

Abstract
Introduction
The promise of the natural immunoregulator, Galectin-1 (Gal1), as an immunomodulatory therapeutic is challenged by its unstable homodimeric conformation. Previously, a Gal1 homodimer stabilized via covalent poly(ethylene glycol) diacrylate (PEGDA) cross-linking demonstrated higher activity relative to the non-covalent homodimer.
Methods
Here, we report Gal1 homodimers formed using an alternative thiol-Michael addition linker chemistry.
Results
Poly(ethylene glycol) bismaleimide (PEGbisMal) reacted with Gal1 at multiple sites with greater efficiency than PEGDA. However, multiple PEGbisMal molecules were conjugated to Gal1 C130, a Gal1 mutant with one surface cysteine (cys-130) and two cysteines thought to be buried in the solvent-inaccessible protein core (cys-42 and cys-60). Site-directed mutagenesis demonstrated that cys-60 was the site at which additional PEGbisMal molecules were conjugated onto Gal1 C130. Compared to WT-Gal1, Gal1 C130 had low activity for inducing Jurkat T cell death, characterized by phosphatidylserine exposure and membrane permeability. PEG cross-linking could restore the function of Gal1 C130, such that at high concentrations Gal1 C130 cross-linked by PEGbisMal had higher activity than both WT-Gal1 and Gal1 C130 cross-linked by PEGDA. Mutating cys-42 and cys-60 to serines in Gal1 C130 did not affect the cell death signaling activity of the Gal1 C130 homodimer cross-linked by PEGbisMal. PEGylated Gal1 C130 variants also eliminated the need for a reducing agent, such as dithiothreitol, which is required to maintain WT-Gal1 signaling activity.
Conclusion
Collectively, these data demonstrate that thiol-Michael addition bioconjugation leads to a PEG-cross-linked Gal1 homodimer with improved extracellular signaling activity that does not require a reducing environment to be functional.
Keywords: Protein engineering, Bioconjugation, Galectin, Protein-polymer conjugate, Protein dimerization
Introduction
Galectin-1 (Gal1), the first lectin to be discovered within the galectin family, is a 14.5 kDa, carbohydrate-binding protein. Through recognition of cell membrane and extracellular matrix glycans, Gal1 can affect various cell processes, such as adhesion, migration, proliferation, and apoptosis.3 These extracellular signaling activities can drive physiological events, such as angiogenesis, metastasis, and embryo implantation.2,16,33 Gal1 is also an important regulator of innate and adaptive immunity.9 By modulating the function of innate immune cells, i.e. promoting dendritic cell migration, inhibition of neutrophil trafficking, and deactivation of macrophages, Gal1 contributes to the resolution of acute inflammation.31 Gal1 can also upregulate the secretion of anti-inflammatory cytokines (i.e. IL-10), while downregulating the secretion of T helper 1 (Th1) related pro-inflammatory cytokines (i.e. IL-2). Through interaction with the cell death machinery, Gal1 is a key regulator of Th1 and T helper 17 (Th17) survival. As a consequence of this bioactivity, Gal1 has demonstrated therapeutic potential in pre-clinical animal models for treatment of inflammatory and autoimmune pathologies mediated by Th1 and Th17 cell dysfunction, such as Crohn’s disease, multiple sclerosis, myasthenia gravis, rheumatoid arthritis, and graft-versus-host-disease.9,10,31
Despite these successes, therapeutic use of Gal1 is hindered by its structural and biochemical properties. Classified as a “prototype” galectin, Gal1 has one carbohydrate recognition domain (CRD) encoded in its primary sequence, as well as a dimerization interface. The CRD facilitates binding to β-galactosides present on extracellular matrix (ECM) glycoproteins (e.g. laminin and collagen IV), as well as various glycoprotein receptors on the cell surface (e.g. CD2, CD3, CD7, CD43, and CD45 on immune cells).3,9,10,31 While monomeric Gal1 can bind carbohydrates and plays a role in promoting axonal regeneration, Gal1 biological activity in the context of the immune system is correlated with homodimer-mediated cross-linking of cell-surface glycoproteins into lattices.20,26 However, Gal1 homodimer formation is reversible with a dissociation constant (KD) of ~ 1–7 μM.3 Thus, Gal1 lacks most of its immunomodulatory signaling activity at concentrations ≤ 1 μM.
Several protein engineering strategies have been reported that stabilize Gal1 dimerization, including fusion to an immunoglobulin Fc domain, polypeptide linkers, and the use of a leucine zipper.4,6,34 As an alternative to amino acid-based modifications, we recently reported a stable Gal1 dimer cross-linked by the synthetic polymer, poly(ethylene glycol) or “PEG”.10 PEG conjugation to proteins (i.e., “PEGylation”) is widely used to improve the pharmacokinetics and pharmacodynamics of protein drugs, as it can prevent degradation by enzymes, decrease phagocytosis by the reticuloendothelial system, and reduce renal elimination.13,17,27,29 Further, conjugation of PEG can improve protein stability, increase protein water-solubility, and decrease protein immunogenicity.13,27 Although it has recently been suggested that PEG may be immunogenic in some circumstances based on clinical evidence for anti-PEG antibodies, ~ 20 PEGylated therapeutics are approved by the FDA, with the long-term safety of PEGylation supported by hundreds of thousands of patient outcomes.13,27 Finally, PEG is easy to synthesize and purify, and a PEG-based cross-linking strategy may be less susceptible to degradation than the use of a polypeptide linker.24,34
Although a stable, PEG cross-linked Gal1 homodimer may provide advantages over previously engineered strategies, conjugation of polymer to protein presents a unique set of challenges and considerations. For example, reaction conditions must be mild enough to prevent denaturation of the target protein while maintaining maximum efficiency. The reaction must also be site-specific to prevent modification of off-target amino acids.17 The thiol-Michael-addition reaction is widely used for site-specific bioconjugation due to the thiol’s native nucleophilicity and low abundance, as free cysteine residues are not often located on the solvent accessible surface area (SASA) of proteins.17,18,28 Gal1 has 6 cysteine residues in its protein structure, 4 of which are proposed to be found on the SASA of the molecule.12,23 In our prior work, 3 of 4 surface cysteines (cys-2, cys-16, cys-88) were mutated to serine; the remaining surface cysteine (cys-130) was left intact, providing a single site for PEGylation. A thiol-Michael addition reaction between the terminal acrylate moieties of PEG diacrylate (“PEGDA”) and cys-130 residues on Gal1 yielded a covalently cross-linked homodimer with higher activity for inducing Jurkat T cell metabolic activity loss than the non-cross-linked protein.10
In the thiol-Michael addition reaction, a nucleophile reacts with an electron-deficient C=C bond acceptor under mild reaction conditions to form stable adducts containing S–C bonds. Generally, the susceptibility of the acceptor to Michael addition becomes greater with increasing electrophilicity of the C=C bond. For example, maleimides, with 2 electron-withdrawing carbonyls, are significantly more reactive than acrylates, which are more reactive than methacrylates.21,36 Due to its fast reaction kinetics and specificity for cysteine in proteins, maleimide is the most widely used thiol-Michael addition acceptor in bioconjugation.18,28 The thiol-maleimide conjugation is also beneficial because the proximal chemical linkages are more stable in physiological conditions than those of acrylates or methacrylates. For example, ester linkages that are present adjacent to a thiol-acrylate adduct in PEGDA are susceptible to hydrolysis at near-neutral pH, with a half-life of ~ 11 days.7,15 This irreversible cleavage would separate a PEG-cross-linked Gal1 dimer into a pair of monomers, thereby diminishing the activity of the construct over time in vivo. The thiosuccinimide ring formed via reaction of a thiol with a maleimide adduct is also susceptible to hydrolysis at physiological pH; however, this hydrolysis serves to stabilize the adduct rather than irreversibly cleave the linkage.11 One consequence of increased Michael acceptor reactivity of the maleimide relative to acrylate is that the thiosuccinimide linkage can be eliminated via retro-Michael reaction or exchange in the presence of excess thiol, diminishing in-vivo stability.1,11,14,30 However, recent developments in stabilizing this linkage can prevent the retro reaction from occurring. For example, the use of a differently substituted maleimide, mechanical force through mild ultrasonication, or selection of a cysteine that is less solvent-accessible for conjugation, are three biocompatible techniques that can promote the hydrolysis reaction and prevent thiol exchange, thereby stabilizing thiol-maleimide adducts in vivo.1,14,30 Thiol-acrylate linkages currently have no such stabilization potential.
