

Abstract
Dynamic full-field optical coherence microscopy (DFFOCM) was used to characterize the intracellular dynamic activities and cytoskeleton of HeLa cells in different viability states. HeLa cell samples were continuously monitored for 24 hours and compared with histological examination to confirm the cell viability states. The averaged mean frequency and magnitude observed in healthy cells were 4.79±0.5 Hz and 2.44±1.06, respectively. In dead cells, the averaged mean frequency was shifted to 8.57±0.71 Hz, whereas the magnitude was significantly decreased to 0.53±0.25. This cell dynamic activity analysis using DFFOCM is expected to replace conventional time-consuming and biopsies-required histological or biochemical methods.
1. Introduction
Cells are the structural and functional units that make up all living creatures and contain information about their composition [1–3]. Previous studies on cell growth, metabolism, and death have provided many clues to various research fields including disease treatment [4–6], early diagnosis [7–10], and drug development [11–14]. As an example, cell death assay studies have been used as early indicators of several incurable diseases such as cancer [8–10,15,16], and Alzheimer's disease [7,17,18]. Because of these great potential applications, various methods have been attempted to observe changes in internal cellular dynamic activity and properties beyond morphology changes [4–18]. Common methods for observing cell viability and/or structural images include: analyzing signals generated by stimulating cells using light or ultrasound from the outside [19,20]; reactivity testing by biological markers [21]; and monitoring the internal structure or spectral properties of cells using light characteristics [22,23]. Imaging techniques using ultrasound can measure objects located in a deep region of a sample using their long-wavelength features but are not suitable for observing small objects of tens of microns or less, such as cells. Although high-frequency ultrasound transduction has been studied and developed to improve resolution, its practical use is limited due to the manufacturing complexity and cost of a transducer having a sub-micron resolution. Also, as the frequency of ultrasound increases, the observable penetration depth is inversely decreased [24]. Biological markers (e.g., fluorophore and quantum dots) have contributed to detect phenomena, which are unobservable with a microscope and depend only on the transmission and reflection of light by labeling proteins, nucleic acids, or small molecules in cells [25,26]. However, the use of biological markers by injecting a foreign substance into the cell can lead to different results rather than cell-specific properties [27]. Though most studies have claimed that biological markers are rarely harmful to cells and humans [28], contradictory results have also been reported [29,30]. With fluorescent proteins, clinical trials are limited due to fluorescence property changes during long-term experiments. Fluorescence imaging can only see the structure of the region where the fluorophores react with a specific excitation laser [31]. Where fluorophores are rarely present, a rather complex imaging system must be constructed to observe the overall structure of the sample [32]. Moreover, the use of specific single-wavelength lasers for fluorescence expression may change sample conditions [30]. In addition, biological markers may exhibit different fluorescence and labeling intensities in similar populations, so the characteristics of the expressing entity must be considered [28,33]. Therefore, in order to analyze the intrinsic characteristics of biological samples, an observational method in which foreign substances are not added is preferred.
Previous studies have attempted to detect dynamic motion inside cells using optical interferometric imaging techniques [22,23]. Optical interferometric-based imaging techniques using lasers can observe signals in a sample in a non-destructive and non-invasive manner but are affected by the distribution of scatterers and absorbers present inside the sample [22,23]. Additionally, most optical interferometric imaging systems use a point-measurement method to observe a sample [22,23,34], which causes a time delay in the entire sample observation and can lead to errors in monitoring its dynamic motion. For that reason, in the studies of Meer et al. and Farhat et al., optical interference signals were analyzed only in a region-of-interest (part of the sample or one point).
In this paper, we demonstrate a method to quantitatively analyze the dynamic activities of cell in different viability states using dynamic full-field optical coherence microscopy (DFFOCM) equipped with a high-speed complementary metal-oxide-semiconductor (CMOS) camera. Since DFFOCM observes the entire area of a cell in a full-field observation method, both optical properties of cells and its dynamic movement can be measured and analyzed quantitatively. When healthy cells undergo the cell death process, the intracellular dynamic activity and cytoskeleton are transformed (such as blebbing, cell swollen), so we hypothesize that large dynamic changes will occur, and these characteristic changes are expected to be observed in the DFFOCM system. To confirm our hypothesis, we used the DFFOCM system to monitor the dynamic activities of HeLa cells, which is an immortal human cell line widely used in cell research, at 15-minute intervals during the first 10 hours of the experiment, and then at larger intervals for the subsequent 14 hours. It was continuously monitored over the course of the experiment. In addition, the histological experiments were conducted to confirm cell viability states. The dynamic activities measured with DFFOCM will be a key indicator of cell death evaluation by visually and numerically analyzing the features that appear when undergoing cell death and these quantitative results are expected to contribute significantly to the existing cytology studies that are subjective to the experts’ experience.
