

Abstract
Clandestine chemists are currently exploiting the pyrrolidinophenone scaffold to develop new designer drugs that carry the risk of abuse and overdose. These drugs promote addiction through the rewarding effects of increased dopaminergic neurotransmission. 3,4-Methylenedioxypyrovalerone (MDPV) and its analogs are illicit psychostimulants of this class that are ~50-fold more potent than cocaine at inhibiting the human dopamine transporter (hDAT). In contrast, MDPV is a weak inhibitor at both the human serotonin transporter (hSERT) and, as it is shown here, the Drosophila melanogaster DAT (dDAT). We studied three conserved residues between hSERT and dDAT that are unique in hDAT (A117, F318, and P323 in dDAT), and one residue that is different in all three transporters (D121 in dDAT). hDAT residues were replaced in the dDAT sequence at these positions using site-directed mutagenesis and stable cell lines were generated expressing these mutant transporters. The potencies of MDPV and two of its analogs were determined using a Ca2+-mobilization assay. In this assay, voltage-gated Ca2+ channels are expressed to sense the membrane electrical depolarization evoked when dopamine is transported through DAT. Each individual mutant slightly improved MDPV’s potency, but the combination of all four increased its potency ~100-fold (2 log units) in inhibiting dDAT activity. Molecular modeling and docking studies were conducted to explore the possible mode of interaction between MDPV and DAT in silico. Two of the studied residues (F318 and P323) are at the entrance of the S1 binding site, whereas the other two (A117 and D121) face the aryl moiety of MDPV when bound to this site. Therefore, these four non-conserved residues can influence MDPV selectivity not only by stabilizing binding, but also by controlling access to its binding site at DAT.
Keywords: hDAT, dDAT, drugs of abuse, structure, monoamine transporters, calcium channels, flakka, bath salts, 3D molecular modeling, α-PVP, MDPPP, synthetic cathinones
Introduction
3,4-Methylenedioxypyrovalerone (MDPV) and a number of related synthetic cathinones are potent inhibitors of the dopamine transporter (DAT) (Baumann et al., 2013; Cameron et al., 2013b; Eshleman et al., 2013; Kolanos et al., 2013; Marusich et al., 2014; Meltzer et al., 2006; Simmler et al., 2013). Accordingly, these compounds inhibit dopamine (DA) reuptake, thereby augmenting levels of this neurotransmitter in the nucleus accumbens and promoting behavioral hallmarks of abused psychostimulants in experimental animals (Bonano et al., 2014; Schindler et al., 2016). As a component of “bath salts” preparations that were sold as “legal highs” in the US and Europe, MDPV has been abused by humans (De Felice et al., 2014; Marinetti and Antonides, 2013; Shanks et al., 2012). Accordingly, the United States Drug Enforcement Administration (DEA) has added MDPV and a few of its analogs including α-pyrrolidinovalerophenone (α-PVP, illegally sold as “flakka”) to the list of U.S. Schedule I substances (federal register: 76 FR 65371 and 82 FR 12171, respectively). New variants of the pyrrolidinophenone scaffold repeatedly emerge in clandestine markets. For example, a new designer drug, 4F-3-methyl-α-PVP, is the latest reported by the National Forensic Laboratory Information System (NFLIS) of the DEA. MDPV is ~50-fold more potent than cocaine in blocking DA uptake through DAT (Baumann et al., 2013; Cameron et al., 2013b; Steele and Eltit, 2019). Furthermore, MDPV presents a strong preference for DAT showing little potency to inhibit the serotonin transporter (SERT) (Baumann et al., 2013). Experiments performed in vitro indicate that the inhibitory action of MDPV at DAT persevere longer than that of cocaine (Cameron et al., 2013a; Cameron et al., 2013b). Consequently, abuse-related effects of MDPV are long-lasting in vivo, a property attributable in part to its slow dissociation kinetics from DAT (Bonano et al., 2014) but differential pharmacokinetic properties between these drugs is an additional factor to consider.
Structural studies of monoamine transporters (MAT) have offered new and powerful insights on ligand-MAT interactions at the atomic level and, although the Drosophila melanogaster DAT (dDAT) and human SERT (hSERT) have been resolved (Coleman et al., 2016; Penmatsa et al., 2013; Wang et al., 2013; Wang et al., 2015), the 3D structure of the human DAT (hDAT) is still outstanding. While these reports have identified a central binding site, the S1 site, the underpinnings of how the structural features of these proteins drive selectivity for certain agents remains less clear. In this work, unique structural features were identified that shape the potent MDPV inhibition at hDAT. To accomplish this, dDAT was used as a reference transporter. dDAT displays a curious pharmacology; dopamine has similar affinity at hDAT and dDAT, but other ligands potent at hDAT, such as amphetamine and cocaine, are much weaker at dDAT (Porzgen et al., 2001). Moreover, dDAT also displays affinity for other reuptake inhibitors that target hSERT but not hDAT (Porzgen et al., 2001). Here we show that MDPV and its analogs are only marginally active at dDAT. Taking an approach akin to previous chimeric monoamine transporter studies (Andersen et al., 2010; Andersen et al., 2015), experiments were designed to identify key non-conserved residues that influence MDPV’s high potency/affinity at hDAT. First, amino acid residues were identified that line the S1 site and that were conserved between hSERT and dDAT (but unique in hDAT). Then, they were mutated to the corresponding hDAT residue in the dDAT template. Permanent cell lines were created expressing these mutant dDAT constructs, and “gain-of-function” experiments were performed evaluating the potency of MDPV and two MDPV analogues (α-PVP and 3,4-methylenedioxy-α-pyrrolidinopropiophenone [MDPPP]) in inhibiting the function of these transporters. α-PVP retains all but the methylenedioxy substituent of MDPV, whereas MDPPP retains this substituent but possesses a truncated α-side chain.
The substrate activity of DA and the inhibitory action of these agents at hDAT, wild-type (wt) dDAT and dDAT mutants were assayed using a previously validated MAT-Ca2+ assay (Battisti et al., 2018; Davies et al., 2020; Ruchala et al., 2021; Solis et al., 2017; Steele and Eltit, 2019). Importantly, dopamine potency in generating Ca2+ signals was comparable on these constructs, suggesting that while the S1-lining residues studied here seemingly modulate selectivity for MDPV and related inhibitors at hDAT, they have limited influence on the response for the natural substrate dopamine.
The simultaneous mutation of four residues was necessary to mimic most of MDPV’s potency at hDAT in the dDAT background. In contrast, three of these mutations were enough to improve the potency of α-PVP and MDPPP at dDAT. Homology modeling and docking studies were performed to interpret the potential effect of these mutations in the overall architecture of the S1 site at dDAT and to help understand which structural features influence the high potency of MDPV at hDAT.
Materials and Methods
Generation of new DNA constructs and production of stable cell lines.
The wt dDAT cDNA was kindly provided by Dr. Satinder Singh (Yale University). This cDNA was subcloned into the KpnI/XbaI sites of the pGEM 7Zf(+) vector to generate an intermediate plasmid. The single- and double-point mutations were generated using the QuickChangeII Mutagenesis kit (Agilent Technologies) using primers described in Table S1, the DNA amplification was performed in a miniPCR thermocycler. Positive clones were identified by sanger sequencing (Genewiz, South Plainfield, NJ) and the coding sequences of interest were subcloned into KpnI/XhoI sites of the pcDNA5/FRT/TO expression vector. The triple P323V/A117S/D121G dDAT mutant was obtained ligating the KasI/XhoI fragment of the pcDNA5/FRT/TO/P323V plasmid into the same sites of pcDNA5/FRT/TO/A117S/D121G. The quadruple P323V/F318C/A117S/D121G dDAT mutant was obtained ligating the KasI/XhoI fragment from pGEM 7Z/P323V/F318C into the same sites of pcDNA5/FRT/TO/A117S/D121G. The full transporter open reading frame (between KpnI and XhoI sites) of all final pcDNA5/FRT/TO plasmids were sequenced using sanger method (Genewiz, South Plainfield, NJ) before the generation of the stable cell lines.
The stable cell lines were generated using the Flp-In T-REx system (Thermo Fisher) as previously reported in Cameron et al. (Cameron et al., 2015). Briefly, Flp-In T-REx 293 cells were co-transfected with a pcDNA/FRT/TO plasmid coding an individual mutant and the pOG44 plasmid coding the Flp recombinase using Fugene6 as transfection reagent. After 3 days, cells were exposed to hygromycin (100 μg/ml) to start the selection process. In this system, Flp produces a targeted recombination of the gene of interest on the cell genome, in addition cells that underwent recombination earn a hygromycin resistance by restoring a first ATG codon of the resistance gene. Cells resistant to hygromycin were expanded and stored in liquid N2 for later use.