We expected that replacing PEGDA with poly(ethylene glycol) bismaleimide (PEGbisMal) would increase reaction efficiency and specificity with Gal1, as well as improve the stability of PEG-cross-linked Gal1 homodimers (Fig. 1). In this study, we first characterized the reactivity of a Gal1 mutant having one surface-accessible cysteine at amino acid 130 (i.e., “Gal1 C130”) with PEGbisMal, versus previously reported PEGDA.10 We also characterized the reactivity of PEGbisMal with Gal1 mutants having a single surface-accessible cysteine at one of three different locations on the protein (cys-2, cys-16, cys-88). Unexpectedly, we observed conjugation of a second PEG molecule onto Gal1 C130, which was not observed with the other proteins. This suggested that one of the cysteines thought to be in the solvent inaccessible core of Gal1 (cys-42 or cys-60) was on the SASA of the Gal1 C130 mutant. To test this, we created new mutants in which cys-42, cys-60, or both were mutated to serine, which was chosen due to the chemical similarity of the hydroxyl and sulfhydryl. This site-directed mutagenesis demonstrated that a second PEG molecule was conjugated to cys-60. Jurkat T cell apoptosis studies indicated that PEG cross-linking restored the activity of Gal1 C130 to induce phosphatidylserine exposure and membrane permeability, and that at high concentrations PEGbisMal-cross-linked Gal1 C130 homodimers had enhanced activity compared to PEGDA-cross-linked Gal1 C130 and non-cross-linked wild-type Gal1 (WT-Gal1) protein.
Figure 1.

Schematic showing covalent cross-linking of Gal1 via (1) PEGDA or (2) PEGbisMal (not drawn to scale). Cross-linking was performed in 1x PBS at pH 7.4 via overnight incubation at 25 °C.
Materials and Methods
Cloning and Mutagenesis of Galectin-1 Variants
Methods were based from previous work.10 A gene encoding recombinant human Galectin-1 (Gal1) was obtained from Origene and inserted into a pET21d vector. Cysteine to serine mutations were made via the QuikChange Multisite Mutagenesis kit (Agilent). The following primers were used:
C2S: 5′-gagatataccatggctagtggtctggtcgccag-3′, 5′-ctggcgaccagaccactagccatggtatatctc-3′, C16S: 5′-ctcaaa-cctggagagagccttcgagtgcgag-3′, 5′-ctcgcactcgaaggctctctccaggtttgag-3′, C42S: 5′-ggttgaagtgcaggctcaggttgtt-gctgtc-3, 5′-gacagcaacaacctgagcctgcacttcaacc-3′, C60S: 5′-gccaacaccatcgtgagcaacagcaaggacg-3, 5′-cgtcct-tgctgttgctcacgatggtgttggc-3′, C88S: 5′-agtgttgcagaggtgagcatcaccttcgacc-3′, 5′-ggtcgaaggtgatgctcacctctgca-acact-3′, and C130S: 5′-tcaaaggccacacttttgatcttgaagtcaccgtca-3′, 5′-tgacggtgacttcaagatcaaaagtgtggcctttga-3′.
Mutagenesis was performed using instructions from the manufacturer. Clones were sampled and used to inoculate 5 mL 100 μg/mL ampicillin-containing LB media, for culture at 37 °C, 220 rpm in an orbital shaker. After overnight incubation, plasmids were isolated from cultures using a plasmid mini-prep kit (Qiagen) according to the manufacturer’s instructions and were sequenced by GeneWiz.
Protein Expression and Purification
Origami B (DE3) cells were transformed with pET21d(+) WT-Gal1, pET21d(+) Gal1 C2, pET21d(+) Gal1 C16, pET21d(+) Gal1 C88, pET21d(+) Gal1 C130, pET21d(+) Gal1 C130 No Internal Cysteines (“No IC”), pET21d(+) Gal1 C60 C130, and pET21d(+) Gal1 C42 C130. Clones were sampled and incubated overnight at 37 °C on 100 μg/mL ampicillin and 50 μg/mL kanamycin A-doped LB/agar plates. Positive clones were selected to inoculate 5mL 100 μg/mL ampicillin and 50 μg/mL kanamycin A-containing LB media, and grown overnight at 37 °C, 220 rpm on an orbital shaker. Clones were subcultured into 100 μg/mL ampicillin and 50 μg/mL kanamycin A-containing 2xTY media (10g/L Yeast, 16g/L Tryptone, and 5g/L NaCl), and grown at 37 °C, 220 rpm on an orbital shaker, until an optical density at 600 nm = 0.6–0.8 was reached. To induce protein expression, cultures were treated with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), and incubated for 18 h at 18 °C, 220 rpm in an orbital shaker. Bacteria were pelleted by centrifugation, culture media was decanted, and bacteria were washed/resuspended in 1x phosphate buffered saline (PBS). Bacteria were lysed with B-PER (Thermo Fischer), ½ protease inhibitor tablet (Thermo Fischer), 300 units DNase I from bovine pancreas (Sigma), and 100 μg lysozyme (Sigma) for 20 min. Lysed bacteria were cleared by centrifugation, and supernatant lysate containing recombinant proteins was loaded onto columns containing α-lactose:Agarose (Thermo Fisher) equilibrated with PBS. Columns were washed with 5–10 column volumes of PBS and bound Gal1 was eluted with 100 mM β-lactose in PBS. β-Lactose was removed via centrifugation with Amicon filter tubes (MWCO 10 kDa) (Millipore). Protein MW and purity were analyzed using SDS-PAGE, and protein concentrations were determined using a bicinchoninic acid assay (Thermo Fischer) calibrated to a bovine serum albumin standard curve. After sterilization by passing through a 0.22 μM syringe filter, all mutant proteins were stored in 1x PBS.
