

ABSTRACT
Heme oxygenase 1 (HO-1) is the key enzyme for heme catabolism and cytoprotection. Whereas HO-1 gene expression in response to various stresses has been investigated extensively, the precise mechanisms by which HO-1 gene expression is regulated by the HO-1 substrate heme remain elusive. To systematically examine whether stress-mediated induction and substrate-mediated induction of HO-1 utilize similar or distinct regulatory pathways, we developed an HO-1–DsRed-knock-in reporter mouse in which the HO-1 gene is floxed by loxP sites and the DsRed gene has been inserted. Myeloid lineage-specific recombination of the floxed locus led to fluorescence derived from expression of the HO-1–DsRed fusion protein in peritoneal macrophages. We also challenged general recombination of the locus and generated mice harboring heterozygous recombinant alleles, which enabled us to monitor HO-1–DsRed expression in the whole body in vivo and ex vivo. HO-1 inducers upregulated HO-1–DsRed expression in myeloid lineage cells isolated from the mice. Notably, analyses of peritoneal macrophages from HO-1–DsRed mice lacking NRF2, a major regulator of the oxidative/electrophilic stress response, led us to identify NRF2-dependent stress response-mediated HO-1 induction and NRF2-independent substrate-mediated HO-1 induction. Thus, the HO-1 gene is subjected to at least two distinct levels of regulation, and the available lines of evidence suggest that substrate induction in peritoneal macrophages is independent of CNC family-based regulation.
KEYWORDS: HO-1, NRF2, stress response, hemin
INTRODUCTION
Heme oxygenase (HO) is an enzyme that degrades heme to ferrous iron (Fe2+), carbon monoxide (CO), and biliverdin (1). Biliverdin is then transformed to bilirubin by biliverdin reductase. HO has been shown to exhibit cytoprotective activity, as biliverdin and bilirubin possess antioxidative, anti-inflammatory and antiapoptotic activities. Additionally, CO plays an important role in vasodilation and antiproliferative processes. There are at least two isoforms of HO, heme oxygenase 1 (HO-1) and heme oxygenase 2 (HO-2) (2, 3). HO-1 and HO-2 are expressed in an inducible and a constitutive manner, respectively.
Expression of the HO-1 gene is induced by various stress stimuli (4). Nuclear factor erythroid 2-related factor 2 (NRF2), a member of the cap'n'collar (CNC) subfamily of basic-region leucine zipper (bZIP) transcription factors (5), is the major regulator of the HO-1 gene, and the NRF2-HO-1 axis appears to be an important system for stress protection (6, 7). NRF2 is regulated by Kelch-like ECH-associated protein 1 (KEAP1), an adaptor component of the CUL3 (Cullin 3)-based ubiquitin E3 ligase complex (8–10). Under basal conditions, NRF2 is efficiently ubiquitinated by the KEAP1-CUL3 complex, after which it is degraded through the proteasome system in the cytoplasm. Upon exposure to toxic chemicals (often electrophiles) or oxidative stressors, the ubiquitin E3 ligase activity of the KEAP1-CUL3 complex is repressed, which leads to NRF2 stabilization (11).
NRF2 then translocates into the nucleus, where it forms a heterodimer with one of the small MAF (sMAF) proteins. The NRF2-sMAF heterodimer binds antioxidant/electrophile response elements (AREs/EpREs), collectively called CNC-sMAF-binding elements (CsMBEs) (12), and upregulates the expression of downstream cytoprotective genes, including the HO-1 gene (13, 14). Diethyl maleate (DEM) and 1-[2-cyano-3,12,28-trioxooleana-1,9(11)-dien-28-yl]-1H-imidazole (CDDO-Im) are representative NRF2 inducers (8, 15, 16). NRF2 inducers have been extensively developed for the treatment of various diseases (13), and dimethyl fumarate (Tecfidera) has already been approved by the FDA (17). Two new analogs of CDDO-Im, 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]-4(-pyridin-2-yl)-1H-imidazole (CDDO-2P-Im) and 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]-4(-pyridin-3-yl)-1H-imidazole (CDDO-3P-Im), exhibit high stability and have been developed for prospective clinical use (18).
HO-1 gene expression is known to be induced by the substrate heme. This induction of HO-1 by heme is an elaborate mechanism of metabolic pathway regulation and prototypical substrate induction (19). Hemin, which consists of protoporphyrin IX containing ferric iron (Fe3+) with a coordinating chloride ligand, is a substrate of HO-1. Hemin has long been used as an experimental HO-1 inducer (3, 20), and BTB and CNC homology 1 (BACH1), another member of the CNC-bZIP family, is a regulator of the HO-1 gene in response to hemin (21).
Under basal conditions, the BACH1-sMAF heterodimer is proposed to bind CsMBEs in two enhancer regions of the HO-1 gene (22, 23). This makes the enhancers inaccessible to NRF2 and other activators, leading to the inhibition of HO-1 expression. Exposure of fibroblasts or erythroleukemia cells to hemin upregulates HO-1 expression by the derepression of BACH1-mediated repression, as the binding of hemin represses the DNA-binding activity of BACH1 (21). The binding of hemin to BACH1 also induces BACH1 nuclear export (24) and degradation (25, 26). Indeed, BACH1-knockout mice display elevated HO-1 expression in several tissues (22), supporting the notion that BACH1 is a critical repressor of the HO-1 gene. Interestingly, however, high-level HO‐1 expression in the absence of BACH1 was mostly abrogated by compound knockout of Nrf2 in the heart but not the thymus (22), suggesting differences in the activators responsible for HO-1 expression between tissues. Considering that macrophages are mainly responsible for removing senescent red blood cells (27, 28), it is important to clarify the mechanisms responsible for substrate-induced HO-1 expression in macrophages.
In addition to hemin and typical NRF2 inducers, there are several unclassified HO-1 inducers. For instance, cannabidiol (CBD), another HO-1 inducer, was reported to target BACH1 and induce HO-1 in an NRF2-independent manner (29). Additionally, hypoxia induces HO-1 expression through the hypoxia-inducible factor (HIF) pathway (30). Cobalt(II) chloride (CoCl2) also induces HO-1 expression (31–34). While CoCl2 can potently activate the HIF pathway by stabilizing the HIF protein and inducing its accumulation (35), CoCl2 is also known to activate the NRF2 pathway by increasing oxidative stress (36, 37). However, the molecular basis by which these different HO-1 inducers activate HO-1 gene expression has not been fully investigated.
In this regard, we hypothesized that the development of a system that can systemically monitor HO-1 gene expression in vivo and ex vivo would help to clarify how various classes of stimuli or chemicals induce HO-1 gene expression. We noticed pioneering work involving the use of a transgenic mouse line carrying a 2.1-kb region of the HO-1 gene, including the basal promoter and proximal enhancer, fused to the luciferase gene. The study showed that luciferase expression was induced in response to cadmium exposure, but this increase in luciferase expression did not seem to recapitulate endogenous HO-1 gene expression (38). As we surmised that this may have been due to a lack of sufficient transcriptional regulatory elements, in this study, we decided to construct a new system to monitor HO-1 gene expression that effectively recapitulates gene regulation. We successfully generated HO-1–DsRed reporter (HO-1DsRed/+) mice and found that the mouse model is quite useful for monitoring HO-1 gene expression both ex vivo and in vivo. By utilizing HO-1–DsRed reporter mice crossed with Nrf2-knockout mice, we discovered that HO-1 induction events triggered by different stimuli converged into at least two categories: those caused by an NRF2-dependent stress response and those induced by substrate heme but independent of NRF2.
RESULTS
Generation of myeloid lineage-specific HO-1-knockout mice.
To generate an elaborate and efficient system to monitor HO-1 expression, we decided to utilize conditional HO-1-knockout mice (Fig. 1A) in which the HO-1 gene is floxed by two loxP sites and the DsRed gene has been inserted into the 3′ region (39). We first examined whether Cre-mediated recombination of the flox allele can induce DsRed expression under regulation of the endogenous HO-1 locus. As many studies have revealed that macrophages express HO-1 (7, 40), we generated HO-1flox/flox::LysMCre mice by mating the HO-1flox/flox mice with LysMCre mice and assessed whether deletion of the HO-1 gene and expression of DsRed occurred in myeloid lineage cells from this compound mutant mouse line (Fig. 1B).
FIG 1.
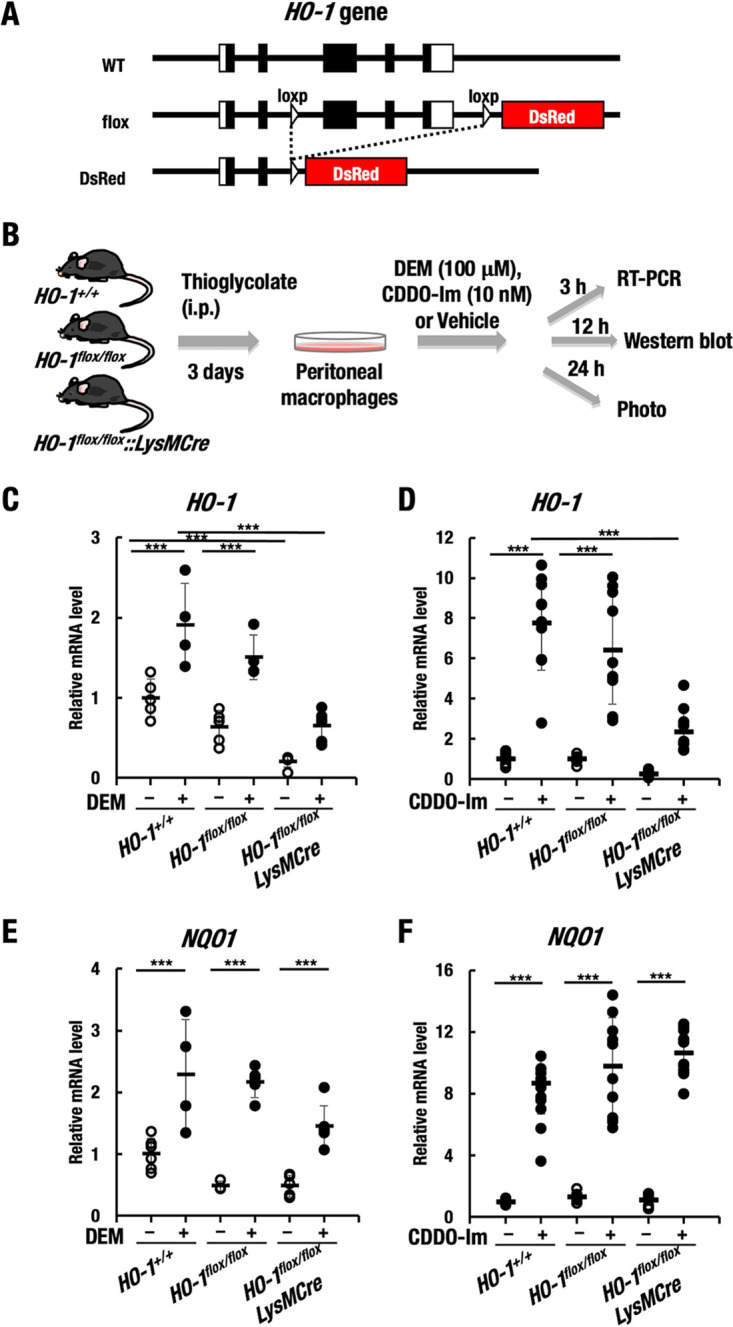
Generation of myeloid lineage-specific HO-1-knockout mice. (A) The HO-1 gene is floxed by two loxP sites, and the DsRed gene has been inserted. i.p., intraperitoneal. (B) Thioglycolate-induced induction of peritoneal macrophages isolated from HO-1+/+, HO-1flox/flox, and HO-1flox/flox::LysMCre mice. The cells were treated with 100 μM DEM, 10 nM CDDO-Im, or DMSO (vehicle). (C and D) Relative mRNA level of HO-1 after 3 h of treatment with DEM, CDDO-Im, or DMSO (vehicle). (E and F) Relative mRNA level of NQO1 after 3 h of treatment with DEM, CDDO-Im, or DMSO (vehicle). ***, P < 0.001.