Intracellular Ca2+ determinations.
Cells were plated in a 96-well imaging plates, and then were transfected with plasmids encoding the voltage-gated Ca2+ channel CaV1.2 and the auxiliary subunits β3 and α2δ as previously described in Cameron et al (Cameron et al., 2015). The transfection mix also contained an EGFP expression plasmid used as a transfection marker. The proportion of plasmids used in the transfection was CaV1.2:β3:α2δ:EGFP=1:0.5:0.75:0.25. After 5–8 hours the media was changed, and cells were exposed to doxycycline (1 μg/ml) to upregulate the expression of the transporters. After 3 days, cells were subjected to intracellular Ca2+ determination using fluorescence microscopy. The composition of the external solution used for Ca2+ imaging was: 135 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 Hepes (in mM, pH was adjusted to 7.3 using NaOH). The day of the experiment cells were loaded with Fura-2AM for 30 min. Then cells were washed once with external solution and after an equilibration period of 20 min cells were ready for Ca2+ measurement. The equipment used consisted of an Olympus IX71 microscope equipped with an 20X Oil 0.80 NA objective and connected to an EasyRatioPro System (Horiba). The illumination of this system is monochromator-based allowing a quick exchange between 340 nm and 380 nm excitation wavelengths. The images were acquired using a sCMOS camera (pco.edge 4.2 LT) and all the system is controlled by its built-in software. The system is equipped with an automatic and pressurized perfusion system (Automate Scientific) with multiple electronic valves (controlled by an analog output) that precisely switches between solutions. In addition, recordings were performed at ~35°C using a ThermoClamp temperature control system (Automate Scientific), and the tip of the perfusion outlet was placed right above cells using a micromanipulator, thus the change in solution is instantaneous.
The experiments are designed as a two-pulse protocol. Cells first are exposed for 10 s to the external solution, followed by a 5 s exposure to DA (10 μM). After a 30 s wash, cells are exposed for 30s to the test compound alone and then for 5 s to DA (10 μM) in combination with the compound, and the protocol ends with a 30 s wash. Fig. 1A and 1B depict the timeline of the perfusion protocol used. Each well is exposed to a single concentration of the tested compound.
Figure 1.
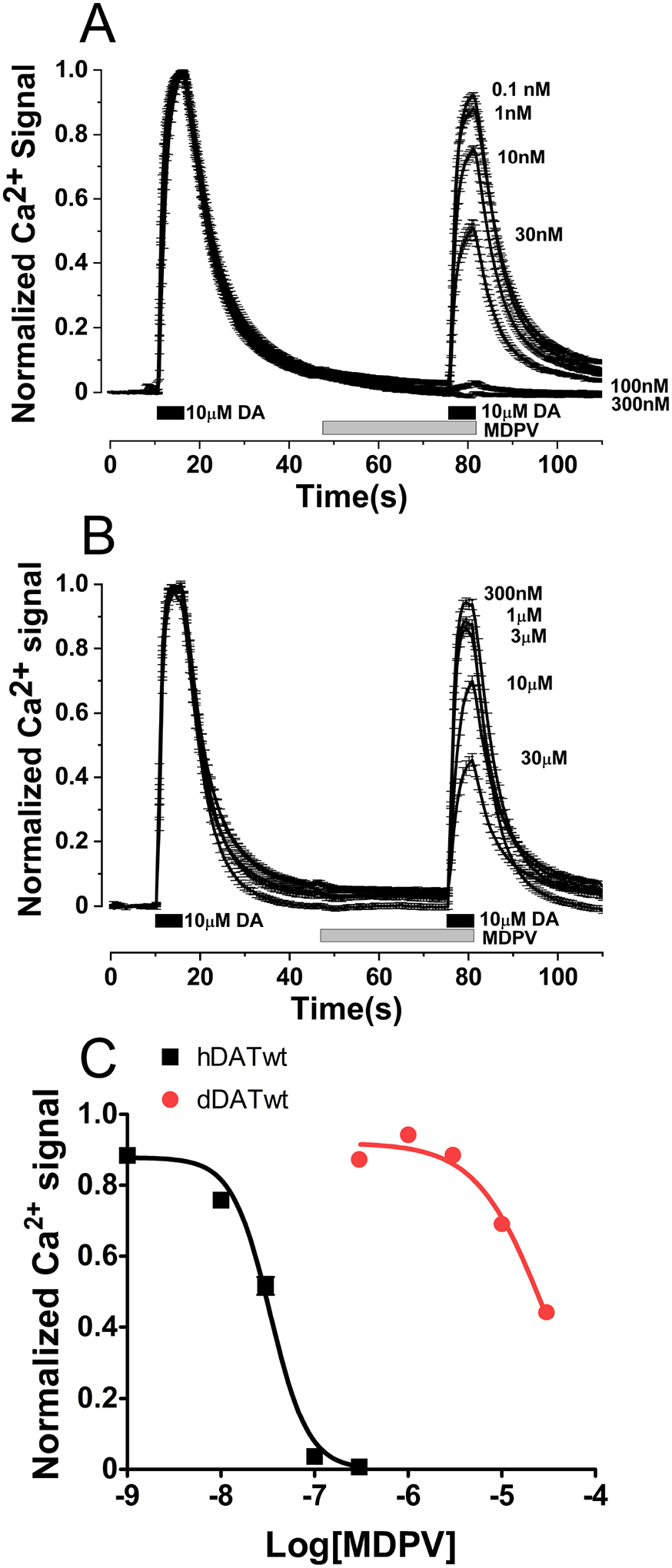
Potency of MDPV in inhibiting DA-induced Ca2+ signals at hDAT and dDAT. Stable cell lines expressing hDAT or dDAT were transfected with CaV1.2 and auxiliary subunits. Intracellular Ca2+ signals were determined using fluorescence microscopy under constant perfusion at 35°C in Fura2-AM loaded cells. A) Ca2+ signals showing the inhibitory effect of MDPV at different concentrations at hDAT, the traces are normalized to the first DA pulse, each trace is mean ± SEM of N ≥ 59 cells analyzed per MDPV concentration. B) Traces showing the inhibitory effect of MDPV at dDAT, the traces are normalized to the first DA pulse, each trace is mean ± SEM of N ≥ 77 cells analyzed per MDPV concentration. C) Dose-response curves obtained from the data shown in A and B. Since MDPV was quite weak at dDAT, the bottom of the curve fitting was constrained to zero to estimate the pIC50. The fitting parameters for MDPV inhibition at hDAT and dDAT are included in Table 2.
We previously described MDPV, α-PVP, and MDPPP in Kolanos et al. (Kolanos et al., 2013). At the beginning of the experimental day, the system is tested by measuring the response of cells to DA using the two-pulse protocol, in absence of inhibitor. For some constructs we observed a small rundown of the signal in the second pulse. This rundown reverses ~3 min after a 5 s DA pre-pulse, and then the two-pulse protocol (described above) produces a consistent consecutive response to DA. pIC50 values obtained in datasets with and without a DA pre-pulse are the same. For example, MDPV’s effect on the triple and quadruple dDAT mutants shown in Table 1 and Table 2 are identical, despite that DA pre-pulse was used in only datasets shown in Table 2.
Table 1.
Potency of MDPV inhibiting DA-induced Ca2+ signals at dDAT mutants.
| Construct | pIC50 ± SEM(M) | IC50 (nM) | # cells analyzed |
|---|---|---|---|
| P323V | 5.01 ± 0.03 | 9,694 | 272 |
| A117S | 5.08 ± 0.04 | 8,391 | 336 |
| F318C | 5.08 ± 0.02 | 8,237 | 369 |
| D121G | 5.33 ± 0.04 | 4,646 | 297 |
| A117S/D121G | 5.66 ± 0.04 | 2,209 | 434 |
| A117S/D121G/P323V | 5.93 ± 0.02 | 1,184 | 747 |
| A117S/D121G/P323V/F318C | 6.60 ± 0.03 | 253 | 459 |
Table 2.
