

ABSTRACT
The global health crisis and economic tolls of COVID-19 necessitate a panoply of strategies to treat SARS-CoV-2 infection. To date, few treatment options exist, although neutralizing antibodies against the spike glycoprotein have proven to be effective. Because infection is initiated at the mucosa and propagates mainly at this site throughout the course of the disease, blocking the virus at the mucosal milieu should be effective. However, administration of biologics to the mucosa presents a substantial challenge. Here, we describe bifunctional molecules combining single-domain variable regions that bind to the polymeric Ig receptor (pIgR) and to the SARS-CoV-2 spike protein via addition of the ACE2 extracellular domain (ECD). The hypothesis behind this design is that pIgR will transport the molecule from the circulation to the mucosal surface where the ACE ECD would act as a decoy receptor for the nCoV2. The bifunctional molecules bind SARS-Cov-2 spike glycoprotein in vitro and efficiently transcytose across the lung epithelium in human tissue-based analyses. Designs featuring ACE2 tethered to the C-terminus of the Fc do not induce antibody-dependent cytotoxicity against pIgR-expressing cells. These molecules thus represent a potential therapeutic modality for systemic administration of neutralizing anti-SARS-CoV-2 molecules to the mucosa.
KEYWORDS: Polymeric Ig receptor, pIgR, sars-cov-2. coronavirus, antibody, Igg, bifunctional, bispecific, transcytosis, mucosa
Introduction
Since its outbreak, the COVID-19 pandemic caused by SARS-CoV-2 virus has resulted in over 230 million infections and over 4.7 million deaths.1 The global health and economic disruptions caused by the spread of COVID-19 necessitate rapid development of treatments. Interventions largely focus on three strategies: vaccines, antiviral drugs, and biologics.2 Although vaccine administration is the most effective way to reduce viral transmission and illness, widespread administration, high compliance, and high effectiveness are required.3 Additionally, patients with compromised immune systems frequently show low antibody titers after vaccine challenge and are prone to hospitalization. To date, a few antibody and anti-viral therapies have been granted emergency use authorization to treat hospitalized COVID-19 patients.4 Biologic therapies offer higher potential specificity, efficacy, and lower toxicities than antiviral therapies. However, for respiratory targets, delivery of biologics has proven a challenge due to poor accessibility of IgG in the mucosal spaces.5,6 Thus, there remains a critical unmet medical need for prophylactic and therapeutic interventions for infected patients, particularly during acute infection.
SARS-CoV-2 is a betacoronavirus featuring a positive-sense single-stranded, 30 kb RNA genome whose envelope is decorated by a homo-trimeric, 140 kDa spike glycoprotein (Uniprot ID P0DTC2). The spike glycoprotein is divided into S1 and S2 subunits, which are cleaved by furin but remain bound together (Supplementary Fig. S1).7 The S1 subunit contains the receptor binding domain (RBD), which binds the host receptor angiotensin-converting enzyme 2 (ACE2) upon proteolytic cleavage and helps to stabilize the “closed”, or pre-fusion state of the spike glycoprotein.8–14 The S2 subunit comprises the helical structure containing the fusion peptide responsible for target cell membrane fusion resulting in viral entry into the host cell. Upon binding to ACE2, the spike glycoprotein undergoes a conformational change to the “open” state, allowing the fusion pore to breach the target cell membrane. Fusion and host cell entry occurs in minutes, followed by an eclipse period of ~10 hr, leading up to a viral burst of ~103 virion particles per cell.15
ACE2 converts angiotensin II into angiotensin 1–7 and is expressed in lung at only 0.8 transcripts per million.16 However, expression of ACE2 is localized to alveolar, oral, and nasal epithelial cells, capillary endothelium cells, and type II pneumocytes, which appear to be the targets of viral entry into the respiratory system.17,18 ACE2 is also highly expressed in intestinal enterocytes, and gut infection represents an additional pathogenic mechanism for SARS-CoV-2.19–21 Vaccine development for anti-SARS-CoV-2 prophylaxis has yielded several successful candidates, but an unmet need remains for treatment of acute and severe COVID-19 cases. Identification of novel anti-SARS-CoV-2 antibody-based therapeutics has been under intense investigation, including characterization of patient-derived antibodies for both convalescent plasma transfer and production of recombinant therapeutics.22
Only one anti-viral antibody (Ab)-based therapeutic (anti-respiratory syncytial virus palivizumab (SYNAGIS)) has been approved for respiratory infection, while a total of 5 anti-SARS-CoV-2 antibodies (bamlanivimab; bamlanivimab/etesevimab; REGEN-COV (casirivimab/imdevimab; sotrovimab) have received emergency use authorizations as monotherapy or part of a mixture. In contrast, anti-influenza antibodies have shown mixed responses in clinical studies, highlighting both the potential and challenges of biologics in viral disease.23–27 Biologics offer long half-lives, high target specificity, and can exploit immune effector functions against targets, although effector function has been a proposed source of antibody-mediated enhancement (ADE) of disease.28 Viral mutation, however, can lead to the emergence of escape mutants. One strategy to overcome viral escape is administration of the recombinant receptor extracellular domain (ECD) recognized by the viral spike protein (e.g., ACE2 for SARS-coronaviruses), and indeed, treatment with recombinant soluble ACE2 protein may have potential therapeutic benefit in SARS-2 infected patients.29–31 Recombinant decoy receptor therapeutics offer two distinct advantages. First, they are unlikely to be affected by escape mutants, which appear to be emerging for SARS-CoV-2.32, 33 Second, recombinant decoy receptors need not be identified de-novo for new viruses within a family. These decoy receptors have also been formatted with various half-life extension moieties, such as Fc fusion to improve therapeutic properties.33,34
The main challenge to both antibody and ACE2-based molecules is that the molecules are not able to traverse from circulation to the lung mucosa, and thus require high doses, in the ranges of 20–50 mg/kg (~3.5 g of antibody per dose), which presents a challenge regarding cost of goods. One anti-influenza antibody, VIS410, achieved ~20 ug/mL nasopharyngeal concentration upon dosing at 50 mg/kg and showed promise in prophylactic settings, while used as a treatment showed maximum efficacy with doses of 2,000 mg, which can lead to a high cost of goods.25,26,35 These results suggest that, given sufficient exposure, biologic treatments can reduce viral burden and limit disease symptoms. However, increasing both the rate and magnitude of mucosal exposure of biologics can lead to substantial improvements in patient outcomes and reduction in cost of commercial manufacturing by reduced dosages.
Innate mucosal immunity is dominated by IgA, and although IgA is the most highly produced Ig type, it is present only at low levels in serum.36 Dimeric IgA is transported efficiently into the mucosa via the polymeric Ig receptor (pIgR).37,38 Indeed, a strong secretory IgA-dominant SARS-CoV-2 immune response in human milk after COVID-19 infection has been observed, suggesting the susceptibility of SARS-CoV-2 to elimination via secreted antibodies.39 Use of dimeric IgA as a therapeutic is complicated by production challenges such as low expression yields and glycan heterogeneity, necessitating alternative strategies to enrich neutralizing antibodies in the mucosa.40 We and others have developed anti-pIgR single-domain antibodies (VHH) that can exploit pIgR-mediated transcytosis to deliver IgG across the endothelium into the lung mucosa.41,42 We have shown that molecules harboring the anti-pIgR domains displayed increased level of transport across Madin-Darby canine kidney (MDCK) cell-derived monolayers and across human-derived lung epithelial layers by >30-fold compared to conventional IgG.41 This platform, which we call BRING-IT (Bi-directional polymeric Ig receptor mediated transport of biologics), has the potential to deliver IgG into the mucosal lumen, where it can target and neutralize mucosal antigens or pathogens such as SARS-CoV-2.
