

Abstract
Alzheimer's Disease (AD) is the leading form of dementia worldwide. Currently, the pathological mechanisms underlying AD are not well understood. Although the glutamatergic system is extensively implicated in its pathophysiology, there is a gap in knowledge regarding the expression of glutamate receptors in the AD brain. This study aimed to characterize the expression of specific glutamate receptor subunits in post‐mortem human brain tissue using immunohistochemistry and confocal microscopy. Free‐floating immunohistochemistry and confocal laser scanning microscopy were used to quantify the density of glutamate receptor subunits GluA2, GluN1, and GluN2A in specific cell layers of the hippocampal sub‐regions, subiculum, entorhinal cortex, and superior temporal gyrus. Quantification of GluA2 expression in human post‐mortem hippocampus revealed a significant increase in the stratum (str.) moleculare of the dentate gyrus (DG) in AD compared with control. Increased GluN1 receptor expression was found in the str. moleculare and hilus of the DG, str. oriens of the CA2 and CA3, str. pyramidale of the CA2, and str. radiatum of the CA1, CA2, and CA3 subregions and the entorhinal cortex. GluN2A expression was significantly increased in AD compared with control in the str. oriens, str. pyramidale, and str. radiatum of the CA1 subregion. These findings indicate that the expression of glutamatergic receptor subunits shows brain region‐specific changes in AD, suggesting possible pathological receptor functioning. These results provide evidence of specific glutamatergic receptor subunit changes in the AD hippocampus and entorhinal cortex, indicating the requirement for further research to elucidate the pathophysiological mechanisms it entails, and further highlight the potential of glutamatergic receptor subunits as therapeutic targets.
Keywords: Alzheimer's disease, AMAPA receptor, entorhinal cortex, glutamate receptor, hippocampus, NMDA receptor, subiculum, superior temporal gyrus
GTC Glutamatergic receptor GluA2, GluN1, and GluN2A subunit expression changes in the Alzheimer's disease hippocampus and entorhinal cortex.
1. INTRODUCTION
Alzheimer's Disease (AD) is the predominant form of dementia worldwide and is associated with various clinical hallmarks including a decline in cognitive function, behavior, and memory (1). As life expectancy increases worldwide, there follows a proportional increase in the aging population and the geriatric ailments that accompany it. Since its discovery, AD has dominated the frontiers of science, yet a possible cure, or at the very least, a suitable management strategy, has yet to be found. At present, therapeutic drugs serve only to provide mild symptomatic relief and do not delay or reverse the progression of AD. Currently, our understanding of the pathogenesis of AD is based on two central dogmas: the amyloid‐beta (Aβ) hypothesis and the tau hypothesis. The exact roles and interactions of these two hypotheses remain controversial, although there is consensus that both Aβ plaques and hyperphosphorylated tau deposits play a central role in AD neurodegeneration. Various other causes underlying the disease have also been proposed, such as cholinergic deficits, mitochondrial dysfunction, and oxidative stress (2, 3, 4). Aside from the predominant tau and Aβ hypotheses, glutamatergic dysfunction has been extensively implicated in the pathogenesis of AD, with repercussions on neuronal and synaptic functioning.
Glutamate is the primary excitatory neurotransmitter in the brain. Glutamate acts on a variety of receptors, traditionally categorized as ionotropic and metabotropic. Ionotropic receptors include the alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid receptor (AMPAR), N‐methyl‐D‐aspartate receptor (NMDAR), and kainate (GluK) receptor classes. The metabotropic class of receptors is subdivided into three functionally distinct groups; group I are coupled with phospholipase C, while groups II and III are coupled with adenylyl cyclase. These receptor subtypes are localized to dendrites of postsynaptic cells, astrocytes, and oligodendrocytes, as well as on glial cells (5). The receptors are formed by multiple subunits which are classified as follows: GluN1‐3 (NMDA), GluA1‐4 (AMPA), GluK1‐5 (kainate), and mGluR1‐5 (metabotropic) (6, 7). Each functonal receptor is composed of different combinations of subunits altering the pharmacological properties of receptors in different areas of the brain (6, 8).
Glutamatergic dysfunction in AD appears to be mediated through a variety of mechanisms, including Aβ binding to glutamate receptors, tau tethering to intrinsic cytoskeletal proteins resulting in overactivation of receptors, and the internalization of glutamate transporters resulting in glutamate accumulation in synaptic and extrasynaptic areas (9, 10). Glutamate is involved in many critical signaling and metabolic functions, but control of the glutamatergic system requires constant moderation to avoid excitotoxicity (11). As yet, glutamatergic signaling changes that contribute to this process, or result because of this process, have not been thoroughly investigated in the human AD brain. Significant effort has been put into investigating Aβ‐associated glutamate excitotoxicity in cell and animal models involving the dysfunction of calcium‐permeable glutamate receptors in the neuron (12, 13). Aβ has been implicated in the inhibition of glutamate uptake in the synaptic cleft (14), overstimulation of NMDA receptors, the subsequent disruption of calcium‐dependent intracellular pathways (15), and cell death (11, 16).