Potency of MDPV and analogs inhibiting DA-induced Ca2+ signals at the indicated transporters.
| Construct, pIC50 ± SEM (M), (IC50 nM; # cells analyzed) | ||||
|---|---|---|---|---|
| wt dDAT | triple dDAT mut | quad dDAT mut | wt hDAT | |
| MDPV | 4.56 ± 0.02 (27,350; 437) | 5.92 ± 0.02 (1,207; 438) | 6.60 ± 0.04 (251; 396) | 7.47 ± 0.01 (34; 459) |
| α-PVP | 4.68 ± 0.03 (20,710; 699) | 5.82 ± 0.03 (1,531; 599) | 5.96 ± 0.03 (1,094; 481) | 7.29 ± 0.01 (51; 569) |
| MDPPP | Inactive (>>30,000; 246) | 4.80 ± 0.02 (15,960; 551) | 4.89 ± 0.01 (12,950; 507) | 5.47 ± 0.02 (3,418; 639) |
Data analysis and potency determinations.
The background-subtracted ratiometric images of each experiment were used for offline analysis. Cells that showed low resting Ca2+, robust response to the first DA control pulse, and returned to baseline after wash were considered for analysis. If there is any cell detachment in the well that produced Ca2+ waves, this disqualified the full well for further analysis. Usually, Ca2+ signals of 5–10 cells per well are quantified, 3 to 4 wells per concentration are tested per experimental day and the experiments are repeated 3 days. Each cell is analyzed independently using a Microsoft Excel premade template, first the signal of each cell is divided by the average of the first 10 frames to get the F/F0 trace, then the peak of the DA- induced Ca2+ signal in the presence of the tested compound is divided by the peak DA signal of the initial control response. Then all normalized Ca2+ responses as a function of the contraction for each agent studied are pulled together, the concentrations of the agents used were transformed to the log10 scale and the data were fitted to a dose-repose curve as described previously using GraphPad Prism software (Battisti et al., 2018; Solis et al., 2017). pIC50 ± SEM are reported in Table 1 and 2, in addition the 95% confidence intervals, IC50 ± SEM, and EC50 ± SEM are shown in Table S2). The total number of cells analyzed varied from 246 to 747 per compound (see Table 1, Table 2, and Table S2).
3D Molecular Modeling.
One hundred homology models of hDAT were generated using MODELLER 9.24 and three crystal structures as a template (Haddad et al., 2016; Sali and Blundell, 1993). The two highest identity dDAT crystal structures (PDB ID: 4XPB, 4XPT) at 55.35% and 55.33% identity, respectively, as well as the greatest query coverage hSERT crystal structure (PDB ID: 6VRH) at 92% coverage were used (Coleman et al., 2020; Haddad et al., 2016; Wang et al., 2015). The alignment of the structures was conducted using BLAST (Altschul et al., 1990). The sequences of both dDATs, hSERT, and hDAT were obtained from genpept (accession codes 4XPB_A, 4XPT_A, 6VRH_A, and BAA22511, respectively). Due to the lack of corresponding residues, the first 54 residues from the N-terminus were not modelled. The homology model with the lowest discrete optimized protein energy (DOPE) score and highest GA341 score was then used for docking (Melo and Sali, 2007; Sali and Blundell, 1993). GA341 is a multivariate scoring function that depends on compactness and combined statistical potential z-score of the model as well as the percentage sequence identity of the target-template alignment that was used to build the model (Melo and Sali, 2007; Sali and Blundell, 1993). The mutant transporter in the present study was generated using SybylX 2.1.1 (TRIPOS Associates, Inc) by replacing the dDAT (PDB ID: 4XP4) amino acids with the corresponding hDAT amino acids D121G, P323V, A117S, and F318C (Wang et al., 2015). The compounds MDPV, α-PVP, and MDPPP (as their S isomers) were sketched using SybylX 2.1.1 and energy minimized using the Tripos Force Field with Gasteiger-Hückel charges. The automated docking program GOLDsuite2020 was used to dock the three compounds one hundred times in each of the three DAT models (wt dDAT, quadruple dDAT mutant, hDAT) using D46 and D79 residues and the surrounding 10-Å radius to define the binding pocket for the dDAT and hDAT models, respectively (Jones et al., 1997). All solutions generated from GOLD were then merged with their respective protein and energy-minimized using the Tripos Force Field with Gasteiger-Hückel charges in SybylX 2.1.1 (Jones et al., 1997; Sakloth et al., 2015). The Hydropathic INTeraction (HINT) analysis software (Kellogg and Abraham, 2000) within SYBYL 8.1 was used to quantify the nature and magnitude of the molecular interactions between the aromatic and alkyl side chains of the compounds and subpockets A, B, and C of the dDAT and hDAT models. For the initial partitioning phase, the dictionary method was used for the protein and the calculation method was used for the ligands (Kellogg and Abraham, 2000; Sakloth et al., 2015). All other parameters were set to their default values. Atom-based interactions involving atoms within subpockets A, B, and C were extracted from the resulting HINT table and tabulated. MOLCAD and BioPolymer suite within SybylX 2.1.1 were used for binding pocket volume calculations and RMSD calculations, respectively.
Results
Creation of mutants.
A series of single point mutations and combinations in the open reading frame of dDAT were created and these new constructs were expressed permanently in HEK-293 Flp-In TRex cells using a targeted recombination system previously described in Cameron et al. (Cameron et al., 2015). These mutations corresponded to three locations of the shared residues between dDAT and hSERT (A117, F318, and P323) plus the unique dDAT residue (D121). In all cases the dDAT residues were mutated to the corresponding hDAT counterpart (see alignment in Fig. S1). The mutations were D121G, A117S, F318C, and P323V (sequence number in dDAT).
Evaluation of agents.
Permanent cell lines expressing hDAT or dDAT and transfected with voltage-gated Ca2+ channels were used as a biosensor for characterizing DAT ligands (Steele and Eltit, 2019). DAT is a Na+/Cl−-dependent electrogenic transporter, when DA interacts with DAT an inward current through DAT depolarizes the cell membrane. This depolarization activates the co-expressed voltage-gated Ca2+ channel producing a reproducible Ca2+ signal (Steele and Eltit, 2019). Using Fura2 as a Ca2+ indicator and fluorescence microscopy for signal detection, DA induces a Ca2+ signal in hDAT-expressing cells (Fig. 1A). This indicates that hDAT-mediated transport is taking place. Untransfected cells expressing transporters (but no Ca2+ channel) do not show such signals, and cells expressing only Ca2+ channels (without transporters) also fail to produce Ca2+ signals (Cameron et al., 2015; Ruchala et al., 2014). Cells expressing dDAT and voltage-gated Ca2+ channels showed Ca2+ responses when exposed to DA (Fig. 1B), indicative that dDAT also generates a depolarizing current during DA transport as previously described by Porzgen et al. (Porzgen et al., 2001).
The inhibitory activity of MDPV at hDAT is well described, but its activity at dDAT has not been reported in vitro. However, MDPV (20 μM) induced hyperlocomotion in flies presumably by acting at dDAT (Shekar et al., 2017). MDPV inhibited the DA-induced signal at wt dDAT-expressing cells with low potency (IC50 = 27.35 μM). Thus, MDPV is 2.9 log units (~800-fold) more potent at hDAT (IC50 = 34 nM) than at wt dDAT in inhibiting DA-induced signals (Fig. 1C). All single dDAT mutants were evaluated using the Ca2+ assay and exhibited a modest increase in MDPV potency in blocking the DA-induced signals vs. wt dDAT (Table 1 and S2). Three single mutations, A117S (IC50 = 8.39 μM), F318C (IC50 = 8.24 μM), or P323V (IC50 = 9.69 μM) produced a half log-unit improvement in potency (~3-fold) and the D121G (IC50 = 4.65 μM) mutant resulted in a 0.8 log-unit increase in potency (~6-fold) compared to the effect of MDPV in cells expressing wt dDAT (IC50 = 27.35 μM). Next, stable cell lines expressing dDAT constructs with combinations of these mutations were made and evaluated using the Ca2+-assay. The inhibitory effect of MDPV at cells expressing the double mutant A117S/D121G (IC50 = 2.21 μM) showed a 1.1 log-unit improvement in potency (~12-fold) at inhibiting DA signals compared to wt dDAT-expressing cells (IC50 = 27.35 μM, Table 1). Cells expressing the triple mutant A117S/D121G/P323V (IC50 = 1.18 μM) showed a 1.4 log-unit increase in potency compared to wt dDAT (IC50 = 27.35 μM, ~23-fold increase in potency). At the quadruple mutant A117S/D121G/P323V/F318C, MDPV (IC50 = 0.25 μM) showed a 2.04 log-unit (~110-fold) better potency at inhibiting the DA-induced signal than at cells expressing wt dDAT (IC50 = 27.35 μM). To illustrate the “jump” in potency between the triple and quadruple dDAT mutants compared to wt dDAT and wt hDAT, the dose-response curves for MDPV inhibition of the DA-induced signals are depicted in Fig. 2A.
Figure 2.
