

Abstract
Background:
Altered postinjury platelet behavior is recognized in the pathophysiology of trauma-induced coagulopathy (TIC), but the mechanisms remain largely undefined. Studies suggest that soluble factors released by injury may inhibit signaling pathways and induce structural changes in circulating platelets. Given this, we sought to examine the impact of treating healthy. platelets with plasma from injured patients. We hypothesized that healthy platelets treated ex-vivo with plasma from injured patients with shock would impair platelet aggregation, while treatment with plasma from injured patients with significant injury burden, but without shock, would enhance platelet aggregation.
Methods:
Plasma samples were isolated from injured patients (pretransfusion) and healthy donors at a Level I trauma center and stored at −80°C. Plasma samples from four separate patients in each of the following stratified clinical groups were used: mild injury/no shock (injury severity score [ISS] 2–15, base excess [BE]>−6), mild injury/with shock (ISS 2–15, BE≤−6), severe injury/no shock (ISS>25, BE>−6), severe injury/with shock (ISS>25, BE≤−6), minimal injury (ISS 0/1, BE>−6), and healthy. Platelets were isolated from three healthy adult males and were treated with plasma for 30 min. Aggregation was stimulated with a thrombin receptor agonist and measured via multiple-electrode platelet aggregometry. Data were normalized to HEPES Tyrode’s (HT) buffer-only treated platelets. Associations of plasma treatment groups with platelet aggregation measures were tested with Mann–Whitney U tests.
Results:
Platelets treated with plasma from patients with shock (regardless of degree of injury) had significantly impaired thrombin-stimulated aggregation compared with platelets treated with plasma from patients without shock (P=0.002). Conversely, platelets treated with plasma from patients with severe injury, but without shock, had amplified thrombin-stimulated aggregation (P=0.030).
Conclusion:
Shock-mediated soluble factors impair platelet aggregation, and tissue injury-mediated soluble factors amplify platelet aggregation. Future characterization of these soluble factors will support development of novel treatments of TIC.
Keywords: Platelet activation, platelet adhesiveness, platelet aggregation, platelet function tests, shock, thrombin, thrombin receptor peptide (1–6)-amide, trauma
INTRODUCTION
Despite advances in resuscitation and treatment strategies, hemorrhage associated with trauma-induced coagulopathy (TIC) remains a significant driver of morbidity and mortality in injured patients (1–3). Our understanding of the pathophysiology of TIC continues to evolve to include increased recognition of cellular-mediated coagulopathy and inflammation. Specifically, alterations in platelet primary and secondary functions are increasingly described in injured patients including ex-vivo evidence of increased circulating activated platelets with paradoxical aggregation impairments, and additionally morphologic derangements, and transcriptomic changes (2–8). Furthermore, evidence suggests that the pathophysiology of TIC involves the release of numerous proteins, damage-associated molecular patterns (DAMPs), and microparticles in response to tissue injury and hypoperfusion due to hemorrhage that may affect platelet function (8–13). However, there is vast heterogeneity in injury patterns resulting in varying degrees of tissue damage and hypoperfusion from hemorrhage, which have distinct effects on platelet hemostatic behaviors (14).
After trauma, tissue injury and endothelial disruption are thought to release large quantities of activating factors including tissue factor, microparticles, and thrombin, activating circulating platelets, even at sites distant from the injury (15, 16). However, hemorrhagic shock has been shown to induce impairments in platelet aggregation, which may be mediated by several factors including histones, soluble fibrin, taurocholic acid, or by depletion of platelet energy substrates such as adenosine triphosphate (ATP) and guanine triphosphate (7, 17–19). Recent studies have also demonstrated it is possible to impair calcium signaling and aggregation in healthy platelets by treating them with injured patient plasma ex-vivo (17, 20). However, the differential effects of tissue injury and shock on ex-vivo platelet aggregation have not been examined. Therefore, in this study, we sought to further characterize the effect of injured patient plasma treatment on healthy donor platelet function, specifically focusing on the degree of injury and presence of shock in the injured patient plasma donors. We hypothesized that healthy platelets treated ex-vivo with plasma from injured patients with shock would impair platelet aggregation, while treatment with plasma from injured patients with significant injury burden, but without shock, would enhance platelet aggregation.
PATIENTS AND METHODS
Healthy donor platelet isolation
Whole blood from three healthy adult males with no known bleeding diathesis or history of antiplatelet or anticoagulant therapy was collected in sodium citrate (0.109 M) (Becton Dickinson, Franklin Lakes, NJ) on six separate days (18 mL total). Blood was immediately centrifuged at 200×g for 20 min at room temperature. The resulting platelet-rich plasma was diluted with an equal volume of modified HEPES-buffered Tyrode’s solution (HT, 137 mM NaCl, 2.7 mM KCl, 2mM MgCl2, 0.42mM NaH2PO4, 5 mM glucose, 10 mM HEPES, pH 7.4), and Prostaglandin-E1 (Cayman Chemicals, Ann Arbor, Mich) was added to a final concentration of 1 μM to prevent premature platelet activation during preparation. Contaminating erythrocytes and leukocytes were then pelleted by centrifugation at 100×g for 20 min at room temperature. The resulting platelet-rich plasma was again supplemented with 1 μM. Prostaglandin-E1 and centrifuged at 800×g for 20 min to pellet platelets. The diluted plasma supernatant was removed, and the platelet pellet was washed twice with HT buffer. Platelets were resuspended in HT buffer supplemented with 3 mg/mL bovine serum albumin (Gold Biotechnology, St. Louis, Mo), counted, and diluted further with HT to a final concentration of 2.5×108/mL. Platelets were used immediately after preparation and not stored.
Injured patient plasma samples
On arrival to the emergency department and prior to any blood product transfusion, whole blood was collected in sodium citrate (0.109 M) from patients with the highest level of trauma activation as a part of a longitudinal, prospective study of coagulation and inflammation following injury (2011–2019), approved by the Committee on Human Research at the University of California, San Francisco. Plasma was prepared by centrifugation of whole blood at 2,960×g for 10 min at room temperature and stored at −80°C. For this study, patients with the following characteristics were excluded: female, under age 18, burns covering more than 20% of body surface area, or use of anticoagulant or antiplatelet therapy. In all, 20 injured patient plasma samples were selected to generate five groups with distinct injury and shock characteristics. Injury severity was stratified using the injury severity score (ISS): a score of 1 or 0 indicating minimal or no injury, 2 to 15 mild injury, and >25 severe injury. Shock was defined by base excess (BE) ≤ −6 meq/L on arrival to the emergency department (21–23). Four plasmas from patients in each of the following injury/shock groups were included: mild injury/no shock (ISS 2–15, BE>−6), mild injury/with shock (ISS 2–15, BE≤−6), severe injury/no shock (ISS>25, BE>−6), severe injury/with shock (ISS>25, BE≤−6), and minimally injured patients or “trauma controls” (ISS of 0 or 1 and BE>−6). Plasma from injured patients with moderate injury (ISS 15–25) was not included to maximize injury/shock effects. Injured patient plasma donor characteristics are shown in Table 1. Healthy donor plasma samples from four adult males (age 30–38) were obtained, processed, and stored in the same fashion to the injured patient plasma samples after obtaining written informed consent according to approval by the Committee on Human Research at the University of California, San Francisco. Prior to use in aggregometry experiments, all plasma samples were thawed and centrifuged at 13,000×g for 5 min at 4°C (to remove residual contaminating cells).
Table 1.
