abarenkov K. 2019. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 47:D259–D264. doi: 10.1093/nar/gky1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McMurdie PJ, Holmes S. 2014. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10:e1003531. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rotthauwe JH, Witzel KP, Liesack W. 1997. The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl Environ Microbiol 63:4704–4712. doi: 10.1128/aem.63.12.4704-4712.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB. 2005. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc Natl Acad Sci USA 102:14683–14688. doi: 10.1073/pnas.0506625102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Herman DJ, Halverson LJ, Firestone MK. 2003. Nitrogen dynamics in an annual grassland: oak canopy, climate, and microbial population effects. Ecol Applic 13:593–604.2.0.CO;2]. doi: 10.1890/1051-0761(2003)013[0593:NDIAAG]2.0.CO;2. [DOI] [Google Scholar]
- 77.Hart SC, Stark JM, Davidson EA, Firestone MK. 1994. Nitrogen mineralization, immobilization, and nitrification, p 985–1018. In Weaver RW, Angle S, Bottomley P, Bezdicek D, Smith S, Tabatabai A, Wollum A (ed), Methods of soil analysis. Soil Science Society of America, Madison, WI. [Google Scholar]
- 78.Dixon P. 2003. VEGAN, a package of R functions for community ecology. https://10.1111/j.1654-1103.2003.tb02228.x.
- 79.Osborne JW. 2010. Improving your data transformations: applying the Box-Cox transformation. Pract Assess Res Eval 15:12. doi: 10.7275/qbpc-gk17. [DOI] [Google Scholar]
- 80.Box GEP, Cox DR. 1964. An analysis of transformations: applying the Box-Cox transformation. J R Stat Soc 26:211–252. [Google Scholar]
- 81.Wessa P. 2015. Box-Cox normality plot (v1.1.11) in Free Statistics Software (v1.1.23-r7).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of cropping systems on bulk soil physicochemical properties. Download Table S1, PDF file, 0.08 MB (81.5KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Analysis of variance of α-diversity indices. Download Table S2, PDF file, 0.1 MB (107.4KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA- and RNA-based family level relative abundance of Deltaproteobacteria (A to C) and Gammaproteobacteria (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance (<0.02%) Delta- and Gammaproteobacteria families are not visible. Download FIG S1, JPG file, 1.1 MB (1.1MB, jpg) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA- and RNA-based family level relative abundance of Actinobacteria (A to C) and Acidobacteria (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance Actinobacteria (<0.08%) and Acidobacteria (<0.05% for soil and rhizosphere; <0.01% for endosphere) families are not visible. Download FIG S2, JPG file, 2.0 MB (2MB, jpg) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA- and RNA-based family level relative abundance of Planctomycetes (A to C) and Verrucomicrobia (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance Planctomycetes (<0.05% for soil and rhizosphere; <0.005% endosphere) and Verrucomicrobia (<0.05% for soil and rhizosphere; <0.02% for endosphere) families are not visible. Download FIG S3, JPG file, 1.4 MB (1.4MB, jpg) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA- and RNA-based family level relative abundance of Bacteroidetes (A to C) and Firmicutes (D to F) in conventional and diversified cropping systems. Red arrows indicate statistically different (LDA, P < 0.05) DNA-based relative abundance between cropping systems. Green arrows indicate statistically different (LDA, P < 0.05) relative abundance between DNA- and RNA-based profiles within a cropping system. Blue arrows indicate statistically different RNA-based relative abundance between cropping systems. An asterisk over two adjacent bars indicates statistically significant different total relative abundance between the RNA- and DNA-based analyses. Low-abundance Bacteroidetes (<0.02%) and Firmicutes (<0.01%) families are not visible. Download FIG S4, JPG file, 1.4 MB (1.4MB, jpg) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Class level relative abundance of fungal community profiles in conventional and diversified cropping systems. Arrows indicate statistically different (LDA, P < 0.05), DNA-based relative abundance of a class between the two cropping systems. Phyla below 1% were summed. Phyla are abbreviated as follows: Ascom, Ascomycota; Basid, Basidiomycota; Morti, Mortierellomycota; Chytr, Chytridiomycota; Glome, Glomeromycota. Download FIG S5, JPG file, 0.5 MB (563.1KB, jpg) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Analysis of variance of ammonia oxidizers and their potential metabolic activity. Download Table S3, PDF file, 0.09 MB (94.6KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Detailed Materials and Methods. Download Text S1, PDF file, 0.2 MB (201.6KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mock community identification at the genus level. Download Table S4, PDF file, 0.1 MB (108.4KB, pdf) .
Copyright © 2021 Bay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
All sequences were deposited into the NCBI Sequence Read Archive (PRJNA686799 and PRJNA685216).