Here we describe bifunctional molecules comprising the anti-pIgR VHH moieties that mediate transcytosis across the epithelium and the ACE2 ECD capable of neutralizing SARS-2 viruses. Both of these moieties are fused to an Fc to extend the serum lifetime and to potentially facilitate effector functions.
Results
Design of anti-pIgR based bifunctional molecules
We sought to generate bifunctional molecules that could engage both pIgR and the RBD of the SARS-CoV-2 spike glycoprotein, which includes all residues required for binding to ACE2 (Supplementary Figure 1). We previously identified a panel of heavy chain only (VHH) antibodies that could bind human pIgR with affinities ranging from ~4 to 500 nM.41 Of these, VHH2 and VHH6 were included in this analysis since VHH2 displayed cross-reactivity to mouse pIgR and both displayed strong transcytosis in an MDCK monolayer-based assay and in a human epithelial airway model. To mediate binding to the SARS-CoV-2 spike glycoprotein, we identified 2 antibodies that were reported to display neutralization activity – D001 (Sino Biological cat. # 40150-D001) and SAD-S35 (Acro Biosystems cat. # SAD-S35). Since the emergence of SARS-CoV-2 spike protein variants may lead to viral escape from neutralizing monoclonal Abs (mAbs), we focused on use of recombinant ACE2, and thus, these two neutralizing mAbs were used as binding controls. Another non-neutralizing mAb, CR3022, was used as a negative control in neutralization studies.43 Bifunctional molecules were generated with either VHH2/6 and the ECD from ACE2 (Supplementary Figure 1, Table 1). Two truncations of the ACE2 ECD were used: residues 18–611 and 18–725, which begin after the native signal peptide. The short truncation was based on analysis of the crystal structure of ACE2 (PDB ID 1R42), which suggests that both constructs could form stable molecules.44 The ACE2 ECD was fused to either the N- or C-terminus of the Fc while the anti-pIgR VHH moieties were formatted only on the N-terminus of the bifunctional molecules to avoid binding with preformed antibodies in human sera.45 All molecules featured a human IgG1-based constant region.
Table 1.
Description of Proteins
| Protein Name | Glyph | Description |
|---|---|---|
| CV19B308 |  |
pIgR-VHH2-Fc-MSCD342-ACE218-611 |
| CV19B307 |  |
pIgR-VHH2-Fc-MSCD342-ACE218-725 |
| CV19B301 |  |
pIgR-VHH6-Fc-MSCD342-ACE218-725 |
| CV19B290 |  |
null-VHH-Fc-MSCD342-ACE218-611 |
| CV19B289 |  |
null-VHH-Fc-MSCD342-ACE218-725 |
| CV19B283 |  |
pIgR-VHH2-MSCD342-ACE218-725-Fc |
| CV19B277 |  |
pIgR-VHH6-MSCD342-ACE218-725-Fc |
| CV19B265 |  |
null-VHH-MSCD342-ACE218-725-Fc |
| CR3022 |  |
pan-SARS-CoV mAb |
| D001 |  |
SARS-CoV-2 neutralizing mAb |
| SAD-S35 |  |
SARS-CoV-2 neutralizing mAb |
Binding properties of antibodies to pIgR and spike glycoprotein
Spike glycoprotein
We measured the abilities of the molecules to bind both the spike glycoprotein and pIgR. In this study, relative KD values, based on surface biolayer interferometry, are reported (Figure 1, Table 2). All control antibodies, including D001 and SAD-S35, along with the ACE218-611 and ACE218-725 fusion proteins, displayed binding to COVID-19 RBD.46 Bifunctional molecules featuring the VHH-ACE2-Fc architecture (CV19B265, CV19B277, CV19B283, and CV19B289) bound to the SARS-CoV-2 spike glycoprotein with similar affinities, each ~2-3-fold weaker than D001 and SAD-S35. Interestingly, of these four antibodies, the two featuring the negative control VHH, targeted against mouse EGFR, (CV19B265 and CV19B289) displayed off-rates of binding ~5-fold faster than the mAbs and the bifunctional molecules featuring VHH2 (CV19B277 and CV19B283). Bifunctional molecules featuring the ACE2 ECD attached to the C-terminus of the Fc (CV19B290, CV19B301, CV19B307, and CV19B308) displayed a wide range of affinities for spike glycoprotein. We note that CV19B290 featured the short truncation of ACE2 and displayed the weakest binding.
Figure 1.
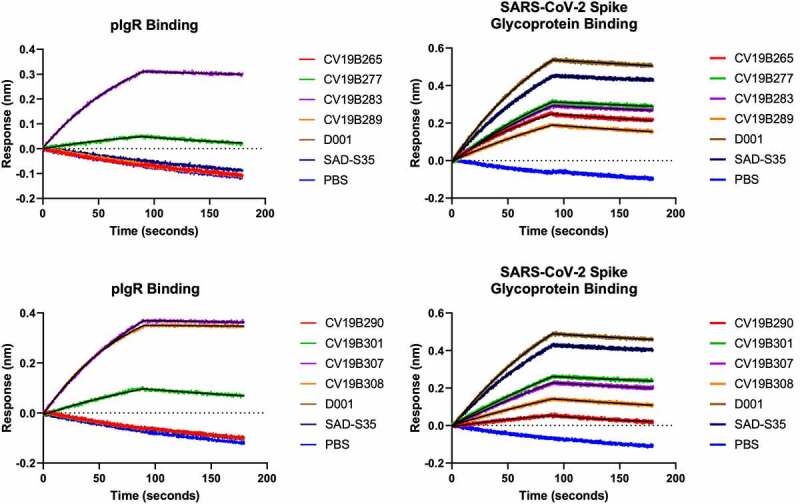
Surface Biolayer Interferometry-based binding of mAbs and bifunctional molecules to spike glycoprotein and to pIgR. Antibodies and antigens are indicated in the graphs. Graphs represent magnitude of response (nm) over time. Association and dissociation are displayed along with fitted curves
Table 2.
Binding kinetic values from surface biolayer interferometry
| Protein | SARS-CoV-2 spike |
|
pIgR |
|
|||
|---|---|---|---|---|---|---|---|
|
kon (min−1 nM−1) |
koff (min−1) |
Kd (nM) |
kon (min−1 nM−1) |
koff (min−1) |
Kd (nM) |
Relative Kd (VHH2) | |
| CV19B277 | 32157 | 0.0008426 | 26.2 | 39.9 | 0.008349 | 2 x 105 | 142857 |
| CV19B308 | 2975 | 0.003050 | 1.025 x 103 | 63861 | 8.646 x 10–5 | 1.4 | 1 |
| CV19B307 | 8646 | 0.001496 | 173.1 | 44364 | 0.0002236 | 5.0 | 4 |
| CV19B301 | 10783 | 0.001092 | 101.2 | 13406 | 0.003557 | 265.3 | 190 |
| CV19B290 | 37.88 | 0.008619 | 2.28 x 105 | NB | NB | NB | NB |
| CV19B289 | 15478 | 0.002270 | 146.7 | NB | NB | NB | NB |
| CV19B283 | 28303 | 0.0009639 | 34.1 | 5.1 x 104 | 0.0004055 | 7.8 | 6 |
| CV19B265 | 37364 | 0.001704 | 45.6 | NB | NB | NB | NB |
| D001 | 43749 | 0.0006574 | 15.0 | NB | NB | NB | NB |
| SAD-S35 | 37484 | 0.0005347 | 14.3 | NB | NB | NB | NB |
The emergence of SARS-CoV-2 variants has led to the question of whether neutralizing monoclonal antibodies would retain activity, but recombinant ACE2 is expected to effectively neutralize all variants because ACE2 is the native receptor for SARS-CoV-2.47,48 Thus, we tested the abilities of the ACE2 bifunctional molecules to bind to the Y435F, N439K, N501Y, and D614G variants, which are associated with increased infectivity of SARS-CoV-2 and can prevent neutralization by some mAbs.49 For example, mutations at N439 were shown to modulate interaction with REGN-COV2, and is thus likely to represent part of an important epitope for neutralization.48 All molecules tested here showed qualitatively similar binding to the spike glycoprotein variants Y435F, N439K, and N501Y as to wild-type spike glycoprotein (Figure 1, Supplementary Fig. S2). The D614G mutation stabilizes the “closed” structure of the spike glycoprotein, and was shown to decrease the ability of ACE2 to bind the spike glycoprotein.50 Consistently, both the ACE2 bifunctional molecule and the mAbs all showed modest decrease in binding to the D614G variant. Interestingly, one bifunctional molecule, CV19B307 displayed similar ability to bind all variants, including the D614G variant.