Despite clear evidence of the glutamatergic system's role in neurodegeneration (9, 10, 17, 18), the pathogenic mechanisms leading to expression alterations of glutamatergic components are yet to be elucidated in AD. Of the few studies performed, results observed remain inconclusive and controversial, with study designs that do not provide quantitative data (19, 20, 21) or information on the region and/or layer specificity of glutamate receptor subunit expression within hippocampal subfields of AD and control brains (22, 23).
In this study, we examined the region‐ and layer‐specific expression of glutamatergic receptor subunits within the hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus (STG), and then compare these expression levels and patterns between control and AD cases to gain a better understanding of how the glutamatergic system is altered. The hippocampal formation and temporal lobe have been the focus of our study because of their central role in memory formation, which is of particular interest as AD is characterized by the loss of memory and cognitive function. We have demonstrated here region‐ and layer‐specific alterations in the expression of AMPAR subunit GluA2, and NMDAR subunits GluN1 and GluN2A which suggest specific compensatory mechanisms or spatial susceptibility of glutamatergic receptor subunits to AD pathology.
2. METHODS
2.1. Human brain tissue preparation and neuropathological analysis
The research was carried out at the University of Auckland, Centre for Brain Research. Donated post‐mortem human brain tissue was obtained from the Neurological Foundation Human Brain Bank. The tissue was acquired through a donor program, the procedures were approved by the University of Auckland Human Participant's Ethics Committee (Approval number: 011654) and the study was not preregistered. Processing of tissue followed the procedure described in Waldvogel et al. (24). The right hemisphere of the brain was fixed by perfusion with 15% formalin, cut into anatomical blocks, cryoprotected with sucrose solutions, and frozen at −80℃. Hippocampal (also containing the subiculum and entorhinal cortex) and STG blocks were used for this study. Nine control (Table 1) and eight AD cases (Table 2), with an average age of 78.5 years and maximum post‐mortem time of 48 h were used for immunohistochemistry (IHC) experiments.
TABLE 1.
Normal human brain case details used for immunohistochemistry
| Case | Age | Sex | PM delay | Cause of death | Weight (g) |
|---|---|---|---|---|---|
| H122 | 72 | F | 9 | Emphysema | 1230 |
| H123 | 78 | M | 7.5 | Aortic aneurysm | 1260 |
| H169 | 81 | M | 24 | Asphyxia | 1225 |
| H180 | 73 | M | 33 | Ischemic heart disease | 1318 |
| H181 | 78 | F | 20 | Aortic aneurysm | 1292 |
| H202 | 83 | M | 14 | Aortic aneurysm | 1245 |
| H226 a | 73 | F | 48 | Mesothelioma | 1279 |
| H239 a | 64 | M | 15.5 | Ischaemic Heart Disease | 1529 |
| H245 | 63 | M | 20 | Asphyxia | 1194 |
Cases used for 3,3′‐diaminobenzidine‐peroxidase immunohistochemistry.
TABLE 2.
Alzheimer's disease human brain case details used for immunohistochemistry
| Case | Age | Sex | PM delay | Cause of death | CERAD classification | Braak and Braak score | Weight (g) |
|---|---|---|---|---|---|---|---|
| AZ45 | 82 | M | 4.5 | Pneumonia | Probable AD | IV | 1230 |
| AZ88 a | 83 | M | 21 | Pneumonia | Definite AD | IV | 1121 |
| AZ90 | 73 | M | 4 | Gastrointestinal haemorrhage | Definite AD | IV | 1260 |
| AZ92 | 93 | F | 11.5 | Bronchopneumonia | Probable AD | IV | 1225 |
| AZ98 | 91 | F | 20.5 | Alzheimer's dementia/atrial fibrillation | Definite AD | VI | 1318 |
| AZ102 | 84 | F | 14.5 | Lower respiratory tract infection & hyaline arteriosclerosis | Definite AD | VI | 1292 |
| AZ103 | 87 | M | <24 | Cerebrovascular accident | Definite AD | VI | 1245 |
| AZ113 a | 77 | M | 3.5 | Alzheimer's dementia/pneumonia | Definite AD | IV | 1261 |
Cases used for 3,3′‐diaminobenzidine‐peroxidase immunohistochemistry.
All control cases included in this study had no history of any primary neurodegenerative, psychiatric disorder, and neurological disease abnormalities, whereas all of the Alzheimer's cases had clinical dementia. Standard sections, including the middle frontal gyrus, middle temporal gyrus, cingulate gyrus, hippocampus, caudate nucleus, substantia nigra, locus coeruleus, and cerebellum were examined from both control and AD groups by a neuropathologist. The distribution and density of tau and Aβ pathology were assessed immunohistochemically. The neuritic plaque density in the AD cases was classified into sparse, moderate, or frequent according to the criteria from the Consortium to Establish a Registry for Alzheimer's disease (25). Only those cases that fit this criterion for definite or probable AD were included in this study.