We collected thioglycolate-induced peritoneal macrophages from HO-1+/+, HO-1flox/flox, and HO-1flox/flox::LysMCre mice and examined whether the HO-1 gene had been deleted in primary peritoneal macrophages from the HO-1flox/flox::LysMCre mice. Since NRF2 is a major regulator of the HO-1 gene (13), we treated the macrophages with the common NRF2 inducers diethyl maleate (DEM) and oleanolic acid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl] imidazole (CDDO-Im) and then examined the mRNA and protein levels of HO-1 and DsRed fluorescence (Fig. 1B).
We found that HO-1 mRNA levels were significantly increased in macrophages from HO-1+/+ and HO-1flox/flox mice under DEM (100 μM) treatment for 3 h compared to those under vehicle control treatment. In contrast, HO-1 mRNA levels under both DEM-treated and untreated conditions were decreased in macrophages from HO-1flox/flox::LysMCre mice compared to those from HO-1+/+ and HO-1flox/flox mice (Fig. 1C). Similarly, while HO-1 mRNA levels were significantly increased in macrophages from HO-1+/+ and HO-1flox/flox mice under CDDO-Im (10 nM) treatment compared to vehicle control treatment, HO-1 mRNA levels under both CDDO-Im-treated and untreated conditions were markedly decreased in macrophages from HO-1flox/flox::LysMCre mice compared to those from HO-1+/+ and HO-1flox/flox mice (Fig. 1D). As a control, we also measured NQO1 mRNA levels and found that the NQO1 mRNA level was upregulated under exposure to DEM or CDDO-Im in macrophages from HO-1+/+, HO-1flox/flox, and HO-1flox/flox::LysMCre mice (Fig. 1E and F). These results indicated that NRF2 was indeed activated in HO-1flox/flox::LysMCre macrophage cells under exposure to DEM or CDDO-Im. We conclude that the HO-1 gene has been specifically knocked out in the macrophages from the HO-1flox/flox::LysMCre mice.
Expression of the HO-1–DsRed fusion protein in macrophages.
We next examined HO-1 protein levels in macrophages (Fig. 2A). We unexpectedly found that, under basal conditions, HO-1 protein expression in the macrophages from HO-1flox/flox mice was decreased compared to that in the macrophages from HO-1+/+ mice. When we measured this decrease, it was less than 40% and reproducible. While treatment with DEM induced HO-1 protein expression in the macrophages from HO-1+/+ mice, the induced HO-1 protein levels were also decreased, albeit not substantially, in the macrophages of HO-1flox/flox mice. These results indicate that the floxed allele into which DsRed had been integrated rendered the HO-1 locus hypomorphic and that HO-1 gene expression from the flox allele is mildly reduced without Cre-mediated recombination.
FIG 2.
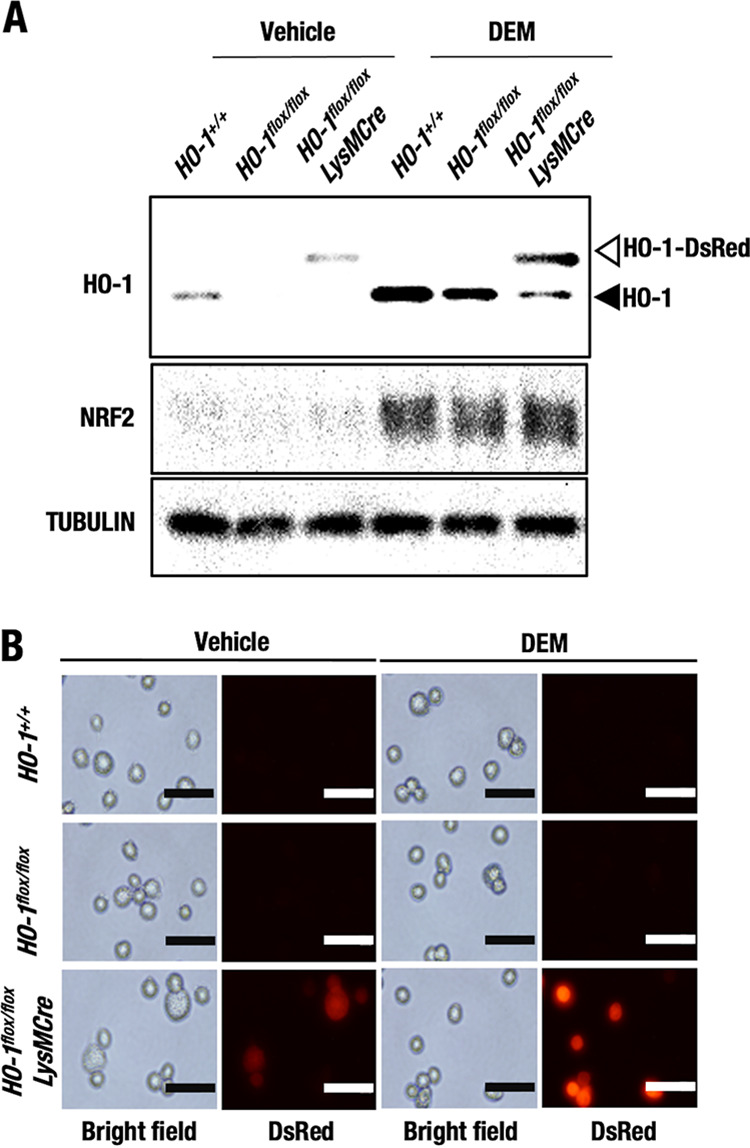
HO-1–DsRed expression in macrophages by Cre-mediated recombination of the HO-1 flox allele. Thioglycolate-induced peritoneal macrophages were isolated from HO-1+/+, HO-1flox/flox, and HO-1flox/flox::LysMCre mice and treated with 100 μM DEM or DMSO (vehicle). (A) HO-1, NRF2, and tubulin protein levels in the macrophages after 12 h of treatment were examined by Western blot analysis. Open and closed arrowheads indicate HO-1-DsRed and endogenous HO-1, respectively. (B) Representative DsRed fluorescence microscopy images of peritoneal macrophages after 24 h of treatment with 100 μM DEM or DMSO (vehicle). Scale bars, 50 μm.
Importantly, in the macrophages from HO-1flox/flox::LysMCre mice, a band corresponding to a higher-molecular-weight species emerged, and the band was more abundant than that corresponding to the endogenous HO-1 protein. Since the anti-HO-1 antibody used in this study recognizes the N terminus of the HO-1 protein, a fusion protein between the HO-1 N-terminal region and DsRed (HO-1–DsRed) was likely to be detected equally. While endogenous HO-1 protein expression in macrophages from the HO-1+/+ and HO-1flox/flox mice was increased by treatment with DEM compared to vehicle control treatment, the HO-1–DsRed fusion protein in macrophages from the HO-1flox/flox::LysMCre mice was also upregulated by treatment with DEM compared to vehicle control treatment (Fig. 2A). This observation further supports our assertion that HO-1–DsRed is expressed under regulation of the endogenous HO-1 gene locus.
NRF2 protein levels were comparably induced by DEM in macrophages from HO-1+/+, HO-1flox/flox, and HO-1flox/flox::LysMCre mice (Fig. 2A), which is consistent with the finding that NQO1 mRNA levels were normally induced in HO-1flox/flox::LysMCre macrophages by DEM (Fig. 1F). These results indicate that NRF2 was activated by DEM in the HO-1-deleted macrophages.
We next investigated whether a DsRed signal would be detected in macrophages from HO-1flox/flox::LysMCre mice. HO-1–DsRed fluorescence expression after 24 h of treatment with DEM was investigated (Fig. 2B). We did not detect any fluorescence in the macrophages from HO-1+/+ or HO-1flox/flox mice under DEM-treated or untreated conditions. DsRed fluorescence could be detected in macrophages from the HO-1flox/flox::LysMCre mice under basal vehicle-treated conditions, but the fluorescence became much stronger upon exposure to DEM than under basal conditions (Fig. 2B). All these data indicate that the HO-1–DsRed fusion protein can be successfully generated in HO-1flox/flox::LysMCre macrophages by Cre-mediated recombination of the HO-1 flox allele and that HO-1–DsRed is expressed under regulation of the endogenous HO-1 gene locus.
In vivo imaging system (IVIS) analysis shows in vivo HO-1–DsRed induction by CDDO-Im or hemin.
To develop mice for HO-1–DsRed imaging to detect the whole-body signal, we next generated HO-1DsRed/+ mice by crossing HO-1flox/+ mice and general deleter AyuI-Cre mice (41) (Fig. 3A). We found that homozygous HO-1DsRed/DsRed mice (i.e., HO-1-null mice) were lethal, which is consistent with a previous report (42). Therefore, we used heterozygous HO-1DsRed/+ mice for further analyses.
FIG 3.

IVIS analyses show in vivo HO-1–DsRed induction by CDDO-Im and hemin in HO-1DsRed/+ mice. (A) The DsRed gene was knocked-in the HO-1 gene locus. (B) HO-1+/+ and HO-1DsRed/+ mice were treated with CDDO-Im, hemin, or vehicle 72 h before IVIS analysis. (C) Representative whole-body DsRed fluorescence images of HO-1+/+ and HO-1DsRed/+ mice orally treated with 30 μmol/kg of BW of CDDO-Im or vehicle. (D) Representative whole-body DsRed fluorescence images of HO-1+/+ and HO-1DsRed/+ mice peritoneally injected with 30 mg/kg hemin. (E and F) Representative DsRed fluorescence images of the organs isolated ex vivo from HO-1+/+ and HO-1DsRed/+ mice after 72 h of treatment with CDDO-Im (E) and hemin (F).