Summary of demographics and clinical characteristics of injured patient plasma donors
| Patient no. | Age | Injury/shock category | ISS | Base excess (mmol/L) | Mechanism | RBCs 0–24 h | Status at discharge | Clinical synopsis |
|---|---|---|---|---|---|---|---|---|
| 1 | 53 | ISS 0 or 1 | 0 | 2.3 | Found down | 0 | Alive | Found down with minor abrasions to head, intoxicated |
| 2 | 23 | ISS 0 or 1 | 1 | −1.6 | Found down | 0 | Alive | Found down, no injuries, intoxicated |
| 3 | 40 | ISS 0 or 1 | 1 | 0.0 | SW | 0 | Alive | Superficial stab wound to back, intoxicated |
| 4 | 40 | ISS 0 or 1 | 1 | −3.8 | SW | 0 | Alive | Superficial stab wounds to shoulder and back |
| 5 | 30 | MI, NS | 5 | 3.0 | Blunt | 0 | Alive | Assault, multiple facial fractures and lacerations |
| 6 | 42 | MI, NS | 5 | −1.6 | SW | 0 | Alive | Multiple superficial penetrating wounds, intoxicated |
| 7 | 37 | MI, NS | 10 | 5.3 | SW | 0 | Alive | Penetrating wound thorax, chest tube for hemothorax |
| 8 | 41 | MI, NS | 13 | −1.8 | Crush | 0 | Alive | Lower extremity crush injury with multiple open fractures |
| 9 | 46 | MI, +S | 3 | −7.6 | SW | 0 | Alive | Abdominal wound, facial lacerations, to OR for laparotomy |
| 10 | 37 | MI, +S | 5 | −18.2 | Blunt | 0 | Alive | Assault, multiple facial fractures and lacerations, intoxicated |
| ii | 28 | MI, +S | 5 | −7.5 | GSW | 2 | Alive | Multiple wounds lower extremity, to OR for exploration and angiogram, minor vascular injury, intoxicated |
| 12 | 19 | MI, +S | 6 | −6.4 | SW | 1 | Alive | Blunt and SW assault with liver injury, to OR for laparotomy |
| 13 | 53 | SI, NS | 29 | −3.0 | GSW | 0 | Alive | Wound to chest and abdomen, to OR for management of diaphragm and gastric injuries, hemothorax |
| 14 | 59 | SI, NS | 35 | −1.7 | Blunt | 0 | Expired | MVC with fractures, SCI, to OR for orthopedic fixation |
| 15 | 38 | SI, NS | 41 | −5.1 | GSW | 0 | Alive | Liver, diaphragm, and pericardial injuries, to OR for laparotomy, median sternotomy and pericardial window, intoxicated |
| 16 | 37 | SI, NS | 42 | −1.3 | Blunt | 0 | Alive | Fall with spinal fractures and cervical SCI, to OR for fixation |
| 17 | 65 | SI, +S | 42 | −6.9 | Blunt | 4 | Alive | MVC w/ spinal fractures and SCI, to OR for fixation |
| 18 | 41 | SI, +S | 57 | −4.7 | Blunt | 5 | Alive | MVC w/ pneumothorax, fractures, grade 5 splenic injury- to OR for laparotomy and splenectomy |
| 19 | 55 | SI, +S | 59 | −8.1 | Blunt | 18 | Expired | MVC w/ splenic and liver lacerations, bladder injury, pelvic fractures to OR for laparotomy and splenectomy |
| 20 | 21 | SI, +S | 75 | −7.9 | GSW | 6 | Expired | Penetrating wounds (neck, leg), SCI, cardiac arrest in field, regained pulse w/ CPR. Declared brain dead in ICU |
ISS indicates injury severity score. Injury/shock categories: ISS 0 or 1—trauma control; MI, NS—mild injury, no shock (ISS 2–15, base excess [BE] > −6); MI, +S—mild injury with shock (ISS 2–15, BE ≤−6); SI, NS—severe injury, no shock (ISS>25, BE > −6); SI, +S—severe injury with shock (ISS>25, BE ≤ −6). GSW indicates gunshot wound; SW, stab wound; MVC, motor vehicle collision; OR, operating room; SCI, spinal cord injury. RBCs 0 to 24 h—number of packed red blood cell units transfused in first 24 h. None of the included patients had traumatic brain injury or were using antiplatelet or anticoagulant medications. “Intoxicated” patients had positive blood alcohol levels (range 93 mg/dL –497 mg/dL).
Aggregometry with plasma-treated platelets
A schematic representation of the experimental workflow for aggregometry with plasma-treated platelets can be found in Figure 1A. Washed platelets suspended in HT buffer were treated with injured patient or healthy donor plasma to a final concentration of 12% plasma by volume, similar to prior studies examining the effects of plasma on platelet function (17, 20). As an additional control, platelets were also treated with HT buffer alone. After incubation for 20 min, the samples were loaded into test cells of a Multiplate impedance-based platelet aggregometer (Roche, Basel, Switzerland). Electrical impedance was then recorded for 10 min with continuous stirring at 37°C to detect effects of plasma on platelet aggregation prior to agonist stimulation. Thrombin Receptor Activating hexapeptide (TRAP, Hart Biologicals, Hartlepool, UK) was then added to a final concentration of 32 μM, and the resulting platelet aggregation responses were recorded for an additional 15 min. Plasma-stimulated aggregation was measured as the electrical impedance just prior to the addition of TRAP agonist (Fig. 1A, plasma stimulated aggregation). Platelet aggregation responses to TRAP stimulation were calculated by integrating impedance over time to generate the area under the curve (Fig. 1A, AUC) in aggregation units. The change from maximum impedance to end impedance was calculated to measure the degree of platelet disaggregation from the electrodes (examples in Fig. 1E and G). To control for differential platelet reactivity between the healthy donors and between blood draws performed on different days from the same donor, platelet aggregometry parameters from platelets with 12% plasma were normalized to those from platelets with HT buffer only, run in parallel. The Multiplate aggregometer has five sample channels, allowing an HT buffer control and four plasma samples of a given injury/shock group to be interrogated simultaneously. This was repeated for each of the injury/shock groups with the freshly prepared platelets from each of the three healthy platelet donors. Platelets from each donor were used immediately after preparation and were only used in one set of assays to prevent any effects of storage on platelet aggregation.
Fig. 1. Healthy platelets treated with injured patient plasma with varying degrees of injury and presence of shock have differential aggregation responses to agonist stimulation.