PEGylation of Galectin-1 Variants
PEGDA (MW = 2 kDa) (Sigma) was added to Gal1 C2, Gal1 C16, Gal1 C88, and Gal1 C130 at a 3 or 25-fold molar excess. PEGbisMal (MW = 3.4 kDa) was added to Gal1 C2, Gal1 C16, Gal1 C88, Gal1 C130, Gal1 C130 no IC, Gal1 C42 C130, and Gal1 C60 C130 at a 3-fold molar excess in 1x phosphate buffered saline (PBS). These solutions were incubated under stirring conditions overnight at room temperature. Unreacted PEGDA and PEGbisMal were separated from the sample using Amicon filter tubes (MWCO 10 kDa) (Millipore). MW and purity were analyzed using SDS-PAGE. Extent of reaction was analyzed indirectly based on free thiol content using Ellman’s reagent (DNTB) (Fisher). The assay was performed according to manufacturer instructions and scaled for use in a 96-well plate. Briefly, a working dilution of Ellman’s reagent was prepared by adding 200 μL reaction buffer to 4 μL Ellman’s reagent. Reaction buffer (10 mL) and Ellman’s reagent solution (200 μL) were mixed, and 204 μL from this mixture, in addition to 20 μL of protein sample, were incubated in 96-well plate for 15 min at room temperature, and then absorbances were measured at 412 nm using a SpectraMax M3 plate reader (Molecular Devices).
Fourier Transform Infrared Spectroscopy (FTIR)
The FTIR spectra were recorded on a Frontier FTIR spectrophotometer (PerkinElmer) equipped with a universal ATR sampling accessory. 3 mg of lyophilized protein powder was spotted onto the ATR accessory. All the samples were blanked to lyophilized PBS powder and scanned 64 times with the average of the spectra reported.
Lactose Affinity Chromatography
An α-lactose-agarose resin (MilliporeSigma) was packed into a TricornTM 5/50 Column (GE Healthcare), according to manufacturer’s instructions. 500 µl of each protein solution was prepared at 30 µM into a leur-lock syringe. Protein was loaded onto the α-lactose-agarose affinity column connected to an ÄKTA pure FPLC system (GE Healthcare). Unbound protein was washed with 10 column volumes of PBS. Bound protein was eluted using a gradient of 0–100 mM soluble β-d-lactose (ThermoFisher) in PBS. Eluted protein was measured at absorbance 280 nm and results were normalized based on the maximum signal intensity.
Flow Cytometry
Methods were based on previous work.8,26 WT-Gal1, Gal1 C130, Gal1 C130 PEGbisMal, Gal1 C130 PEGDA, Gal1 C130 no IC, and Gal1 C130 no IC PEGbisMal were all sterilized via passage through a 0.22 μm syringe filter. Jurkat T-cells were expanded in complete media (RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 1% penicillin–streptomycin, l-glutamine 2 mM, 10 mM HEPES buffer) at 37 °C, 5% CO2. Cells were aliquoted at 100 μL into sterile 12 × 75 mm flow cytometry tubes at 2,000,000 cells/mL (200,000 cells/tube). Gal1 protein variants and buffer controls (PBS or 1 mM DTT in PBS) were added at a 1:1 volume ratio (100 μL protein or buffer control : 100 μL cells) at n = 3 per group. Cells were then incubated for 24 hours at 37 °C, 5% CO2. Protein concentrations used in this experiment refer to the molar concentrations of individual Gal1 molecules, with [PEGylated Gal1] = ½ * [total Gal1]. Positive single stain controls for Annexin-V and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit (Invitrogen) were produced by treating cells with either 250 ng/mL anti-Fas antibody (clone CH11, MilliporeSigma) or 1 μM camptothecin (MilliporeSigma) and allowed to incubate for 24 hours at 37 °C, 5% CO2. After incubation, half the volume of the camptothecin controls were heated at 56 °C for 5 min, followed by a 5 min incubation on ice. The cells were recombined with the non-heat-treated cells. Further, 1 mL of ice-cold PBS with 100 mM β-lactose was added to all tubes. Tubes were centrifuged at 500 x g for 5 min at 4 °C. The supernatant was removed, and cells were washed with 900 µL of ice-cold PBS at 500 x g for 5 min at 4 °C. 100 μL of 1x Annexin V Binding Buffer (BD Biosciences) was added to each tube along with 5 μL BV421 Annexin V (BD Biosciences) to stain for phosphatidylserine exposure, and 1 μL LIVE/DEAD™ Fixable Near-IR Dead Cell Stain (Invitrogen), to stain for membrane permeability, except in the case of single stain controls where only one dye was added, respectively. The tubes were gently swirled and incubated for 15 min in the absence of light. 200 μL of 1x Annexin V Binding Buffer (BD Biosciences) were added to each tube and the tubes were placed on ice for flow cytometric analysis. Data was acquired on a BD FACSCelestaTM flow cytometer, and data were analyzed and graphed as scatter plots using BD FlowJoTM software. Cell death was calculated as follows: [(% annexin V − and LIVE/DEAD® − cells in untreated group) − (% annexin V − and LIVE/DEAD® − cells in treated group)]/(% annexin V − and LIVE/DEAD® − cells in untreated group). * 100. Values less than zero were reported as zero.
