

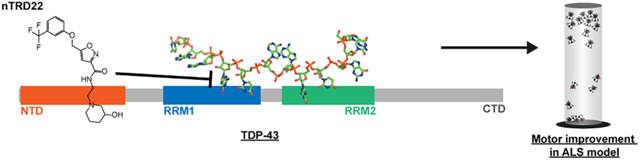
Abstract
In this study, we targeted the N-terminal domain (NTD) of transactive response (TAR) DNA binding protein (TDP-43), which is implicated in several neurodegenerative diseases. In silico docking of 50K compounds to the NTD domain of TDP-43 identified a small molecule (nTRD22) that is bound to the N-terminal domain. Interestingly, nTRD22 caused allosteric modulation of the RNA binding domain (RRM) of TDP-43, resulting in decreased binding to RNA in vitro. Moreover, incubation of primary motor neurons with nTRD22 induced a reduction of TDP-43 protein levels, similar to TDP-43 RNA binding-deficient mutants and supporting a disruption of TDP-43 binding to RNA. Finally, nTRD22 mitigated motor impairment in a Drosophila model of amyotrophic lateral sclerosis. Our findings provide an exciting way of allosteric modulation of the RNA-binding region of TDP-43 through the N-terminal domain.
Graphical Abstract
Transactive response (TAR) DNA binding Protein-43 (TDP-43), which is a critical factor in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD), is involved in almost all aspects of RNA metabolism.1,2 TDP-43 consists of an N-terminal domain (NTD), two RNA recognition motifs (RRM1 and RRM2), and an unstructured glycine-rich domain.
The NTD is responsible for TDP-43 subcellular localization, since it contains a nuclear localization signal (NLS)3-6 and one mitochondrial targeting sequence (M1).7 TDP-43 NTD has been shown to form dimers that can assemble into reversible higher-order oligomers, required for splicing activity8-11 and contributing to liquid–liquid phase separation.8,11 Several studies have hinted at the ability of TDP-43 NTD to serve as a scaffold for nucleic acid binding and to contribute to specificity toward certain nucleic acid sequences,12-14 although a nucleotide binding interface of the NTD remains to be determined.
In a previous study,15 we targeted the RNA-binding domain (RRM1) of TDP-43 and found a compound, rTRD01, that was bound to TDP-43, partially disrupted RNA binding, and improved locomotor defects in a Drosophila model of ALS. The current study is distinct because it targets the N-terminal domain and identifies an indirect effect on the RNA-binding domain. Based on the potential role of TDP-43 NTD in nucleic acid binding, as well as protein aggregation, we sought to target the NTD with small molecules using in silico docking. Neither X-ray structures (PDB 5mdi8) nor NMR structures (PDB 2n4p12) of TDP-43 reveal any obvious surface pocket(s) or large cleft(s) capable of accommodating small molecules. The program SiteMap15,16 was used to determine if residues directly implicated in dimerization or residues nearby could form a druggable pocket for binding of small molecules. A druggable pocket was identified by Sitemap on the NMR structure, pointing to the involvement of residues Ser48, Ala66, and Asn70 (Supplementary Figure 1A in the Supporting Information). A slightly different site was found on the X-ray structure and surrounded the following residues: Tyr43, Leu56, and Asp65 (see Supplementary Figure 1B in the Supporting Information).
A 50 000-compound library was then docked onto a grid of 6 Å surrounding the Sitemap pockets, using Glide’s virtual screening workflow. Even though the initial pockets were different on the NMR and crystal structure, compounds were docked around Ser48, Ala66, and Asn70 in both structures (Figure 1). A total of 20 compounds were chosen based on score and other energy-related terms: nTRD09–nTRD18 for the X-ray structure and nTRD19–nTRD28 for the NMR structure (Supplementary Table 1 in the Supporting Information). Visual inspection of the docking poses was implemented to remove unrealistic poses as they might result in high scoring.16
Figure 1.

Docking of small molecules on N-terminal domain of TDP-43. Top 10 compounds (green, sticks and balls representation) from in silico docking on TDP-43-NTD: (A) NMR structure (PDB ID: 2n4p12) and (B) crystal structure (PDB ID: 5mdi8).