To verify the inducible nature of HO-1–DsRed, IVIS analyses were conducted using HO-1+/+ and HO-1DsRed/+ mice. The mice were orally administered 30 μmol/kg body weight (BW) CDDO-Im or peritoneally injected with 30 mg/kg BW hemin. HO-1–DsRed fluorescence was measured 72 h after the treatment (Fig. 3B). We found that the difference in basal HO-1–DsRed fluorescence detected by whole-body IVIS analysis was only marginal in vehicle-treated HO-1DsRed/+ mice compared to HO-1+/+ mice (Fig. 3C). Unexpectedly, slight CDDO-Im-mediated induction of HO-1–DsRed fluorescence was detected in a restricted area corresponding to the liver. In contrast, and to our surprise, we observed a significant hemin-mediated increase in HO-1–DsRed fluorescence in the whole-body surface or skin of HO-1DsRed/+ mice (Fig. 3D). HO-1 was reported to be expressed in keratinocytes (43), so we surmise that this expressed HO-1–DsRed might also have been in keratinocytes.
As the fluorescence patterns induced by CDDO-Im and hemin were unique and unexpected, we decided to examine HO-1–DsRed signals ex vivo in the organs. To this end, we sacrificed the mice and analyzed HO-1–DsRed fluorescence in their organs. Under basal, vehicle-treated conditions, HO-1–DsRed fluorescence was very weak but reproducibly detected in the spleen and skin of the HO-1DsRed/+ mice (Fig. 3E and F, middle panels), but HO-1–DsRed fluorescence was not detected in HO-1+/+ mice (left panels).
Notably, we found a clear and significant increase in HO-1–DsRed fluorescence in the liver, spleen, stomach, brown adipose tissue (BAT), kidney, and small intestine of HO-1DsRed/+ mice upon CDDO-Im treatment (Fig. 3E, right panel). In contrast, we detected the distinct induction of HO-1–DsRed fluorescence in hemin-treated mouse tissues and organs. In the mice, HO-1–DsRed expression was induced in the liver, spleen, stomach, kidney, and skin (Fig. 3F, right panel). In particular, quite strong HO-1–DsRed induction was observed in the skin. We surmise that this is why hemin treatment appeared to strongly induce fluorescence upon whole-body IVIS examination (Fig. 3D). It might be hard for CDDO-Im to reach the skin, probably because of its unstable nature in plasma (18). These results thus demonstrate that the induction of HO-1–DsRed fluorescence by electrophilic stress (CDDO-Im) or substrate (hemin) treatment can be monitored in vivo by means of IVIS, making the HO-1DsRed/+ mouse model an excellent in vivo/ex vivo system in which to monitor HO-1 gene expression.
Immunostaining results support HO-1–DsRed induction by CDDO-Im and hemin.
As a significant increase in HO-1–DsRed was reproducibly detected in the spleen and liver from HO-1DsRed/+ mice treated with the electrophile CDDO-Im and substrate hemin by IVIS analyses, we next examined which type of cells express HO-1–DsRed in the spleen and liver by immunohistochemistry (Fig. 4).
FIG 4.
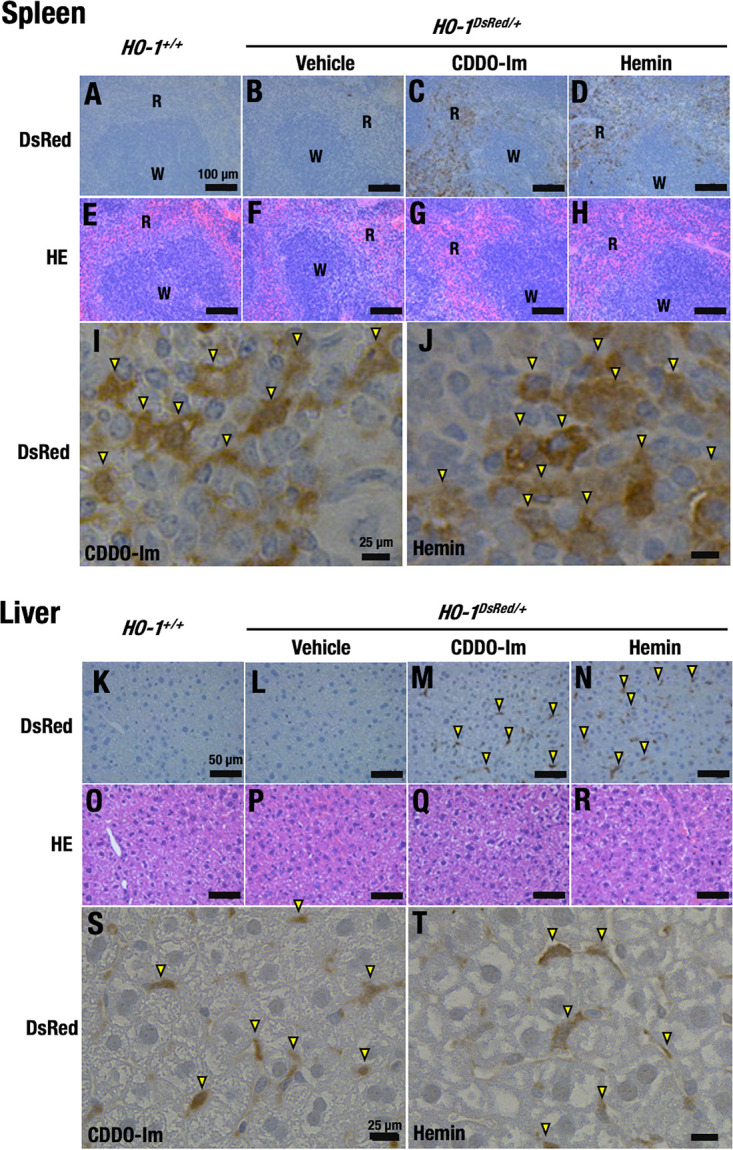
CDDO-Im and hemin induce HO-1–DsRed expression in the spleens and livers of HO-1DsRed/+ mice. Shown are representative DsRed immunostaining images (A to D, I, J, K to N, S, and T) and HE staining images (E to H and O to R) of the spleens (A to J) and livers (K to T) of HO-1+/+ mice (A, E, K, and O) and HO-1DsRed/+ mice (B to D, F to H, I, J, L to N, P to R, S, and T) treated with 10 μmol/kg BW CDDO-Im (C, G, I, M, Q, and S), 30 mg/kg hemin (D, H, J, N, R, and T), or vehicle (B, F, L, and P) for 72 h. Scale bars, 100 μm (A to H), 50 μm (K to R), and 25 μm (I, J, S, and T). W and R indicate white pulp and red pulp of the spleen, respectively. Note that red pulp macrophages in the spleen and Kupffer cells in the liver are immunostaining positive. Arrowheads indicate DsRed-positive cells in spleen and liver.
For this purpose, HO-1DsRed/+ mice were treated with CDDO-Im, hemin, or vehicle, and the spleens and livers were collected from HO-1+/+ and HO-1DsRed/+ mice 72 h after treatment. The results of immunostaining with anti-DsRed antibody revealed that DsRed-positive cells accumulated in the red pulp (R), but not the white pulp (W), of the spleens of CDDO-Im-treated HO-1DsRed/+ mice (Fig. 4A to C). The red and white pulp could be clearly distinguished by hematoxylin-eosin (HE) staining (Fig. 4E to G). Similarly, under hemin treatment conditions, DsRed expression was clearly induced in the red pulp in the spleens of the HO-1DsRed/+ mice (Fig. 4D and H). We observed very weak brown signals in the spleens of vehicle-treated HO-1DsRed/+ mice, but such signals were not detected in the spleens of the HO-1+/+ mice, indicating the specificity of the induction.
We surmise that the DsRed-positive cells observed in the red pulp of the spleen were macrophages/monocytes (Fig. 4I and J), as macrophages in splenic red pulp act to remove senescent red blood cells (27) and harbor the ability to induce HO-1 gene expression to enforce the defense against heme.
We also examined livers from HO-1+/+ and HO-1DsRed/+ mice treated with CDDO-Im or hemin (Fig. 4K to T). Immunostaining with anti-DsRed antibody revealed an increase in DsRed-positive cells in the livers of CDDO-Im- or hemin-treated mice (Fig. 4K to N). We found that the DsRed-positive cells were scarce in the liver (Fig. 4S and T), suggesting that the positive cells were Kupffer cells, local macrophages in the liver. Taken together, these results support our assertion that treatment of the mice with electrophilic stress or substrate induced HO-1 gene expression in myeloid lineage cells localized in various tissues and organs.
Representative NRF2 inducers activate HO-1–DsRed expression in macrophages.
As our analyses strongly supported the notion that the HO-1DsRed/+ mouse model is an excellent in vivo/ex vivo system to monitor HO-1 gene expression and that local macrophages appeared to inducibly express HO-1–DsRed fluorescence, we decided to address the unanswered question of whether various HO-1 inducers activate HO-1 gene expression by a common regulatory pathway or multiple different regulatory pathways utilizing peritoneal macrophages isolated ex vivo from HO-1DsRed/+ model mice. This system presents an advantage that we are able to intercross various mouse lines in which critical genes have been modified to the HO-1–DsRed-monitoring mouse line and to assess the contributions of various regulators to HO-1 gene expression.
To validate the approach with macrophages, we compared the expression of endogenous HO-1 gene and knocked-in DsRed gene in peritoneal macrophages from wild-type and HO-1DsRed/+ mice under DEM-treated or -untreated conditions (Fig. 5A). We found that the DEM treatment significantly induced the expression of HO-1 gene in wild-type macrophages (Fig. 5B). DEM-mediated induction of the HO-1 gene expression in HO-1DsRed/+ macrophages was approximately half of that of wild-type macrophages, which is consistent with haplodeficiency of the endogenous HO-1 gene in HO-1DsRed/+ macrophages. Showing very good agreement with this observation, the expression of DsRed knocked-in the HO-1 gene locus was markedly induced by DEM treatment only in HO-1DsRed/+ macrophages to the same extent as the endogenous HO-1 gene expression (Fig. 5C). These results support the use of this mouse line as a monitoring system of HO-1 gene expression.
FIG 5.