Results and Discussion
Comparing Thiol-Acrylate vs. Thiol-Maleimide Reaction in PEG Cross-Linking of Gal1 Homodimers
To compare the relative efficiencies of the thiol-maleimide and thiol-acrylate reactions, two different conjugations were carried out. In previous work, reacting a 25-fold excess of PEGDA with Gal1 having one surface-exposed cysteine at amino acid 130 (“Gal1 C130”), resulted in efficient cross-linking of Gal1 monomers into a stable homodimer.10 We hypothesized that due to the increased reactivity of surface thiol with maleimide relative to acrylate, a lower excess of PEGbisMal to protein would drive the reaction to completion.21 No unreacted Gal1 C130 was detected with SDS-PAGE when the protein was reacted with a 3x excess of PEGbisMal, whereas a band corresponding to Gal1 C130 was observed when the protein was reacted with the same molar excess of PEGDA (Fig. 2a). The major band in the lane corresponding to the product of the reaction of Gal1 C130 with PEGDA migrated to a distance equivalent to a molecular weight (MW) of approximately 30 kDa, which was consistent with the MW expected for a Gal1 homodimer cross-linked by a 2 kDa PEGDA molecule. The major band in the lane corresponding to the product of the reaction of Gal1 C130 with PEGbisMal migrated to a distance equivalent to a MW of approximately 40 kDa, which was ~ 6 kDa larger than that expected for a Gal1 homodimer cross-linked by a 3.4 kDa PEGbisMal molecule. Further analysis of the SDS-PAGE gel identified a second band at a higher MW than what was predicted for the Gal1 homodimer cross-linked by PEGDA (~ 35 kDa), consistent with a prior report.10 Likewise, smearing was observed over a higher MW range than what was predicted for a homodimer cross-linked by PEGbisMal, which could be an artifact due to the flexibility and hydrophobicity of PEG.25,37 Alternatively, this smearing could indicate formation of constructs with more than 2 Gal1 domains. This will be returned to later in the paper. Ellman’s assay indicated that 97.1 ± 4.37% of available cysteines were consumed when Gal1 C130 was reacted with a 3x excess of PEGbisMal, whereas only 78.6 ± 8.08% of available cysteines were consumed following reaction with PEGDA. Collectively, these data demonstrate that maleimide has greater reactivity for the surface thiol on Gal1 C130 than acrylate.
Figure 2.
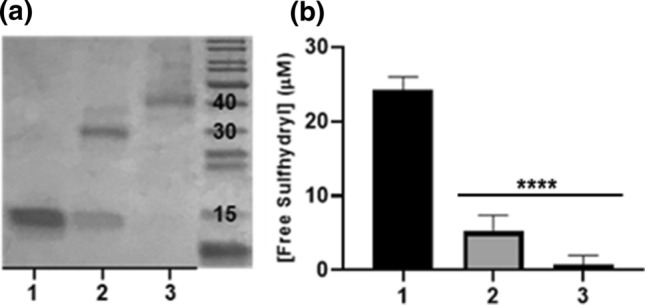
PEGDA and PEGbisMal cross-link Gal1 via thiol-Michael addition with different reaction efficiencies. (a) SDS-PAGE gel scan of (1) unreacted Gal1 C130 (control) and the products of the reaction of (2) Gal1 C130 with 3-fold molar excess PEGDA or (3) Gal1 C130 with 3-fold molar excess PEGbisMal after 25 °C overnight reaction in 1x PBS (pH 7.4). (b) Plot of unreacted thiol, determined using the Ellman’s assay, of (1) unreacted Gal1 C130 (control) and the products of the 25 °C overnight reaction in 1x PBS (pH 7.4) of (2) Gal1 C130 with 3-fold molar excess PEGDA or (3) Gal1 C130 with 3-fold molar excess PEGbisMal.
To further verify the conjugation of the PEGbisMal linker onto Gal1 C130, FTIR was performed on lyophilized powders of both Gal1 C130 and Gal1 C130 PEGbisMal after removal of unreacted PEGbisMal via centrifugal filtration (Fig. 3). A peak at 1450 cm−1 corresponding to the imide unit of the thiosuccinimide moiety of PEGbisMal was present in the Gal1 C130 PEGbisMal spectrum and absent in the Gal1 C130 spectrum. Additionally, there were broad peaks over the range of 1000-1150 cm−1 corresponding to the ether stretch of PEGbisMal in the Gal1 C130 PEGbisMal spectrum that were absent in the Gal1 C130 spectrum. These data along with the PAGE gel and Ellman’s assay from Fig. 2, together demonstrate the conjugation of PEGbisMal onto Gal1 C130 via thiol-Michael addition reaction chemistry.
Figure 3.

PEGbisMal is incorporated into the cross-linked Gal1 C130 construct. (a) FTIR spectra of Gal1 C130 (black line) and Gal1 C130 PEGbisMal (red line). (b) FTIR spectra of the amide bond stretch. (c) FTIR spectra of the imide and ether bond stretches. Gal1 C130 was reacted with PEGbisMal overnight at 25 °C in 1x PBS (pH 7.4), after which unreacted PEGbisMal was removed by centrifugal filtration.
Evaluating Site-Specificity of Cross-Linking
Cys-130 was initially chosen as the PEGylation site due to its proximity to the native dimerization domain of Gal1. We evaluated the reactivity of each of the 3 additional surface-exposed cysteine residues on native Gal1 by generating a set of variants with different cysteine-to-serine mutations: C16S/C88S/C130S (“Gal1 C2”), C2S/C88S/C130S (“Gal1 C16”), and C2S/C16S/C130S (“Gal1 C88”) (Fig. 4a). The Ellman’s reagent assay indicated that the reaction efficiency of Gal1 and PEGbisMal was similar across all point mutants (Fig. 4b). Additionally, no unreacted protein was detected with SDS-PAGE after reacting Gal1 C2, Gal1 C16, and Gal1 C88 with a 3-fold excess of PEGbisMal (Fig. 4c). In contrast, reacting Gal1 C2 with a 25-fold molar excess of PEGDA yielded some homodimer product, whereas even less homodimer product was detected after reacting Gal1 C16 or Gal1 C88 with a 25-fold molar excess of PEGDA (data not shown).
Figure 4.

Gal1 can be cross-linked via PEGbisMal at different surface cysteines, although the products differ with regard to stoichiometry. (a) Schematic showing the 4 cysteine-to-serine Gal1 mutants “Gal 1 C2” (C16S/C88S/C130S), “Gal1 C16” (C2S/C88S/C130S), “Gal1 C88” (C2S/C16S/C130S), and “Gal1 C130” (C2S/C16S/C88S). Dark grey residues indicate cysteine-to-serine mutations while light grey residues indicate location of the surface cysteine. (b) Plot of measured reaction efficiency between Gal1 C2 + PEGbisMal, Gal1 C16 + PEGbisMal, Gal1 C88 + PEGbisMal, or Gal1 130 + PEGbisMal, determined via Ellman’s reagent, after overnight reaction at 25 °C in 1x PBS (pH 7.4). (c) SDS-PAGE gel scan of the product of the reaction of Gal1 C2 + PEGbisMal, Gal1 C16 + PEGbisMal, Gal1 C88 + PEGbisMal, or Gal1 130 + PEGbisMal after overnight reaction at 25 °C in 1x PBS (pH 7.4). (d) Plot of free sulfhydryl of Gal1 C2, Gal1 C16, Gal1 C88, and Gal1 C130 in the absence of PEGbisMal (“-PEGbisMal) in 1x PBS (pH 7.4), detected with Ellman’s reagent (n = 5). **** indicates p < 0.001, ANOVA with Tukey’s post-hoc.