pIgR
All the mAbs and bifunctional molecules featuring the null VHH (CV19B265, CV19B289, and CV19B290) failed to bind pIgR, as expected (Figure 1, Table 2). Bifunctional molecules containing VHH2 (CV19B283, CV19B307, and CV19B308) bound to pIgR with Kd values ~5 nM, consistent with our previous findings.41 Bifunctional molecules containing VHH6 (CV19B277 and CV19B301) bound with Kd ~ 0.2 mM or 265 nM, respectively. The significantly weaker binding by CV19B277 may have been due to its being fused in tandem on the N-terminus of ACE2, although the mechanism for its reduced binding was unknown.
Bifunctional molecules display anti-SARS-CoV-2 activity and design-dependent ADCC activity
Antibodies were then tested for their abilities to compete binding between SARS-CoV-2 S-protein and ACE2 using an MSD-based surrogate neutralization assay modified from a previous report (Figure 2a, Table 3, Supplementary Fig. S3).51 This immunoassay-based surrogate neutralization assay has been shown good correlation with the infectivity-based neutralization assay. D001 displayed inhibitory activity with IC50 = 0.2 nM, while CR3022, an anti-SARS-CoV antibody known to lack the ability to neutralize SARS-CoV-2,52,53 failed to compete. All bifunctional molecules featuring the ACE2 ECD displayed surrogate neutralization ability, with IC50 ranging from 1–3 nM, with maximum activity within 4–10-fold to that of D001. Thus, competition with SARS-CoV-2 S-protein appeared to be universal to all bifunctional molecules featuring the ACE2 moiety. The bifunctional molecules were designed to target pIgR on mucosal epithelial cells and co-transcytose across epithelial layers. However, targeting pIgR with an antibody featuring an active Fc region posed the risk of inducing antibody-dependent cell-mediated cytotoxicity (ADCC) against the epithelial cells, resulting in potential undesirable toxicity. We assessed the ability of the bifunctional molecules to mediate ADCC against pIgR-expressing MDCK cells (Figure 2b). Each bifunctional molecule was designed using either an active, wild-type IgG1 Fc or a silenced Fc, designated “silent” (containing mutations to disrupt binding to Fcγ receptors). Peripheral blood mononuclear cell (PBMC) samples were composed of ~ 7% CD19 + B cells, 60% CD3 + T cells, and 6% CD56+, CD16+ natural killer (NK) cells (Supplementary Fig. S4). Each bifunctional molecule was formatted on both an active and “silent” Fc region to disrupt interaction with Fcγ receptors.54 Two control molecules harboring the ACE2 ECD and a non-binding control VHH failed to mediate ADCC, as expected since they lack binding to pIgR. Bifunctional molecules featuring anti-pIgR VHH moieties on a silent Fc also failed to mediate ADCC, due to their inability to bind Fcγ receptors on NK cells. Interestingly, of the bifunctional molecules having an active Fc, only those in which the anti-pIgR VHH was fused in tandem with the ACE2 ECD on the N-terminus of the Fc (CV19B277 and CV19B283) displayed modest ADCC activity. Other bifunctional molecules in which the anti-pIgR VHH was fused onto the N-terminus of the Fc with the ACE2 ECD on the C-terminus of the Fc (CV19B301 and CV19B307) failed to mediate ADCC, despite binding pIgR on the MDCK cells and having an active Fc. This suggested that either the presence of the C-terminal ACE2 ECD inhibited ADCC or that this architecture allowed transcytosis rates to exceed the binding rate for Fcγ receptor engagement.
Figure 2.
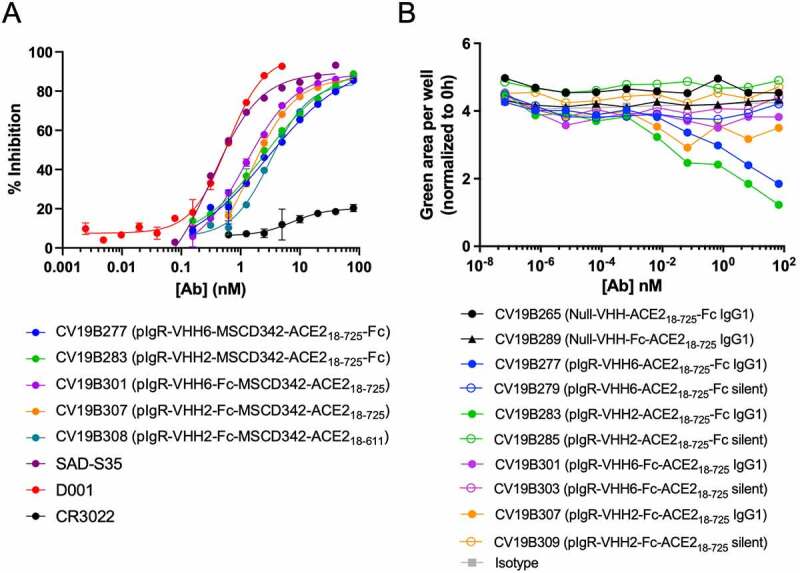
Bifunctional molecules Display Specific Functional Activity Against SARS-CoV-2. (a) Functional competition ability is plotted vs. antibody concentration. Antibodies are indicated in the graph. (b) PBMC-mediated ADCC of MDCK-pIgR cells is plotted as green area per well (normalized to 0 h) vs concentration of bifunctional molecule (nM). Antibodies are indicated in the legend
Table 3.