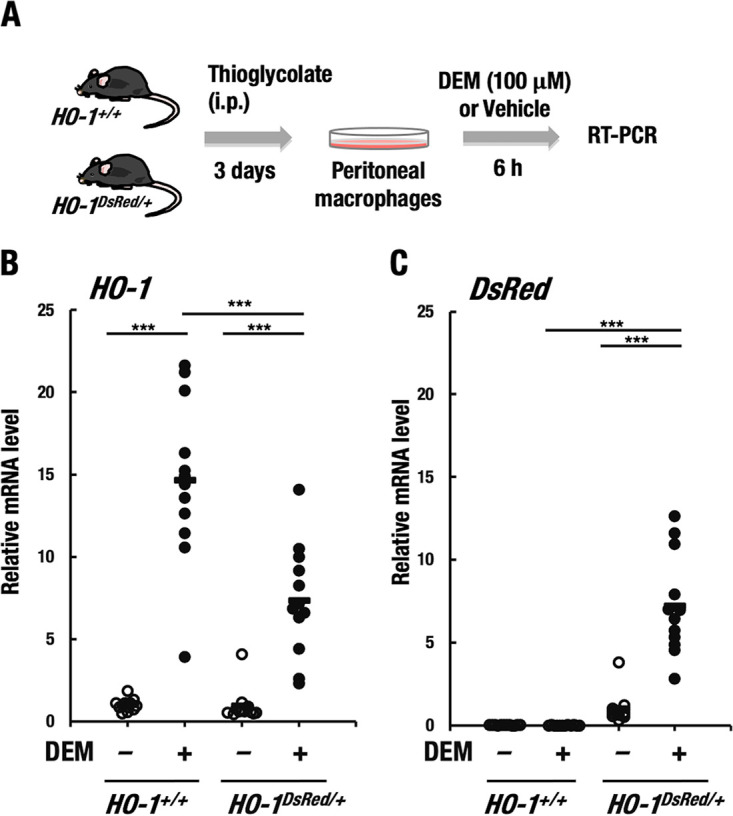
HO-1–DsRed expression recapitulates endogenous HO-1 gene expression. (A) Thioglycolate-induced peritoneal macrophages were isolated from HO-1+/+ and HO-1DsRed/+ mice. After the macrophages were treated with 100 μM DEM or DMSO (vehicle) for 6 h, the expression of HO-1 and DsRed genes was examined by RT-PCR. (B and C) Relative mRNA level of HO-1 (B) and DsRed (C) of HO-1+/+ and HO-1DsRed/+ macrophages treated with DEM or DMSO (vehicle). *, P < 0.01; ***, P < 0.001.
Utilizing this system, we applied this advantage to our assessment of NRF2 contributions to HO-1 gene expression as summarized in Fig. 6A. We examined NRF2 contributions to the regulation of HO-1 gene expression using peritoneal macrophages isolated from Nrf2−/−::HO-1DsRed/+ mice. We first validated this model system using the representative NRF2 inducers DEM, CDDO-Im, and CDDO-Im derivatives. We found that DEM treatment increased DsRed fluorescence in HO-1DsRed/+ macrophages in a dose-dependent manner, but this induction was not observed in the Nrf2−/−::HO-1DsRed/+ mice (Fig. 6B and C). These results demonstrate that DEM activates HO-1 expression through an NRF2-dependent mechanism.
FIG 6.

NRF2-dependent HO-1–DsRed induction by representative NRF2 inducers. (A) Thioglycolate-induced peritoneal macrophages were isolated from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice. After the macrophages were treated with several HO-1 inducers for 48 h, DsRed fluorescence images were obtained, and DsRed fluorescence intensities were measured with a plate reader. (B to I) Representative DsRed fluorescence microscopy images (B, D, F, and H) and relative DsRed fluorescence intensity (C, E, G, and I) in macrophages after 48 h of treatment with DEM (B and C), CDDO-Im (D and E), CDDO-2P-Im (F and G), or CDDO-3P-Im (H and I). Scale bar, 200 μm.
We also examined the effects of CDDO-Im and two derivatives known to show improved activity over that of CDDO-Im. CDDO-Im challenge markedly increased HO-1–DsRed fluorescence in HO-1DsRed/+ cells in a dose-dependent manner, but this induction was abrogated in the Nrf2−/−::HO-1DsRed/+ macrophages (Fig. 6D and E). As seen from the vertical axis (relative fluorescence level) and horizontal axis (concentration) of the corresponding figures, CDDO-Im showed 5 × 103-fold stronger efficacy for HO-1 induction than DEM. These results thus demonstrate that the induction of HO-1 expression by DEM and CDDO-Im can be quantitatively monitored by the HO-1–DsRed monitoring system and that these electrophilic inducers indeed utilize the NRF2 regulatory pathway.
We tested two new analogs of CDDO-Im, i.e., CDDO-2P-Im and CDDO-3P-Im, which were developed to exhibit high stability in human plasma (18). CDDO-2P-Im and CDDO-3P-Im have been shown to induce HO-1 expression, but their dependence on NRF2 has not been verified. We found that CDDO-2P-Im (Fig. 6F and G) and CDDO-3P-Im (Fig. 6H and I) increased HO-1–DsRed fluorescence in HO-1DsRed/+ macrophages but not Nrf2−/−::HO-1DsRed/+ macrophages in a dose-dependent manner. The efficacies of CDDO-2P-Im and CDDO-3P-Im appeared comparable to that of CDDO-Im. These results demonstrate that CDDO-2P-Im and CDDO-3P-Im are NRF2-dependent HO-1 inducers, as are DEM and CDDO-Im.
The HO-1 substrate hemin activates HO-1–DsRed in an NRF2-independent manner.
HO-1 gene expression can be induced by the substrate heme, but hemin has been used as an inducer of this gene as well (3, 20). This phenomenon has been referred to as substrate induction, but the precise mechanism underlying substrate induction remains to be clarified. One mechanistic hypothesis is that hemin regulates HO-1 gene expression through a dynamic exchange between the repressor BACH1 and the activator NRF2 (23), but this mechanism has not been rigorously tested. Therefore, in this study, we investigated whether hemin-mediated induction of HO-1 gene expression in macrophages depends on the NRF2 pathway by using the HO-1–DsRed fluorescence system.
To this end, we utilized the same system used to evaluate the electrophilic inducers as described in the previous section and treated peritoneal macrophages isolated from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice with hemin. We found that hemin treatment increased HO-1–DsRed fluorescence in macrophages from the HO-1DsRed/+ mice in a dose-dependent manner (Fig. 7A and B). However, in contrast to the electrophilic NRF2 inducers, the loss of NRF2 did not influence the hemin-mediated induction of HO-1–DsRed fluorescence in peritoneal macrophages. HO-1–DsRed fluorescence in macrophages from Nrf2−/−::HO-1DsRed/+ mice was almost comparable to that in macrophages from HO-1DsRed/+ mice even after hemin challenge.
FIG 7.
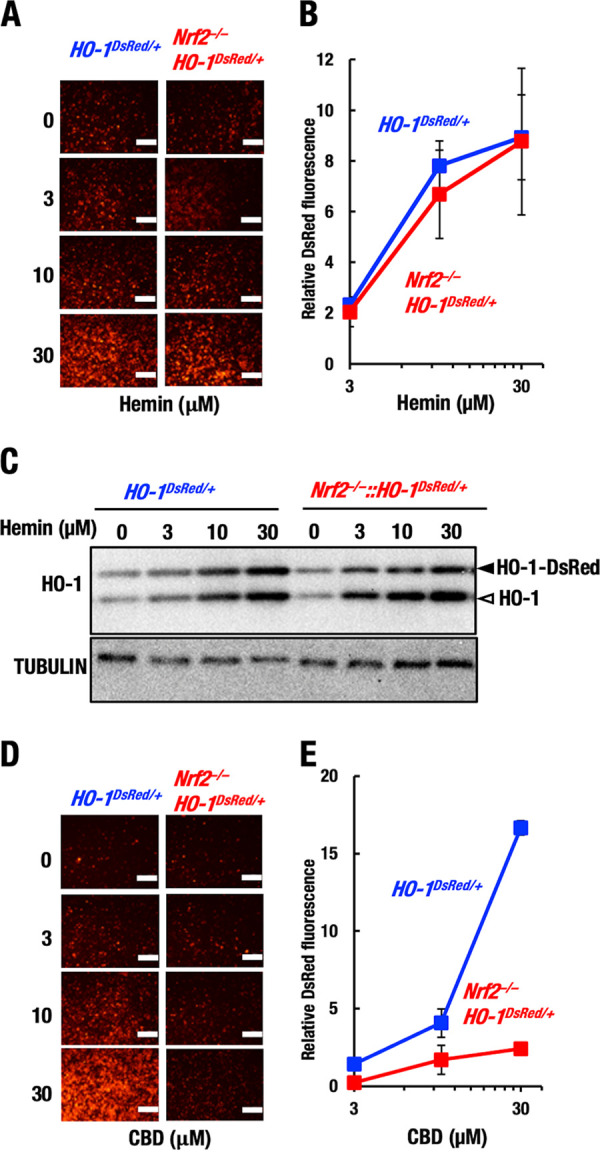
NRF2-independent HO-1–DsRed induction by hemin. Thioglycolate-induced peritoneal macrophages were isolated from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice. (A and B) Representative images (A) and relative DsRed fluorescence intensity (B) in macrophages after 48 h of treatment with hemin. Note that Nrf2 knockout did not affect the induction of HO-1 gene expression by hemin. Scale bars, 200 μm. (C) Protein levels of HO-1 and HO-1–DsRed in macrophages after 3 h of treatment with hemin. Open and closed arrowheads indicate HO-1-DsRed and endogenous HO-1, respectively. (D and E) Representative images (D) and relative DsRed fluorescence intensity (E) in macrophages after 48 h of treatment with CBD. Scale bars, 200 μm.
To further verify the results of the HO-1–DsRed fluorescence study, we conducted a Western blot analysis. We found that hemin indeed increased HO-1 and HO-1–DsRed protein levels in a dose-dependent manner, and these increases were comparable in HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+macrophages (Fig. 7C). These results unequivocally demonstrate that hemin upregulates HO-1 gene expression in an NRF2-independent manner. Thus, at least two regulatory pathways for HO-1 gene induction have emerged, i.e., NRF2-dependent (electrophilic inducers or the stress response) and NRF2-independent (hemin) pathways.
CBD induces HO-1 expression in an NRF2-dependent manner.
CBD was reported to induce HO-1 expression in keratinocytes by regulating BACH1 activity, but this induction is independent of NRF2 (29). BACH1 activity has also been shown to be linked to the intracellular heme concentration (44), suggesting that CBD and hemin act through the same pathway. Therefore, we next tested whether CBD induces HO-1 gene expression in an NRF2-independent manner in macrophages as well.
For this purpose, we treated peritoneal macrophages from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice with CBD by utilizing the same protocol used for hemin treatment. We found that the DsRed fluorescence level was indeed upregulated in HO-1DsRed/+ macrophages upon CBD treatment (Fig. 7D and E), indicating that CBD indeed induced HO-1 in the macrophages. However, in contrast to that upon treatment with hemin, the DsRed fluorescence level was not upregulated in the Nrf2−/−::HO-1DsRed/+ macrophages, even upon challenge with 30 μM CBD. These results demonstrate that CBD induces HO-1 expression in an NRF2-dependent manner in peritoneal macrophages. The molecular mechanism by which CBD activates NRF2 needs to be clarified.
Differential effects of CDDO-Im and hemin on NRF2 and BACH1 in macrophages.