Evaluating the SDS-PAGE gel migration patterns, we noted that the products of the reaction of PEGbisMal with Gal1 C2, Gal1 C16, or Gal1 C88 migrated to a distance equivalent to a MW of ~ 35 kDa, which was consistent with the expected MW of two Gal1 molecules cross-linked by one 3.4 kDa PEGbisMal molecule. In contrast, the major band for the product of the reaction of Gal1 C130 with PEGbisMal migrated to a distance equivalent to a MW of ~ 40 kDa, which was ~ 6 kDa higher than the MW predicted for a homodimer of Gal1 C130 cross-linked by one 3.4 kDa PEGbisMal molecule. This higher-than-expected MW was also observed when the reaction of PEGbisMal with Gal1 C130 was compared to the reaction of PEGDA with Gal1 C130 (Fig. 2), demonstrating that this shift was reproducible and likely a property inherent to Gal1 C130. This molecular weight shift suggested that a second or third 3.4 kDa PEGbisMal molecule were conjugated onto the Gal1 C130 variant, but not Gal1 C2, Gal 1 C16, or Gal1 C88. We hypothesized that one or both of the “internal” Gal1 cysteine residues thought to be in the hydrophobic core of the wild-type protein, cys-42 and cys-60, may be more solvent-accessible on the Gal1 C130 mutant relative to the other variants.12 To test this hypothesis, Ellman’s assay was used to quantify the amount of free thiol on each of the Gal1 point mutants. These data indicated that Gal1 C130 had more available free thiol than an equivalent concentration of Gal1 C2, Gal1 C16, and Gal1 C88 (Fig. 4d), demonstrating that indeed one of the “internal cysteine” residues is on the SASA of Gal1 C130.
To determine whether cys-42, cys-60, or both were accessible on the surface of Gal1 C130, we characterized the reaction of PEGbisMal with new points mutants, Gal1 C2S/C16S/C42S/C88S (“Gal1 C60 C130”), Gal1 C2S/C16S/C60S/C88S (“Gal1 C42 C130”), and Gal1 C2S/C16S/C42S/C60S/C88S (“Gal1 C130 No IC”) (Fig. 5a). No unreacted protein was detected after reacting Gal1 C60 C130, Gal1 C42 C130, Gal1 C130 No IC, or Gal1 C130 with a 3-fold excess of PEGbisMal (Fig. 5b). One major band, which migrated to a distance equivalent to a MW of ~ 35 kDa, was identified in each of the lanes with the product of the reaction of Gal1 C42 C130 or Gal1 C130 No IC with PEGbisMal. This migration pattern was consistent with the expected molecular weight of two Gal1 molecules cross-linked by one 3.4 kDa PEGbisMal molecule. In contrast, three bands, at MW of ~ 35, 40, and 50 kDa, were observed in the lane for the product of the reaction of Gal1 C60 C130 with PEGbisMal. We estimate that the ~ 40 kDa band corresponds to a construct consisting of two Gal1 molecules coupled to two PEGbisMal molecules, while the ~ 50 kDa band corresponds to a construct consisting of two or three Gal1 molecules coupled to two or three PEGbisMal molecules (we note that PEG conjugation can perturb protein migration during SDS-PAGE).25,37 Likewise, a distinct band at ~ 40 kDa, as well as smearing at higher MWs, was again observed in the lane for the product of the reaction of Gal1 C130 with PEGbisMal. Collectively, these data suggested that cys-60 becomes surface accessible on Gal1 when cys-2, cys-16, and cys-88 are mutated to serine, which leads to conjugation of two or more PEG molecules onto each Gal1 homodimer (Fig. 5c).
Figure 5.
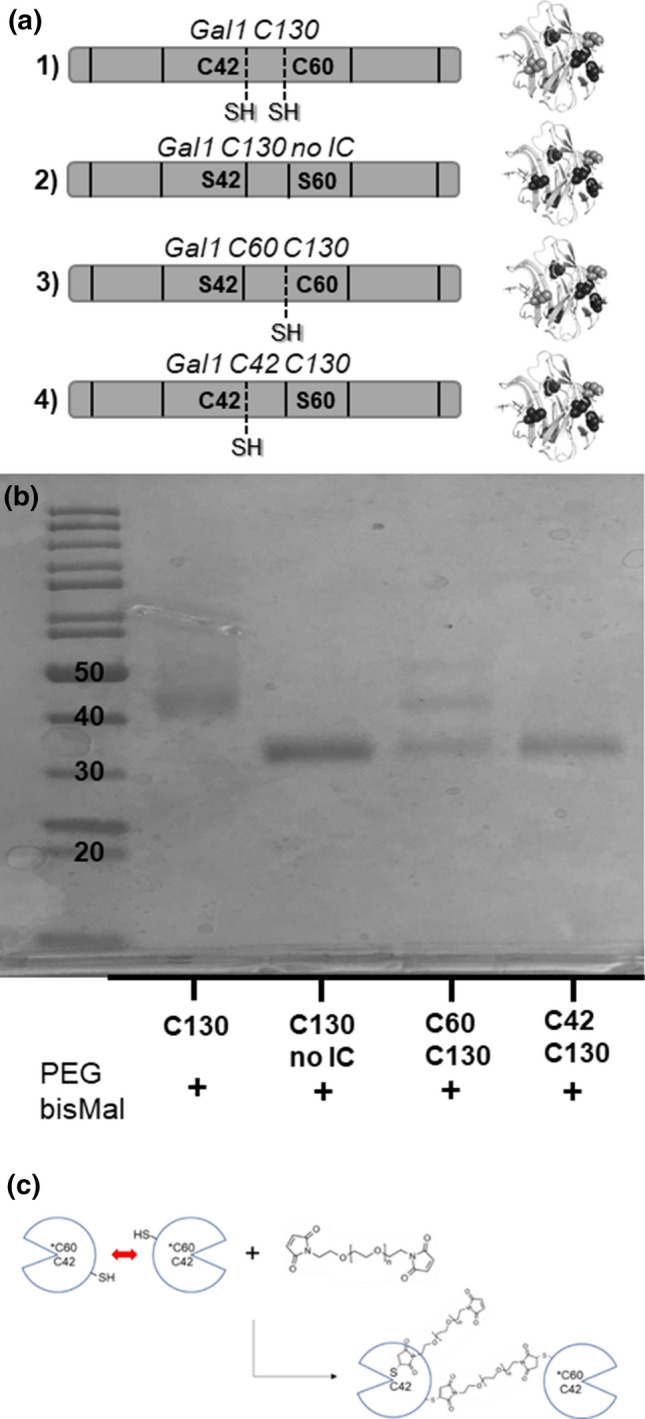
Internal cysteines are the sites for alternative product formation via PEGbisMal reaction with Gal1 C130. (a) Schematic outlining 3 new cysteine-serine variants, “C130 no IC” (C2S/C16S/C42S/C60S/C88S), “Gal1 C60 C130” (C2S/C16S/C42S/C88S), and “Gal1 C42 C130” (C2S/C16S/C60S/C88S). Dark grey residues indicate cysteine-to-serine mutation while light grey residues indicate locations of present surface and internal cysteines. (b) Scan of the SDS-PAGE gel of the products of reaction of Gal1 C130 + PEGbisMal, C130 No IC + PEGbisMal, Gal1 C60 C130 + PEGbisMal, and Gal1 C42 C130 + PEGbisMal after overnight reaction at 25 °C in 1x PBS (pH 7.4). (c) Schematic detailing thiol-maleimide side-reaction at C60 (not drawn to scale).