2. Materials and methods
2.1. Cell culture and membrane preparation
The HeLa cell line was obtained from the American Type Culture Collection (Manassas, VA) and cultured in DMEM growth media supplemented with 10% Fetal Bovine Serum, 2 mM Glutamine, and Pen-Strep antibiotic mixture. The flask containing cells were placed in an incubator which was maintained at a temperature of 37°C and 5% concentration of carbon dioxide (CO2) gas. When preparing samples for the experiment, cells were detached from the flask by using 0.05% Trypsin. Approximately 30,000 cells were plated on a Snapwell insert membrane fabricated out of Polyethylene (3801, Corning Inc., Corning, NY). The membrane containing cells were placed back in the incubator for 24 hours prior to imaging. Three Snapwell membranes were prepared for cell imaging and viability checks over time.
2.2. DFFOCM system
The dynamic full-field optical coherence imaging system was developed and reported [35] to present an endogenous approach to reveal intracellular metabolic contrasts. DFFOCM is designed by mounting a CMOS camera capable of acquiring high-speed images in a Linnik interferometer system and constructed based on full-field illumination, as a usual full-field optical coherence microscopy (FFOCM) [35,36]. The representative difference between FFOCM and DFFOCM is the method of acquiring an interfering signal that contains sample information. In a typical FFOCM, when the reference arm and sample arm are within a coherence-gating, the reference arm is modulated with a specific phase distance for each depth of the sample to obtain information about the interferometer amplitude and phase [36]. On the other hand, DFFOCM, with the contribution of a high-speed CMOS camera, collects continuous interfering signals at the same depth of a sample without modulation of the reference arm and observes only the movement of the scatterers inside the sample. In other words, during a short recording time, the sample is stationary, hence only inner cell movements show up. In addition, high sensitivity measurement is required to observe objects with a small backscattering level, such as cells. CMOS cameras can improve the signal-to-noise ratio for samples with low backscattering levels by time oversampling. As a result, only motion inside the sample contributes to the modulation of the interfering signal [35]. The DFFOCM system used in the experiment is a product of a commercialized Light-CT scanner (LLTech SAS, France). The system schematic of the scanner was reported in our previous paper [35]. Based on the full-field observation method, the system is equipped with a 10x objective lens as standard and provides a field of view of 1.2 mm2. The combination of a low-coherence broadband light source and a high-precision stage allows the DFFOCM system (Light-CT scanner) used in our experiment to detect signals in the sample with an axial resolution of 1 µm and a transverse resolution of 1.5 µm. The axial and transverse resolution can be improved by replacing the light source, mechanical stage, and/or objective lens. In addition, the system is equipped with a high-speed CMOS camera with a frame rate of 300 Hz, which allows it to achieve a temporal resolution of up to approximately 6.7 ms (150 Hz).
2.3. Experimental procedure
Prior to imaging, the three Snapwell membranes were removed from the incubator and transferred to optical sample holders. The sample holders were filled with warmed cell culture media (37°C) and two Snapwell membranes (Sample 1 and 2) were placed upside down. At the same time, a mixture of culture media and Trypan blue (1:1 ratio) was added to the third Snapwell membrane (Sample 3). The total time for the sample preparation was approximately 10 minutes. After that, all three samples were brought to an experiment room, where the temperature was maintained at 22°C. Sample 1 was then placed into the DFFOCM system for optical imaging for 24 hours and Sample 2 was kept near the DFFOCM system to have a similar environmental condition for both samples. When the optical imaging started, Sample 3 was placed under a microscope to check cell viability. The same cell viability test was performed on Sample 2 after 6 hours and on Sample 1 when it was removed from the DFFOCM system after 24 hours.
2.4. Data analysis
2.4.1. Image acquisition
The DFFOCM system used in the experiment exhibited a drift of less than about 1 µm in the axial direction during 24 hours stability measurements. We observed the cell dynamic movement inside the cell at a height of about 5 µm from the Snapwell membrane surface, taking into account the stability of the system. A single image acquired with the DFFOCM system without modulation of the reference arm does not clearly provide information on scatter signals of the sample (Fig. 1(a)). Figure 1(a) is an actual bright-field image continuously acquired through our DFFOCM system, and the black rectangle represents the same single pixel in each image. However, by calculating the standard deviation (STD) for each image pixel as shown in Eq. (1) [35] for a number of interfering images acquired at a high speed by the CMOS camera, it is possible to distinguish between random noise and signals caused by scatterers.
| (1) |
Fig. 1.