Thus far, our results demonstrated at least two modes of HO-1 gene induction: NRF2-dependent stress response-mediated and NRF2-independent substrate induction. In this regard, hemin was proposed to induce HO-1 expression by means of derepression of the repression mediated by BACH1. This derepression by hemin has been suggested to be attained through the repression of DNA-binding activity (21), inducing the nuclear export (24) and degradation (25, 26) of BACH1. To address whether these mechanisms are also operational in peritoneal macrophages, we examined the protein levels of NRF2 and BACH1 in HO-1DsRed/+ macrophages upon induction by CDDO-Im (10 nM) and hemin (30 μM) through time course studies by means of Western blot analyses (Fig. 8A).
FIG 8.
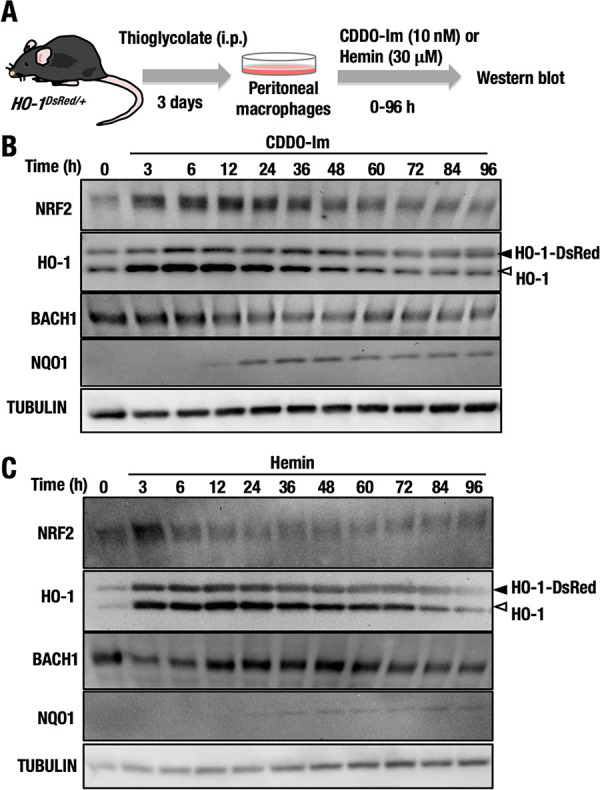
NRF2 and BACH1 expression over time in HO-1DsRed/+ macrophages treated with CDDO-Im and hemin. (A) Thioglycolate-induced peritoneal macrophages were isolated from HO-1DsRed/+ mice, and the macrophages were treated with 10 nM CDDO-Im or 30 μM hemin for 0 to 96 h. (B and C) The protein levels of NRF2, HO-1, BACH1, NQO1, and tubulin in macrophages treated with CDDO-Im (B) or hemin (C) for 0 to 96 h were examined by Western blot analysis. Open and closed arrowheads indicate HO-1-DsRed and endogenous HO-1, respectively.
We found that the NRF2 protein level was increased as early as 3 h after CDDO-Im treatment and started to decrease 48 h after treatment (Fig. 8B). Showing very good correlation with NRF2 accumulation, the protein levels of both endogenous HO-1 and HO-1–DsRed were simultaneously increased after 3 h of CDDO-Im treatment and returned to basal levels approximately 60 h after treatment. These results suggested that NRF2 immediately and quickly activated HO-1 gene expression in response to CDDO-Im. In contrast, the BACH1 level increased slightly and gradually decreased 24 h after treatment. The protein level of NQO1, another representative NRF2 target gene, began to increase 24 h after CDDO-Im treatment, indicating that the NQO1 induction by NRF2 is much slower than HO-1 induction.
Similarly, we executed a time course study to determine the protein levels of NRF2, BACH1, HO-1, and NQO1 after hemin treatment (Fig. 8C). Unlike the case following CDDO-Im treatment, NRF2 was transiently increased 3 h after hemin treatment, but the increased level of NRF2 immediately returned to normal after 6 h of hemin treatment. Expression of the endogenous HO-1 and HO-1–DsRed proteins was concomitantly increased after 3 h of treatment, but these increases had not been maintained at 84 h after treatment. Hemin treatment did not substantially induce NQO1 expression, further supporting the notion that NRF2 does not contribute to the hemin-mediated induction of HO-1. The BACH1 protein level was transiently and slightly decreased 3 h after hemin treatment, but the level returned to normal 12 h after treatment. These results support the conclusion that the hemin-mediated induction of HO-1 gene expression is independent of the contribution of NRF2.
Differential effects of CDDO-Im and hemin on the expressions of SPIC and NQO1 genes in macrophages.
To gain further insights into the heme-mediated and NRF2-mediated regulations of HO-1 gene, we examined the expressions of other target genes of CDDO-Im and hemin. At first, we examined induction of HO-1 mRNA by CDDO-Im or hemin treatment. Consistent with the expression profiles of HO-1–DsRed fluorescence and HO-1 protein, we found that the CDDO-Im-mediated induction of HO-1 mRNA was highly dependent on NRF2 (Fig. 9A), while the hemin-mediated induction of HO-1 mRNA expression was independent of NRF2 (Fig. 9B).
FIG 9.

Differential effects of CDDO-Im and hemin on the expression of HO-1, SPIC, and NQO1 genes. Thioglycolate-induced peritoneal macrophages were isolated from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice, and the macrophages were treated with 10 nM CDDO-Im (A, C, and E) or 30 μM hemin (B, D, and F) for 6 h. Relative expressions of HO-1 (A and B), SPIC (C and D), and NQO1 (E and F) of HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ macrophages were examined. ***, P < 0.001.
It has been shown that the expression of the SPIC gene is induced by hemin (45). SPIC is a transcription factor required for the development of red pulp macrophage in the spleen. Therefore, we examined expression of the SPIC gene in peritoneal macrophages isolated from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice. We found that CDDO-Im treatment did not induce the expression of the SPIC gene in either HO-1DsRed/+ or Nrf2−/−::HO-1DsRed/+ macrophages (Fig. 9C), while hemin induced expression of the SPIC gene in both HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ macrophages (Fig. 9D). These results indicate that the SPIC gene is regulated by hemin in an NRF2-independent manner.
We also examined expression of the NQO1 gene, a representative NRF2 target gene. We found that the expression of NQO1 mRNA was induced by both CDDO-Im (Fig. 9E) and hemin (Fig. 9F). However, in contrast to the case for the HO-1 gene, these inductions were abrogated almost completely in the Nrf2−/−::HO-1DsRed/+ macrophages, indicating that CDDO-Im- and hemin-mediated inductions of the NQO1 gene are both dependent on NRF2. These results thus clearly demonstrate that the regulatory pathways activated by hemin and CDDO-Im are distinct and the former activates specifically the SPIC gene, whereas the latter is operating specifically for the NQO1 gene. An intriguing finding in this study is that these two regulatory pathways act for the HO-1 gene simultaneously but independently.
Cobalt induces HO-1–DsRed expression through the NRF2 pathway but not the HIF pathway.
Another well-known HO-1 inducer is CoCl2 (31–34). While CoCl2 can potently activate the hypoxia response system by stabilizing the HIF protein, causing its accumulation (35), CoCl2 is also known to activate the NRF2 pathway by increasing oxidative stress (36, 37). However, its genuine contribution remains debatable. To address this issue, we tested the effects of CoCl2 using HO-1–DsRed reporter macrophages in the presence or absence of NRF2. We found that CoCl2 exposure elevated HO-1–DsRed expression in the HO-1DsRed/+ macrophages but not the Nrf2−/−::HO-1DsRed/+ macrophages (Fig. 10A and B). This result supports the notion that CoCl2 activates HO-1 gene expression through the NRF2 pathway.
FIG 10.
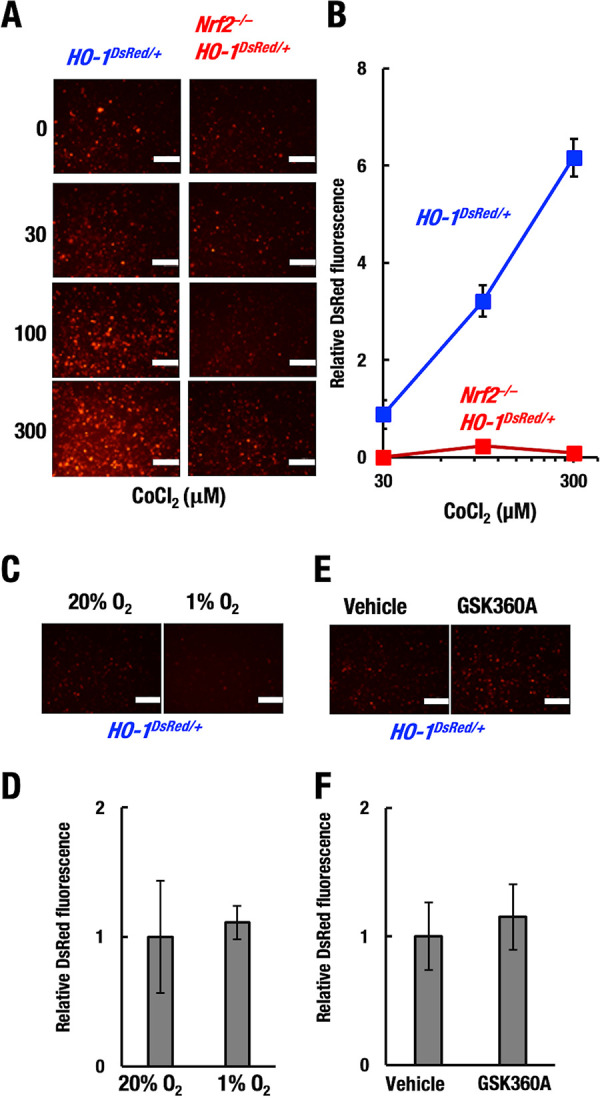
CoCl2 induces HO-1–DsRed expression through the NRF2 pathway. Thioglycolate-induced peritoneal macrophages were isolated from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice. Representative images (A, C, and E) and relative DsRed fluorescence intensity (B, D, and F) in macrophages after 48 h of treatment with CoCl2 (A and B), hypoxia (1% O2) (C and D), or the PHD inhibitor 50 μM GSK360A (E and F). Scale bar, 200 μm.
Considering that CoCl2 is known to activate the hypoxia response system (35), we cultured HO-1–DsRed macrophages under hypoxic conditions (1% O2) for 48 h. We found that hypoxia induced hardly any HO-1–DsRed expression in the HO-1DsRed/+ macrophages (Fig. 10C and D). In addition, to test whether the prolyl hydroxylase domain-containing protein (PHD)-HIF pathway contributes to HO-1 expression, we treated the HO-1–DsRed macrophages with the chemical PHD inhibitor GSK360A, an activator of the HIF pathway (46). GSK360A treatment induced hardly any HO-1–DsRed expression in the HO-1DsRed/+macrophages (Fig. 10E and F). These results are consistent with a previous report showing that cobalt induced HO-1 expression by a HIF-independent mechanism in Chinese hamster ovary cells (37). Thus, these results indicate that CoCl2-mediated HO-1 induction depends on the NRF2 pathway but not the HIF pathway.