Together, these observations demonstrate an important advantage of the thiol-maleimide chemistry over thiol-acrylate chemistry for PEGylation of Gal1. Namely, the high reactivity of PEGbisMal with each of the native cysteine residues on the SASA affords selectivity in the choice of cross-linking site, which may be advantageous for optimizing Gal1 homodimer activity. Furthermore, the desired product, with a 2:1 protein to polymer ratio and site-specific conjugation, can be prepared in high yield, under mild conditions with only a slight molar excess of polymer, using commercially available reagents. This contrasts with other instances of successful polymer-mediated protein homodimerization, in which lengthy, in-house polymeric syntheses are often required to achieve the desired product.19,32,35
Comparing Bioactivity of the PEG-Cross-Linked Gal1 C130 Variants
WT-Gal1 can induce death of Jurkat T cells,26 characterized by phosphatidylserine exposure and membrane permeability, in a concentration dependent manner over the range of 0.5–10 μM, which is near the monomer-dimer dissociation constant. However, this signaling activity requires the presence of a reducing agent, such as dithiothreitol, to prevent oxidative inactivation of WT-Gal1.26 Here we compared the activity of Gal1 C130 and the PEGylated variants relative to WT-Gal1 using phosphatidylserine exposure and membrane permeability as markers of cell death (Fig. 6). The cell-death inducing activity of Gal1 C130 was drastically reduced compared to WT-Gal1 (Fig. 6a). Likewise, the PEGylated Gal1 C130 variants had weaker activity than WT-Gal1 at a galectin concentration of 0.5 μM (Fig. 6a). However, at a galectin concentration of 5 μM, the PEGylated Gal1 C130 variants had higher activity than WT-Gal1, irrespective of the conjugation chemistry (Fig. 6a). At a galectin concentration of 10 μM, PEGbisMal cross-linked Gal1 C130 had significantly higher activity than both WT-Gal1 and Gal1 C130 cross-linked by PEGDA (Fig. 6a). At a galectin concentration of 10 μM, WT-Gal1, PEGbisMal cross-linked Gal1 C130, and PEGDA cross-linked Gal1 C130 demonstrated similar preference for inducing phosphatidylserine exposure (Annexin V+), associated with early apoptosis, and the combination of phosphatidylserine exposure plus membrane permeability (LIVE/DEAD® +), associated with late apoptosis, with late apoptosis being the dominant phenotype (Fig. 6b). Notably, the cell death dose responses observed here contrasted with our prior report which demonstrated that Gal1 C130 and the PEGDA cross-linked variant can decrease metabolic activity of Jurkat T cells, with the PEGylated variant having a 10-fold lower effective dose.10 Collectively, these observations suggest that the signaling threshold and/or receptors required for Gal1 to signal Jurkat T cell metabolic activity loss may differ relative to the receptors and or signaling threshold required to induce phosphatidylserine exposure and membrane permeability.
Figure 6.

PEG cross-linking increases the cell death signaling activity of Gal1 C130 (a) Percentage of Jurkat T cells positive for phosphatidylserine and membrane permeability (i.e., “dead cells”) when treated with 10 μM, 5 μM or 0.5 μM WT-Gal1, Gal1 C130, Gal1 C130 + PEGDA, or Gal1 C130 + PEGbisMal. (b) Flow cytometry scatter plots of Jurkat T cells treated with 10 μM WT-Gal1, 10 μM Gal1 C130, 10 μM Gal1 C130 + PEGDA, or 10 μM Gal1 C130 + PEGbisMal. Values refer to molar concentrations of individual Gal1 molecules. * indicates significance relative to Gal1 C130, ^ indicates significance relative to WT-Gal1, # indicates significance relative to Gal1 C130 + PEGDA, and & indicates significance relative to Gal1 C130 + PEGbisMal. **, ***, **** indicate p < 0.01, p < 0.001, or p < 0.0001, respectively, ANOVA with Tukey’s post-hoc.
A plausible explanation for the difference in activity of WT-Gal1, Gal1 C130, and the PEG cross-linked variants could be the relative binding affinity of each protein for β-galactosides. Competitive-binding immobilized lactose-affinity chromatography demonstrated that Gal1 C130 and Gal1 C130 cross-linked by PEGbisMal had comparable binding affinity (Fig. 7), which was consistent with previously reported lactose-binding affinities for WT-Gal1 and other cysteine-to-serine point mutants.10 These observations suggested collectively that the increased signaling activity of the PEG-cross-linked homodimer, as well as the diminished activity of Gal1 C130 relative to WT-Gal1, were not due to differences in glycan-binding affinity.
Figure 7.

Competitive inhibition lactose-affinity chromatography to compare the binding properties of Gal1 C130 and Gal1 C130 + PEGbisMal. Gal1 C130 was reacted with PEGbisMal overnight at 25 °C in 1x PBS (pH 7.4).
To determine if conjugation of additional PEG chains onto Gal1 C130 affected its biological activity, we used the Jurkat cell death assay to compare the activity of Gal1 C130 cross-linked by PEGbisMal to Gal1 C130 no IC cross-linked by PEGbisMal. Both Gal1 C130 cross-linked with PEGbisMal and Gal1 No IC C130 cross-linked with PEGbisMal induced T cell death characterized by phosphatidylserine exposure and membrane permeability (Fig. 8a). These PEGylated Gal1 homodimers induced a similar extent of T cell death as a function of galectin concentration (Fig. 8b). Together, these data demonstrate that mutating Gal1 C130 to vary the extent of PEGylation does not affect the biological activity of the homodimer.
Figure 8.

“Internal” cysteine-to-serine mutations do not affect the cell death signaling activity of PEG cross-linked Gal1 C130. (a) Flow cytometry scatterplots of Jurkat T cells treated with 3.75 μM Gal1 C130 + PEGbisMal or 3.75 μM Gal1 C130 no IC + PEGbisMal. (b) Percentage of dead Jurkat T cells when treated with 0.5, 2.5 or 3.75 μM Gal1 C130 + PEGbisMal or Gal1 C130 no IC + PEGbisMal.