Saturation transfer difference nuclear magnetic resonance (STD-NMR) spectroscopy was then used to screen the binding of the compounds to TDP-431–260. As a negative control, binding to TDP43102–269, a construct lacking the NTD, was also determined (Supplementary Figure 2A in the Supporting Information). We observed nTRD12–nTRD14 and nTRD22, nTRD25–nTRD28 as positive hits on TDP-431–260 that did not bind to TDP43102–269 (see Supplementary Figures 2B-2D in the Supporting Information).
The 2D 15N-HSQC NMR spectrum of each positive hit was determined to further define the binding sites of the small molecule interactions with TDP-43 (Supplementary Figure 3 in the Supporting Information). Using TDP-431–260, small molecules were added at a 4:1 molar ratio. The addition of 5 out of 6 of the compounds either caused protein aggregation or showed little to no binding, possibly because of weak binding, and were not further characterized. However, the addition of nTRD022 (Figures 2A and 2B) caused visible chemical shift perturbations but no protein aggregation and, hence, was chosen for further study.
Figure 2.

Biophysical characterization of nTRD22. (A) Two-dimensional (2D) representation of nTRD22 binding pocket in TDP-43 NTD obtained from Glide docking. nTRD22 is predicted to make (i) hydrogen bonds (shown in pink) with Glu3, Tyr4, Gly69, and Ala66, (ii) Π–Π stacking (shown in green) with Tyr43, (iii) a salt bridge (shown in blue) with Glu3, and (iv) a Π-cation interaction (shown in red) with Tyr4. (B) Structure of N-(2-(3-hydroxypiperidin-1-yl)ethyl)-5-((3-(trifluoromethyl)phenoxy)methyl)isoxazole-3-carboxamide (nTRD22). (C) MST values from thermographs of NT-647-labeled TDP431–260 in the presence of increasing concentrations (from 0.03 μM to 1 mM) of nTRD22 were used to determine dissociation constant for binding of nTRD22 to TDP431–260. Apparent Kd = 145 ± 3 μM. Data are presented as mean ± SD (n = 3). (D) Superposition of 1H–15N heteronuclear single quantum correlation spectroscopy (HSQC) spectra of 15N-labeled human TDP431–260 (100 μM), free (blue) and in complex with nTRD22 with different ratios. (E–H) Close-up of shifts around TDP43 residues from TDP431–260 Cys173 (panel (E)) and Gly148 and Gly110 (panel (F)) or from TDP43102–269 Cys173 (panel (G)) and Gly148 and Gly110 (panel (H)). (I) Chemical shift changes for assigned residues of the 15N-labeled TDP-431–260 (RRM portion only) upon complex formation with nTRD22. The average chemical shift changes of cross-peaks were calculated as described in the Methods section in the Supporting Information. The horizontal lines are thresholds (calculated as previously described17) above which a shift is considered significant with 2σ (back line). nTRD22 is able to induce shifts of residues localized in the ribonucleotide interacting motifs (RNP) of RRM1 and RRM2. (J) The chemical shift difference observed on TDP431–260 were mapped on TDP43102–269 (PDB ID: 4bs217). (K) A concentration-dependent curve was obtained for nTRD22’s disruption of nucleic acid-TDP-431–260 interaction at a single RNA concentration (0.6 nM). Data are represented as mean ± SEM (n = 3). (L) Representative SPR sensograms showing the signal for binding of 1 nM TDP-431–260 and immobilized (UG)6 RNA in the presence of nTRD22 dissolved in DMSO (0.1–500 μM). 150 RU of (UG)6 RNA is immobilized on a SA chip. (M) A concentration-dependent curve was obtained for nTRD22’s disruption of nucleic acid-TDP-431–260 interaction at a single RNA concentration showing IC50 = 124 μM. Data are represented as mean ± SEM (n = 3). Some error bars are smaller than the symbols.