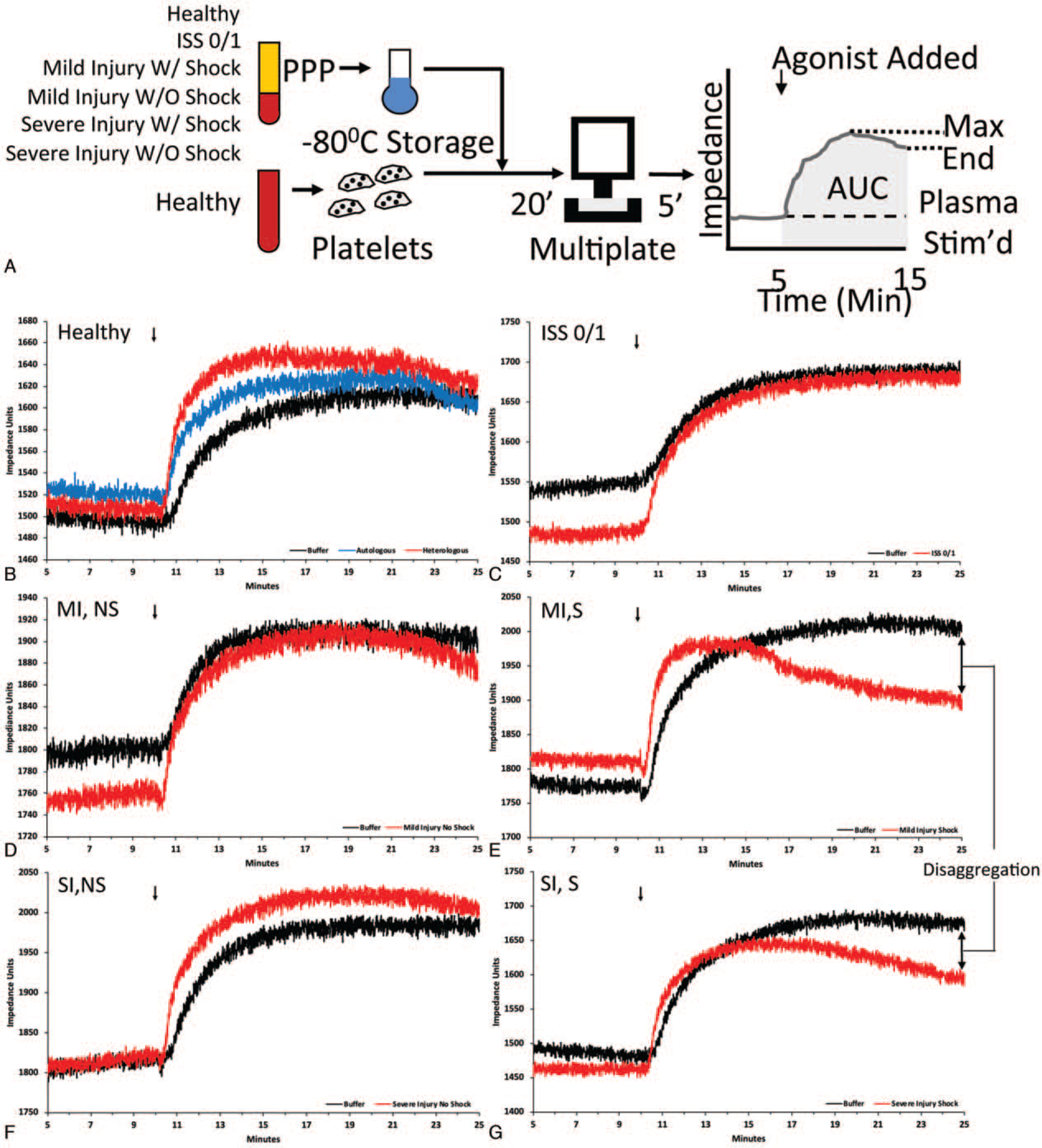
A, Diagram of experimental workflow: Healthy platelets were treated with plasma (PPP), then loaded onto the impedance aggregometer (Multiplate), and the change in impedance was recorded prior to the addition of agonist (plasma-stimulated aggregation), and after the addition of agonist (AUC, area under the curve, stimulated platelet aggregation). Disaggregation calculated as the change from max to end impedance. B–G, Representative multiplate tracings of healthy platelets treated with different plasmas. Platelets were treated with HT Buffer (black), autologous (blue), or heterologous (red) healthy plasma (B); plasma from patients with Injury Severity Score (ISS) of 0 or 1 (C); mild injury without shock (MI, NS) (D); mild injury with shock (MI, S) (E); severe injury without shock (SI, NS) (F); severe injury with shock (SI, S) (G).
Statistical analysis
The platelet aggregometry measurements after treatment with each of the plasmas were averaged across the three healthy platelet donors. These average values were summarized using mean, median, and standard deviations across the injury/shock groups (n=4 plasmas per group). Our total sample size of n=20 represents the number of individual plasmas tested (each with repeated measures across the three healthy platelet donors to assess reproducibility). The bivariate associations between the average platelet aggregometry measurements and the injury or shock group of the injured patient plasma donors versus healthy control plasmas were examined using non-parametric Wilcoxon tests (Mann–Whitney tests), with n=4 per group. A two-sided P value of <0.05 was considered statistically significant. Statistical analyses were performed using STATA version 15 (StataCorp, Tex).
RESULTS
Characteristics of injured patient plasma donors
The 20 male adult injured patient plasma donors were generally young, included those with blunt and penetrating mechanisms, and were without traumatic brain injury (Table 1). When comparing characteristics and outcomes across our injury/shock groups, ISS and base excess were significantly different according to our stratification of injury (by ISS) and shock (by base excess), with median ISS scores of 1 in the ISS 0/1 group, 5 and 8 in the mild injury/no shock and mild injury/with shock groups respectively, and 38 and 58 in the severe injury/no shock and severe injury/with shock groups respectively (<0.01; Table 2). Base excess was similar and close to even in groups without shock, ranging from −0.8 to −2.4, while it was much lower in the groups with shock at −7.6 and −7.9 for mild injury/with shock and severe injury/with shock respectively (P<0.01; Table 2). While there was a trend toward worse shock indices in the patients categorized in the mild injury/with shock and severe/injury with shock groups, this was not statistically significant (Table 2). Only patients in the groups with severe injuries died or suffered a major complication (acute respiratory distress syndrome, multiple organ failure, or venous thromboembolism), though this was not statistically significant due to our small group sizes (Table 2).
Table 2.
Characteristics and outcomes of injured patient plasma donors grouped by injury/shock categories*
| Variables† | ISS 0 or 1 | Mild injury, no shock | Mild injury, with shock | Severe injury, no shock | Severe injury, with shock | P value |
|---|---|---|---|---|---|---|
| Age | 40 (32–46) | 39 (34–42) | 33 (24–42) | 46 (38–56) | 48 (31 –60) | <0.53 |
| ISS | 1 (0–1) | 8 (5–12) | 5 (4–6) | 38 (32–42) | 58 (50–67) | <0.01 |
| Blunt Mechanism | 2 (50%) | 2 (50%) | 1 (25%) | 3 (75%) | 3 (75%) | <0.85 |
| Base excess (mmol/L) | −0.8 (−2.7–1.2) | 0.7 (−1.7–4.2) | −7.6 (−12.9–7.0) | −2.4 (−4.1–1.5) | −7.9 (−8.1–6.9) | <0.01 |
| Admit Shock index | 0.69 (0.57–0.88) | 0.73 (0.64–0.76) | 0.84 (0.68–1.15) | 0.57 (0.49–0.71) | 0.74 (0.55–0.89) | <0.61 |
| Worst shock index in ED | 0.85 (0.60–1.01) | 0.94 (0.81–1.34) | 1.01 (0.83–1.34) | 0.78 (0.67–0.90) | 1.17 (0.92 1.45) | <0.29 |
| Hematocrit (%) | 44 (42–48) | 42 (41 –44) | 42 (41–45) | 41 (39–43) | 38 (36–39) | <0.04 |
| Platelet count (×109) | 232 (164–313) | 248 (186–372) | 291 (278–325) | 248 (180–265) | 225 (208–276) | <0.63 |
| TRAP AUC | 73 (59–86) | 78 (68–116) | 89 (76–98) | 105 (71–108) | 41 (8–86) | <0.40 |
| Extem MCF | 60 (52–68) | 66 (54–67) | 64 (60–66) | 67 (62–70) | 56 (46–62) | <0.68 |
| CK MA | 59 (54–64) | 67 (58–71) | 66 (61–68) | 68 (65–70) | 70 (70–70) | <0.36 |
| 24 h RBCs | 0 (0, 0) | 0 (0–0) | 0.5 (0–1.5) | 0 (0–0) | 5.5 (4.5–12) | <0.01 |
| ARDS | 0 (0%) | 0 (0%) | 0 (0%) | 1 (25%) | 1 (33%) | <0.44 |
| VTE | 0 (0%) | 0 (0%) | 0 (0%) | 1 (25%) | 0 (0%) | <1.00 |
| MOF | 0 (0%) | 0 (0%) | 0 (0%) | 1 (25%) | 3 (75%) | <0.07 |
| Mortality at discharge | 0 (0%) | 0 (0%) | 0 (0%) | 1 (25%) | 2 (50%) | <0.44 |
Injury/shock categories: ISS 0 or 1—minimally injured patient, trauma control; mild injury no shock (ISS 2–15, base excess [BE] > −6); mild injury with shock (ISS 2–15, BE < −6); severe injury no shock (ISS > 25, BE > −6); severe injury, with shock (ISS > 25, BE ≤ −6). Continuous variables presented as median (IQR) and binary variables as number (n) and percent (%) in each group. P values calculated based on Kruskal–Wallis test for continuous variables and Fisher exact test for binary variables.