IC50 Values for SARS-CoV-2 competition
| Antibody | IC50 (nM) |
|---|---|
| CV19B277 | 2.876 |
| CV19B283 | 1.512 |
| CV19B301 | 1.195 |
| CV19B307 | 1.424 |
| CV19B308 | 3.206 |
| CR3022 | N/A |
| SAD-S35 | 0.3233 |
| D001 | 0.2991 |
pIgR Engagement Led to Serum Clearance and Mucosal Enrichment
The anti-pIgR VHH modules could mediate transcytosis across both MDCK cell bilayers and in a human epithelial microtissue model, and we thus asked whether this pIgR-mediated transcytosis could occur in vivo.41 We selected VHH2, and VHH6 for pharmacokinetic analysis in either monovalent or bivalent format in C57BL/6 mice (Supplementary Fig. S4, Supplementary Table S1). Although VHH2 and VHH6 were used in the bifunctional molecules, only VHH2 could cross-react with mouse pIgR. As expected, a monovalent VHH6 displayed a serum half-life of ~ 8.2 d, consistent with that of the null VHH-Fc (terminal half-life (T1/2) ~ 8.2 d). Conversely, molecules harboring a single copy of the mouse pIgR cross-reactive VHH2 module displayed rapid serum clearance, with T1/2 ~ 3.2 d, demonstrating a target-mediated drug disposition effect. A bivalent antibody featuring two copies of VHH2 displayed even faster serum clearance, having a T1/2 value of 0.55 d. Distinct from primates, mice display high pIgR expression in the liver, and this could explain the faster clearance of the anti-pIgR VHH2 domains.55 As a result of pIgR expression in mouse liver, we cannot study the mucosal transport of VHH2-containing bifunctional molecules in a mouse model.
To assess the ability of the VHH domains to transcytose in a human, we analyzed the bifunctional molecules featuring VHH2 and VHH6 using the EpiAirway 3D model (MatTEK Life Sciences), an established lung tissue model engineered from primary human tracheal bronchial cells, to test the transcytosis activity of bifunctional molecules to the mucosal lumen (Figure 3a). As expected, bifunctional molecules featuring the null-VHH, which does not bind pIgR, did not effectively transcytose (CV19B289, CV19B290, and CV19B265). Conversely, bifunctional molecules that could bind pIgR displayed higher apical concentration, indicating higher transcytosis. Indeed, protein recovery from the mucosal extract appeared to result through cross-tissue transport, as shown by confocal imaging (Figure 3b-c). Staining for both pIgR and anti-VHH at 24 hr showed that CV19B307 (VHH2) was localized throughout the tissue while CV19B290 (null VHH) failed to transcytose through the tissue at all. Interestingly, bifunctional molecules in which the anti-pIgR VHH domains were fused directly to the N-terminus of the Fc (CV19B301, CV19B307, and CV19B308) appeared to transcytose more effectively than bifunctional molecules in which the anti-pIgR VHH was fused in tandem with the ACE2 ECD on the N-terminus of the Fc (CV19B277 and CV19B283). In the case of CV19B301 and CV19B307, approximately 6% of the total protein was recovered in the mucociliary milieu, compared to ~0.3% for the bifunctional molecules featuring the null VHH, or ~16-fold enrichment in the mucosa. Although the amount of protein retained in the basal chamber was not measured, a large amount of bifunctional molecule was retained within the tissue space and non-recoverable from the mucosal extract, and it is possible that this bifunctional molecule would ultimately be fated for mucosal secretion (Figure 3b). The binding affinity of the anti-pIgR VHH had less effect on transcytosis, although the bifunctional molecule featuring the shorter ACE2 fragment (CV19B308) displayed somewhat weaker transcytosis. Based on transcytosis ability, the two most optimal bifunctional molecules were CV19B301 and CV19B307, having mucosal enrichment >16-fold compared to CD19B265 (null VHH).
Figure 3.
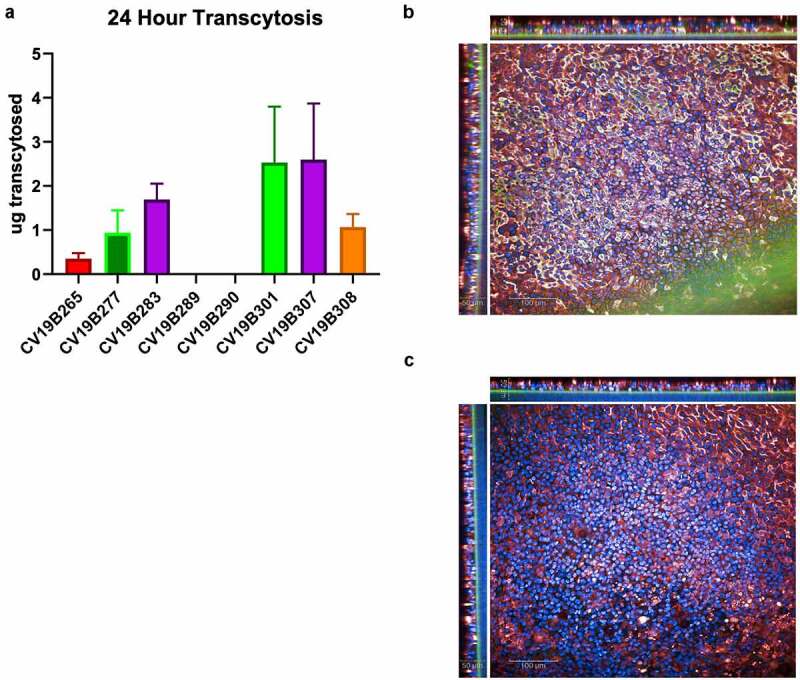
Transcytosis in lung microtissues. A) Transcytosis of bifunctional molecules in EpiAirway tissue model in 24 h post application. For each sample, 20 mg of protein was added to the basolateral well, and after 24 hr, the mucosal surface was washed and the levels of transcytosed antibodies were quantified. Levels are shown in total micrograms transcytosed in a 24 h period. Error bars represent standard error and are representative of at least 2 independent experiments. B-C) Confocal image showing the human EpiAirway microtissue for CV19B307 (b) and CV19B290 (c). Staining shows: blue (nuclei), green (anti-VHH), and red (pIgR). Scale bars shows 50 mm (insets) and 100 mm (main image)
Discussion
Here we describe a panel of bifunctional molecules that bind both pIgR and the SARS-CoV-2 spike glycoprotein. These bifunctional molecules can be efficiently transcytosed across the lung epithelium into the mucosa in an ex vivo human epi-airway model and can compete with SARS-CoV-2 for binding to ACE2-expressing cells (Figure 4). Antibodies were designed using the ACE2 ECD to avoid escape mutants of SARS-CoV-2. Bifunctional molecules featuring N-terminal anti-pIgR VHH2 and a C-terminal ACE2 fusion were preferred, based on maintenance of spike protein and pIgR binding, functional activity, and transcytosis. The design of CV19B301 and CV19B307, having the anti-pIgR VHH fused to the N-terminus of the Fc and ACE2 fused to the C-terminus (Table 1), were most advantageous since they displayed ~ 16-fold enrichment in the mucosa, compared to null bifunctional molecules.
Figure 4.