In silico docking of nTRD22 predicted interactions with Glu3, Tyr43, Ser48, Gly59, and Ala66 (see Figure 2A). 1H NMR of nTRD22 was used to evaluate the on-resonance energy transfer from STD-NMR of nTRD22 with TDP-431–260. The peaks seen in the STD-NMR spectrum were in the region of the benzyl group ((ii), 6.9–7.2 ppm) (see Figure 2C), confirming the Π-stacking interaction of the benzene group of nTRD22 with Tyr43 that was predicted by virtual screening. Microscale thermophoresis (MST) measured an apparent Kd value of 145 ± 3 μM (Figure 2C) for TDP-431–260 and 96 ± 36 μM for TDP-431–102 (NTD only) (Supplementary Figure 3 in the Supporting Information).
To further characterize where nTRD22 binds on TDP-43, we measured perturbations using a 2D 15N-HSQC NMR with increasing concentrations of nTRD22, up to an 8:1 molar ratio of nTRD22:TDP-431–260 (Figures 2D-J). We observed chemical shifts in the 2D 15N-HSQC of TDP-431–260, the majority of them occurring in the RRM region of the protein. To determine if these shifts were due to direct binding of nTRD22 to TDP-43102–269 (RRM region), we collected a 2D 15HSQC-NMR on TDP43102–269 with nTRD22 at a 4:1 molar ratio (see Figures 2G and 2H). We observed no shifts in the spectra including the residues seen for the TDP-431–260 titration. nTRD22 was synthesized in-house for further experiments (see the Methods section in the Supporting Information) and was submitted to 1H NMR and mass spectrometry for validation (see the supplementary figures provided in the Supporting Information).
To define which residues in the RRM domains were affected, we mapped the chemical-shift perturbations (CSPs) induced by nTRD22 onto the known structure of TDP-43102–269 (PDB: 4bs217) using a color gradient (Figures 2I and 2J). Interestingly, in addition to peak broadening, several of the shifted residues are involved in RNA binding. More precisely, Ile107 is part of the ribonucleotide interacting motif 1 and Lys145 is close to the ribonucleotide interacting motif 2, highly conserved short sequences known as RNP-1 (octameric sequence: KGFGFVRF in RRM1 and RAFAFVTF in RRM2) and RNP-2 (hexameric sequence: LIVLGL in RRM1 and VFVGRC in RRM2). Arg227 is part of RNP-1 in RRM2.18 Moreover, Cys173 is also shown to be affected in the presence of RNA.17 Replicates of this experiment showed a similar profile of residues affected by nTRD22 (Supplementary Figure 5 in the Supporting Information). Although the shifts are small, RNP-2 remains consistently shifted, even in different CSPs (Supplementary Figure 5); in addition, since RNP sequences are highly conserved sequence motifs on TDP-43 required for nucleic acid recognition, CSP data suggests that nTRD22 might indirectly modulate TDP-43 RNA binding. To test this, we used an amplified luminescent proximity homogeneous (alpha) assay recently developed for another compound targeting TDP-4315. TDP-431–260 binding to its canonical RNA sequence (UG6) was measured with varying concentrations of compound and nTRD22 was able to inhibit 50% of the interaction with an IC50 value of ~100 μM (Figure 2K). We also used surface plasmon resonance (SPR) to measure disruption of TDP-43-RNA binding (Figures 2L and 2M) and calculated an IC50 value of ~145 μM and ~30% inhibition of TDP43-RNA interaction.
TDP-43 NTD can be responsible for dimer formation of the protein.11 To test the effect of nTRD22 on TDP-43 dimerization, we immobilized NTD (TDP-431–102) on a CM5 chip and injection of TDP-431–260 (500 nM) resulted in binding (56 RU), because of self-interaction between TDP-43 NTD and TDP-431–260 (Supplementary Figure 6A in the Supporting Information). The experiment was repeated in the presence of 200 μM nTRD22 or DMSO. Based on quantification of the response signals, there was no significant effect of nTRD22 on the interaction of TDP-43 NTD and TDP-431–260 (Supplementary Figure 6B in the Supporting Information). Thus, nTRD22 did not affect NTD dimerization.