ISS indicates injury severity score; ED, emergency department. Shock Index—heart rate/systolic blood pressure. Admit shock index=on arrival to ED. Lowest shock index in ED=lowest shock index during initial resuscitation in ED. TRAP AUC—thrombin-stimulated platelet aggregation in aggregation units (AUC). Extern MCF: maximum clot firmness (mm) by extrinsic pathway channel, rotational thromboelastometry CK MA—maximum clot amplitude (mm) in citrated kaolin thromboelastography channel. RBCs 0 to 24 h—number of packed red blood cell units transfused in first 24 h. ARDS indicates acute respiratory distress syndrome; VTE, venous thromboembolism; MOF, multiple organ failure.
Treatment of healthy donor platelets with injured patient plasma
Treatment of healthy donor platelets with plasma from injured patients by injury/shock group resulted in qualitative and quantitative differences in platelet aggregation responses to TRAP stimulation (Figs. 1 and 2). Qualitatively, treatment of healthy platelets with plasma from injured patients potentiated the initial phase of TRAP stimulated platelet aggregation, compared with treatment of healthy platelets with buffer solution alone (Fig. 1, B–G). However, we observed important differences during the middle and later phases of aggregation. Platelets treated with severe injury/no shock plasma exhibited increased TRAP stimulated aggregation (AUC), and platelets treated with plasma from patients with shock (mild injury/with shock or severe injury/with shock) demonstrated slowed aggregation at middle phases evidenced by early plateauing of the impedance curves, and subsequent decreases in impedance at later phases consistent with a disaggregation effect (Fig. 1, E and G).
Fig. 2. Effect of injured patient plasma on healthy donor platelet aggregation.
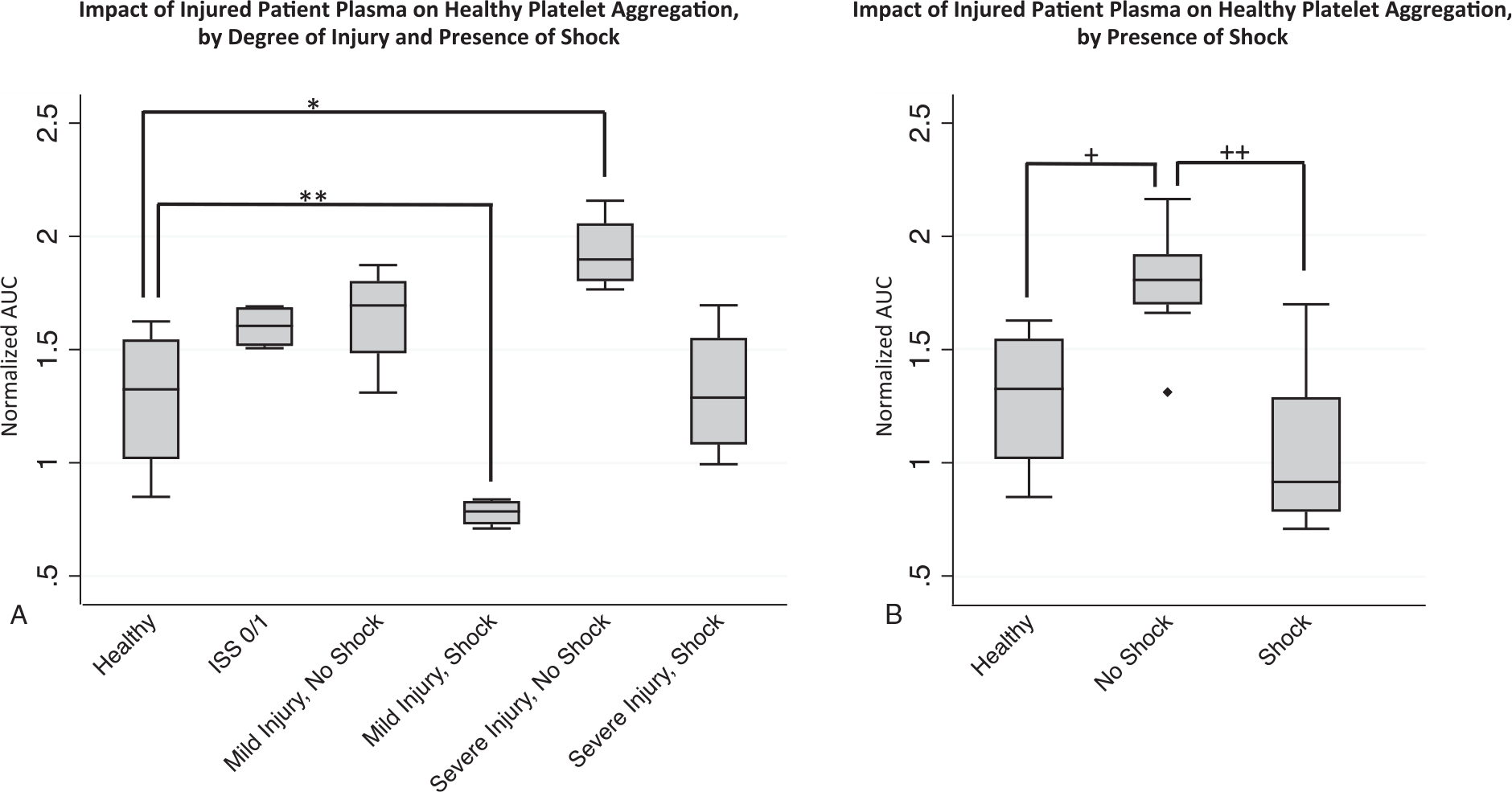
Healthy donor platelets were treated with individual injured patient plasmas and platelet aggregation responses to TRAP (thrombin receptor activating peptide) were then recorded by impedance aggregometry over 15 min and the stimulated platelet aggregation (AUC, area under the aggregation curve) was calculated and normalized to healthy platelet aggregation in buffer solution (normalized AUC—y axis) to control for differences in platelet reactivity between platelet donors. A, Differential effects of injured patient plasma treatment, grouped by patient degree of injury and presence of shock, on healthy platelet aggregation, with *P=0.030 for the pairwise comparison of healthy to severe injury without shock and **P=0.030 for comparison of healthy to mild injury with shock group. n=4 plasmas (with each n representing the average value across the three healthy platelet donors). B, Differential effects of injured patient plasmas grouped by presence or absence of shock, with +P=0.017 for the pairwise comparison of healthy controls to no shock group and ++P=0.002 for comparison of shock to no shock grouping. For other pairwise comparisons with healthy all P>0.05. n=4 plasmas for healthy group and n=8 plasmas for the trauma group (with each n representing the average value across the three healthy platelet donors). Statistical significance was assessed by pairwise Mann–Whitney U tests. All data are presented as box and whisker plots (line—median, box—interquartile range, whiskers— adjacent values, ♦—outside value).