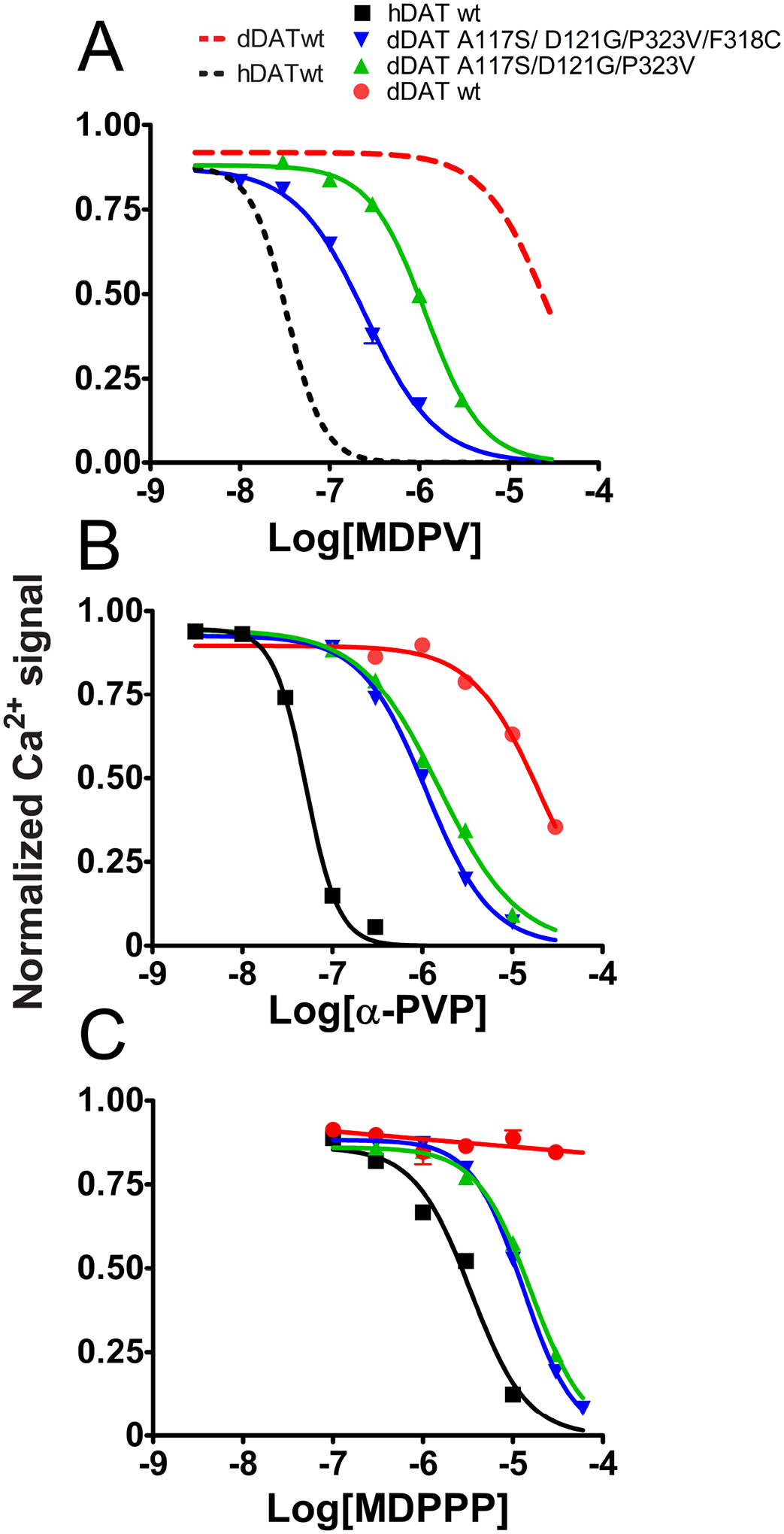
Potency of MDPV, α-PVP, and MDPPP in inhibiting the activity of hDAT, wt dDAT, and the triple and quadruple dDAT mutants. Stable cell lines expressing the indicated transporters were transfected with CaV1.2 and auxiliary subunits. Intracellular Ca2+ signals were determined using fluorescence microscopy under constant perfusion at 35°C in Fura2-AM loaded cells. A) Dose-response curves of the inhibitory effect that MDPV has at the A117S/D121G/P323V dDAT and A117S/D121G/P323V/F318C dDAT mutants. The curves for MDPV at hDAT and wt dDAT shown in Fig. 1C are included in the graph for reference (dashed lines). Each data point represents the mean ± SEM of N ≥ 60 cells analyzed per concentration. B) Dose-response curves of the inhibitory effect of α-PVP at hDAT, wt dDAT, and A117S/D121G/P323V dDAT and A117S/D121G/P323V/F318C dDAT mutants. Each data point represents the mean ± SEM of N ≥ 66 cells analyzed per concentration. C) Dose-response curves of the inhibitory effect of MDPPP at hDAT, wt dDAT, and A117S/D121G/P323V dDAT and A117S/D121G/P323V/F318C dDAT mutants. Each data point represents the mean ± SEM of N ≥ 53 cells analyzed per concentration. The pIC50 values are described in Table 2.
When the des-methylendioxy version of MDPV, α-PVP (IC50 = 51 nM), was tested, it showed a similar potency to MDPV (IC50 = 34 nM) at hDAT when tested using the Ca2+ assay (Fig. 2B and Table 2). This is consistent with previous reports showing that the methylenedioxy ring is not a major factor regarding MDPV’s high potency in inhibiting hDAT-mediated DA uptake (Kolanos et al., 2013; Marusich et al., 2014). α-PVP (IC50 = 20.71 μM) had low potency at wt dDAT similar to the value obtained for MDPV (IC50 = 27.35 μM) at this transporter, see Fig. 2. In the triple dDAT mutant (A117S/D121G/P323V), α-PVP (IC50 = 1.53 μM) showed a 1.1 log-unit (~14-fold) improvement in potency compared to its action at wt dDAT (IC50 = 20.71 μM) inhibiting Ca2+-signals. α-PVP (IC50 = 1.09 μM) showed an improvement of 1.3 log units (~20-fold) at the quadruple mutant (A117S/D121G/P323V/F318C) compared to its effect at wt dDAT (IC50 = 20.71 μM, Fig. 2B and Table 2). The substitution of α-PVP (IC50 = 20.71 μM) for MDPV (IC50 = 27.35 μM) produced a very small decrease in potency at wt dDAT of −0.12 log units (0.76-fold). Conversely, this substitution produced a tiny increase in potency at hDAT of 0.18 log units (~1.5-fold, α-PVP [IC50 = 51 nM] vs MDPV [IC50 = 34 nM], Table 2). At the dDAT triple mutant (A117S/D121G/P323V), the α-PVP (IC50 = 1.53 μM) to MDPV (IC50 = 1.21 μM) substitution produced a similar small increase in potency of 0.10 log units (~1.3-fold), whereas the substitution of α-PVP (IC50 = 1.09 μM) for MDPV (IC50 = 0.25 μM) produced a larger 0.6 log-unit (4-fold) increase in potency in the quadruple (A117S/D121G/P323V/F318C) dDAT mutant (Table 2). These data suggest that the methylendioxy ring in MDPV has a weak but overall positive contribution to its recognition at hDAT and the dDAT mutants, but not at wt dDAT.
The α-propyl aliphatic chain of MDPV is required for its high potency inhibiting DA uptake through hDAT. In a previous study, MDPV inhibited DA uptake in hDAT- expressing cells with an IC50 = 135 nM, and MDPPP, which only differs from MDPV in the shorter α-methyl substitution, had a potency of 3,500 nM (Kolanos et al., 2013). Here, a similar result was obtained: MDPV inhibited DA-induced Ca2+ signals in hDAT-expressing cells co-expressing Ca2+ channels with an IC50 = 34 nM and MDPPP showed an IC50 = 3.42 μM (Fig. 2 and Table 2). Conversely, MDPPP was inactive when assayed at several concentrations up to 30 μM at wt dDAT (Fig. 2C). Both the triple and quadruple mutations increased the MDPPP activity at the dDAT transporter; the potency progressed from virtually inactive at wt dDAT to an IC50 = 15.96 μM and 12.95 μM at A117S/D121G/P323V dDAT- and A117S/D121G/P323V/F318C dDAT-expressing cells, respectively (Fig. 2C and Table 2). Inhibiting with MDPV (IC50 = 34 nM) instead of MDPPP (IC50 = 3.42 μM) produces a 2 log-unit (100-fold) increase in potency on the Ca2+ assay at wt hDAT (Table 2), illustrating the importance of the α-propyl aliphatic chain in the inhibitory effect of MDPV. In A117S/D121G/P323V dDAT, MDPPP (IC50 = 15.96 μM) to MDPV (IC50 = 1.21 μM) transition produces only a 1.1 log-unit (~13-fold) increase in potency, and in A117S/D121G/P323V/F318C dDAT this transition increases potency by 1.7 log units (~51-fold, MDPPP [IC50 = 12.95 μM] vs MDPV [IC50 = 0.25 μM]), as shown in Table 2. These results strongly suggest that A117S/D121G/P323V residues are important in DAT recognition of MDPV and MDPV analogs at the central binding site, and the addition of F318C in the quadruple mutant better accommodates the bulkier MDPV molecule.