(a) Experimental raw images of HeLa cells observed through the DFFOCM system. To reconstruct the dynamic image, 1000 frames of raw images were acquired continuously. (b) Image of HeLa cells reconstructed by taking the average of sub-data STDs. The HeLa cell analyzed for dynamic changes were enlarged and inserted.
The STD structural image of DFFOCM can be reconstructed by dividing the total raw images continuously acquired using a high-speed CMOS camera into a sub-data set which have N number of frames and calculating the average of the STDs of each sub-data set over time (). is the mean standard deviation calculated from the total raw interference images and represents each pixel of the DFFOCM image (shown in Fig. 1(b)). The number of sub-data images can be efficiently adjusted according to the object of interest. For example, for a small number of sub-stack images, it is appropriate to observe an object with relatively fast dynamic motion, and a large number of sub-stack images are required to observe an object with relatively slow dynamic motion [35]. To get the dynamic motion inside a cell in our experiment, 1000 images of the interfering signal inside the cell were acquired without changing the depth position, then they were divided into 10 sub-data (each sub-data has 100 frames) and the DFFOCM image was restored. No averaging was conducted on image acquisition, and it took around 3.3 seconds to acquire 1000 frames of raw images. Identical data acquisitions were performed for 24 hours.
2.4.2. Dynamic activity calculation
By using more than two DFFOCM images continuously observed faster than the cell movement, dynamic motion inside and outside of the cell can be analyzed with frequency spectrum. To remove the external noise caused by the experimental environment in the intracellular dynamic frequency spectrum, the extracellular dynamic motion was simultaneously observed. However, since the cell’s intrinsic dynamic motion is very small, it is difficult to observe this motion using the commonly used frequency analysis method called the full-time Fast Fourier Transform (FFT). Full-time FFT analysis is sensitive to non-stationary noise (e.g., mechanical, and environmental noise) but it is not suitable for identifying small signals such as dynamic cell movements. To alleviate this problem, we used the Welch’s method to analyze frequency information. Welch’s method [37] partially calculates frequency characteristics of raw data according to the size of a window. By overlapping each window, the spectral characteristic of the entire signal is maintained and smoothed over non-systematic noise. The frequency spectrum was calculated for each pixel as shown in Fig. 1(a). We used a Hamming window and selected the window size to match the number of sub-data frames defined to obtain the DFFOCM image. To compensate for the signal loss due to frequency analysis using the windowing method, each window was calculated by overlapping 50%. The calculated frequency spectrum of each pixel was used to reconstruct dynamic images of the cells (shown in Results Section 3.1), mean frequency and magnitude values (shown in Results Section 3.2).
2.4.3. Mean frequency and magnitude calculation
Images of cells observed with DFFOCM equipped with a high-speed CMOS camera can quantify changes within cells (cytoplasmic migration, changes in membrane state, cell swelling, and others) by frequency and magnitude values. To numerically observe activities changed inside a cell, a mean frequency () was calculated by using Eq. (2) with a frequency inside a cell and a power spectral density. M is the number of frequency bins of the observation range frequency spectrum (in our case, from 0 Hz to 30 Hz), and is the frequency of spectrum at bin i of M. Then, is the power density of the spectrum at bin i of M. To minimize the influence of external noise, the cell area is segmented, and the calculated background frequency is subtracted from the cell area before calculating . The dynamic activity magnitude of cells was calculated by summing the of the observed frequency spectra. The and the magnitude of dynamic activity of the cell over time is shown in Results Section 3.
| (2) |
2.5. Cell viability measurement
To check cell viability, a mixture of cell culture media and Trypan blue (1:1 ratio) was added to the Snapwell membranes at the beginning of the experiment, after 6 hours, and after 24 hours (Sample 3, 2, and 1, respectively). Trypan blue selectively stains dead tissues or cells and does not stain cells with intact cell membranes that have not undergone a cell death process. The Trypan blue staining method makes it difficult to explore the metabolic processes occurring inside the cell, but it can be compared with the results observed over time using the DFFOCM system. The stained HeLa cells in three Snapwell membranes were observed through a bright-field microscope (EVOS XL, Thermo fisher Scientific, USA).