DISCUSSION
HO-1 is the key enzyme in the heme catabolic pathway. HO-1 also plays critical roles in various cytoprotective responses, especially those against oxidative stress, inflammation, and apoptotic stimuli (1). HO-1 is also known to be involved in the pathogenesis of various diseases, including neurodegeneration, diabetes, and cancer (47). Considering these broad contributions, regulation of the HO-1 gene seems to be complex and multimodal. However, the levels at which the HO-1 gene is regulated and details of the related transcriptional control mechanisms have been largely unexplored. In this study, we developed a new system to monitor HO-1 gene expression by using HO-1–DsRed-knock-in mice. We found that HO-1–DsRed fluorescence could be induced by treatment with a variety of HO-1 inducers in vivo and ex vivo. The latter analysis, which utilized peritoneal macrophages derived from HO-1–DsRed mice lacking NRF2, a major regulator of the oxidative/electrophilic stress response, led us to identify two distinct types of HO-1 gene expression regulation: regulation through the NRF2-dependent stress response and that through substrate-mediated induction independent of NRF2.
HO-1 gene expression can be induced by various stressors (4), and NRF2 has been shown to be the major regulator of stress-mediated gene expression (7, 13). In this study, we efficiently and reliably monitored the dependence of HO-1 induction on NRF2 by comparing gene expression in peritoneal macrophages derived from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice. In fact, our study revealed that DEM, CDDO-Im, CDDO-2P-Im, CDDO-3P-Im, CBD, and CoCl2 all induced HO-1 expression in a manner dependent on NRF2. In contrast, the HO-1 substrate hemin has emerged as an NRF2-independent inducer in peritoneal macrophages. As there are reports that suggest that NRF2 contributes to hemin-mediated induction of the HO-1 gene in certain tissues (22), we designed experiments from multiple angles and rigorously tested reproducibility. As summarized in Fig. 11, we conclude that hemin-mediated induction of the HO-1 gene in peritoneal macrophages is regulated by a pathway(s) independent of NRF2.
FIG 11.
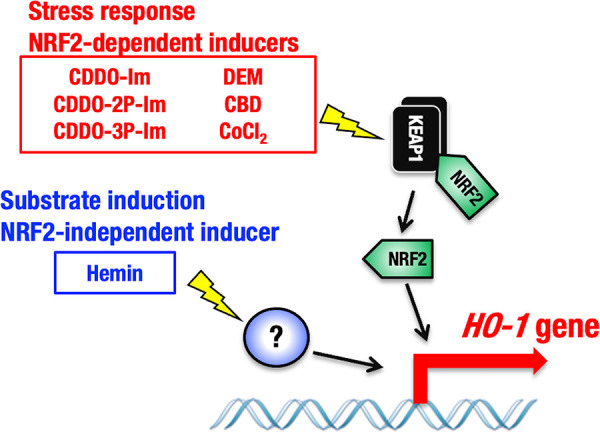
Distinct levels of HO-1 gene regulation by the NRF2-mediated stress response and NRF2-independent substrate induction. The HO-1–DsRed reporter system revealed two levels of HO-1 gene expression regulation by different HO-1 inducers. Whereas CBD, CoCl2, and representative electrophilic NRF2 inducers (DEM, CDDO-Im, CDDO-2P-Im, and CDDO-3P-Im) activate the HO-1 gene in an NRF2-dependent manner, the HO-1 substrate hemin induces HO-1 expression independent of NRF2. The activating transcription factors that contribute to hemin-mediated HO-1 induction are currently unknown.
The molecular basis of HO-1 gene regulation has been extensively studied, especially in relation to NRF2. One of the salient findings along this line is that NRF2-mediated HO-1 upregulation requires the interaction of NRF2 with Brahma-related gene 1 (BRG1), a catalytic subunit of SWI2/SNF2-like chromatin-remodeling complexes (48). Knockdown of BRG1 in SW480 cells selectively decreased inducible expression of the HO-1 gene by treatment with the electrophile DEM but did not affect the expression of other NRF2 target genes, such as NQO1. Deletion of the Neh5 domain in NRF2 markedly repressed the association of cAMP response element-binding protein (CREB)-binding protein (CBP) and BRG1 with NRF2, diminishing their cooperative enhancement of HO-1 promoter activity (49). NRF2-induced BRG1 recruitment to the HO-1 promoter enhances the formation of left-handed DNA or Z-DNA and recruitment of RNA polymerase II, resulting in HO-1 transcriptional activation (50). In addition, RNA polymerase II binding is modulated by noncoding RNA derived from the distal enhancer region (E2) of the HO-1 gene (51). These mechanisms seem to nicely explain the more rapid onset of HO-1 gene induction than induction of the other NRF2 target genes. The onset of induction by electrophilic inducers is typically 3 to 6 h for HO-1 and 12 to 24 h for the other NRF2 target genes.
It has been proposed that hemin-mediated HO-1 gene induction is under the regulation of the heme-binding transcription factor BACH1 (22), which possesses four functional heme-binding motifs, i.e., cysteine-proline motifs (52). In this scenario, the binding of hemin to BACH1 would weaken the DNA-binding activity of BACH1, leading to the nuclear exclusion and rapid degradation of BACH1 (25, 26). As BACH1 binds the DNA motif known as ARE/EpRE/CsMBE, which also hosts NRF2, upon the binding of hemin to BACH1, the motif would be vacant, allowing NRF2 to bind (23). In fact, various organs of Bach1 gene knockout mice, including the thymus, heart, and small intestine, showed upregulated HO-1 expression, but this upregulation was weak in the spleen and liver (22). Compound-knockout mice lacking Nrf2 and Bach1 still showed the induction of HO-1 gene expression in the thymus, heart, lung, and liver, and the levels were substantially higher than those in Nrf2 single-knockout mice. This observation suggests that BACH1 indeed acts to repress constitutive HO-1 gene expression and contributes to the determination of basal HO-1 expression level (22). In contrast, we found in the time course study by Western blotting that changes in the BACH1 protein level did not correlate with changes in HO-1 protein induction. These results suggest that CNC-sMAF regulation, including the switch from BACH1 to NRF2, is unlikely to be involved in the substrate-mediated induction of HO-1 in peritoneal macrophages. One remaining possibility is that DNA-free BACH1 protein charged with hemin is present within cells, but the observation that such BACH1 degrades rapidly within fibroblasts and erythroleukemia cells does not support this possibility (25).
Regarding the molecular basis underlying the hemin-mediated and NRF2-independent induction of HO-1 gene expression, we do not have solid mechanistic insights. However, we surmise that the following observations in the literature may be pertinent. Transactivation of the HO-1 gene involves activator protein-1 (AP-1) (53), nuclear factor-kappa B (NF-κB) (54), heat shock factor (HSF) (55), and HIF (30). The involvement of AP-1 in HO-1 gene regulation is supported by pharmacological inhibition of AP-1 activity in human endothelial cells (56). Since the NRF2-binding site ARE/EpRE/CsMBE includes the AP-1-binding 12-O-tetradecanoylphorbol-13-acetate (TPA) response element (TRE) motif, specific cross talk among bZIP transcription factors may occur, but the relationship between hemin challenge and AP-1 activity has not been tested rigorously. Similarly, an increase in NF-κB DNA binding in response to heme has been reported (57), but the participation of NF-κB in heme-mediated HO-1 gene activation has not been rigorously explored. MAPK cascades have been suggested to play a role in signal-mediated HO-1 gene activation (58), but such regulation seems to be indirect, requiring the cooperation of sequence-specific DNA-binding transcription factors.
In pioneering work for the use of IVIS, the generation of transgenic mice carrying a 2.1-kb region including the basal promoter and proximal enhancer of the HO-1 gene fused to the luciferase gene and the results of analyses of the mice were reported (38). This transgenic mouse line showed the induction of luciferase expression in response to cadmium exposure, but this luciferase expression did not seem to recapitulate endogenous HO-1 gene expression due to the lack of sufficient transcriptional regulatory elements. In contrast, in our HO-1DsRed/+ mouse system, the DsRed gene is under regulation by the endogenous HO-1 locus. Therefore, HO-1–DsRed fluorescence appears to recapitulate endogenous HO-1 gene expression. The HO-1DsRed/+ mouse system will be useful for examining the expression profile of the HO-1 gene. Recently, based on a similar concept, the LacZ, hCG, and luciferase genes were inserted into the HO-1 locus, and mouse lines were developed to facilitate elaborate monitoring of HO-1 gene expression (59). Showing very good agreement, induction of HO-1 gene expression by hemin was also demonstrated in these monitoring mouse lines. One of the advantages of the HO-1DsRed/+ mouse system over this previously reported system is that this system can detect HO-1–DsRed fluorescence in vivo without the administration of any inducing substances.
Chemical NRF2 inducers attract wide-ranging pharmacological interest as antioxidative and anti-inflammatory drugs. In fact, dimethyl fumarate (Tecfidera) has been approved for multiple sclerosis (17), and bardoxolone methyl (Bardoxolone) is in phase-3 clinical trials for diabetic nephropathy (60). We surmise that the NRF2-mediated induction of HO-1 underlies their efficacy. In this regard, the HO-1–DsRed reporter system clearly demonstrates that CDDO-2P-Im and CDDO-3P-Im, two new analogs of CDDO-Im, activate HO-1 expression via the NRF2 pathway. As CDDO-Im is known to inactivate the ubiquitin E3 ligase activity of KEAP1 through modification of Cys151 of KEAP1, leading to the stabilization of NRF2 (61), our present results suggest that CDDO-2P-Im and CDDO-3P-Im induce NRF2 accumulation through the cysteine modification of KEAP1, as is the case for CDDO-Im. Since CDDO-2P-Im and CDDO-3P-Im show promise for prospective clinical use because of their better bioavailability than that of CDDO-Im (18), the ex vivo results indicating that these compounds in fact induce HO-1 gene expression in an NRF2-dependent manner provide important mechanistic support. We surmise that the use of the present monitoring system should be fruitful in further drug development.
In addition to well-known NRF2-activating chemicals, in this study, we examined several HO-1 inducers that have been suggested to exploit pathways other than the NRF2 pathway by capitalizing on the developed monitoring system. CBD has been reported to induce HO-1 expression in keratinocytes by regulating BACH1 independently of NRF2, as observed for hemin (29). Our results revealed that CBD-mediated HO-1 induction was diminished in Nrf2−/−::HO-1DsRed/+ macrophages, indicating that CBD utilizes the NRF2 pathway to induce HO-1 expression. We infer that CBD might modify the cysteine residues of KEAP1 and activate NRF2, similar to electrophilic NRF2 inducers in macrophages. Whether CBD induces HO-1 gene expression via distinct mechanisms in keratinocytes and macrophages remains to be clarified. Multiple mechanisms have been reported for the HO-1 inducer CoCl2. CoCl2 appears to activate the HIF pathway by inhibiting PHD (35) and to activate the NRF2 pathway by increasing oxidative stress (36, 37). We found that CoCl2 induced HO-1 gene expression, but neither hypoxia nor the PHD inhibitor GSK360A induced HO-1 gene expression. Our results show that CoCl2-mediated HO-1 induction is dependent on NRF2, thus providing compelling evidence that CoCl2 induces HO-1 expression through the NRF2 pathway in peritoneal macrophages.