Effect of dopamine.
Given that the apparent potency of MDPV in the Ca2+-assay may be correlated to the DA response at the respective transporter variant, the potency of DA in eliciting Ca2+ signals was determined in cells expressing the triple mutant, quadruple mutant, and wt dDAT transporters. The potencies measured for DA to produce Ca2+ signals were similar. For these constructs: pEC50 = 5.84 ± 0.03 (EC50 = 1.43 μM), 5.69 ± 0.03 (2.04 μM), and 5.49 ± 0.02 (3.21 μM) for wt dDAT, A117S/D121G/P323V dDAT, and A117S/D121G/P323V/F318C dDAT, respectively (Table S2). Previously, we reported an pEC50 = 5.89 ± 0.04 (EC50 = 1.30 μM) using the same technique for DA at hDAT (Steele and Eltit, 2019) that is virtually equipotent with wt dDAT (Table S2). The DA concentration of 10 μM used in the inhibitory assays described above was saturating even for the quadruple dDAT mutant that was the weakest reactive construct to DA. The maximal difference in potency observed was between wt dDAT and the quadruple dDAT mutant that was 0.35 log units (2.2-fold) weaker at the latter. The triple and quadruple dDAT mutants showed a difference of 0.20 log units (1.6-fold) to produce DA-induced signals (Table S2); in these dDAT mutants IC50 values observed for α-PVP or for MDPPP were very similar (Table 2). These results indicate that the substantial improvement in potency of MDPV at dDAT mutants, especially at the quadruple mutant, is not a manifestation of altered DA activity at these transporters.
3D Molecular modeling.
The functional data described above clearly show that the substitution of four non-conserved residues from hDAT into a dDAT backbone (A117S/D121G/P323V/F318C) enhanced recognition of MDPV by at least 100-fold compared to wt dDAT (Table 2). These residues are either part of or very close to the S1 binding site previously described as the main interacting site for substrates and inhibitors in dDAT and hSERT crystal structures (Wang et al., 2015). The available dDAT and hSERT crystal structures were used as templates to generate 3D homology models of the quadruple (A117S/D121G/P323V/F318C) dDAT mutant and wt hDAT, and docking studies were then conducted to better understand potential molecular interactions with MDPV, α-PVP, and MDPPP at these transporters.
The overall structure of these energy-minimized models was similar; the RMSD was 2.02 Å between hDAT and the quadruple dDAT mutant homology models, and 2.16 Å between hDAT and the wt dDAT structures. As expected, wt dDAT and the quadruple dDAT mutant models were very similar (RMSD = 0.29 Å). When the binding pockets were compared, the RMSD between hDAT and the quadruple dDAT mutant was 2.11 Å, and between hDAT and wt dDAT models, it was 2.12 Å. The RMSD of the binding pockets of the wt dDAT and quadruple dDAT mutant models was 0.36 Å. The S1 binding site has an architecture that can be divided in three subpockets (A, B, and C) (Cheng and Bahar, 2019; Wang et al., 2015). The total volume of the pockets increased from 2425 Å3 for wt dDAT, to 2533 Å3 for the quadruple dDAT mutant, to 3170 Å3 for hDAT.
In our docking experiments the only criteria used to define the central binding pocket was a 10-Å radius surrounding the conserved D79/D46, for hDAT and dDAT, respectively. Using these criteria, all generated solutions were oriented to form a salt bridge interaction between the protonated N atom of the tested agent and the carboxylate oxygen atom of D79/D46. Accordingly in almost all 900 total solutions generated by GOLD, the pyrrolidine ring of the ligands is exclusively situated within subpocket A of the S1 site (Figure S2). All 100 solutions (i.e., poses) for each ligand docked at each transporter model were clustered using an RMSD of 1.5 Å as a cutoff (Table S3) and the presented final orientations of the three compounds in each DAT model were based on a combination of the highest Hydropathic INTeraction (HINT) scores and lowest energy conformations for the ligands (Fig 3). Indeed, the difference in the GOLD score for the highest GOLD-score pose and the highest HINT-score pose (using the GOLD scoring function) is only ~5 points, but the difference of HINT scores between these two poses is much larger. HINT scores account for favorable and unfavorable interactions of the docked agents in the 3D model, thus the pose with higher HINT score would represent stronger ligand-protein interactions (see Table S4). In addition, the differences in ligand energy of the highest GOLD-score solution within the largest population and the highest HINT-score pose in the S1 central binding site were greater than 3 kcals/mol suggesting that the ligands are unable to reorient to different conformations upon protein interaction and most likely bind in the lower-energy conformation (Table S4). The differences in the overall ligand-protein complex energies are insignificant as the maximum energy difference is only ~1% (Table S4). With the combination of these two parameters (i.e., HINT score and energy calculations) the specific poses we selected for presentation correlate well with each other within the S1 central binding pocket (Fig. 3) and mimicked the pose of cocaine bound to dDAT in the crystal structure (PDB ID: 4XP4; Fig.S3).
Figure 3.
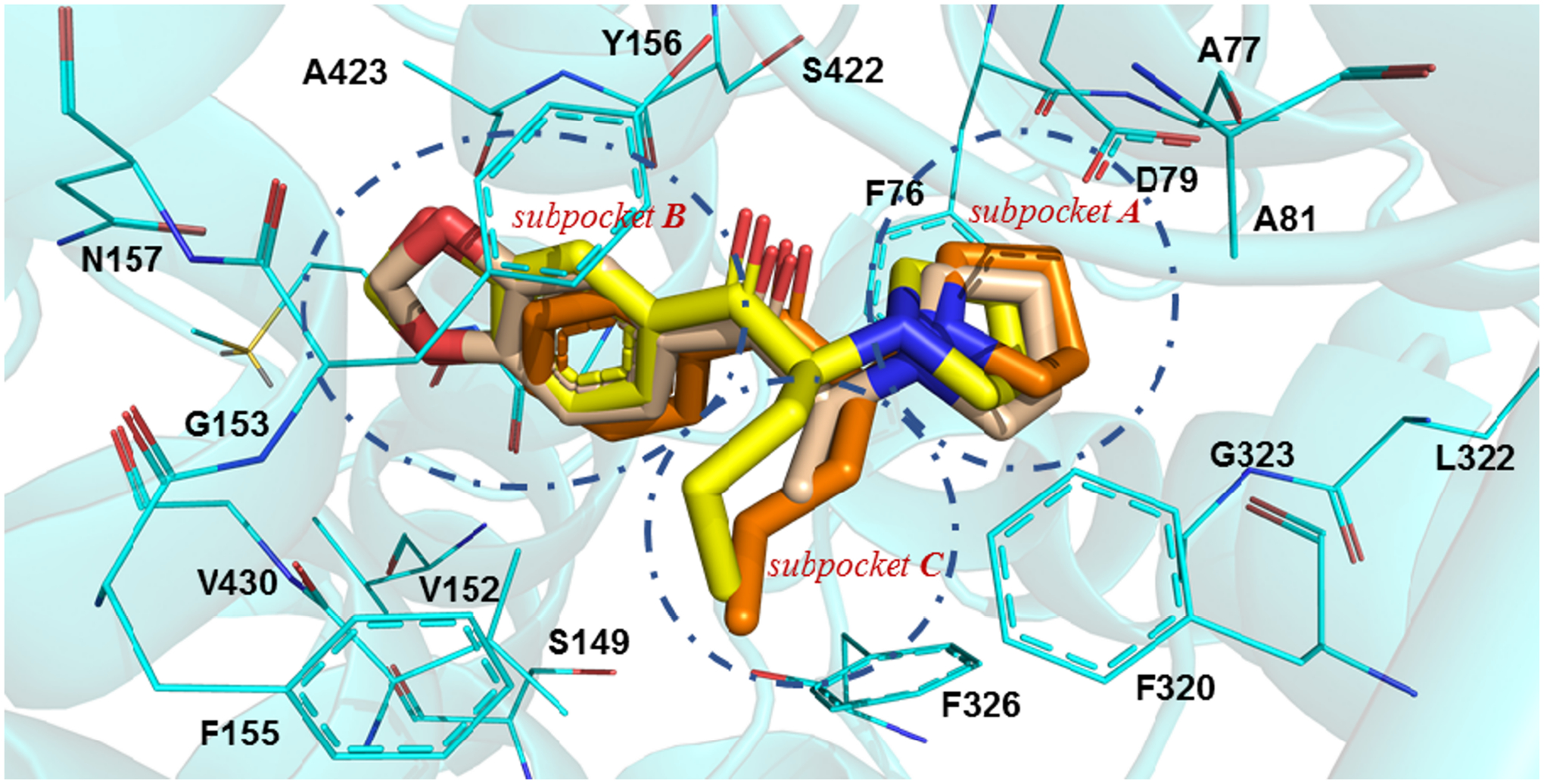
In silico interaction of MDPV and its analogs at the S1 binding site of hDAT. Possible binding mode of MDPV (capped sticks rendering; yellow carbon atoms), MDPPP (wheat carbon atoms), and α-PVP (orange carbon atoms) at the hDAT homology model (amino acid residues depicted as wire; cyan carbon atoms).