2.6. Statistical analysis
A two way-ANOVA test was conducted to confirm the justification of the difference in the dynamic activity of living and dead cells. Data for statistical analysis was acquired by 3 times repeating the cell dynamic activity observation for 24 hours performed in Methods Sections 2.3 to 2.4. At time t = 0h, HeLa cells were considered to be alive, while they were assumed to be dead at time t = 24h. Cell dynamic activities ( and magnitude) of HeLa cells observed at t = 0h and t = 24h were used as the main comparator for the ANOVA test. In the test, cell viability states (live and dead) and cell samples (three samples) were used as the two factors for the two-way ANOVA test. The results analyzed for and magnitude are shown in Results Section 3.4.
3. Results
3.1. Dynamic intracellular motion analysis
Major scatterers of cells that can be observed through optical imaging techniques are nucleus, membrane, mitochondria, and cytoplasm [36–38]. Figure 2(a) shows the changes in intracellular migration and structure monitored for 24h of a single cell enlarged in Fig. 1(b) (Observations at 15-minute intervals are shown in Visualization 1 (17.5MB, avi) ). Figure 2(a) represents results of the reconstructed intracellular motion through frequency and power spectrum density. Each pixel contains several frequency spectra, but the dominant frequency and its power density make up the image. Figure 2(b) is a segmentation mask of the cell through image processing to calculate only frequency inside the cell, and the external structure change of the cell can also be clearly observed. In Fig. 2(a), when t = 0h, relatively uniform scatterers were spread throughout the cell. Considering that there is no clearly distinct region, we carefully assumed that the observed scatterers are cytoplasm including mitochondria. At t = 4h, the dominant movement of the scatterers was concentrated in the center of the cell, and the transformation of the cytoskeleton was observed. At t = 6h, the dynamic image indicates that the scatterers moved toward the cell membrane. Considering the characteristics of blebbing and swelling when cells die, it appears that cell death is progressing through the segmentation image in Fig. 2(b). At t = 24h, most scatterers with active dynamic activity inside the cell were moved to the outside and maintained only the swollen cytoskeleton. Small spots were observed in the swollen cytoskeleton even after the scatterers were released from the cells, which we carefully considered as vesicles, taking into account the nature of their morphology and dynamic activity [39]. The cell then decomposed into small fragments and, the disappearance of the dynamic movement inside the cell was clearly observed in dynamic and segmentation images (i.e., The power density for the movement of scatterers inside the cell decreases).
Fig. 2.

Dynamic and structural changes in HeLa cell over time. (a) The frequency component with the dominant dynamic motion of the scatterers is prominently displayed in the image. The intracellular migration of main dynamic active elements is indicated by white arrows. The power density was normalized. (b) The segmentation mask image of the cell. The mask is segmented by using STD images and frequency spectral information.
3.2. Mean frequency, frequency and power density spectrum of cells
Figure 3(a) shows the spectrum of dynamic activity in cells at 15-minute intervals during the first 10 hours of the experiment, and then at large intervals for the subsequent 14 hours. In the early stage of the experiment, the main scatterers inside the healthy cells had remarkable movements below 6 Hz. As time passed, the magnitude of scatterers’ movement gradually decreased, and after about 4 hours the movement within the cell clearly and remarkably disappeared. The spectral change after 4 hours in Fig. 3(a) is consistent with the dynamic and segmentation images at 4h in Fig. 2 when the cytoskeleton is transformed. Figure 3(b) is the that numerically represents the spectrum of Fig. 3(a), and Fig. 3(c) indicates the magnitude of the dynamic movement of the cell. After 4 hours, (the red dashed line in Fig. 3(b)) had a sharp increase in frequency temporarily due to the movement of the scatterer inside the cell and the change of the skeleton as shown in Fig. 2. After 6 hours (red solid line), the scatterers inside the cell moved outside and lost the intrinsic dynamic motion. Thereafter, a tendency was observed for the of cells to converge to a relatively high frequency by vesicles (as shown Fig. 2(a)). Following a similar trend, the magnitude also decreases at the same time point after 6 hours and then converges to the vesicles level.
Fig. 3.

Dynamic motion analysis of single HeLa cell measured for 24 hours. Shows a pronounced dynamic change after 4 hours (red dash line). (a) The frequency spectrum of the cell. (b) Mean frequency of the cell frequency spectrum. (c) Magnitude of the dynamic movement of the cell.