Previous studies indicated that disruption of TDP-43 RNA binding in neuronal cells via the expression of RNA binding-deficient TDP-43 elicits the formation of intranuclear droplet-like structures and higher TDP-43 protein turnover.19 Most importantly, the authors showed neuroprotective effects of an RNA binding-deficient form of TDP-43. Because nTRD22 is able to reduce TDP-43 binding to RNA, we hypothesized that nTRD22 may be able to recapitulate some of the effects described with RNA binding-deficient TDP-43. To test this, we expressed TDP-43 fused to enhanced green fluorescent protein (TDP-43-EGFP) in rodent primary cortical neurons, applied nTRD22 at concentrations ranging from 5 μM to 100 μM, and imaged GFP positive cells by automated fluorescence microscopy.19,20 As expected, control cells incubated with dimethylsulfoxide (DMSO) exhibited a diffuse GFP signal, mainly in the nucleus, consistent with normal TDP-43 localization (Figure 3A). We observed the formation of intranuclear TDP-43-EGFP droplet-like structures in nTRD22 treated neurons, similar to those formed by an RNA binding-deficient variant TDP-43-EGFP19 (see Figures 3B and 3C, as well as Supplementary Figures 7A-7C in the Supporting Information). This was accompanied by a dose-dependent reduction in TDP-43-EGFP steady-state levels in treated neurons (Figure 3D). No change in the fluorescence of control cells transfected with EGFP was observed (Figure 3E). At higher doses, we noted toxicity upon application of nTRD22 to primary neurons (Supplementary Figure 7D in the Supporting Information), potentially because of interference with the endogenous TDP-43 function, other essential RNA binding proteins, or off-target effects. Consistent with the disruption experiments demonstrating the effects of nTRD22 on the RNA binding property of TDP-43 (Figures 2K-M), these data suggest that nTRD22 likely elicits TDP-43 phase separation and TDP-43 degradation by blocking RNA binding.
Figure 3.

nTRD22 effect on primary cortical neurons. Confocal images of rodent primary cortical neurons with transfected TDP43-EGFP with (A) DMSO or (B) 100 μM nTRD22, or (C) transfected with TDP-43-FL (RNA binding-deficient mutant). Quantitation of GFP signal in TDP-43-EGFP (panel (D)) or EGFP (panel (E)) transfected primary neurons treated with increasing concentration of nTRD22 or DMSO. Data are presented as mean ± SEM (n ≥ 183 neurons, from eight technical and three biological replicates). Statistical difference was assessed by using a Kruskal–Wallis test [legend: (***) p < 0.001; (**) p = 0.01].
We also tested nTRD22 in a Drosophila line overexpressing human TDP-43, which is a well-known model of ALS.15 This model was shown to recapitulate important neuropathological and clinical features of the human proteinopathy, including TDP-43 aggregate formation, neuronal loss in an age-dependent manner, motor defects and reduced survival.21 Moreover, because of the advantages endowed by its small size, a short generation time, rapid propagation, and relatively small costs associated with stock maintenance, Drosophila melanogaster is an excellent animal model system to test compounds.
We used the climbing assay, which is a behavior that measures motor strength and coordination in adult flies. Briefly, when flies are placed on a vial, their innate behavior is to attempt to climb to the top of the vial, which is a behavior called negative geotaxis. A sensitized version of the negative geotaxis assay that allows for earlier detection of milder defects over time was used according to ref 22 (see Figure 4A).
Figure 4.

nTRD22 mitigates motor defects in a Drosophila model of ALS overexpressing TDP-43. (A) Principle of the sensitized version of the negative geotaxis assay.22 Flies are transferred without anesthesia to a wax-sealed glass graduated cylinder. Flies are tapped to the bottom, and their subsequent climbing activity is quantified for 2 min. The number of flies crossing the target line (red) at each time point chosen (every 10 s) is recorded. (B) Fly food supplemented with nTRD22 (50 μM) resulted in increased motor performance, compared to naïve or DMSO-treated flies (n = 60, (****) p < 0.0001, two-way ANOVA).