HINT scores showed low scores for MDPPP in all models, whereas MDPV and α-PVP showed much higher scores at hDAT (Table S5). The number of positive and negative interactions between a docked molecule at each protein model showed that the total favorable interactions were highest at hDAT and the more unfavorable interactions were highest at wt dDAT for each agent (Table S6). These results are consistent with the functional data (Table 2).
A description of the potential interaction of MDPV and its analogs at each subpocket is provided below.
Interactions at subpocket A.
MDPV and its analogs placed the pyrrolidine ring in subpocket A. This subpocket is comprised of residues F43, A44, D46, A48, Y124, F319, L321, G322, and S421 for the dDAT proteins or residues F76, A77, D79, A81, Y156, F320, L322, G323, and S422 in hDAT (Figs. 4A and S4A). Subpocket A had the highest scoring interaction compared to the other subpockets. This is mainly driven by the salt bridge formed between D46 (dDAT)/ D79 (hDAT) and the pyrrolidine nitrogen atom present in all the evaluated ligands.
Figure 4.
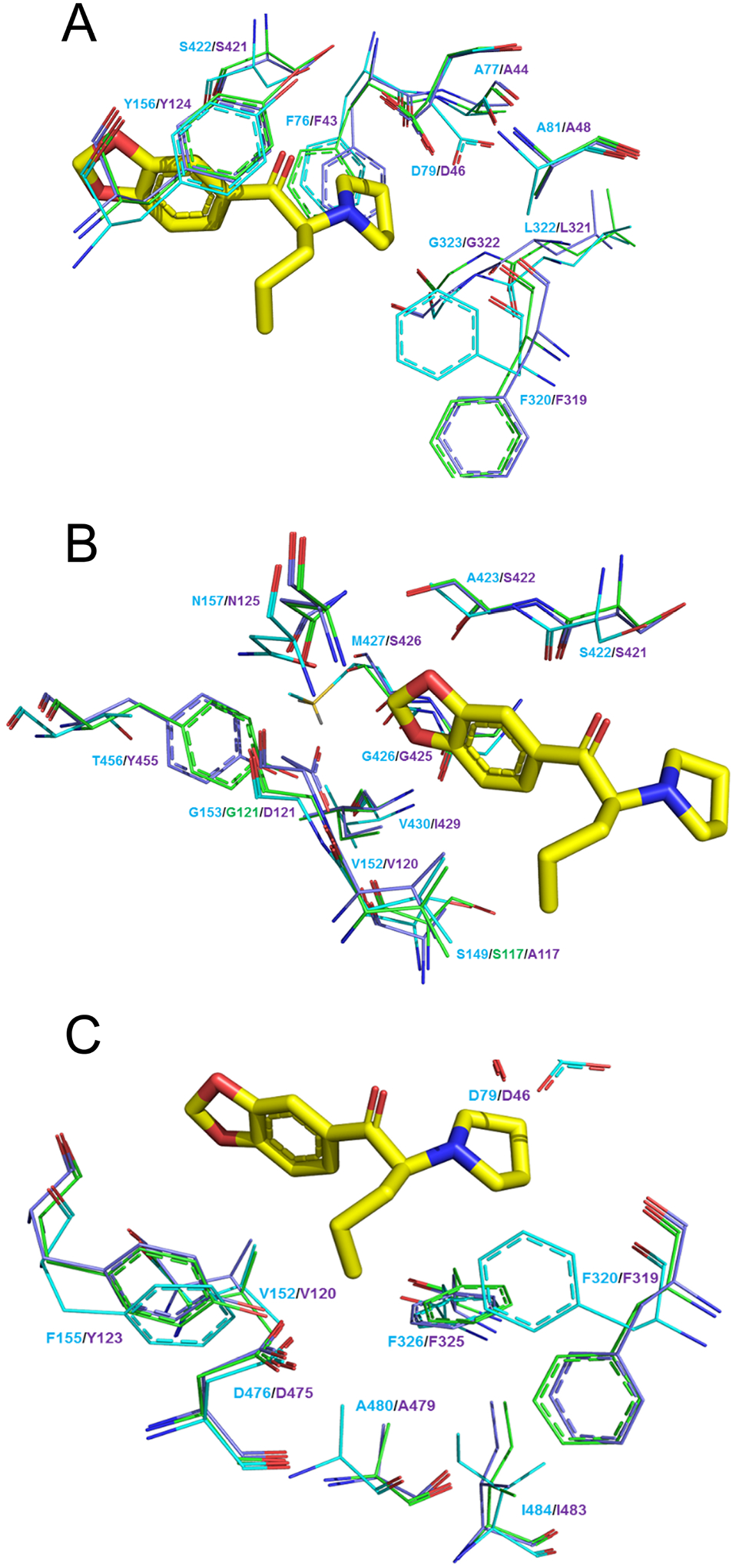
Amino acid residues that make up subpocket A, B, and C. For each panel hDAT is shown in cyan, quadruple dDAT mutant in green, and wt dDAT in purple (amino acid residues depicted as wire). MDPV as a reference is depicted as capped sticks rendering, yellow carbon atoms.
Interactions at subpocket B.
The aromatic portion of the evaluated agents was oriented within subpocket B of the transporters. This subpocket consisted of the residues A117, V120, D121, S421, S422, G425, I429, Y455, S426, and N125 at wt dDAT or residues S149, V152, G153, S422, A423, G426, V430, T456, M427, and N157 at wt hDAT (Figs. 4B and S4B). hDAT displayed the most favorable interactions with the compounds at residues V152, S422, A423, G426, and M427; in contrast, wt dDAT only showed favorable interactions with residues V120, G425, I429. Two of the mutations evaluated in this study, A117S and D121G, made up a portion of subpocket B and corresponded to residues S149 and G153 in hDAT. The major clashes observed were between the docked agents (the methylenedioxy ring for MDPV/MDPPP and the phenyl ring for α-PVP) with A117 and D121 in wt dDAT. These interactions are more favorable with the cognate residues S149 and G153 in hDAT (see discussion).
The quadruple dDAT mutant, having the D121G mutation, exchanges one negative interaction, comprised of the clash of the bulkier size and the charged nature of D121 with the aryl portion of the ligands, for a positive one, that is the increased space and nonpolar clashing with the evaluated agents. The A117S mutation in the quadruple mutant favors interactions with the tested ligands by possible hydrogen bonding with the methylenedioxy ring for MDPV/MDPPP and/or by bonding with the protein backbone, thereby increasing the size of the pocket (see discussion). In addition, the quadruple dDAT mutant earned a negative interaction at G425 with the aryl moiety of the ligands compared to wt dDAT.
Interactions at the subpocket C.
The alkyl side chains, methyl for MDPPP and n-propyl for MDPV and α-PVP, occupied subpocket C with favorable hydrophobic interactions. This subpocket presents interactions between the α-propyl chain of MDPV and α-PVP with residues D46, V120, F319, F325, D475, A479, I483, and Y123 at wt dDAT, and residues D79, V152, F320, F326, D476, A480, I484, and F155 at hDAT (Figs. 4C and S4C). The methyl substituent of MDPPP did not display any overall negative interactions, only fewer favorable interactions within subpocket C compared to the other two compounds. That is, in the hDAT model the α-methyl substituent of MDPPP is not long enough to produce positive hydrophobic interactions with residues (I484 and F155), or residues (I483 and A479) in the mutant and wt dDAT models, respectively.