In summary, we have established a new monitoring system that enables us to elaborately analyze HO-1 gene expression. Utilizing this system, we demonstrate that the HO-1 gene is subjected to regulation at two distinct levels through NRF2-dependent stress response-mediated induction and NRF2-independent substrate-mediated induction.
MATERIALS AND METHODS
Mice.
HO-1flox/flox::LysMCre mice lacking the HO-1 gene in which DsRed is expressed in myeloid lineage cells were generated by mating HO-1flox/flox mice (39) and LysMCre mice (62). For the HO-1 flox allele, the HO-1 gene is floxed by loxP sites with the DsRed gene inserted into the 3′ region (39). HO-1DsRed/+ mice were generated by mating HO-1flox/+ mice with Ayu1Cre mice (41). Nrf2−/−::HO-1DsRed/+ mice were generated by mating Nrf2−/− mice (6) with HO-1DsRed/+ mice. Mice with a C57BL/6J background were maintained according to the regulations of the standards for human care and use of laboratory animals of Tohoku University (Sendai, Japan). All animal experiments were executed with the approval of the Tohoku University Animal Care Committee.
Chemical reagents.
The compounds used during the experiment are listed in Table 1.
TABLE 1.
Compounds
| Compound | Company | Catalog no. |
|---|---|---|
| CDDO-Im | Namiki Co., Ltd. | HY-15725 |
| CDDO-2P-Im | Michael B. Sporn lab | None |
| CDDO-3P-Im | Michael B. Sporn lab | None |
| Diethylmaleate (DEM) | Wako Chemicals | 059-02052 |
| Hemin | Alfa Aesar | A11165 |
| Cannabidiol (CBD) | Cayman Chemical | 13956-29-1 |
| Cobalt(II) chloride hexahydrate (CoCl2) | Nacalai | 09206-92 |
Mouse treatments.
CDDO-Im was dissolved in dimethyl sulfoxide (DMSO). This concoction was then mixed with Cremophor-EL–PBS (phosphate-buffered saline) at a 1:1:8 ratio to a final concentration of 3 μM. Ten milliliters of 3 μM CDDO-Im per kg of body weight (BW) was orally administered to the mice at 8 to 12 weeks of age. Hemin was dissolved in 0.2 M NaOH, and the pH was adjusted to 7.4 with 1 M HCl. Hemin (30 mg/kg BW) was intraperitoneally injected into the mice at 8 to 12 weeks of age.
Isolation of peritoneal macrophages.
Mice were intraperitoneally injected with 2.0 ml of 4% thioglycolate (BD). Four days later, macrophages were isolated from the peritoneal cavity in PBS and then immediately placed on ice. The cells were then suspended in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and penicillin (10 U)/streptomycin (0.1 mg/ml). A total of 1 × 106 cells per well were plated in 6-well plates and placed in a 37°C humidified incubator under 5% CO2.
RNA extraction and RT-PCR.
Total RNA was extracted from peritoneal macrophages with a Sepazol-RNA I Super G RNA extraction kit (Nacalai Tesque) and reverse transcribed with ReverTra Ace qPCR RT master mix with gDNA remover (Toyobo). Reverse transcription PCR (RT-PCR) was performed using TaqMan or SYBR with a QuantStudio 6 real-time PCR analyzer (Thermo Fisher Scientific). Gene-specific primer and probe sequences are provided in Table 2.
TABLE 2.
Sequences of primers and probes used for RT-PCRa
| Assay and gene | Sequence |
|---|---|
| RT-PCR (TaqMan) | |
| NQO1 | F: AGCTGGAAGCTGCAGACCTG |
| R: CCTTTCAGAATGGCTGGCA | |
| P: ATTTCAGTTCCCATTGCAGTGGTTTGGG | |
| HPRT | F: CTGGTGAAAAGGAACCTCTCG |
| R: TGAAGTACTCATTATAGTCAAGGG | |
| P: ATCCAACAAAGTCTGGCCTGTATCCAAC | |
| RT-PCR (SYBR) | |
| HO-1 CT | F: CACGCATATACCCGCTACCT |
| R: CCAGAGTGTTCATTCGAGCA | |
| SPIC | F: TGGTAGACAGCATTTACCCTCA |
| R: CAGGCCACCATCCTGTTCTG | |
| DsRed | F: CCCCGTAATGCAGAAGAAGACT |
| R: GATTGACTTGAACTCCACCAGGT |
F, forward primer; R, reverse primer; P, probe.
Western blotting.
Peritoneal macrophages were washed with PBS and lysed in 300 μl of sample buffer (0.1 M Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate [SDS], 20% glycerol, 12% 2-mercaptoethanol, and 0.001% bromophenol blue). The lysates were stored at −80°C. The lysates were then thawed, subjected to 20 s of sonication, and heated at 98°C for 3 min. The proteins were separated by electrophoresis on an 8% or 12% SDS-polyacrylamide gel using the Bio-Rad gel electrophoresis system with SDS-containing running buffer (25 mM Tris-HCl [pH 7.4], 192 mM glycine, and 0.1% SDS) and then electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes by a Trans-Blot Turbo transfer system (Bio-Rad). After 30 min of blocking with 10% nonfat milk at room temperature, the membranes containing the proteins were incubated with primary antibodies for 1 h at room temperature or overnight at 4°C. The membranes were then incubated with secondary antibodies for 1 to 2 h at room temperature. The primary antibodies used for Western blotting are described in Table 3.
TABLE 3.
Antibodies used for Western blotting
| Antibody | Catalog no. | Company/reference | Dilution |
|---|---|---|---|
| Anti-HO-1 | ADI-SPA-985 | Enzo | 1/1,000 |
| Anti-NRF2 | D1Z9C | Cell Signaling Technology | 1/1,000 |
| Anti-NQO1 | ab2346 | Abcam | 3/10,000 |
| Anti-α-tubulin | T9026 | Sigma | 1/1,000 |
| Anti-BACH1 | AF5777 | R&D Systems | 1/1,000 |
In vivo HO-1–DsRed imaging.
In vivo HO-1–DsRed imaging was conducted utilizing an IVIS (PerkinElmer). Male wild-type HO-1DsRed/+ mice at 8 to 12 weeks of age were used in the experiments. The mice were orally administered vehicle or 10 ml of 3 μM CDDO-Im per kg of BW or intraperitoneally injected with vehicle or 30 mg/kg hemin. Seventy-two hours after administration, the mice were anesthetized with isoflurane and immediately placed in a sealed chamber, and fluorescence activity was imaged for 1 s. After measuring whole-body fluorescence, the mice were sacrificed, and the fluorescence activity of several organs was imaged for 1 s. Photons emitted from various regions in the mice were quantified using Living Image software (PerkinElmer).
Histological analysis.
Livers and spleens harvested from HO-1+/+ and HO-1DsRed/+ mice were fixed with Mildform 10N (Wako Pure Chemical Industries). The fixed tissues were then embedded in paraffin, cut into 4-μm-thick sections, and mounted on slides. Tissues were stained with HE. Immunostaining was performed using rabbit polyclonal anti-DsRed antibody (1:1,000; no. 632496, TaKaRa). Images were captured with a DM2500 LED instrument (Leica) using Leica Application Suite version 4.8.0 software (Leica).
HO-1–DsRed fluorescence analysis by plate reader.
Thioglycolate-induced peritoneal macrophages were isolated from HO-1DsRed/+ and Nrf2−/−::HO-1DsRed/+ mice. The macrophages were plated in 96-well plates (black with a flat, transparent bottom) at 5 × 105 cells per well and placed in a 37°C humidified incubator with 5% CO2 in air. The DsRed fluorescence level was detected with a plate reader (PHERAStar).
Statistics.
Data are presented as mean values ± standard errors (SEs). Statistical significance was evaluated by two-way analysis of variance (ANOVA). P values of less than 0.01 were considered to indicate statistical significance.
ACKNOWLEDGMENTS
We thank Fumiki Katsuoka for discussion and advice. We also thank the Biomedical Research Core of Tohoku University Graduate School of Medicine for technical support.
This work was supported in part by MEXT/JSPS KAKENHI (grant no. 19H05649 to M.Y. and grant no. 19K07340 and 17KK0183 to T.S.), the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from AMED under grant no. JP20am0101095 (M.Y.), P-CREATE from AMED under grant no. JP20cm0106101 (M.Y.), and the Takeda Science Foundation (M.Y. and T.S.).
A.Z., T.S., and M.Y. designed the research and analyzed the data. A.Z., S.A., and E.N. conducted the experiments. N.S. synthesized GSK360A compound. M.B.S. synthesized compounds of CDDO-2P-Im and CDDO-3P-Im. T.H., K.I., and M.Y. generated the HO-1 flox mice. A.Z., T.S., and M.Y. wrote the paper.
M.B.S. is an employee and shareholder of Triterpenoid Therapeutics. The other authors declare no competing financial interests.
Contributor Information
Takafumi Suzuki, Email: taka23@med.tohoku.ac.jp.
Masayuki Yamamoto, Email: masiyamamoto@med.tohoku.ac.jp.