3.3. Cell viability at t = 0, 6, and 24 hours
In parallel with the DFFOCM experiment, viability of the HeLa cells cultured under the same conditions were observed using Trypan blue. As shown in Fig. 4(a), the membrane of HeLa cells was intact at the beginning of the experiment and live cells were not stained with Trypan blue. After 6 hours, the HeLa cells reacted to Trypan blue and were partially stained (Fig. 4(b)). By 24 hours, some cells were degraded and disappeared, resulting in an overall decrease in the number of cells, and the remaining cells were stained with Trypan blue (Fig. 4(c)). This result does not provide information on the changes inside a cell, but it is consistent with the results shown in Results 3.1 and 3.2. The skeletal structure of the cell has changed, and the decomposed shape can be observed at 6 and 24 hours.
Fig. 4.

Observation of the histological cell death process using Trypan blue (a) at the beginning of the experiment, (b) after 6 hours, and (c) after 24 hours. The inset white scale bar represents 100 µm.
3.4. ANOVA analysis of live and dead cells
Figure 5 shows the dynamic activity parameters of live (t = 0h) and dead (t = 24h) HeLa cells of three membrane samples. The was significantly different between live and dead cells (F(1,72) = 752.1, p = 0). However, there was no significant difference in the between observed membrane samples (F(2,72) = 1.2, p = 0.3) and no interaction between samples and viability (F(2,72) = 2.4, p = 0.1). In dynamic magnitude, there was no significant difference between membrane samples (F(2,72) = 1.2, p = 0.3), but there was a slight interaction between samples and viability (F(2,72) = 3.5, p = 0.04). On the other hand, the dynamic magnitude of live and dead cells was significantly different (F(1,72) = 135.4, p = 0).
Fig. 5.
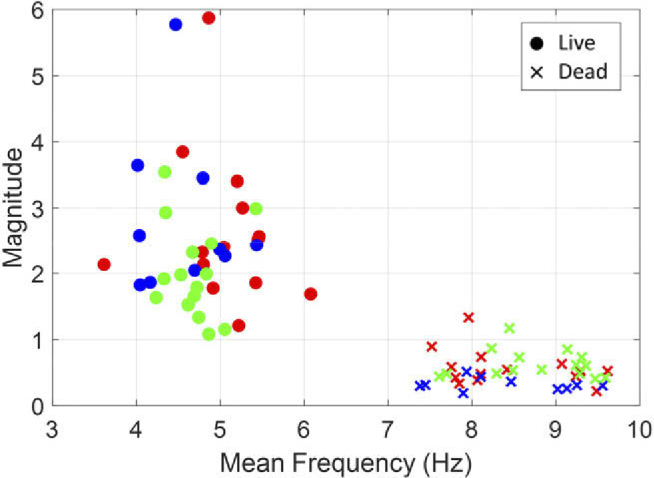
Dynamic activities of HeLa cells in three samples were observed for 24 hours using the DFFOCM system, and mean frequency and magnitude of live (t = 0h) and dead (t = 24h) are shown. The three samples are each marked with different colors (red, green, blue).
In addition, the numerical results (frequency and magnitude of dynamic movement) after cell deaths in Fig. 5 confirmed that the properties of vesicles were similarly observed in all three different membrane samples.
4. Discussion
This study presents a method to evaluate intracellular dynamic activities quantitatively. Experiments were conducted on HeLa cells and their internal dynamic movements were continuously monitored for 24 hours using a DFFOCM system. Movements of scatterer insides of live cells were observed at an averaged of 4.790.5 Hz and dynamic magnitude of 2.441.06. In the process of cell death, the movement of scatterers inside a cell and the transformation of the cytoskeleton (such as blebbing of the cell membrane) changed cell dynamic motility. As a result, dynamic and magnitude shifted to 8.570.71 Hz and 0.530.25, respectively. At a first glance, an increase in the during a cell death process seems to be contradictory because dynamic activities are expected to decrease when the cell is dead. However, in the current study, we used the , which represents the frequencies with high weights. As can be seen in Fig. 3(a), dynamic activity in viable cells at the beginning of the experiment is mostly in low frequency spectrum (< 6 Hz). With a dominance of low frequency bands, the is shifted toward a lower frequency. As cell death progresses, cell dynamic activity is suppressed and low-frequency dominance vanishes (around after 4 hours), hence the is shifted toward a higher frequency considered by vesicles. In addition, reduction of cell dynamic activities during the cell death process has been demonstrated through a significant decrease in magnitude value. Two-way ANOVA test with dynamic activity parameters analyzed through our method showed that live and dead cells can be clearly distinguished by F(1,72) = 752.1, p = 0 in mean frequency and F(1,72) = 135.4, p = 0 in magnitude. Our experimental results have proved that the developed DFFOCM system can visually and quantitively monitor changes in the dynamic movement of the scatterers inside the cells.