Discussion
It is well established that MDPV and some of its analogs are potent inhibitors of mammalian DAT (Baumann et al., 2013; Eshleman et al., 2013; Meltzer et al., 2006; Simmler et al., 2013). The effect of MDPV at monoamine transporters was reported in several studies using diverse techniques. Electrophysiological determinations in Xenopus oocytes expressing hDAT showed that MDPV produces an apparent outward hyperpolarizing current typical of inhibitors of DAT (Cameron et al., 2013b). Inhibitors of monoamine transporters hinder a constitutive inward “leak” current through the transporters and this blockade can be used for the early identification of MAT inhibitors (Mager et al., 1994; Sonders et al., 1997). MDPV inhibited DA transport mediated through DAT at nanomolar concentrations in rat brain membrane preparations (synaptosomes) (Marusich et al., 2014), a result that was reproduced measuring the blockade of DA-mediated inward current or DA transport through DAT in cells expressing hDAT (Cameron et al., 2013b; Steele and Eltit, 2019). DA has comparable potency at both dDAT and hDAT (shown here and in Porzgen et. al.) (Porzgen et al., 2001). These transporters show distinctive selectivity profiles to inhibitors. For example, MAT inhibitors that are virtually inactive at hDAT (but potent at hSERT), such as amitriptyline, desipramine, imipramine, fluoxetine, and paroxetine, are much more potent at dDAT (Porzgen et al., 2001). Moreover, the situation for cocaine is the opposite. Cocaine has higher potency at hDAT than at dDAT in blocking DA uptake (Porzgen et al., 2001), and MDPV, α-PVP and MDPPP follow the same trend, and are much more potent at hDAT than at dDAT. MDPPP is a unique case, in that it shows low potency inhibiting hDAT, while it is virtually inactive at dDAT. These observations strongly suggest that MDPV and the MDPV analogs assayed here have much higher affinity at hDAT than at dDAT.
hNET is the most similar monoamine transporter to hDAT, having an identity of 67% (hSERT and dDAT have an identity of 48% and 52% compared to hDAT, respectively, Fig S1). Thus, it is not surprising that several ligands potent at hDAT are also very potent at hNET, in fact, dopamine is equipotent at these transporters when tested in rat brain synaptosomes (Rothman and Baumann, 2003). As a note, rat and human monoamine transporters have > 91% interspecies identity and the sites studied here by mutagenesis are conserved between human and rat transporters. Gannon et al. performed experiments using rat brain synaptosomes showing that MDPV is active and potent at NET and it is only ~ 4-fold weaker inhibiting uptake than at DAT. In the same study it was shown that MDPPP is active but weaker than MDPV at NET, and it is ~ 5-fold weaker inhibiting uptake at NET than at DAT, and α-PVP is strong and equipotent inhibiting uptake at both NET and DAT (Gannon et al., 2018). Regarding its primary structure, hNET has a glycine at position 149 (position 121 at dDAT) which is also a glycine in hDAT. hNET does not have a proline at position 321 having an alanine instead, that is similar to the valine present in hDAT at the cognate position (position 323 at dDAT). Unexpectedly, hNET is identical to hSERT and dDAT at positions 117 and 318 (of dDAT). As discussed in more detail below, serine at position 117 in the mutant dDAT (and in hDAT) may have a role accommodating the methylenedioxy ring of the aryl substituent of these compounds at subpocket B. The alanine instead of a serine in that equivalent position would explain the weaker effect of MDPV and MDPPP at NET vs DAT and the equipotency of α-PVP at these transporters in the rat synaptosome experiments described above; further experimentation would be necessary to test the role of these sites controlling MDPV binding at human (and rat) NET.
The high-resolution 3D structures of dDAT, hSERT, and the more distant bacterial ortholog leucine transporter (LeuT) were previously resolved (Coleman et al., 2016; Penmatsa et al., 2013; Wang et al., 2013; Wang et al., 2015; Yamashita et al., 2005). The overall architecture of these transporters is very similar. These structures exhibit a central “S1” binding site that can accommodate substrates and inhibitors of these transporters. Although the hDAT 3D structure has not yet been resolved, through primary sequence alignments of the segments forming the S1 site from dDAT and hSERT structures and using a 3D homology model of hDAT we identified residues lining S1 that were identical between dDAT and hSERT, but unique in hDAT. These included residues present in the S1 site as well as neighboring residues. This analysis identified four residues matching this criterion. These were P323, F318, A117, and Y123 at the dDAT sequence. Additionally, D121 was identified as unique in dDAT. As described above, from this list we studied A117, D121, P323, and F318. The S1 primary binding site is comprised of the central portions of the transmembrane helices TM1, TM3, TM6, and TM8 (Penmatsa et al., 2013). Note that while helices TM3 and TM8 are uninterrupted, helices TM1 and TM6 are subdivided into two helices (1a and 1b, and 6a and 6b, respectively) connected by a non-helical segment in the middle (Penmatsa et al., 2013). D121 and A117 are an integral part of the S1 binding site located in TM3 and they can directly interact with ligands. In contrast, F318 and P323 are part of TM6 but they are not directly facing the ligands. F318 is adjacent to the highly conserved F319 in the TM6a helix. The latter residue is considered the extracellular gate controlling access to the central cavity from the extracellular vestibule. This gate is “open” in the outward facing conformation, and F319 rotates to a “closed” conformation in the occluded state of the transporter (Wang et al., 2015). The F318C mutation might influence F319 operation in such a way that it allows better access of the bulky MDPV molecule to the S1 site in the outward facing state, this positive effect of F318C is less evident for the smaller α-PVP and MDPPP molecules. P323 is in the TM6 central connecting loop where its mutation to valine might increase structural flexibility towards the entrance of the S1 binding site, also helping the access of bulky ligands to the S1 site. The two other mutations evaluated in dDAT, D121G and A117S, lie within the binding pocket at subpocket B and have a direct effect on ligand/protein interactions at the S1. The acidic aspartate is bulkier and more polar compared to the small and neutral glycine. Thus, glycine leaves more room for the large aromatic portion of the ligands to fit within subpocket B, especially the aryl moiety containing the bulky methylendioxy ring of MDPV. Moreover, the neutral character of this residue produces no electronegative repulsion from the oxygen atoms of the methylenedioxy ring of MDPV, an expected effect when the aspartate carboxylate is located at the 121 position. In fact, the D121G dDAT single mutant was the most effective at increasing MDPV’s potency compared to any other dDAT single mutant evaluated in this study.
The hydrophobic alanine at position 117 of dDAT is occupied by serine in the hDAT cognate position. This position is part of subpocket B of the S1 site and the hydroxyl group of serine theoretically could form a hydrogen bond with one of the oxygen atoms of the methylenedioxy ring of MDPV. However, the measured distances indicate a weak, but more likely, electrostatic interaction in the quadruple dDAT mutant model (3.9 Å) than in the hDAT model (7.53 Å) with MDPV. This weak but favorable interaction would explain why that in all transporters evaluated in Table 2, α-PVP (MDPV without the methylendioxy ring) was slightly weaker than MDPV except for wt dDAT, which lacks a serine at that position. Instead, S117 forms a stronger hydrogen bond with the backbone amide oxygen atom of F325 or F326 in the dDAT mutant and hDAT models, respectively. The measured distances of these hydrogen bonds are 2.6 Å and 2.8 Å for the dDAT mutant and hDAT, respectively. The possible S117 and F325/F326 hydrogen bond would pull the S117 residue away from the binding pocket creating more space for the ligand to bind.
In summary, MDPV is a dangerous illicit psychostimulant, a potent and selective inhibitor of hDAT, and a template for the creation of new psychoactive substances. MDPV is 3 log units weaker at dDAT than at hDAT in functional studies. Four non-conserved hDAT residues when mutated onto the dDAT backbone boost MDPV potency by at least 2 log units. Two of these residues (position 318 and 323 in dDAT) are in the entrance of the S1 binding site, whereas the other two residues (position 117 and 121 at dDAT) face the aryl moiety of MDPV when bound to the S1 site. This study suggests that the non-conserved residues identified can influence MDPV selectivity not solely by stabilizing binding, but also by controlling access to the S1 binding site.
Supplementary Material
Highlights.
Analogs of MDPV are frequent-appearing designer drugs.
Four non-conserved residues of hDAT account for MDPV high potency.
These four residues are marginally implicated in dopamine recognition.
Access and interaction with the S1 site might control MDPV selectivity at DAT.
Acknowledgements
We would like to thank our late mentor, colleague, and friend Dr. Louis De Felice for his support at the initial stages of this project.