Given these observations, we then quantified the intergroup differences in platelet aggregation after treatment with injured patient plasmas. Compared with treatment with healthy donor plasma, our findings demonstrate significantly decreased TRAP stimulated platelet aggregation (AUC) after treatment with injured patient plasma from patients with mild injury/with shock (normalized-AUC 1.28±0.34 vs. 0.78±0.062, P=0.030) (Fig. 2A). Conversely, we found significantly increased TRAP stimulated platelet aggregation after treatment with plasma from patients with severe injury/no shock (normalized-AUC 1.93±0.17vs. 1.28±0.34, P=0.030), and non-significant trends for the comparisons of the ISS 0/1, mild injury/no shock, and severe injury/with shock groups to the healthy donor plasma group (Fig. 2A).
Given these data suggest shock is a key factor in mediating impairments in platelet aggregation, we then examined our results according to the presence or absence of shock in the injured patient plasma donors. These results showed that treatment of platelets with plasma from injured patients with shock (whether mild or severe injury) resulted in significantly impaired aggregation compared with the groups without shock (normalized-AUC 1.05±0.35 vs. 1.79±0.25, P=0.002), and treatment with plasma from injured patients without shock resulted in significantly increased aggregation compared with treatment with healthy donor plasma (normalized-AUC 1.79±0.25 vs. 1.28±0.34, P=0.017) (Fig. 2B).
We also examined whether there were differences in healthy platelet aggregation according to plasma treatment group prior to TRAP stimulation and in the early phase of TRAP stimulated aggregation. To do so, we compared the impedance (Ω) of platelets after incubation with plasma, but just prior to TRAP stimulation. We found a significant decrease in impedance for the platelets treated with injured patient plasma with mild injury/no shock compared with healthy plasma (normalized-Ω 4.8±42.1 vs. 36.8±37.0, P= 0.043), but no other significant trends (Fig. 3). We also found an increase in early TRAP stimulated aggregation compared with healthy for the severe injury/no shock, ISS0/1, and no shock (mild and severe injury without shock) treatment groups (Table 3). There was no difference across the groups with respect to the time it took to reach maximum impedance, however (Table 3).
Fig. 3. Effect of injured patient plasma treatment of healthy donor platelet aggregation prior to TRAP stimulation.
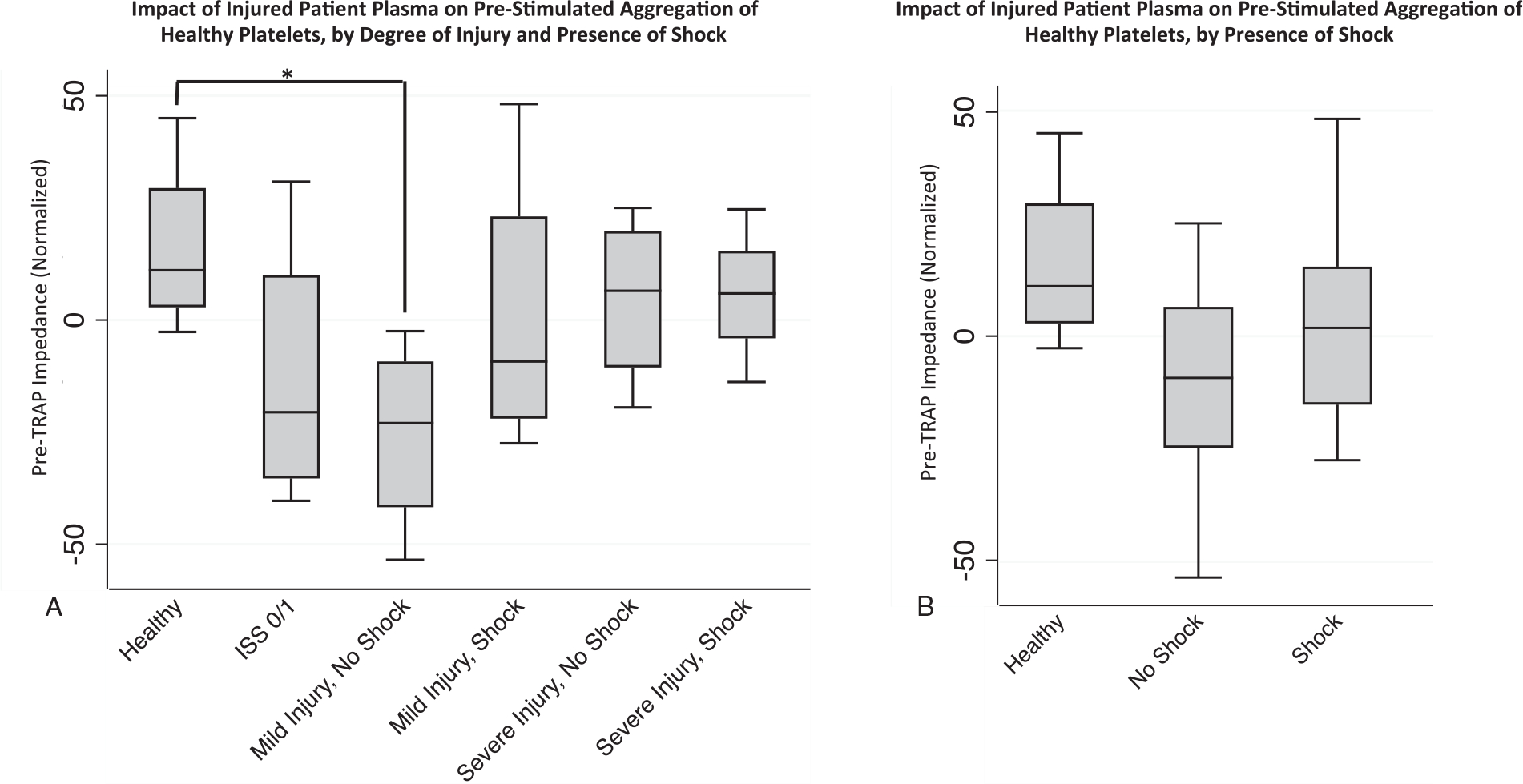
Healthy donor platelets were treated with individual injured patient plasmas for an incubation period of 30 min , and electrical impedance (Ω) was recorded prior to thrombin receptor activating peptide (TRAP) stimulation (y-axis , pre-TRAP impedance) , and was normalized to impedance of healthy donor platelets incubated in buffer to control for inter-donor differences in platelet reactivity. A, Trends toward decreased impedance compared with healthy controls only reached significance for comparison of healthy to mild injury/no shock group, *denoting P=0.043. n=4 plasmas for healthy group and n=8 plasmas for the trauma group (with each n representing the average value across the three healthy platelet donors). B, In aggregate, no significant differences between healthy, shock, and no shock groups (all P>0.05). For other pairwise comparisons with healthy, all P>0.05. Statistical significance was assessed by pairwise Mann–Whitney U tests between healthy and injured plasma groups. n=4 plasmas for healthy group and n=8 plasmas for the trauma group (with each n representing the average value across the three healthy platelet donors). Data are presented as box and whisker plots (line—median, box—interquartile range, whiskers—adjacent values).
Table 3.