Previous studies have used the optical interference imaging technique of a point-measurement method [22,23,38] to analyze the cell death process using optical properties. Though optical interference imaging techniques could observe movement inside cells in a non-invasive and non-destructive manner, without staining or chemical treatment, it was difficult to measure the intrinsic cellular signals due to a weak scattering and absorption property of intracellular scatterers. In addition, when scanning a sample by the point-measurement method, there exists a time delay difference in each area, making it difficult to analyze a wide area. The DFFOCM system can analyze optical properties of a sample as well as dynamic movements of scatterers inside the sample over an entire field, enabling a clear visualization and numerical analysis in real-time. In the future, we plan to apply deep learning technology to enable real-time apoptosis and necrosis analysis through repeated experiments. We observed different morphology and dynamic activity trends in cells considered apoptosis and necrosis, and we will use these characteristics to train deep learning models. Although apoptosis and necrosis can be morphologically distinguished after the cell death process has progressed, early diagnosis is expected to be more helpful for medical development. In addition, if the structure of the DFFOCM system is simplified and technologies such as fiber-optic probe endoscopes [40] are combined, it is expected to be used in combination during clinical trials and surgery, contributing to the development of medical technology.
5. Conclusion
We demonstrated a method of using DFFOCM to capture dynamic intracellular motion and analyze state changes inside cells with dynamic images and numerical values. In a DFFOCM experiment that monitored dynamic activities of HeLa cells for 24 hours, it was confirmed that when cells were alive (early in the experiment), intercellular scatterers were evenly spread throughout the interior and showed uniform movement with a low and high magnitude of dynamic movement. After that, we observed that cell blebbing occurred at the point where the skeleton of the cell was deformed (after 4 hours in the experiment), and the cell's changed sharply, while the magnitude of the dynamic movement was significantly reduced. In addition to cell death, this method can also be applied in cytology, stem cell, cancer research, and various medical fields, and is expected to replace conventional time-consuming and biopsies-required histological or biochemical methods.
Acknowledgement
We would like to thank Intramural Research Program in Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health to support us performing this study. We also thank Alicia Lillich and the NIH Library Editing Service for manuscript editing assistance.
Funding
National Institutes of Health10.13039/100000002.
Disclosures
The authors declare no conflicts of interest
Data Availability
Data underlying the results presented in this paper are not publicly available at this time but may be obtained from the authors upon request.
References
- 1.LeGrys V. A., Leinbach S. S., Silverman L. M., “Clinical applications of DNA probes in the diagnosis of genetic diseases,” Crit. Rev. Clin. Lab. Sci. 25(4), 255–274 (1987). 10.3109/10408368709105885 [DOI] [PubMed] [Google Scholar]
- 2.Meyer S. C., Signature in the Cell: DNA and the Evidence for Intelligent Design (Zondervan, 2009). [Google Scholar]
- 3.Ehrenfeucht A., Harju T., Petre I., Prescott D. M., Rozenberg G., Computation in Living Cells: Gene Assembly in Ciliates (Springer Science & Business Media, 2003). [Google Scholar]
- 4.Thompson C. B., “Apoptosis in the pathogenesis and treatment of disease,” Science 267(5203), 1456–1462 (1995). 10.1126/science.7878464 [DOI] [PubMed] [Google Scholar]
- 5.Wollert K. C., Drexler H., “Cell therapy for the treatment of coronary heart disease: a critical appraisal,” Nat. Rev. Cardiol. 7(4), 204–215 (2010). 10.1038/nrcardio.2010.1 [DOI] [PubMed] [Google Scholar]
- 6.Barata P. C., Rini B. I., “Treatment of renal cell carcinoma: current status and future directions,” CA Cancer J. Clin. 67(6), 507–524 (2017). 10.3322/caac.21411 [DOI] [PubMed] [Google Scholar]