Funding
This work was partially funded by NIH grants: R01 DA033930, P30 DA033934, and R01 AR067738.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing interests.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, 1990. Basic local alignment search tool. J Mol Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andersen J, Olsen L, Hansen KB, Taboureau O, Jorgensen FS, Jorgensen AM, Bang-Andersen B, Egebjerg J, Stromgaard K, Kristensen AS, 2010. Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J Biol Chem 285, 2051–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J, Ringsted KB, Bang-Andersen B, Stromgaard K, Kristensen AS, 2015. Binding site residues control inhibitor selectivity in the human norepinephrine transporter but not in the human dopamine transporter. Sci Rep 5, 15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battisti UM, Sitta R, Harris A, Sakloth F, Walther D, Ruchala I, Negus SS, Baumann MH, Glennon RA, Eltit JM, 2018. Effects of N-Alkyl-4-Methylamphetamine Optical Isomers on Plasma Membrane Monoamine Transporters and Abuse-Related Behavior. ACS Chem Neurosci 9, 1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M, Rothman RB, Goldberg SR, Lupica CR, Sitte HH, Brandt SD, Tella SR, Cozzi NV, Schindler CW, 2013. Powerful cocaine-like actions of 3,4-methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology 38, 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonano JS, Glennon RA, De Felice LJ, Banks ML, Negus SS, 2014. Abuse-related and abuse-limiting effects of methcathinone and the synthetic “bath salts” cathinone analogs methylenedioxypyrovalerone (MDPV), methylone and mephedrone on intracranial self-stimulation in rats. Psychopharmacology (Berl) 231, 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron K, Kolanos R, Vekariya R, De Felice L, Glennon RA, 2013a. Mephedrone and methylenedioxypyrovalerone (MDPV), major constituents of “bath salts,” produce opposite effects at the human dopamine transporter. Psychopharmacology (Berl) 227, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron KN, Kolanos R, Solis E Jr., Glennon RA, De Felice LJ, 2013b. Bath salts components mephedrone and methylenedioxypyrovalerone (MDPV) act synergistically at the human dopamine transporter. Br J Pharmacol 168, 1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron KN, Solis E Jr., Ruchala I, De Felice LJ, Eltit JM, 2015. Amphetamine activates calcium channels through dopamine transporter-mediated depolarization. Cell Calcium 58, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MH, Bahar I, 2019. Monoamine transporters: structure, intrinsic dynamics and allosteric regulation. Nat Struct Mol Biol 26, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Green EM, Gouaux E, 2016. X-ray structures and mechanism of the human serotonin transporter. Nature 532, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Navratna V, Antermite D, Yang D, Bull JA, Gouaux E, 2020. Chemical and structural investigation of the paroxetine-human serotonin transporter complex. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies RA, Baird TR, Nguyen VT, Ruiz B, Sakloth F, Eltit JM, Negus SS, Glennon RA, 2020. Investigation of the Optical Isomers of Methcathinone, and Two Achiral Analogs, at Monoamine Transporters and in Intracranial Self-Stimulation Studies in Rats. ACS Chem Neurosci 11, 1762–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice LJ, Glennon RA, Negus SS, 2014. Synthetic cathinones: chemical phylogeny, physiology, and neuropharmacology. Life Sci 97, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A, 2013. Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol 85, 1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon BM, Baumann MH, Walther D, Jimenez-Morigosa C, Sulima A, Rice KC, Collins GT, 2018. The abuse-related effects of pyrrolidine-containing cathinones are related to their potency and selectivity to inhibit the dopamine transporter. Neuropsychopharmacology 43, 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad Y, Heger Z, Adam V, 2016. Guidelines for Homology Modeling of Dopamine, Norepinephrine, and Serotonin Transporters. ACS Chem Neurosci 7, 1607–1613. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R, 1997. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Kellogg GE, Abraham DJ, 2000. Hydrophobicity: is LogP(o/w) more than the sum of its parts? Eur J Med Chem 35, 651–661. [DOI] [PubMed] [Google Scholar]
- Kolanos R, Solis E Jr., Sakloth F, De Felice LJ, Glennon RA, 2013. “Deconstruction” of the abused synthetic cathinone methylenedioxypyrovalerone (MDPV) and an examination of effects at the human dopamine transporter. ACS Chem Neurosci 4, 1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager S, Min C, Henry DJ, Chavkin C, Hoffman BJ, Davidson N, Lester HA, 1994. Conducting states of a mammalian serotonin transporter. Neuron 12, 845–859. [DOI] [PubMed] [Google Scholar]
- Marinetti LJ, Antonides HM, 2013. Analysis of synthetic cathinones commonly found in bath salts in human performance and postmortem toxicology: method development, drug distribution and interpretation of results. J Anal Toxicol 37, 135–146. [DOI] [PubMed] [Google Scholar]
- Marusich JA, Antonazzo KR, Wiley JL, Blough BE, Partilla JS, Baumann MH, 2014. Pharmacology of novel synthetic stimulants structurally related to the “bath salts” constituent 3,4-methylenedioxypyrovalerone (MDPV). Neuropharmacology 87, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo F, Sali A, 2007. Fold assessment for comparative protein structure modeling. Protein Sci 16, 2412–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer PC, Butler D, Deschamps JR, Madras BK, 2006. 1-(4-Methylphenyl)-2-pyrrolidin1-yl-pentan-1-one (Pyrovalerone) analogues: a promising class of monoamine uptake inhibitors. J Med Chem 49, 1420–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E, 2013. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porzgen P, Park SK, Hirsh J, Sonders MS, Amara SG, 2001. The antidepressant-sensitive dopamine transporter in Drosophila melanogaster: a primordial carrier for catecholamines. Mol Pharmacol 59, 83–95. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, 2003. Monoamine transporters and psychostimulant drugs. Eur J Pharmacol 479, 23–40. [DOI] [PubMed] [Google Scholar]
- Ruchala I, Battisti UM, Nguyen VT, Chen RY, Glennon RA, Eltit JM, 2021. Functional characterization of N-octyl 4-methylamphetamine variants and related bivalent compounds at the dopamine and serotonin transporters using Ca(2+) channels as sensors. Toxicol Appl Pharmacol 419, 115513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchala I, Cabra V, Solis E Jr., Glennon RA, De Felice LJ, Eltit JM, 2014. Electrical coupling between the human serotonin transporter and voltage-gated Ca(2+) channels. Cell Calcium 56, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakloth F, Kolanos R, Mosier PD, Bonano JS, Banks ML, Partilla JS, Baumann MH, Negus SS, Glennon RA, 2015. Steric parameters, molecular modeling and hydropathic interaction analysis of the pharmacology of para-substituted methcathinone analogues. Br J Pharmacol 172, 2210–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL, 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Schindler CW, Thorndike EB, Goldberg SR, Lehner KR, Cozzi NV, Brandt SD, Baumann MH, 2016. Reinforcing and neurochemical effects of the “bath salts” constituents 3,4-methylenedioxypyrovalerone (MDPV) and 3,4-methylenedioxy-N-methylcathinone (methylone) in male rats. Psychopharmacology (Berl) 233, 1981–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks KG, Dahn T, Behonick G, Terrell A, 2012. Analysis of first and second generation legal highs for synthetic cannabinoids and synthetic stimulants by ultra-performance liquid chromatography and time of flight mass spectrometry. J Anal Toxicol 36, 360–371. [DOI] [PubMed] [Google Scholar]
- Shekar A, Aguilar JI, Galli G, Cozzi NV, Brandt SD, Ruoho AE, Baumann MH, Matthies HJG, Galli A, 2017. Atypical dopamine efflux caused by 3,4-methylenedioxypyrovalerone (MDPV) via the human dopamine transporter. J Chem Neuroanat 83–84, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME, 2013. Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168, 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis E Jr., Partilla JS, Sakloth F, Ruchala I, Schwienteck KL, De Felice LJ, Eltit JM, Glennon RA, Negus SS, Baumann MH, 2017. N-Alkylated Analogs of 4-Methylamphetamine (4-MA) Differentially Affect Monoamine Transporters and Abuse Liability. Neuropsychopharmacology 42, 1950–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG, 1997. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci 17, 960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele TWE, Eltit JM, 2019. Using Ca(2+)-channel biosensors to profile amphetamines and cathinones at monoamine transporters: electro-engineering cells to detect potential new psychoactive substances. Psychopharmacology (Berl) 236, 973–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E, 2013. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 503, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KH, Penmatsa A, Gouaux E, 2015. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E, 2005. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 437, 215–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.