Conclusions
Cross-linking of Gal1 into a covalent homodimer via PEGDA through thiol-acrylate Michael addition has been shown in prior work to decrease its effective dose in vitro.10 Here, we replaced the thiol-acrylate chemistry with thiol-maleimide chemistry, and demonstrated that the increased reactivity of maleimide relative to acrylate increased reaction efficiency. Reaction between PEGbisMal and Gal1 monomers formed homodimers at a roughly 10-fold lower polymer molar excess than that required for equivalent cross-linking via PEGDA. Gal1 homodimers could be formed via PEGbisMAL reaction with any of the 4 cysteine residues on the SASA to a comparable extent; however, SDS-PAGE gels suggested that an additional PEG molecule was conjugated onto the Gal1 C130 variant. Additional point mutants suggested that cys-60, which was thought be buried in the solvent-inaccessible protein interior, was accessible on the Gal1 C130 mutant. Recent NMR studies demonstrate that WT-Gal1 can undergo a change in conformation upon binding to carbohydrate ligands, such as lactose.5,22 For the Gal1 C130 mutant, we propose that there are two possible ways by which cys-60 can become solvent accessible. On one hand, mutating cys-2, cys-16, and cys-88 to serine may change the tertiary conformation of Gal1 C130 relative to WT-Gal1, leading to exposure of cys-60 on the protein surface. On the other hand, grafting PEG onto cys-130 may induce a change in the tertiary conformation of Gal1 C130 that leads to exposure of cys-60 on the protein surface. Future studies will seek to understand how the structure of Gal1 C130 differs relative to that of WT-Gal1. Nonetheless, mutating cys-60 to serine resulted in a Gal1 homodimer with the desired 2:1 protein-to-polymer stoichiometry. Introducing cysteine-to-serine point mutations reduced the activity of Gal1 C130 to induce phosphatidylserine exposure and membrane permeability, two markers of apoptosis; however, PEGbisMal cross-linking of Gal1 C130 led to a construct with greater potency than WT-Gal1, and mutating cys-60 and cys-42 to serine to ensure site-specific grafting at cys-130 did not diminish cross-linked Gal1 C130 homodimer activity. Collectively, these observations demonstrate that a Gal1 variant having only a single cysteine residue at the 130 position is a favorable candidate for homodimer synthesis, as it demonstrates high reaction efficiency with PEGbisMal, affords site-specificity of polymer conjugation, provides a product with an apparent Gal1:PEG stoichiometry of 2:1, and has enhanced activity relative to the unmodified protein. We envision the PEGbisMal cross-linked Gal1 homodimer reported here may lead to an immunotherapeutic with more reliable and reproducible efficacy than native Gal1 or other polymer-cross-linked variants reported previously.
Acknowledgments
This research was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under University of Florida Clinical and Translational Science Award TL1TR001428, the National Science Foundation grant DMR-1455201, and start-up funds from the University of Florida. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, nor the National Science Foundation.
Conflict of interest
Bryant J. Kane, Margaret M. Fettis, Shaheen A. Farhadi, Renjie Liu declare that they have no conflicts of interest. Gregory A. Hudalla is a founder of Anchor Biologics, Inc.
Ethical standards
No human studies were carried out by the authors for this article. No animal studies were carried out by the authors for this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Baldwin AD, Kiick KL. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjug. Chem. 2011;22:1946–1953. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrientos G, Freitag N, Tirado-González I, Unverdorben L, Jeschke U, Thijssen VLJL, Blois SM. Involvement of galectin-1 in reproduction: past, present and future. Hum. Reprod. Update. 2014;20:175–193. doi: 10.1093/humupd/dmt040. [DOI] [PubMed] [Google Scholar]
- 3.Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–157R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- 4.Cedeno-Laurent F, Barthel SR, Opperman MJ, Lee DM, Clark RA, Dimitroff CJ. Development of a nascent galectin-1 chimeric molecule for studying the role of leukocyte galectin-1 ligands and immune disease modulation. J. Immunol. 2010;185:4659–4672. doi: 10.4049/jimmunol.1000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chien C-TH, Ho M-R, Lin C-H, Hsu S-TD. Lactose binding induces opposing dynamics changes in human galectins revealed by NMR-based hydrogen–deuterium exchange. Molecules. 2017;22(8):1357. doi: 10.3390/molecules22081357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Earl LA, Bi S, Baum LG. Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology. 2011;21:6–12. doi: 10.1093/glycob/cwq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elbert DL, Hubbell JA. Conjugate addition reactions combined with free-radical cross-linking for the design of materials for tissue engineering. Biomacromolecules. 2001;2:430–441. doi: 10.1021/bm0056299. [DOI] [PubMed] [Google Scholar]
- 8.Farhadi SA, Fettis MM, Liu R, Hudalla GA. A synthetic tetramer of galectin-1 and galectin-3 amplifies pro-apoptotic signaling by integrating the activity of both galectins. Front. Chem. 2020;7:898. doi: 10.3389/fchem.2019.00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farhadi SA, Hudalla GA. Engineering galectin–glycan interactions for immunotherapy and immunomodulation. Exp. Biol. Med. 2016;241:1074–1083. doi: 10.1177/1535370216650055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fettis MM, Hudalla GA. Engineering reactive oxygen species-resistant galectin-1 dimers with enhanced lectin activity. Bioconjug. Chem. 2018;29:2489–2496. doi: 10.1021/acs.bioconjchem.8b00425. [DOI] [PubMed] [Google Scholar]
- 11.Fontaine SD, Reid R, Robinson L, Ashley GW, Santi DV. Long-term stabilization of maleimide–thiol conjugates. Bioconjug. Chem. 2015;26:145–152. doi: 10.1021/bc5005262. [DOI] [PubMed] [Google Scholar]