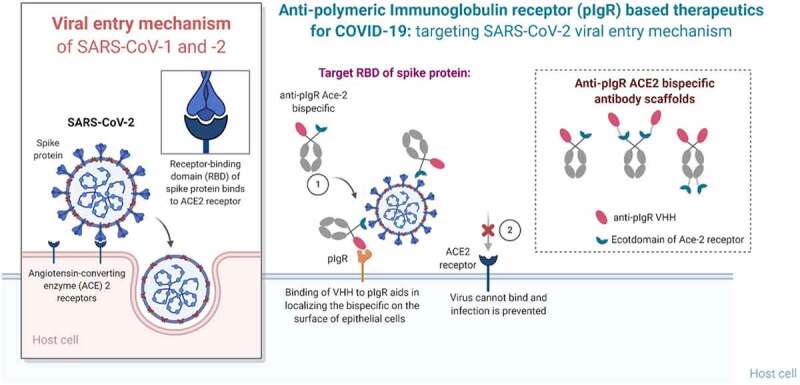
Summary of the mechanism of pIgR-based targeted transport. SARS-CoV-2 viral entry occurs upon binding of the RBD domain of its spike glycoprotein to the ACE2 receptor on target cells. Bifunctional molecules gain access to the lung mucosa through pIgR-mediated transport and bind/neutralize SARS-CoV-2 by binding the RBD domain through their ACE2 ECD moiety in a steric mechanism
Several groups have suggested use of the ACE2 ECD-Fc as a therapeutic, and some have showed that ACE2 can neutralize SARS-CoV-2.31–33,56 The bifunctional molecules tested in this study could compete with the viral spike glycoprotein for ACE2 ECD binding in a surrogate neutralization assay, indicating that the addition of the anti-pIgR VHH moieties did not disrupt interactions between the spike glycoprotein and ACE2. Although ACE2 can compete with SARS-CoV-2 S-protein, biologic-based therapies for mucosal targets present challenges for development. In particular, IgG-based antibodies delivered intravenously or subcutaneously often fail to efficiently cross endothelial boundaries. Thus, expected efficacious doses for IgG targeted to some mucosal antigens such as influenza can be as high as 50 mg/kg, which can be prohibitive for commercial development.26 Addition of the anti-pIgR moieties is expected to increase transcytosis across endothelial layers by ~30-fold, which suggests that efficacious doses could be lowered by a similar level, resulting in more economical commercial manufacturing. Additionally, ACE2-Fc was tested clinically for its ability to decrease inflammation levels in acute respiratory distress syndrome, but was unsuccessful largely due to low exposure in the mucosa. pIgR is highly expressed in lung and gut endothelium, and these layers comprise ~78 m2 and 32 m2 of surface area, respectively, in humans, suggesting that the efficiency of pIgR-mediated transcytosis is a bulk transport mechanism and that bifunctional molecules would be transported primarily to lung mucosa where it could neutralize SARS-CoV-2 and potentially target infected cells and reduce inflammation.57,58 Although SARS-CoV-2 presents a potentially wider therapeutic window than influenza, based on the longer time-course of viral shedding, the cascade of inflammatory cytokines associated with severe COVID-19, brought on by the release of damage-associated molecular patterns by infected cells suggests that early treatment would be favored to limit viral burden in patients.59 Although the potential benefit of therapeutic ACE2 administration after severe infection are unknown, the expected benefit in viral clearance suggests that at least the time-course of inflammatory dysregulation could be decreased. The potential gut localization of the bifunctional molecules could provide a critical advantage for SARS-CoV-2 infection in this mucosal space, since SARS-2 is known to infect enterocytes and elicit pathologic effects in the gut, providing additional benefit to patients.19–21
Although the ACE2 fusion molecules displayed neutralizing activity, mAbs are often preferred due to higher affinity binding and ease of manufacturing. Indeed, neutralizing Abs D001 and SAD-S35 displayed more potent neutralizing activity compared with ACE2, and these may present another potential therapeutic design. However, one limitation of mAb-based antiviral therapeutics is the ability of viruses, particularly fast-mutating RNA viruses, to mutate and escape neutralization by mAbs. Recently characterized SARS-CoV-2 variants, including the B.1.135 and B.1.1.7 lineages, appear to spread more readily and possess mutations that can decrease the ability of mAbs to neutralize virus.49 Escape mutants are not expected to evade ACE2-based therapeutics, and indeed, the three S-protein variants tested here, Y435F, N439K, and N501Y, displayed binding to ACE2 similar to the wild-type spike glycoprotein. Additional SARS-CoV-2 spike glycoprotein variants are emerging, and these may further enhance the ability of SARS-CoV-2 to escape antibody-based neutralization, supporting the development of ACE2-based therapeutics. The D614G variant, which stabilizes the closed structure of the spike glycoprotein, displayed weaker binding to both neutralizing Abs and to ACE2, although unexpectedly CV19B307 displayed similar abilities to bind all variants. A corollary advantage to ACE2-based anti-SARS-CoV-2 therapeutics will be the ability to exploit naturally occurring ACE2 polymorphisms, some of which were shown to enhance binding to the spike glycoprotein, to generate versions of the recombinant receptor that can outcompete the endogenous cell-surface ACE2 in the patient.60
A major cause of clinical complications in COVID-19 patients is inflammation. Antibodies featuring an IgG1 Fc can recruit NK cells, macrophages, and neutrophils to mediate effector functions, and this can further inflame infected areas, exacerbating the symptoms, in ADE of the disease, which is observed with COVID-19, and several other viral diseases.28,61,62 Future designs could feature an Fc that lacks the ability to recruit effector cells (silent Fc). Bifunctional molecules featuring a silent Fc may have less cytotoxic activity against infected cells, but may still provide sufficient neutralizing abilities to result in clinical response. Influenza represents another respiratory infection with unmet medical need; however, antibody therapeutics against influenza viruses have shown mixed results in clinical studies. In addition to the potential for ADE, anti-influenza antibody therapeutics suffer from short therapeutic windows for treatment. Influenza viral shedding peaks ~1-2 days after the onset of symptoms, allowing a maximum treatment window of ~24 hr. This short window strongly favors treatment with inexpensive antiviral drugs, such as oseltamivir. SARS-CoV and SARS-CoV-2 display viral shedding significantly longer, with peaks ~9−10 days after onset of symptoms.62 The longer viral shedding period associated with severe COVID-19 suggests that treatment with biotherapeutics could be more effective compared to influenza viral infection.
In this work, we present a set of bifunctional molecules that overcome several challenges for treatment of COVID-19, including 1) an unmet medical need for hospitalized COVID-19 patients, 2) the potential for ADE in COVID-19 patients by use of a silent Fc, 3) difficulty in obtaining high mucosa exposure of IgG-based biologics, and 4) the potential of RBD variants to escape antibody-based neutralization (by use of the ACE2 ECD). These advantages, along with both the extended period of SARS-CoV-2 viral shedding and the lack of other effective treatment options suggests a path to treatment with the anti-pIgR-ACE2-Fc biologics.
Materials and Methods
Construct designConstructs were designed fusing anti-pIgR VHHs to the N-terminus of the heavy chain of each mAb with a 2(G4S) linker. Null controls were designed in IgG1 mAbs as well as N-terminal VHH fusions bearing a null VHH binder. Additionally, constructs were designed using human ACE2 in conjunction with anti-pIgR VHHs. Human ACE2 ECD (725 AA) as well as a truncated variant (611 AA) were designed. These molecules consisted of two formats: VHH-2(G4S)-ACE2-Fc, and VHH-Fc-2(G4S)-ACE2. DNA sequences for variable regions and ACE2 were codon-optimized for CHO expression and cloned into Lonza-pEE6.4 (heavy chains) or Lonza-pEE12.4 (light chains).
ProteinsExpression plasmids encoding bifunctional molecules were transfected into ExpiCHO cells according to the manufacturer’s instructions. Cell supernatants were harvested after 6–7 days by centrifugation at 4,000 X g for 15 min, passed through a 0.45 mm filter, and purified by MabSelectTM SuRe (ThermoFisher) chromatography on an ÄKTA express system using phosphate-buffered saline (PBS) as running buffer and 0.1 M sodium acetate, pH 3.5 as elution buffer. Eluted fractions were immediately neutralized using 25% (v/v) 2 M Tris-HCl pH 7.0, dialyzed to PBS, sterilized by 0.22 mm filtration and stored at 4°C. Protein concentration was determined by UV-visible spectroscopy. Final yields ranged from 7 mg to 22 mg protein after 35 mL expression.