Flies expressing TDP-43 in motor neurons using the GAL4-UAS bipartite expression system exhibited locomotor defects in negative geotaxis (Figure 4 and Supplementary Figure 8 in the Supporting Information). nTRD22 was significantly able to rescue climbing defects compared to DMSO-treated or naive flies (Figure 4B). This rescue effect was increased in aged flies (see Supplementary Figure 8).
This study illustrates that the ability of nTRD22 to modulate TDP-43 RNA binding in vitro has neuroprotective properties in a Drosophila model of ALS. However, the exact mechanism by which the compound modulates TDP-43 binding to RNA is still unclear. We hypothesized that nTRD22 could influence residues important in RNA binding and allosterically modulate the RRM domain by (i) disrupting the binding of NTD to the RRM1 domain, leading to a modification of an open/closed conformation suggested in ref 23 or (ii) modifying the orientation of RRM1 and RRM2 toward each other, as previously discussed,19 or (iii) stabilizing a RNA-free form of TDP-43. The first hypothesis was tested by measuring chemical shift perturbations using 2D 15N-HSQC NMR. The NTD portion of TDP-43, residues 1–102, was added to TDP-43102–269 in a NTD: TDP-43102–269 molar ratio of 8:1. Data showed few significant perturbations related to NTD interacting with RRM domain (Supplementary Figure 9 in the Supporting Information). Hence, we do not think there is significant interaction between TDP-43 NTD and the RRM. Unless the loop region between NTD and the RRM domains influence binding, as this was not included in this experiment. Therefore, all hypotheses remain open to further investigation.
The compound nTRD22 offers new opportunities as a tool to further study allostery within TDP-43 and validate reduction of RNA binding by chemical modulation as a possible neuroprotective avenue. Furthermore, nTRD22, by its unique property of allosteric modulation, makes it possible to target TDP-43 with potentially less off-targeting of RRM domains, which are present in other RNA-binding proteins.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from University of Arizona-sponsored training grant, T32 grant from the National Institute of Health (No. GM008804) and the National Institutes of Health (No. 1R01AG063409-01) to M. Khanna. We appreciate access to instrumentation at the UCLA-DOE NMR facility granted by R. Clubb.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00494.
Supplementary Table 1 (XLSX)
Description of the methods, spectral data, and all supplementary figures (PDF)
The authors declare no competing financial interest.
Contributor Information
Niloufar Mollasalehi, Department of Pharmacology, College of Medicine and Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States.
Liberty Francois-Moutal, Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States.
David D. Scott, Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States
Judith A. Tello, Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States
Haley Williams, Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States.
Brendan Mahoney, Department of Chemistry and Biochemistry, University of California, Los Angeles (UCLA), Los Angeles, California 90095, United States.
Jacob M. Carlson, Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States
Yue Dong, Arizona Center for Drug Discovery, College of Pharmacy and Pharmacology and Toxicology Department, College of Pharmacy, University of Arizona, Tucson, Arizona 85721, United States.
Xingli Li, Department of Neurology, University of Michigan Health System, Ann Arbor, Michigan 48109, United States.
Victor G. Miranda, Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States
Vijay Gokhale, Bio5 Institute, University of Arizona, Tucson, Arizona 85721, United States.
Wei Wang, Arizona Center for Drug Discovery, College of Pharmacy and Pharmacology and Toxicology Department, College of Pharmacy, University of Arizona, Tucson, Arizona 85721, United States.
Sami J. Barmada, Department of Neurology, University of Michigan Health System, Ann Arbor, Michigan 48109, United States
May Khanna, Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona 85724, United States; Center of Innovation in Brain Science, Tucson, Arizona 85721, United States.