- 12.Guardia CM, Caramelo JJ, Trujillo M, Méndez-Huergo SP, Radi R, Estrin DA, Rabinovich GA. Structural basis of redox-dependent modulation of galectin-1 dynamics and function. Glycobiology. 2014;24:428–441. doi: 10.1093/glycob/cwu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong L, Wang Z, Wei X, Shi J, Li C. Antibodies against polyethylene glycol in human blood: a literature review. J. Pharmacol. Toxicol. Methods. 2020;102:106678. doi: 10.1016/j.vascn.2020.106678. [DOI] [PubMed] [Google Scholar]
- 14.Huang W, Wu X, Gao X, Yu Y, Lei H, Zhu Z, Shi Y, Chen Y, Qin M, Wang W, Cao Y. Maleimide–thiol adducts stabilized through stretching. Nat. Chem. 2019;11:310–319. doi: 10.1038/s41557-018-0209-2. [DOI] [PubMed] [Google Scholar]
- 15.Hudalla GA, Eng TS, Murphy WL. An approach to modulate degradation and mesenchymal stem cell behavior in poly(ethylene glycol) networks. Biomacromolecules. 2008;9:842–849. doi: 10.1021/bm701179s. [DOI] [PubMed] [Google Scholar]
- 16.Ito K, Stannard K, Gabutero E, Clark AM, Neo S-Y, Onturk S, Blanchard H, Ralph SJ. Galectin-1 as a potent target for cancer therapy: role in the tumor microenvironment. Cancer Metastasis Rev. 2012;31:763–778. doi: 10.1007/s10555-012-9388-2. [DOI] [PubMed] [Google Scholar]
- 17.Ko JH, Maynard HD. A guide to maximizing the therapeutic potential of protein–polymer conjugates by rational design. Chem. Soc. Rev. 2018;47:8998–9014. doi: 10.1039/C8CS00606G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koniev O, Wagner A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015;44:5495–5551. doi: 10.1039/C5CS00048C. [DOI] [PubMed] [Google Scholar]
- 19.Lorenzo MM, Decker CG, Kahveci MU, Paluck SJ, Maynard HD. Homodimeric protein-polymer conjugates via the tetrazine-trans-cyclooctene ligation. Macromolecules. 2016;49:30–37. doi: 10.1021/acs.macromol.5b02323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miura T, Takahashi M, Horie H, Kurushima H, Tsuchimoto D, Sakumi K, Nakabeppu Y. Galectin-1β, a natural monomeric form of galectin-1 lacking its six amino-terminal residues promotes axonal regeneration but not cell death. Cell Death Differ. 2004;11:1076–1083. doi: 10.1038/sj.cdd.4401462. [DOI] [PubMed] [Google Scholar]
- 21.Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN. The thiol-michael addition click reaction: a powerful and widely used tool in materials chemistry. Chem. Mater. 2014;26:724–744. doi: 10.1021/cm402180t. [DOI] [Google Scholar]
- 22.Nesmelova IV, Ermakova E, Daragan VA, Pang M, Menéndez M, Lagartera L, Solís D, Baum LG, Mayo KH. Lactose binding to galectin-1 modulates structural dynamics, increases conformational entropy, and occurs with apparent negative cooperativity. J Mol Biol. 2010;397:1209–1230. doi: 10.1016/j.jmb.2010.02.033. [DOI] [PubMed] [Google Scholar]
- 23.Nishi N, Abe A, Iwaki J, Yoshida H, Itoh A, Shoji H, Kamitori S, Hirabayashi J, Nakamura T. Functional and structural bases of a cysteine-less mutant as a long-lasting substitute for galectin-1. Glycobiology. 2008;18:1065–1073. doi: 10.1093/glycob/cwn089. [DOI] [PubMed] [Google Scholar]
- 24.Nishi N, Itoh A, Fujiyama A, Yoshida N, Araya S, Hirashima M, Shoji H, Nakamura T. Development of highly stable galectins: truncation of the linker peptide confers protease-resistance on tandem-repeat type galectins. FEBS Lett. 2005;579:2058–2064. doi: 10.1016/j.febslet.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 25.Odom OW, Kudlicki W, Kramer G, Hardesty B. An effect of polyethylene glycol 8000 on protein mobility in sodium dodecyl sulfate–polyacrylamide gel electrophoresis and a method for eliminating this effect. Anal. Biochem. 1997;245:249–252. doi: 10.1006/abio.1996.9993. [DOI] [PubMed] [Google Scholar]
- 26.Pace KE, Hahn HP, Baum LG. Preparation of recombinant human galectin-1 and use in T cell death assays. Methods Enzymol. 2003;363:499–518. doi: 10.1016/S0076-6879(03)01075-9. [DOI] [PubMed] [Google Scholar]
- 27.Pelegri-O’Day EM, Lin E-W, Maynard HD. Therapeutic protein–polymer conjugates: advancing beyond PEGylation. J. Am. Chem. Soc. 2014;136:14323–14332. doi: 10.1021/ja504390x. [DOI] [PubMed] [Google Scholar]
- 28.Ravasco JMJM, Faustino H, Trindade A, Gois PMP. Bioconjugation with maleimides: a useful tool for chemical biology. Chem. Eur. J. 2019;25:43–59. doi: 10.1002/chem.201803174. [DOI] [PubMed] [Google Scholar]
- 29.Schellekens H, Hennink WE, Brinks V. The immunogenicity of polyethylene glycol: facts and fiction. Pharm. Res. 2013;30:1729–1734. doi: 10.1007/s11095-013-1067-7. [DOI] [PubMed] [Google Scholar]
- 30.Shen B-Q, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu S-F, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012;30:184–189. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 31.Sundblad V, Morosi LG, Geffner JR, Rabinovich GA. Galectin-1: a jack-of-all-trades in the resolution of acute and chronic inflammation. J. Immunol. 2017;199:3721–3730. doi: 10.4049/jimmunol.1701172. [DOI] [PubMed] [Google Scholar]
- 32.Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HD. Synthetic approach to homodimeric protein-polymer conjugates. Chem. Commun. 2009;16:2148–2150. doi: 10.1039/b822799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thijssen VL, Griffioen AW. Galectin-1 and -9 in angiogenesis: a sweet couple. Glycobiology. 2014;24:915–920. doi: 10.1093/glycob/cwu048. [DOI] [PubMed] [Google Scholar]
- 34.van der Leij J, van den Berg A, Harms G, Eschbach H, Vos H, Zwiers P, van Weeghel R, Groen H, Poppema S, Visser L. Strongly enhanced IL-10 production using stable galectin-1 homodimers. Mol. Immunol. 2007;44:506–513. doi: 10.1016/j.molimm.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 35.White CJ, Bode JW. PEGylation and dimerization of expressed proteins under near equimolar conditions with potassium 2-pyridyl acyltrifluoroborates. ACS Cent. Sci. 2018;4:197–206. doi: 10.1021/acscentsci.7b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Chakma P, Shulman MP, Ke J, Digby ZA, Konkolewicz D. Probing the mechanism of thermally driven thiol-michael dynamic covalent chemistry. Org. Biomol. Chem. 2001;16:2725–2734. doi: 10.1039/C8OB00397A. [DOI] [PubMed] [Google Scholar]
- 37.Zheng CY, Ma G, Su Z. Native PAGE eliminates the problem of PEG–SDS interaction in SDS-PAGE and provides an alternative to HPLC in characterization of protein PEGylation. Electrophoresis. 2007;28:2801–2807. doi: 10.1002/elps.200600807. [DOI] [PubMed] [Google Scholar]