- 7.Humpel C., “Identifying and validating biomarkers for Alzheimer's disease,” Trends Biotechnol. 29(1), 26–32 (2011). 10.1016/j.tibtech.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stylianou A., Lekka M., Stylianopoulos T., “AFM assessing of nanomechanical fingerprints for cancer early diagnosis and classification: from single cell to tissue level,” Nanoscale 10(45), 20930–20945 (2018). 10.1039/C8NR06146G [DOI] [PubMed] [Google Scholar]
- 9.Silverman S., Jr., “Early diagnosis of oral cancer,” Cancer 62(S1), 1796–1799 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Sozzi G., Musso K., Ratcliffe C., Goldstraw P., Pierotti M. A., Pastorino U., “Detection of microsatellite alterations in plasma DNA of non-small cell lung cancer patients: a prospect for early diagnosis,” Clin. Cancer Res. 5(10), 2689–2692 (1999). [PubMed] [Google Scholar]
- 11.Rimann M., Graf-Hausner U., “Synthetic 3D multicellular systems for drug development,” Curr. Opin Biotechnol. 23(5), 803–809 (2012). 10.1016/j.copbio.2012.01.011 [DOI] [PubMed] [Google Scholar]
- 12.Edmondson R., Broglie J. J., Adcock A. F., Yang L., “Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors,” Assay Drug Dev. Technol. 12(4), 207–218 (2014). 10.1089/adt.2014.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breslin S., O’Driscoll L., “Three-dimensional cell culture: the missing link in drug discovery,” Drug Discov. Today 18(5-6), 240–249 (2013). 10.1016/j.drudis.2012.10.003 [DOI] [PubMed] [Google Scholar]
- 14.Khetani S. R., Bhatia S. N., “Microscale culture of human liver cells for drug development,” Nat. Biotechnol. 26(1), 120–126 (2008). 10.1038/nbt1361 [DOI] [PubMed] [Google Scholar]
- 15.Bold R. J., Termuhlen P. M., McConkey D. J., “Apoptosis, cancer and cancer therapy,” Surg. Oncol. 6(3), 133–142 (1997). 10.1016/S0960-7404(97)00015-7 [DOI] [PubMed] [Google Scholar]
- 16.Yu J., Zhang L., “Apoptosis in human cancer cells,” Curr. Opin Oncol. 16(1), 19–24 (2004). 10.1097/00001622-200401000-00005 [DOI] [PubMed] [Google Scholar]
- 17.Shearman M. S., Ragan C. I., Iversen L. L., “Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death,” Proc. Natl. Acad. Sci. U. S. A. 91(4), 1470–1474 (1994). 10.1073/pnas.91.4.1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calvo-Rodriguez M., Hou S. S., Snyder A. C., Kharitonova E. K., Russ A. N., Das S., Fan Z., Muzikansky A., Garcia-Alloza M., Serrano-Pozo A., Hudry E., Bacskai B. J., “Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease,” Nat. Commun. 11(1), 2146 (2020). 10.1038/s41467-020-16074-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strohm E. M., Moore M. J., Kolios M. C., “High resolution ultrasound and photoacoustic imaging of single cells,” Photoacoustics 4(1), 36–42 (2016). 10.1016/j.pacs.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hundt W., O’Connell-Rodwell C. E., Bednarski M. D., Steinbach S., Guccione S., “In vitro effect of focused ultrasound or thermal stress on HSP70 expression and cell viability in three tumor cell lines,” Acad. Radiol. 14(7), 859–870 (2007). 10.1016/j.acra.2007.04.008 [DOI] [PubMed] [Google Scholar]
- 21.Lukinavicius G., Umezawa K., Olivier N., Honigmann A., Yang G., Plass T., Mueller V., Reymond L., Correa I. R., Jr., Luo Z. G., Schultz C., Lemke E. A., Heppenstall P., Eggeling C., Manley S., Johnsson K., “A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins,” Nat. Chem. 5(2), 132–139 (2013). 10.1038/nchem.1546 [DOI] [PubMed] [Google Scholar]
- 22.van der Meer F. J., Faber D. J., Aalders M. C., Poot A. A., Vermes I., van Leeuwen T. G., “Apoptosis- and necrosis-induced changes in light attenuation measured by optical coherence tomography,” Lasers Med. Sci. 25(2), 259–267 (2010). 10.1007/s10103-009-0723-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farhat G., Czarnota G. J., Kolios M. C., Yang V. X., Miriampillai A., “Detecting apoptosis using dynamic light scattering with optical coherence tomography,” J. Biomed. Opt. 16(7), 070505 (2011). 10.1117/1.3600770 [DOI] [PubMed] [Google Scholar]
- 24.Lawrence J. P., “Physics and instrumentation of ultrasound,” Crit. Care Med. 35(Suppl), S314–S322 (2007). 10.1097/01.CCM.0000270241.33075.60 [DOI] [PubMed] [Google Scholar]