Bio-layer interferometry
Binding kinetics were measured between Abs and either pIgR or spike glycoprotein by immobilizing his-tagged pIgR or SARS-CoV-2 RBD to anti-His (HIS2) biosensors. Antigen-coated tips were then exposed to each protein for a 90 second association step, followed by a 90 second dissociation step. Association and dissociation rates were measured by the shift in wavelength (nm). All proteins and antigens were diluted to a concentration of 10 mg/mL in 1X PBS at 25°C. Data were collected with Octet Data Acquisition (ForteBio) and analyzed using Octet Data Analysis (ForteBio). Data were process using Graphpad Prism software.
Functional Surrogate Neutralization Assay analysis
A surrogate neutralization assay was used because it does not require the use of active virus and was validated extensively using patient samples – showing high sensitivity and specificity for SARS-CoV-2 spike protein.51 Abs were prepared by serial dilution in assay buffer (3% bovine serum albumin (BSA) + PBS). Samples were incubated with 0.1 mg/mL of biotinylated spike glycoprotein at 1:1 (v/v) for 1 hr with shaking. After incubation, 25 µL of the mixture was added to Streptavidin Gold multi-array 96-well plates (Meso Scale Discovery®) and incubated for 1 hr with shaking. After incubation, 25 mL of ruthenium labeled ACE2 at 2 mg/mL were added and incubated for 1 hr with shaking. Plates were washed with PBS prior to addition of 150 mL of 2 X MSD read buffer to read the plate on Mesoscale Sector S 600 reader. MSD data was analyzed and IC50 values were calculated using Graphpad Prism software.
ADCC analysis
MDCK cells (ATCC) were transfected with human pIgR and Nuclight green and used as target cells. MDCK cells were cultured in EMEM supplemented with 10% fetal bovine serum (FBS) and 5 µg/ml puromycin. PBMC effector cells were obtained from Hemacare. Percentages of T cells (CD3+), B cells (CD19+), and NK cells (CD56 + 16+) were enumerated by flow cytometry (Supplementary Fig. S2). PBMCs were incubated overnight in RPMI-1640 supplemented with 10% FBS and 1 x NEAA at a density of 1 × 106 cells/mL. MDCK cells were plated at 10,000 cells/well in 100 µL of assay media and incubated 1 hr at 37°C with 5% CO2. Bifunctional molecules were added at 10 mg/mL and diluted 10-fold per well. Equal volumes of PBMCs and MDCK cells were then incubated with antibody for 72 hr at 37°C, 5% CO2 inside an IncuCyte. Cell lysis was measured by total green area per well after incubation.
Pharmacokinetic analysis
Female C57BL/6 mice were injected with VHH-Fc test antibodies intravenously via tail vein at a dose of 5 mg/kg into 5 animals per group. Time points were taken at 0.02, 0.04, 0.08, 0.25, 1, 2, 3, 7, and 14 days. At each time point, 20 μl of whole blood will be obtained via tail snip into micro EDTA tubes. Blood collected was diluted 10 x in LowCross buffer (Candor; cat# 100500), inverted to mix, and kept on wet ice during collection. Immediately after all samples were collected at a specific timepoint, diluted samples were centrifuged at 1500 X g for 2 minutes and supernatants collected and frozen until analysis. The PK study was approved by the Institutional Animal Care and Use Committee at Janssen Research & Development, LLC. All experiments were performed in compliance with the guidelines of the committee.
For detection of the VHH-Fc test antibodies in mouse diluted whole blood, an electro-chemiluminescent immunoassay (ECLIA) was used. Streptavidin Gold multi-array 96-well plates (MSD®) were blocked with 1% BSA in 1x dPBS for 30 minutes. The capture reagent, biotinylated rabbit anti-camelid VHH mAb (GenScript®; cat# A01995), was diluted to 0.5 µg/mL and 40 μL combined with 10 μL of diluted standards, controls, and samples in the assay plate for 60 minutes. Plates were washed in PBS-buffered saline with Tween 20 (PBST) and 50 μL/well of ruthenium-labeled anti-human Fc mAb (Janssen R&D) diluted to 0.5 µg/mL was added and incubated for 60 minutes. Following another wash step, 150 mL/well of 1x Read Buffer T (MSD; cat# R92TD) was added and plates were read in a MSD Sector Imager 600 plate reader. Final sample concentrations of the VHH-Fc antibodies were back-calculated from representative standard curves using a 5-parameter non-linear regression with 1/y2 weighting in Watson LIMS 7.6 analysis software.
T1/2 calculations of the elimination phase (β phase) for PK studies were determined using the 1-phase exponential decay model fitted by non-linear regression of natural log concentration versus time using Prism version 8.0 software. The least squares nonlinear decay model was weighted by the inverse of the fitted concentration. Half-life calculations of the elimination phase (β phase) were determined using the formula T1/2 = ln2/β, where β is the – slope of the line fitted by the least square regression analysis starting after first dose. The T1/2 value for an Ab was determined by taking the average of the T1/2 values calculated for each animal within the test group.
Transcytosis activity in EpiAirway Tissue System
Tissue models were obtained from Mattek Corporation and maintained according to manufacturer’s instructions. 40 mg of test and control VHH-mFc molecules were added to 2 mL of EpiAirway media in the basolateral chamber and 100 μL of samples were collected from the basolateral and apical chambers at 0 and 24 hr. EpiAirway TEER buffer (120 mL) was used to collect the mucus from the apical chambers. The amount of VHH present in basolateral media and apical mucus was quantified by electrochemiluminescence. Streptavidin-coated MSD plates were bound up with a biotinylated anti-VHH antibody (Genscript A01995) at 2 μg/ml in PBS for 2 h at room temperature at 1000 rpm, washed 3 X with PBT, incubated with blocking buffer for 1 h at RT, incubated with VHH-mFc containing media/mucus (at different dilutions) for 1 h at RT at 1000 rpm, washed 3 X with PBT, incubated with ruthenylated anti-human-Fc antibody (Clone R10Z8E9, labeled in-house) at 2 ug/ml in PBS for 1 h at RT with 1000 rpm, washed 3X with PBT and read plates in 150 μL reading buffer using the MSD imager. The amount of VHH in basolateral and apical chambers were calculated by plotting ECLU against VHH-mFc standard curves in Prism (GraphPad).
Confocal Imaging
Immunofluorescence and confocal microscopy were used to track the amount of pIgR and VHH retained across the EpiAirway microtissue 24 h post-treatment. Tissue samples were rinsed 3X with PBS prior to fixing to remove unbound antibodies and mucus. A 10% formalin solution was added to a final volume of 0.4 mL to the apical chamber and samples were fixed for 20 min at room temperature. The fixing reagent was removed by aspiration and chamber was washed 3X with 2 mL PBS supplemented with 1% Triton-X100 (v/v) (PBST) at room temperature. Primary antibodies against pIgR (R&D Systems, MAB27171) and VHH domains (Genscript, A01995) were diluted to 5 mg/mL final concentration in PBST supplemented with 10% FBS and 500 mL was applied to the apical and basolateral chamber for 2 h at room temperature. Both chambers were washed 2 X with 2 mL PBST at room temperature, and incubated with secondary antibodies (100 mL apical, 500 mL basolateral) diluted in PBST for 2 h. The secondary antibody mix contained Alexa-Fluor 647-labeled anti-mouse antibody (Invitrogen A28181, 1:1,000 dilution), Alexa-Flour 488-labeled streptavidin (Invitrogen, S32357, 1:100 dilution) and DAPI (GeneTex GTX16206, 1:1,000 dilution) diluted in PBST with 10% FBS. Transwells were washed 2 X with PBST and placed in 6 well glass bottom plates for imaging with PBS added to each chamber to prevent desiccation of sample. Fixed, permeabilized and stained tissues were imaged at 20 X resolution (40 planes, 1.6 mm z-slice) using an Opera Phenix confocal laser microscope. Image analysis was performed using the Harmony suite, fluorescence readouts were corrected for membrane auto-fluorescence, normalized by mean fluorescence intensity for each color channel. Representative images from a total of 3 separate experiments per condition were reported.