REFERENCES
- 1.Kikuchi G, Yoshida T, Noguchi M. 2005. Heme oxygenase and heme degradation. Biochem Biophys Res Commun 338:558–567. 10.1016/j.bbrc.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 2.Cruse I, Maines MD. 1988. Evidence suggesting that the two forms of heme oxygenase are products of different genes. J Biol Chem 263:3348–3353. 10.1016/S0021-9258(18)69078-7. [DOI] [PubMed] [Google Scholar]
- 3.Shibahara S. 2003. The heme oxygenase dilemma in cellular homeostasis: new insights for the feedback regulation of heme catabolism. Tohoku J Exp Med 200:167–186. 10.1620/tjem.200.167. [DOI] [PubMed] [Google Scholar]
- 4.Ryter SW, Alam J, Choi AM. 2006. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86:583–650. 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 5.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. 1995. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol 15:4184–4193. 10.1128/MCB.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 7.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. 2000. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 275:16023–16029. 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 8.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86. 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M. 2003. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35:238–245. 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24:7130–7139. 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baird L, Yamamoto M. 2020. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol 40:e00099-20. 10.1128/MCB.00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otsuki A, Yamamoto M. 2020. Cis-element architecture of Nrf2-sMaf heterodimer binding sites and its relation to diseases. Arch Pharm Res 43:275–285. 10.1007/s12272-019-01193-2. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki T, Motohashi H, Yamamoto M. 2013. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci 34:340–346. 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Kensler TW, Motohashi H. 2018. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 98:1169–1203. 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB. 2005. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res 65:4789–4798. 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 16.Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, Guilarte TR, Yamamoto M, Sporn MB, Kensler TW. 2007. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther 6:154–162. 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 17.Bomprezzi R. 2015. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Ther Adv Neurol Disord 8:20–30. 10.1177/1756285614564152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao M, Onyango EO, Williams CR, Royce DB, Gribble GW, Sporn MB, Liby KT. 2015. Novel synthetic pyridyl analogues of CDDO-imidazolide are useful new tools in cancer prevention. Pharmacol Res 100:135–147. 10.1016/j.phrs.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 19.Kikuchi G, Yoshida T. 1983. Function and induction of the microsomal heme oxygenase. Mol Cell Biochem 53–54:163–183. 10.1007/BF00225252. [DOI] [PubMed] [Google Scholar]
- 20.Christodoulides N, Durante W, Kroll MH, Schafer AI. 1995. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclase-stimulatory carbon monoxide. Circulation 91:2306–2309. 10.1161/01.cir.91.9.2306. [DOI] [PubMed] [Google Scholar]
- 21.Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, Hayashi N, Yamamoto M, Shibahara S, Fujita H, Igarashi K. 2001. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J 20:2835–2843. 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. 2004. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA 101:1461–1466. 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K. 2002. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J 21:5216–5224. 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki H, Tashiro S, Hira S, Sun J, Yamazaki C, Zenke Y, Ikeda-Saito M, Yoshida M, Igarashi K. 2004. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J 23:2544–2553. 10.1038/sj.emboj.7600248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, Igarashi K. 2007. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol 27:6962–6971. 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, Pass HI, Bhutkar AJ, Tsirigos A, Ueberheide B, Sayin VI, Papagiannakopoulos T, Pagano M. 2019. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 178:316–329.e318. 10.1016/j.cell.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, Frazier WA, Murphy TL, Murphy KM. 2009. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 457:318–321. 10.1038/nature07472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gozzelino R, Soares MP. 2014. Coupling heme and iron metabolism via ferritin H chain. Antioxid Redox Signal 20:1754–1769. 10.1089/ars.2013.5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casares L, García V, Garrido-Rodríguez M, Millán E, Collado JA, García-Martín A, Peñarando J, Calzado MA, de la Vega L, Muñoz E. 2020. Cannabidiol induces antioxidant pathways in keratinocytes by targeting BACH1. Redox Biol 28:101321. 10.1016/j.redox.2019.101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. 1997. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 272:5375–5381. 10.1074/jbc.272.9.5375. [DOI] [PubMed] [Google Scholar]
- 31.Taketani S, Kohno H, Yoshinaga T, Tokunaga R. 1988. Induction of heme oxygenase in rat hepatoma cells by exposure to heavy metals and hyperthermia. Biochem Int 17:665–672. [PubMed] [Google Scholar]
- 32.Christova TY, Gorneva GA, Taxirov SI, Duridanova DB, Setchenska MS. 2003. Effect of cisplatin and cobalt chloride on antioxidant enzymes in the livers of Lewis lung carcinoma-bearing mice: protective role of heme oxygenase. Toxicol Lett 138:235–242. 10.1016/s0378-4274(02)00416-2. [DOI] [PubMed] [Google Scholar]
- 33.Kaliman PA, Nikitchenko IV, Sokol OA, Strel'chenko EV. 2001. Regulation of heme oxygenase activity in rat liver during oxidative stress induced by cobalt chloride and mercury chloride. Biochemistry (Mosc) 66:77–82. 10.1023/a:1002889814723. [DOI] [PubMed] [Google Scholar]
- 34.Loboda A, Jazwa A, Wegiel B, Jozkowicz A, Dulak J. 2005. Heme oxygenase-1-dependent and -independent regulation of angiogenic genes expression: effect of cobalt protoporphyrin and cobalt chloride on VEGF and IL-8 synthesis in human microvascular endothelial cells. Cell Mol Biol (Noisy-le-grand) 51:347–355. [PMC free article] [PubMed] [Google Scholar]
- 35.Muñoz-Sánchez J, Chánez-Cárdenas ME. 2019. The use of cobalt chloride as a chemical hypoxia model. J Appl Toxicol 39:556–570. 10.1002/jat.3749. [DOI] [PubMed] [Google Scholar]
- 36.Tomaro ML, Frydman J, Frydman RB. 1991. Heme oxygenase induction by CoCl2, Co-protoporphyrin IX, phenylhydrazine, and diamide: evidence for oxidative stress involvement. Arch Biochem Biophys 286:610–617. 10.1016/0003-9861(91)90088-z. [DOI] [PubMed] [Google Scholar]
- 37.Gong P, Hu B, Stewart D, Ellerbe M, Figueroa YG, Blank V, Beckman BS, Alam J. 2001. Cobalt induces heme oxygenase-1 expression by a hypoxia-inducible factor-independent mechanism in Chinese hamster ovary cells: regulation by Nrf2 and MafG transcription factors. J Biol Chem 276:27018–27025. 10.1074/jbc.M103658200. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Feng JQ, Harris SE, Contag PR, Stevenson DK, Contag CH. 2001. Rapid in vivo functional analysis of transgenes in mice using whole body imaging of luciferase expression. Transgenic Res 10:423–434. 10.1023/a:1012042506002. [DOI] [PubMed] [Google Scholar]
- 39.Mamiya T, Katsuoka F, Hirayama A, Nakajima O, Kobayashi A, Maher JM, Matsui H, Hyodo I, Yamamoto M, Hosoya T. 2008. Hepatocyte-specific deletion of heme oxygenase-1 disrupts redox homeostasis in basal and oxidative environments. Tohoku J Exp Med 216:331–339. 10.1620/tjem.216.331. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki T, Kelly VP, Motohashi H, Nakajima O, Takahashi S, Nishimura S, Yamamoto M. 2008. Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. J Biol Chem 283:2021–2030. 10.1074/jbc.M708352200. [DOI] [PubMed] [Google Scholar]
- 41.Niwa H, Araki K, Kimura S, Taniguchi S, Wakasugi S, Yamamura K. 1993. An efficient gene-trap method using poly A trap vectors and characterization of gene-trap events. J Biochem 113:343–349. 10.1093/oxfordjournals.jbchem.a124049. [DOI] [PubMed] [Google Scholar]
- 42.Poss KD, Tonegawa S. 1997. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci USA 94:10925–10930. 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Numata I, Okuyama R, Memezawa A, Ito Y, Takeda K, Furuyama K, Shibahara S, Aiba S. 2009. Functional expression of heme oxygenase-1 in human differentiated epidermis and its regulation by cytokines. J Invest Dermatol 129:2594–2603. 10.1038/jid.2009.119. [DOI] [PubMed] [Google Scholar]
- 44.Igarashi K, Sun J. 2006. The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal 8:107–118. 10.1089/ars.2006.8.107. [DOI] [PubMed] [Google Scholar]
- 45.Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseño CG, Satpathy AT, Kretzer NM, Arase H, Rajasekaran NS, Wang L, Egawa T, Igarashi K, Baltimore D, Murphy TL, Murphy KM. 2014. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 156:1223–1234. 10.1016/j.cell.2014.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki N, Matsuo-Tezuka Y, Sasaki Y, Sato K, Miyauchi K, Kato K, Saito S, Shimonaka Y, Hirata M, Yamamoto M. 2018. Iron attenuates erythropoietin production by decreasing hypoxia-inducible transcription factor 2α concentrations in renal interstitial fibroblasts. Kidney Int 94:900–911. 10.1016/j.kint.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 47.Waza AA, Hamid Z, Ali S, Bhat SA, Bhat MA. 2018. A review on heme oxygenase-1 induction: is it a necessary evil. Inflamm Res 67:579–588. 10.1007/s00011-018-1151-x. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Ohta T, Maruyama A, Hosoya T, Nishikawa K, Maher JM, Shibahara S, Itoh K, Yamamoto M. 2006. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol Cell Biol 26:7942–7952. 10.1128/MCB.00700-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Hosoya T, Maruyama A, Nishikawa K, Maher JM, Ohta T, Motohashi H, Fukamizu A, Shibahara S, Itoh K, Yamamoto M. 2007. Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes. Biochem J 404:459–466. 10.1042/BJ20061611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maruyama A, Mimura J, Harada N, Itoh K. 2013. Nrf2 activation is associated with Z-DNA formation in the human HO-1 promoter. Nucleic Acids Res 41:5223–5234. 10.1093/nar/gkt243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maruyama A, Mimura J, Itoh K. 2014. Non-coding RNA derived from the region adjacent to the human HO-1 E2 enhancer selectively regulates HO-1 gene induction by modulating Pol II binding. Nucleic Acids Res 42:13599–13614. 10.1093/nar/gku1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lathrop JT, Timko MP. 1993. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science 259:522–525. 10.1126/science.8424176. [DOI] [PubMed] [Google Scholar]
- 53.Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. 2010. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol 80:1895–1903. 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 54.Alam J, Cook JL. 2007. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol 36:166–174. 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 55.Koizumi S, Gong P, Suzuki K, Murata M. 2007. Cadmium-responsive element of the human heme oxygenase-1 gene mediates heat shock factor 1-dependent transcriptional activation. J Biol Chem 282:8715–8723. 10.1074/jbc.M609427200. [DOI] [PubMed] [Google Scholar]
- 56.Alam J, Den Z. 1992. Distal AP-1 binding sites mediate basal level enhancement and TPA induction of the mouse heme oxygenase-1 gene. J Biol Chem 267:21894–21900. 10.1016/S0021-9258(19)36696-7. [DOI] [PubMed] [Google Scholar]
- 57.Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. 1994. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA 91:5987–5991. 10.1073/pnas.91.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peterson SJ, Rubinstein R, Faroqui M, Raza A, Boumaza I, Zhang Y, Stec D, Abraham NG. 2019. Positive effects of heme oxygenase upregulation on adiposity and vascular dysfunction: gene targeting vs. pharmacologic therapy. Int J Mol Sci 20:2514. 10.3390/ijms20102514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McMahon M, Ding S, Acosta-Jimenez LP, Frangova TG, Henderson CJ, Wolf CR. 2018. Measuring in vivo responses to endogenous and exogenous oxidative stress using a novel haem oxygenase 1 reporter mouse. J Physiol 596:105–127. 10.1113/JP274915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanda H, Yamawaki K. 2020. Bardoxolone methyl: drug development for diabetic kidney disease. Clin Exp Nephrol 24:857–864. 10.1007/s10157-020-01917-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saito R, Suzuki T, Hiramoto K, Asami S, Naganuma E, Suda H, Iso T, Yamamoto H, Morita M, Baird L, Furusawa Y, Negishi T, Ichinose M, Yamamoto M. 2016. Characterizations of three major cysteine sensors of Keap1 in stress response. Mol Cell Biol 36:271–284. 10.1128/MCB.00868-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8:265–277. 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]