- 25.Stepanenko O. V., Stepanenko O. V., Shcherbakova D. M., Kuznetsova I. M., Turoverov K. K., Verkhusha V. V., “Modern fluorescent proteins: from chromophore formation to novel intracellular applications,” BioTechniques 51(5), 313–314, 316, 318 passim (2011). 10.2144/000113765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patterson G. H., Lippincott-Schwartz J., “A photoactivatable GFP for selective photolabeling of proteins and cells,” Science 297(5588), 1873–1877 (2002). 10.1126/science.1074952 [DOI] [PubMed] [Google Scholar]
- 27.Kalyanaraman B., Darley-Usmar V., Davies K. J., Dennery P. A., Forman H. J., Grisham M. B., Mann G. E., Moore K., Roberts L. J., Ischiropoulos H., “Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations,” Free Radic. Biol. Med. 52(1), 1–6 (2012). 10.1016/j.freeradbiomed.2011.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brazelton T. R., Blau H. M., “Optimizing techniques for tracking transplanted stem cells in vivo,” Stem Cells 23(9), 1251–1265 (2005). 10.1634/stemcells.2005-0149 [DOI] [PubMed] [Google Scholar]
- 29.Liu H.-S., Jan M.-S., Chou C.-K., Chen P.-H., Ke N.-J., “Is green fluorescent protein toxic to the living cells?” Biochem. Biophys. Res. Commun. 260(3), 712–717 (1999). 10.1006/bbrc.1999.0954 [DOI] [PubMed] [Google Scholar]
- 30.Jensen E. C., “Use of fluorescent probes: their effect on cell biology and limitations,” Anat. Rec. 295(12), 2031–2036 (2012). 10.1002/ar.22602 [DOI] [PubMed] [Google Scholar]
- 31.Jensen E. C., “Types of imaging, Part 2: an overview of fluorescence microscopy,” Anat. Rec. 295(10), 1621–1627 (2012). 10.1002/ar.22548 [DOI] [PubMed] [Google Scholar]
- 32.Andreana M., Sentosa R., Erkkilä M. T., Drexler W., Unterhuber A., “Depth resolved label-free multimodal optical imaging platform to study morpho-molecular composition of tissue,” Photochem. Photobiol. Sci. 18(5), 997–1008 (2019). 10.1039/C8PP00410B [DOI] [PubMed] [Google Scholar]
- 33.Swenson E. S., Price J. G., Brazelton T., Krause D. S., “Limitations of green fluorescent protein as a cell lineage marker,” Stem Cells 25(10), 2593–2600 (2007). 10.1634/stemcells.2007-0241 [DOI] [PubMed] [Google Scholar]
- 34.Park S., Rim S., Kim Y., Lee B. H., “Noncontact photoacoustic imaging based on optical quadrature detection with a multiport interferometer,” Opt. Lett. 44(10), 2590–2593 (2019). 10.1364/OL.44.002590 [DOI] [PubMed] [Google Scholar]
- 35.Apelian C., Harms F., Thouvenin O., Boccara A. C., “Dynamic full field optical coherence tomography: subcellular metabolic contrast revealed in tissues by interferometric signals temporal analysis,” Biomed. Opt. Express 7(4), 1511–1524 (2016). 10.1364/BOE.7.001511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beaurepaire E., Boccara A. C., Lebec M., Blanchot L., Saint-Jalmes H., “Full-field optical coherence microscopy,” Opt. Lett. 23(4), 244–246 (1998). 10.1364/OL.23.000244 [DOI] [PubMed] [Google Scholar]
- 37.Rahi P. K., Mehra R., “Analysis of power spectrum estimation using welch method for various window techniques,” Int. J. Emerging Technol. and Eng. 2, 106–109 (2014). [Google Scholar]
- 38.Oldenburg A. L., Yu X., Gilliss T., Alabi O., Taylor R. M., 2nd, Troester M. A., “Inverse-power-law behavior of cellular motility reveals stromal-epithelial cell interactions in 3D co-culture by OCT fluctuation spectroscopy,” Optica 2(10), 877–885 (2015). 10.1364/OPTICA.2.000877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu X., Hu C., Zhang W., Zhou J., Ding Q., Sadiq M. T., Fan Z., Yuan Z., Liu L., “Feasibility evaluation of micro-optical coherence tomography (muOCT) for rapid brain tumor type and grade discriminations: muOCT images versus pathology,” BMC Med. Imaging 19(1), 102 (2019). 10.1186/s12880-019-0405-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park S., Rim S., Kim J. W., Park J., Sohn I. B., Lee B. H., “Analysis of design and fabrication parameters for lensed optical fibers as pertinent probes for sensing and imaging,” Sensors 18(12), 4150 (2018). 10.3390/s18124150 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data underlying the results presented in this paper are not publicly available at this time but may be obtained from the authors upon request.