Supplementary Material
Acknowledgments
We thank Dr. Tun Liu (JBIO, Janssen R&D) for assistance with de novo sequencing of D001 mAb. We thank Dr. Ted Kwaks (Viral Vaccines Discovery, Janssen R&D) for providing the sequence of CR3022 and for technical advice and Dr. Joost Kolkman for helpful discussions. We thank members of JBIO for their technical help.
Funding Statement
This study was funded by Janssen Research & Development, LLC.
Notes on Contributions
AZ, IW, SS, RG, MT, and PC conceived the studies. IW and AZ designed the constructs. IW, PH and NT expressed and purified bi-functional molecules. NT and JB performed the transcytosis assays. JH performed ADCC analyses. RD and TY performed the functional activity assays. BG and TJ performed pharmacokinetic studies. All authors were involved in drafting and revising the manuscript.
Abbreviations
: SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; COVID-19, coronavirus disease 2019; IgG, immunoglobulin G; ACE2, angiotensin-converting enzyme 2; ECD, extra-cellular domain; IgA, immumoglobulin A; VHH, variable domain of the heavy chain antibody; MDCK, Madin-Darby canine kidney; BRING-IT, Bi-directional polymeric Ig receptor mediated transport of biologics; pIgR, polymeric Ig receptor; PBMC, peripheral blood mononuclear cell; RBD, receptor-binding domain; NK, natural killer; ADCC, antibody-dependent cell-mediated cytotoxicity
Disclosure statement
All authors are employees of Janssen Research & Development, LLC.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Dong E, Du H, Gardner L.. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20(5):533–11. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hossein-Khannazer N, Shokoohian B, Shpichka A, Aghdaei HA, Timashev P, Vosough M. Novel therapeutic approaches for treatment of COVID-19. J Mol Med (Berl). 2020;98(6):789–803. doi: 10.1007/s00109-020-01927-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peeples L. News Feature: avoiding pitfalls in the pursuit of a COVID-19 vaccine. Proc Natl Acad Sci U S A. 2020;117(15):8218–21. doi: 10.1073/pnas.2005456117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Administration USFaD . CDER Scientific Review Documents Supporting Emergency Use Authorizations for Drug and Biological Therapeutic Products | COVID-19. September 25, 2021. https://wwwfdagov/drugs/coronavirus-covid-19-drugs/cder-scientific-review-documents-supporting-emergency-use-authorizations-drug-and-biological
- 5.Peebles RS Jr., Liu MC, Lichtenstein LM, Hamilton RG, IgA I, IgM. quantification in bronchoalveolar lavage fluids from allergic rhinitics, allergic asthmatics, and normal subjects by monoclonal antibody-based immunoenzymetric assays. J Immunol Methods. 1995;179(1):77–86. doi: 10.1016/0022-1759(94)00275-2. [DOI] [PubMed] [Google Scholar]
- 6.Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368(3):652–65. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 7.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Structure VD. Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281–92 e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A. 2009;106(14):5871–76. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosch BJ, Van Der Zee R, de Haan CA, Rottier PJ. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003;77(16):8801–11. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burkard C, Verheije MH, Wicht O, van Kasteren SI, Van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJ, Bosch BJ. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014;10(11):e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirchdoerfer RN, Cottrell CA, Wang N, Pallesen J, Yassine HM, Turner HL, Corbett KS, Graham BS, McLellan JS, Ward AB. Pre-fusion structure of a human coronavirus spike protein. Nature. 2016;531(7592):118–21. doi: 10.1038/nature17200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A. 2014;111(42):15214–19. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JE, Li K, Barlan A, Fehr AR, Perlman S, McCray PB Jr., Gallagher T. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc Natl Acad Sci U S A. 2016;113(43):12262–67. doi: 10.1073/pnas.1608147113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walls AC, Tortorici MA, Bosch BJ, Frenz B, Rottier PJM, DiMaio F, Rey FA, Veesler D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature. 2016;531(7592):114–17. doi: 10.1038/nature16988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bar-On YM, Flamholz A, Phillips R, Milo R. SARS-CoV-2 (COVID-19) by the numbers. Elife 2020; 9. [DOI] [PMC free article] [PubMed]
- 16.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 17.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–37. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H, Zhong L, Deng J, Peng J, Dan H, Zeng X, Li T, Chen Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12(1):8. doi: 10.1038/s41368-020-0074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding S, Liang TJ . Is SARS-CoV-2 Also an Enteric Pathogen With Potential Fecal-Oral Transmission? A COVID-19 Virological and Clinical Review. Gastroenterology. 2020;159(1):53–61. doi: 10.1053/j.gastro.2020.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, Ravelli RBG, Paul van Schayck J, Mykytyn AZ, Duimel HQ, et al. SARS-CoV-2 productively infects human gut enterocytes. Science. 2020;369(6499):50–54. doi: 10.1126/science.abc1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology. 2020;158(6):1831–3 e3. doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L, Xiong J, Bao L, Shi Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect Dis. 2020;20(4):398–400. doi: 10.1016/S1473-3099(20)30141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim JJ, Nilsson AC, Silverman M, Assy N, Kulkarni P, McBride JM, et al. A Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial of MHAA4549A, a Monoclonal Antibody, plus Oseltamivir in Patients Hospitalized with Severe Influenza A Virus Infection. In: Antimicrob Agents Chemother. Cesar A. Arias. eds. 2020. p. 64. ASM: Washington DC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali SO, Takas T, Nyborg A, Shoemaker K, Kallewaard NL, Chiong R, et al. Evaluation of MEDI8852, an Anti-Influenza A Monoclonal Antibody, in Treating Acute Uncomplicated Influenza. Antimicrob Agents Chemother. 2018;62(11):e00694-18.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hershberger E, Sloan S, Narayan K, Hay CA, Smith P, Engler F, Jeeninga R, Smits S, Trevejo J, Shriver Z, et al. Safety and efficacy of monoclonal antibody VIS410 in adults with uncomplicated influenza A infection: results from a randomized, double-blind, phase-2, placebo-controlled study. EBioMedicine. 2019;40:574–82. doi: 10.1016/j.ebiom.2018.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nachbagauer R, Krammer F. Universal influenza virus vaccines and therapeutic antibodies. Clin Microbiol Infect. 2017;23(4):222–28. doi: 10.1016/j.cmi.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palivizumab. a Humanized Respiratory Syncytial Virus Monoclonal Antibody, Reduces Hospitalization From Respiratory Syncytial Virus Infection in High-risk Infants. Pediatrics. 1998;102:531–37. [PubMed] [Google Scholar]
- 28.Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. 2020;20(6):339–41. doi: 10.1038/s41577-020-0321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, Penninger J, Krahenbuhl S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52(9):783–92. doi: 10.1007/s40262-013-0072-7. [DOI] [PubMed] [Google Scholar]