REFERENCES
- (1).Lagier-Tourenne C, Polymenidou M, and Cleveland DW (2010) TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet 19, R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ratti A, and Buratti E (2016) Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem 138, 95–111. [DOI] [PubMed] [Google Scholar]
- (3).Pinarbasi ES, Cağatay T, Fung HYJ, Li YC, Chook YM, and Thomas PJ (2018) Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci. Rep 8, 7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, and Lee VMY (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem 283, 13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo JM, Hortobágyi T, Shaw CE, and Rogelj B (2010) Nuclear import impairment causes cytoplasmic transactivation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 133, 1763–1771. [DOI] [PubMed] [Google Scholar]
- (6).Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EMH, Li X, Miguez R, Dayton RD, Tamir S, Klein RL, and Barmada SJ (2018) TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep 8, 4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, Jiang S, Ma X, Jiang Z, Da Rocha EL, Sheng M, Choi H, Lerou PH, Li H, and Wang X (2016) The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med 22, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Afroz T, Hock EM, Ernst P, Foglieni C, Jambeau M, Gilhespy LAB, Laferriere F, Maniecka Z, Plückthun A, Mittl P, Paganetti P, Allain FHT, and Polymenidou M (2017) Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun 8, 45 DOI: 10.1038/s41467-017-00062-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jiang LL, Xue W, Hong JY, Zhang JT, Li MJ, Yu SN, He JH, and Hu HY (2017) The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep 7, 6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mompeán M, Romano V, Pantoja-Uceda D, Stuani C, Baralle FE, Buratti E, and Laurents DV (2017) Point mutations in the N-terminal domain of transactive response DNA-binding protein 43 kDa (TDP-43) compromise its stability, dimerization, and functions. J. Biol. Chem 292, 11992–12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang A, Conicella AE, Schmidt HB, Martin EW, Rhoads SN, Reeb AN, Nourse A, Ramirez Montero D, Ryan VH, Rohatgi R, Shewmaker F, Naik MT, Mittag T, Ayala YM, and Fawzi NL (2018) A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 37, No. e97452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mompeán M, Romano V, Pantoja-Uceda D, Stuani C, Baralle FE, Buratti E, and Laurents DV (2016) The TDP-43 N-terminal domain structure at high resolution. FEBS J. 283, 1242–1260. [DOI] [PubMed] [Google Scholar]
- (13).Qin H, Lim LZ, Wei Y, and Song J (2014) TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. U. S. A 111, 18619–18624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chang C. ke, Wu TH, Wu CY, Chiang M.-h., Toh EKW, Hsu YC, Lin KF, Liao Y.-h., Huang T.-h., and Huang JJ-T (2012) The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem. Biophys. Res. Commun 425, 219–224. [DOI] [PubMed] [Google Scholar]
- (15).François-Moutal L, Felemban R, Scott DD, Sayegh MR, Miranda VG, Perez-Miller S, Khanna R, Gokhale V, Zarnescu DC, and Khanna M (2019) Small Molecule Targeting TDP-43’s RNA Recognition Motifs Reduces Locomotor Defects in a Drosophila Model of Amyotrophic Lateral Sclerosis (ALS). ACS Chem. Biol 14, 2006–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lionta E, Spyrou G, Vassilatis D, and Cournia Z (2014) Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem 14, 1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, and Allain FHT (2013) Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol 20, 1443–1449. [DOI] [PubMed] [Google Scholar]
- (18).Kuo PH, Chiang CH, Wang YT, Doudeva LG, and Yuan HS (2014) The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res. 42, 4712–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Flores BN, Li X, Malik AM, Martinez J, Beg AA, and Barmada SJ (2019) An Intramolecular Salt Bridge Linking TDP43 RNA Binding, Protein Stability, and TDP43-Dependent Neurodegeneration. Cell Rep. 27, 1133–1150.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Weskamp K, Safren N, Miguez R, and Barmada S (2019) Monitoring neuronal survival via longitudinal fluorescence microscopy. J. Visualized Exp 2019, DOI: 10.3791/59036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, Woodruff EA, Fushimi K, and Wu JY (2010) A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U. S. A 107, 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Madabattula ST, Strautman JC, Bysice AM, O’Sullivan JA, Androschuk A, Rosenfelt C, Doucet K, Rouleau G, and Bolduc F (2015) Quantitative analysis of climbing defects in a drosophila model of neurodegenerative disorders. J. Visualized Exp 2015, DOI: 10.3791/52741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wei Y, Lim L, Wang L, and Song J (2016) Inter-domain interactions of TDP-43 as decoded by NMR. Biochem. Biophys. Res. Commun 473, 614–619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.