Impact of injured patient plasma treatment on early platelet aggregation
| Delta impedance 1 min (normalized) |
Time to max impedance (normalized) |
|||
|---|---|---|---|---|
| Injury/shock category* | Mean ± SD | P value (pairwise comparison to healthy)† | Mean ± SD | P value (pairwise comparison to healthy) |
| Severe injury, shock | 1.69 ± 0.53 | 0.773 | 0.89 ± 0.26 | 1.000 |
| Severe injury, no shock | 1.78 ± 0.18 | 0.021 | 1.03 ± 0.12 | 0.386 |
| Mild injury, shock | 1.08 ± 0.33 | 0.083 | 0.70 ± 0.08 | 0.149 |
| Mild injury, no shock | 2.69 ± 1.45 | 0.149 | 0.87 ± 0.10 | 1.000 |
| ISS 0/1 | 1.85 ± 0.21 | 0.021 | 0.88 ± 0.08 | 0.773 |
| Shock (with mild or severe injury) | 1.39 ± 0.52 | 0.396, (0.059 compared with no shock) | 0.80 ± 0.20 | 0.396, (0.141 compared with no shock) |
| No shock (with mild or severe injury) | 2.24 ± 1.07 | 0.027, (0.059 compared with shock) | 0.95 ± 0.14 | 0.610, (0.141 compared with shock) |
| Healthy | 1.45 ± 0.16 | — | 0.91 ± 0.19 | — |
Injury shock categories: ISS 0 or 1—minimally injured patient, trauma control; mild injury, no shock (ISS 2–15, base excess [BE] > −6); mild injury with shock (ISS 2–15, BE < −6); severe injury, no shock (ISS>25, BE> −6); severe injury with shock (ISS>25, BE< −6).
P values listed for the pairwise comparisons of each injury/shock category to healthy (or comparing shock to no shock where specified), using the non-parametric Wilcoxon test (Mann–Whitney test). n=4 per group except for shock (n=8) and no shock (n=8). Delta impedance 1 min (normalized)=the change in electrical impedance in the first minute after the addition of thrombin receptor activating peptide (TRAP), corresponding to early aggregation. Time to max impedance (normalized)=time after addition of TRAP to the achievement of maximum electrical impedance. Normalization: All values were normalized to platelets treated only with buffer to control for baseline difference in platelet reactivity between donors.
Finally, we examined differences in the late phase of platelet aggregation responses to thrombin stimulation, by calculating the difference between maximum impedance and impedance at the end of the aggregometry assay, with larger positive values indicative of platelet disaggregation from the electrodes. We observed significantly increased disaggregation when comparing the mild injury/with shock to healthy groups (delta Ω- 70.2±6.7 vs. 28.3±29.3, P=0.021) and when comparing shock to no shock groups (delta Ω- 57.4±18.6 vs. 37.6±16.3, P=0.046), and a non-significant trend toward increased disaggregation for the comparison of shock to healthy plasma treatment (delta Ω- 57.4±18.6 vs. 28.3±29.3, P=0.089) (Fig. 4, A and B).
Fig. 4. Effect of injured patient plasma treatment on healthy platelet disaggregation.
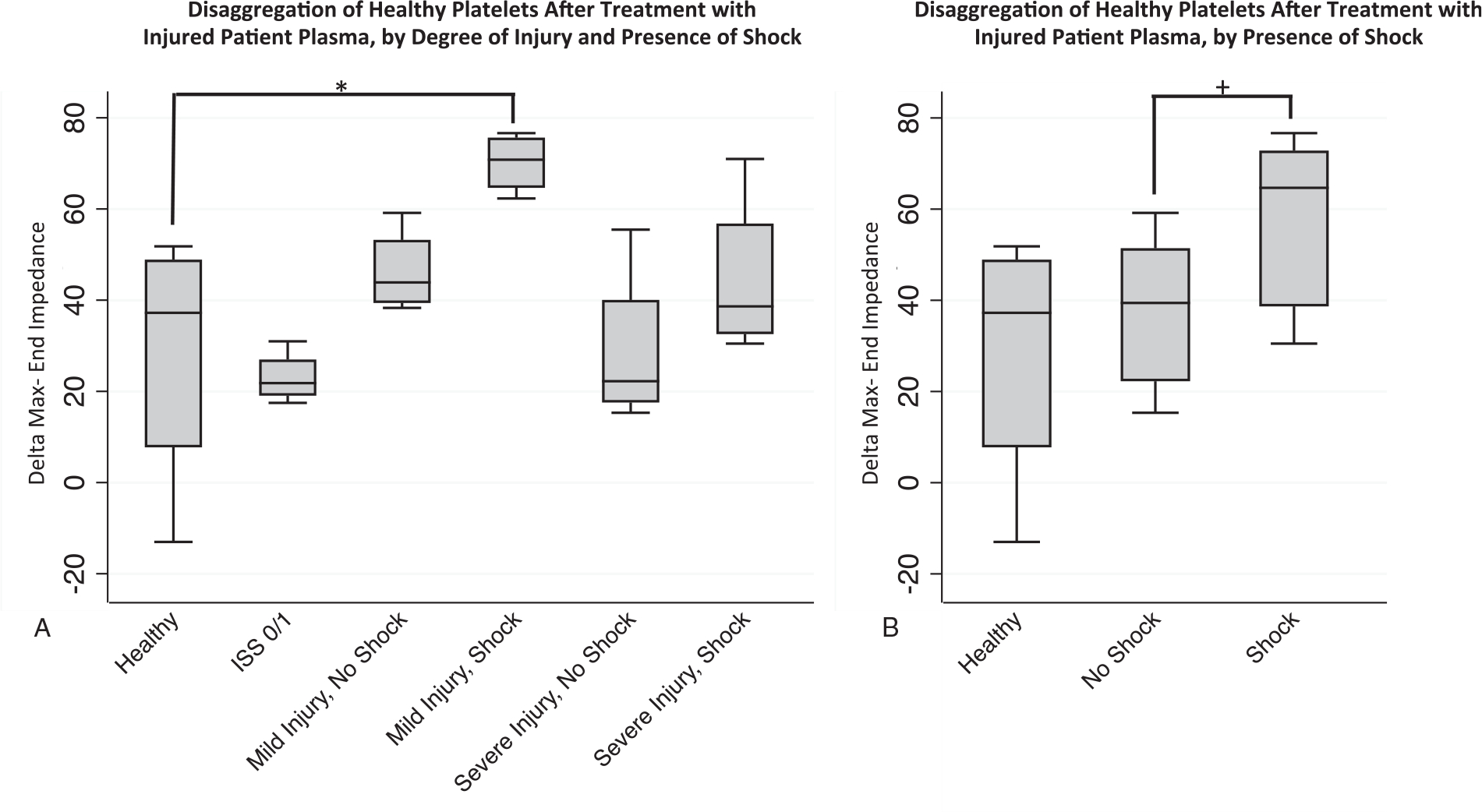
Healthy donor platelets were treated with individual injured patient plasmas and platelet aggregation responses to thrombin receptor activating peptide (TRAP) were then recorded by impedance aggregometry over 15 min. The end-impedance was subtracted from the maximum impedance as a measure of platelet disaggregation (y axis—delta max-end impedance), with higher delta indicative of increased disaggregation of platelets from the electrode. A, Trends toward increased disaggregation shown for injured patient plasmas compared with healthy controls, but only reached statistical significance for the comparison of healthy to mild injury/no shock group, *denoting P=0.021. n=4 plasmas for healthy group and n=8 plasmas for the trauma group (with each n representing the average value across the three healthy platelet donors). B, In aggregate, a significant difference in disaggregation between shock and no shock groups was observed, +denoting P=0.046. For other pairwise comparisons with healthy, all P>0.05. Statistical significance was assessed by pairwise Mann–Whitney U tests. n=4 plasmas for healthy group and n=8 plasmas for the trauma group (with each n representing the average value across the three healthy platelet donors). Data are presented as box and whisker plots (line—median, box—interquartile range, whiskers—adjacent values).