- 30.Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, Hall R, Poirier G, Ronco JJ, Tidswell M, et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21(1):234. doi: 10.1186/s13054-017-1823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zoufaly A, Poglitsch M, Aberle JH, Hoepler W, Seitz T, Traugott M, Grieb A, Pawelka E, Laferl H, Wenisch C, et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir Med. 2020;8(11):1154–58. doi: 10.1016/S2213-2600(20)30418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu NC, Wilson IA. A Perspective on the Structural and Functional Constraints for Immune Evasion: insights from Influenza Virus. J Mol Biol. 2017;429(17):2694–709. doi: 10.1016/j.jmb.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei C, Qian K, Li T, Zhang S, Fu W, Ding M, et al . Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun. 2020;11(1):2070. doi: 10.1038/s41467-020-16048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu P, Wysocki J, Souma T, Ye M, Ramirez V, Zhou B, Wilsbacher LD, Quaggin SE, Batlle D, Jin J. Novel ACE2-Fc chimeric fusion provides long-lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney Int. 2018;94(1):114–25. doi: 10.1016/j.kint.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 35.Wollacott AM, Boni MF, Szretter KJ, Sloan SE, Yousofshahi M, Viswanathan K, Bedard S, Hay CA, Smith PF, Shriver Z, et al. Safety and Upper Respiratory Pharmacokinetics of the Hemagglutinin Stalk-Binding Antibody VIS410 Support Treatment and Prophylaxis Based on Population Modeling of Seasonal Influenza A Outbreaks. EBioMedicine. 2016;5:147–55. doi: 10.1016/j.ebiom.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.France MM, Turner JR . The mucosal barrier at a glance. J Cell Sci 2017;130:307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woof JM, Kerr MA. The function of immunoglobulin A in immunity. J Pathol. 2006;208(2):270–82. doi: 10.1002/path.1877. [DOI] [PubMed] [Google Scholar]
- 38.Lewis MJ, Pleass RJ, Batten MR, Atkin JD, Woof JM. Structural Requirements for the Interaction of Human IgA with the Human Polymeric Ig Receptor. The J of Immunology. 2005;175(10):6694–701. doi: 10.4049/jimmunol.175.10.6694. [DOI] [PubMed] [Google Scholar]
- 39.Fox A, Marino J, Amanat F, Krammer F, Hahn-Holbrook J, Zolla-Pazner S, et al. Evidence of a significant secretory-IgA-dominant SARS-CoV-2 immune response in human milk following recovery from COVID-19. iScience 2020; 23:101735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lombana TN, Rajan S, Zorn JA, Mandikian D, Chen EC, Estevez A, Yip V, Bravo DD, Phung W, Farahi F, et al. Production, characterization, and in vivo half-life extension of polymeric IgA molecules in mice. MAbs. 2019;11(6):1122–38. doi: 10.1080/19420862.2019.1622940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maruthachalam BV, Zwolak A, Lin-Schmidt X, Keough E, Tamot N, Venkataramani S, Geist B, Singh S, Ganesan R . Discovery and characterization of single-domain antibodies for polymeric Ig receptor-mediated mucosal delivery of biologics. MAbs. 2020;12(1):1708030. doi: 10.1080/19420862.2019.1708030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emmerson CD, van der Vlist EJ, Braam MR, Vanlandschoot P, Merchiers P, de Haard HJ, Verrips CT, van Bergen en Henegouwen PM, Dolk E. Enhancement of polymeric immunoglobulin receptor transcytosis by biparatopic VHH. PLoS One. 2011;6(10):e26299. doi: 10.1371/journal.pone.0026299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prabakaran P, Zhu Z, Xiao X, Biragyn A, Dimitrov AS, Broder CC, Dimitrov DS. Potent human monoclonal antibodies against SARS CoV, Nipah and Hendra viruses. Expert Opin Biol Ther. 2009;9(3):355–68. doi: 10.1517/14712590902763755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004;279(17):17996–8007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossotti MA, Belanger K, Henry KA, Tanha J. Immunogenicity and humanization of single-domain antibodies. FEBS J. 2021. doi: 10.1111/febs.15809. [DOI] [PubMed] [Google Scholar]
- 46.Tan X, Lin C, Zhang J, Khaing OMK, Fan X. Rapid and quantitative detection of COVID-19 markers in micro-liter sized samples. bioRxiv 2020:2020.04.20.052233. [Google Scholar]
- 47.Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking Changes in SARS-CoV-2 Spike: evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182(4):812-827.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, Dingens AS, Li JZ, Bloom JD. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science. 2021;371(6531):850–54. doi: 10.1126/science.abf9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, Dingens AS, et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021;371:850-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Juraszek J, Rutten L, Blokland S, Bouchier P, Voorzaat R, Ritschel T, Bakkers MJG, Renault LLR, Langedijk JPM. Stabilizing the closed SARS-CoV-2 spike trimer. Nat Commun. 2021;12(1):244. doi: 10.1038/s41467-020-20321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan CW, Chia WN, Qin X, Liu P, Chen MI, Tiu C, Hu Z, Chen VC, Young BE, Sia WR, et al. A SARS-CoV-2 surrogate virus neutralization test based on antibody-mediated blockage of ACE2-spike protein-protein interaction. Nat Biotechnol. 2020;38(9):1073–78. doi: 10.1038/s41587-020-0631-z. [DOI] [PubMed] [Google Scholar]
- 52.Huo J, Zhao Y, Ren J, Zhou D, Duyvesteyn HME, Ginn HM, Carrique L, Malinauskas T, Ruza RR, Shah PNM, et al. Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host Microbe. 2020;28(3):497. doi: 10.1016/j.chom.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.ter Meulen J, van den Brink EN, Poon LL, Marissen WE, Leung CS, Cox F, Cheung CY, Bakker AQ, Bogaards JA, van Deventer E, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006;3(7):e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saunders KO. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snoeck V, Peters IR, Cox E. The IgA system: a comparison of structure and function in different species. Vet Res. 2006;37(3):455–67. doi: 10.1051/vetres:2006010. [DOI] [PubMed] [Google Scholar]
- 56.Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020;181(4):905–13 e7. doi: 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frohlich E, Mercuri A, Wu S, Salar-Behzadi S. Measurements of Deposition, Lung Surface Area and Lung Fluid for Simulation of Inhaled Compounds. Front Pharmacol. 2016;7:181. doi: 10.3389/fphar.2016.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Helander HF, Fandriks L. Surface area of the digestive tract - revisited. Scand J Gastroenterol. 2014;49(6):681–89. doi: 10.3109/00365521.2014.898326. [DOI] [PubMed] [Google Scholar]
- 59.Jamal M, Bangash HI, Habiba M, Lei Y, Xie T, Sun J, Wei Z, Hong Z, Shao L, Zhang Q. Immune dysregulation and system pathology in COVID-19. Virulence. 2021;12(1):918–36. doi: 10.1080/21505594.2021.1898790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suryamohan K, Diwanji D, Stawiski EW, Gupta R, Miersch S, Liu J, Chen C, Jiang YP, Fellouse FA, Sathirapongsasuti JF, et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun Biol. 2021;4(1):475. doi: 10.1038/s42003-021-02030-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eroshenko N, Gill T, Keaveney MK, Church GM, Trevejo JM, Rajaniemi H. Implications of antibody-dependent enhancement of infection for SARS-CoV-2 countermeasures. Nat Biotechnol. 2020;38(7):789–91. doi: 10.1038/s41587-020-0577-1. [DOI] [PubMed] [Google Scholar]
- 62.He X, Lau EHY, Wu P, Deng X, Wang J, Hao X, Lau YC, Wong JY, Guan Y, Tan X, et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat Med. 2020;26(5):672–75. doi: 10.1038/s41591-020-0869-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.