DISCUSSION
In this study, we demonstrate that treatment of healthy platelets with plasma from injured patients significantly alters platelet aggregation responses to stimulation with the thrombin analogue, TRAP. Furthermore, our results suggest that the specific effects of injured patient plasma treatment on healthy platelet aggregation may differ depending on the degree of tissue injury and presence of shock in the injured patient donor, presumably due to specific soluble factors released into circulation in these clinical contexts. This is supported by the finding that treatment of healthy donor platelets with plasma from injured patients without shock (mild or severe injury) resulted in significantly increased aggregation responses to TRAP compared with treatment with both the healthy plasmas and injured patient plasmas from patients without shock (with mild or severe injury). This finding was most pronounced for the severe injury/no shock plasma-treated platelets, again suggesting higher degrees of tissue injury may release mediators into plasma that activate platelets. Our findings of a lack of significant difference in platelet aggregation between treatment with healthy plasma versus severe injury/with shock plasma may be explained by the opposing effects of shock and tissue damage present in these samples: we hypothesize the activating effects of factors released by severe tissue damage are negated by inhibitory effects of shock on platelet aggregation.
Two previous studies have examined the impact of injured patient plasma on healthy platelet function ex-vivo (17, 20), though to our knowledge, this is the first study to explore the differential effects of tissue injury and shock on healthy donor platelets. In a study by Mitchell et al. (17), the effects of hemothorax blood on ex-vivo platelet aggregation and viscoelastic clot formation were measured to examine impacts on coagulation if hemothorax blood were to be used for autotransfusions. They separated injured patient plasma from hemothorax blood from 17 patients with significant hemorrhage (>500 cc of hemothorax) into microparticle-rich and microparticle-free components. While both impaired healthy platelet aggregation, this inhibition was much stronger in the microparticle-rich plasma (17). While we did not examine microparticle content in our study, this certainly deserves investigation, although recent studies suggest that microparticles released in the setting of trauma (by platelets, endothelial cells, and monocytes) may in fact activate platelets and increase their contribution to viscoelastic clot strength, rather than impair them (15, 16, 24). Furthermore, studies of surface marker expression have shown increased (4), unchanged (25), or decreased (26) levels of platelet activation after injury across a range of injury severities, illustrating the heterogeneity across patients and studies, and the need to better understand both the patient and injury characteristics as well as biological mediators involved.
It is also notable that the impairments in platelet aggregation we observed with injured patient plasma treatment were primarily driven by a later disaggregation effect, rather than an overall blunting of the response to TRAP stimulation. To our knowledge, this has not been previously described but merits further investigation to understand the mechanisms leading to such rapid dissolution of platelet aggregates, as this may reveal new treatment targets. This is also relevant because while platelet transfusions are associated with improved mortality as part of balanced resuscitation practices (27), they have not been shown to correct impairments in platelet aggregation (28, 29). Future studies should continue to focus on determining the potential mediators of these impairments, which recent studies suggest include DAMPs such as histones, fibrin, and taurocholic acid (8, 18, 20, 30).
Our study also provides indirect evidence that the mechanisms of impaired platelet aggregation are not solely due to “platelet exhaustion”—a term used to describe platelets that have activated and degranulated due to the injury stimulus, and therefore cannot further aggregate when measured in ex-vivo assays (31). Impaired postinjury platelet aggregation may in fact be due to a depletion of platelet intracellular energy substrates, as demonstrated in a recent rat model of traumatic injury in which platelet aggregation was initially enhanced, but then impaired at later timepoints in association with decreased ATP (19). However, because we used healthy donor platelets that had not been stimulated prior to the experiments, an exhausted platelet phenotype or a loss of platelet intracellular energy substrates are less likely explanations for our results. Our observed impairments in aggregation were only seen after treatment with plasma from shock groups, while plasma from patients with extensive tissue injury enhanced the ex-vivo aggregation function of healthy platelets.
Limitations
We acknowledge several limitations of this study, including a relatively small sample size limiting statistical power for our comparisons across groups. However, despite some expected variability between platelet donors, the trends we observed between the injured patient plasma injury/shock groups were overall replicable across platelets from three healthy adult male donors in response to stimulation with TRAP. Further experiments are needed to determine if platelet aggregation stimulated by other surface receptor agonists such as adenosine diphosphate and collagen leads to similar results. We additionally did not observe a significant association between platelet aggregation responses to TRAP that were measured in the whole blood from the trauma plasma donors at the time of admission with the aggregation responses of the treated healthy donor platelets observed in our experiments. This is likely related to our small sample size, and due to the large standard deviation in the aggregation values in the injured patients, though it is notable that severe injury/no shock patients had the strongest aggregation responses to TRAP while the severe injury/with shock group had the lowest, similar to our experiment’s findings. Additionally, to avoid further confounding factors, we only selected plasmas from male patients without traumatic brain injury, which may limit the applicability of our results to these groups given the unique features of platelet behavior in traumatic brain injury and the important sex-based differences in coagulation and platelet function (32–34). We also chose to perform these experiments on isolated platelets suspended in buffer (rather than using healthy platelet rich plasma) to prevent any unintended confounding effects of mixing healthy plasma with trauma plasma. However, it will be important to characterize whether the alterations in healthy platelet aggregation due to trauma plasma can be prevented or even reversed by treatment with healthy plasma, as this may have clinical implications for transfusion practices. Lastly, multiple electrode impedance aggregometry is an ex-vivo assay that does not replicate several aspects of vascular physiology that affect platelet function, including flow, shear stress, and the endothelium. However, there are microfluidic models that replicate these factors and could be adapted to test the effects of injured patient plasma on healthy donor platelets (35, 36).
CONCLUSIONS AND FUTURE DIRECTIONS
Similar to recent studies (17, 20), we demonstrated that injured patient plasma significantly altered ex-vivo healthy platelet aggregation. Critically, we found that shock impaired platelet aggregation, while tissue injury enhanced it. These relationships add to our knowledge of the pathophysiology of TIC and imply that transfused platelets may be less effective in the setting of hypoperfusion due to circulating platelet inhibitory factors. Future studies must systematically evaluate the plasma of injured patients to identify the presence of specific proteins, microparticles, or other molecular platelet inhibitors to develop novel treatments for platelet dysfunction in hemorrhaging injured patients.
ACKNOWLEDGMENTS
The authors thank Jin Chengshi, PhD, MS; Erin Ross, BS; and John Park, BS.
Dr LZK is supported by NIH 1K23GM130892-01, Dr RAC is supported by NIH R01—HL149670 and DoD W911QY-15-C-0044, Dr ZAM is supported by The National Center for Advancing Translational Sciences of the NIH 5TL1TR001871-04. Statistical support was provided by the Biostatistics Core which was funded by the UCSF Department of Surgery.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Callcut RA, Kornblith LZ, Conroy AS, Robles AJ, Meizoso JP, Namias N, Meyer DE, Haymaker A, Truitt MS, Agrawal V, et al. : The why and how our trauma patients die: a prospective Multicenter Western Trauma Association study. J Trauma Acute Care Surg 86(5):864–870, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang R, Cardenas JC, Wade CE, Holcomb JB: Advances in the understanding of trauma-induced coagulopathy. Blood 128(8):1043–1049, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kornblith LZ, Moore HB, Cohen MJ: Trauma-induced coagulopathy: the past, present, and future. J Thromb Haemost 17(6):852–862, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG: Platelet activation and function after trauma. J Trauma Acute Care Surg 51(4):639–647, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ: Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg 73(1):13–19, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kornblith LZ, Bainton CMV, Fields AT, Matthay ZA, Magid NT, Nunez-Garcia B, Prakash A, Kurien PA, Callcut RA, Cohen MJ, et al. : A journey upstream: fluctuating platelet-specific genes in cell free plasma as proof-of-concept for using RNA sequencing to improve understanding of post-injury platelet biology. J Trauma Acute Care Surg 88:742–751, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vulliamy P, Kornblith LZ, Kutcher ME, Cohen MJ, Brohi K, Neal MD: Alterations in platelet behavior after major trauma: adaptive or maladaptive? Platelets; 2020;1–10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vulliamy P, Gillespie S, Armstrong PC, Allan HE, Warner TD, Brohi K: Histone H4 induces platelet ballooning and microparticle release during trauma hemorrhage. Proc Natl Acad Sci U S A 116(35):17444–17449, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, Schäffer TE, Bohn E, Frick JS, Borst O, et al. : Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest 125(12):4638–4654, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen MJ, Carles M, Brohi K, Calfee CS, Rahn P, Call MS, Chesebro BB, West MA, Pittet JF: Early release of soluble receptor for advanced glycation end-products after severe trauma in humans. J Trauma Acute Care Surg 68(61273–1278):1273–1278, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deguchi H, Sinha RK, Marchese P, Ruggeri ZM, Zilberman-Rudenko J, McCarty OJT, Cohen MJ, Griffin JH: Prothrombotic skeletal muscle myosin directly enhances prothrombin activation by binding factors Xa and Va. Blood 128(14):1870–1878, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiener G, Moore H, Moore EE, Gonzalez E, Diamond S, Zhu S, D’Alessandro A, Banerjee A: Shock releases bile acid inducing platelet inhibition and fibrinolysis. J Surg Res 195(2):390–395, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kutcher ME, Xu J, Vilardi RF, Ho C, Esmon CT, Cohen MJ: Extracellular histone release in response to traumatic injury: implications for a compensatory role of activated protein C. J Trauma Acute Care Surg 73(6):1389–1394, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starr NE, Matthay ZA, Fields AT, Nunez-Garcia B, Callcut RA, Cohen MJ, Kornblith LZ: Identification of injury and shock driven effects on ex-vivo platelet aggregometry: a cautionary tale of phenotyping. J Trauma Acute Care Surg 89:20–28, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park MS, Xue A, Spears GM, Halling TM, Ferrara MJ, Kuntz MM, Dhillon SK, Jenkins DH, Harmsen WS, Ballman KV, et al. : Thrombin generation and procoagulant microparticle profiles after acute trauma: a prospective cohort study. J Trauma Acute Care Surg 79(5):726–731, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park MS, Owen BA, Ballinger BA, Sarr MG, Schiller HJ, Zietlow SP, Jenkins DH, Ereth MH, Owen WG, Heit JA: Quantification of hypercoagulable state after blunt trauma: microparticle and thrombin generation are increased relative to injury severity, while standard markers are not. Surgery 151(6):831–836, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell TA, Herzig MC, Fedyk CG, Salhanick MA, Henderson AT, Parida BK, Prat NJ, Dent DL, Schwacha MG, Cap AP: Traumatic hemothorax blood contains elevated levels of microparticles that are prothrombotic but inhibit platelet aggregation. Shock 47(6):680–687, 2017. [DOI] [PubMed] [Google Scholar]
- 18.Wiener G, Moore HB, Moore EE, Gonzalez E, Diamond S, Zhu S,D’Alessandro A, Banerjee A: Shock releases bile acid inducing platelet inhibition and fibrinolysis. J Surg Res 195(2):390–395, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darlington DN, Wu X, Keesee JD, Cap AP: Severe trauma and hemorrhage leads to platelet dysfunction and changes in cyclic nucleotides in the rat. Shock 53(4):468–475, 2020. [DOI] [PubMed] [Google Scholar]
- 20.Verni CC, Davila AJ, Balian S, Sims CA, Diamond SL: Platelet dysfunction during trauma involves diverse signaling pathways and an inhibitory activity in patient-derived plasma. J Trauma Acute Care Surg 86(2):250–259, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tisherman SA, Barie P, Bokhari F, Bonadies J, Daley B, Diebel L, Eachempati SR, Kurek S, Luchette F, Carlos Puyana J, et al. : Clinical practice guideline: endpoints of resuscitation. J Trauma Acute Care Surg 57(4):898–912, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF: Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg 245(5):812–818, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen MJBK, Ganter MT, Manley GT, Mackersie RC, Pittet J-F: Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the protein C pathway. J Trauma Acute Care Surg 63(6):1261–1262, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Caspers M, Schafer N, Frohlich M, Bouillon B, Mutschler M, Bauerfeind U, Maegele M: Microparticles profiling in trauma patients: high level of microparticles induce activation of platelets in vitro. Eur J Trauma Emerg Surg 46(1):43–51, 2020. [DOI] [PubMed] [Google Scholar]
- 25.St John AE, Newton JC, Martin EJ, Mohammed BM, Contaifer D, Saunders JL, Brophy GM, Spiess BD, Ward KR, Brophy DF, et al. : Platelets retain inducible alpha granule secretion by P-selectin expression but exhibit mechanical dysfunction during trauma-induced coagulopathy. J Thromb Haemost 17(5):771–781, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramsey MT, Fabian TC, Shahan CP, Sharpe JP, Mabry SE, Weinberg JA, Croce MA, Jennings LK: A prospective study of platelet function in trauma patients. J Trauma Acute Care Surg 80(5):726–732, 2016. [DOI] [PubMed] [Google Scholar]
- 27.Cardenas JC, Zhang X, Fox EE, Cotton BA, Hess JR, Schreiber MA, Wade CE, Holcomb JB, Group PS: Platelet transfusions improve hemostasis and survival in a substudy of the prospective, randomized PROPPR trial. Blood Adv 2(14):1696–1704, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vulliamy P, Gillespie S, Gall LS, Green L, Brohi K, Davenport RA: Platelet transfusions reduce fibrinolysis but do not restore platelet function during trauma hemorrhage. J Trauma Acute Care Surg 83(3):388–397, 2017. [DOI] [PubMed] [Google Scholar]
- 29.Kornblith LZ, Decker A, Conroy AS, Hendrickson CM, Fields AT, Robles AJ, Callcut RA, Cohen MJ: It’s about time: transfusion effects on post-injury platelet aggregation over time. J Trauma Acute Care Surg 87:1042–1051, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee MY, Verni CC, Herbig BA, Diamond SL: Soluble fibrin causes an acquired platelet glycoprotein VI signaling defect: implications for coagulopathy. J Trauma Acute Care Surg 15(12):2396–2407, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pareti FI, Capitanio A, Mannucci L, Ponticelli C, Mannucci PM: Acquired dysfunction due to the circulation of “exhausted” platelets. Am J Med 69(2):235–240, 1980. [DOI] [PubMed] [Google Scholar]
- 32.Samuels JM, Moore EE, Silliman CC, Banerjee A, Cohen MJ, Ghasabyan A, Chandler J, Coleman JR, Sauaia A: Severe traumatic brain injury is associated with a unique coagulopathy phenotype. J Trauma Acute Care Surg 86(4):686–693, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis PKMH, Walsh M, Cassady R, Yount R, Losiniecki A, Moore EE, Wohlauer MV, Howard J, Ploplis VA, Castellino FJ, et al. : Platelet dysfunction is an early marker for traumatic brain injury-induced coagulopathy. Neurocritical Care 18(2):201–208, 2019. [DOI] [PubMed] [Google Scholar]
- 34.Coleman JR, Moore EE, Kelher MR, Samuels JM, Cohen MJ, Sauaia A, Banerjee A, Silliman CC, Peltz ED: Female platelets have distinct functional activity compared with male platelets: implications in transfusion practice and treatment of trauma-induced coagulopathy. J Trauma Acute Care Surg 87(5):1052–1060, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Elmongy H, Sims C, Diamond SL: Ex vivo recapitulation of trauma-induced coagulopathy and preliminary assessment of trauma patient platelet function under flow using microfluidic technology. J Trauma Acute Care Surg 80(3):440–449, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Six KR, Devloo R, Van Aelst B, Vandekerckhove P, Feys HB, Compernolle V: A microfluidic flow chamber model for platelet transfusion and hemostasis measures platelet deposition and fibrin formation in real-time. J Vis Exp; 2017;(120):55351, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]