2.2. Immunohistochemistry
Coronal sections of the hippocampus, subiculum, entorhinal cortex, and STG were cut on a freezing microtome at 60 μm and stored at 4℃ in phosphate‐buffered saline (PBS) containing 0.1% sodium azide. Two hippocampal and two STG sections, starting from the midpoint of the anterior commissure +21.2 mm for the hippocampal block (containing the hippocampus, subiculum, and entorhinal cortex, plate 38–41) and +9.3 mm for the STG block (plate 29–33) according to the Mai, Paxinos and Voss brain atlas (26), for each control and AD case, were immunostained with glutamate receptor subunits specific antibodies. Free‐floating 3,3′‐diaminobenzidine (DAB)‐peroxidase and fluorescent IHC were utilized for the visualization of AMPAR GluA2 subunit, NMDAR GluN1, and GluN2A subunits, using a method published previously (24, 27). The specificity of the primary antibodies has been reported previously for each antibody GluA2 (28), GluN1 (29, 30, 31, 32), and GluN2A (31, 33, 34, 35, 36, 37, 38, 39). All antibody dilutions were optimized. Primary antibodies and dilutions are described in Table 3. The use of the blocking peptide (BLP‐GC002) and omission of the primary antibodies resulted in a complete absence of immunoreactivity except for a small amount of background lipofuscin staining (Figure 1B,C). Antibodies against glutamate receptor subunits and NeuN were diluted in 1% normal goat serum, and 0.04% merthiolate in PBS (immunobuffer).
TABLE 3.
Primary antibodies used in this study
| Antigen | Immunogen | Source, host, species, catalogue number | Dilution (fIHC) | Dilution (DAB IHC) | Dilution (WB) |
|---|---|---|---|---|---|
| GFAP | Full‐length native protein of cow glial fibrillary acidic protein | Abcam, Chicken, ab4674, RRID:AB_304558 | 1:10,000 | 1:2000 | – |
| GluA2 | KLH‐conjugated linear peptide corresponding to the cytoplasmic domain of rat GluR2 | Millipore, Rabbit, AB1768, RRID:AB_ 2247874 | 1:200 | – | 1:100 |
| GluN1 | Recombinant protein corresponding to AA 660 to 811 from rat GluN1 | Synaptic Systems, Mouse, 114‐011, RRID:AB_887750 | 1:500 | 1:200 | 1:100 |
| GluN2A | Peptide GHSHDVTERELRN(C), corresponding to amino acid residues 41–53 of rat NMDAR 2A | Alamone, Rabbit, AGC‐002, RRID:AB_2040025 | 1:200 | 1:200 | 1:1000 |
| Anti‐Neuronal Nuclei (NeuN) | Purified cell nuclei from mouse brain | Millipore, Rabbit, ABN78, RRID:AB_10807945 | 1:1000 | – | – |
| Anti‐Neuronal Nuclei (NeuN) | Purified cell nuclei from mouse brain | Millipore, Mouse, MAB377 RRID:AB_2298772 | 1:1000 | – | – |
FIGURE 1.
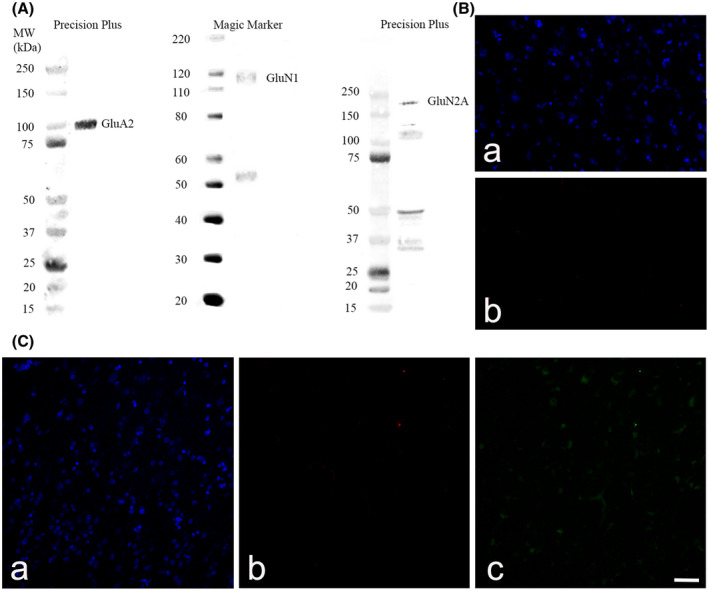
Western blot against human hippocampus homogenates probed with glutamatergic receptor subunit GluA2, GluN1 and GluN2A antibodies (A). The use of the GluN2A blocking peptide (BLP‐GC002) (B) and omission of the primary antibodies (C) resulted in a complete absence of immunoreactivity except for a small amount of background lipofuscin staining. Section were stained with goat anti‐rabbit Alexa Fluor 647 (B,b; C,b) and goat anti‐mouse Alexa Fluor 488 (C,c). Nuclei were counterstained with Hoechst dye (blue) (B,a; C,a). Scale bars B–C = 50 µm
2.3. DAB‐peroxidase immunohistochemistry
DAB‐peroxidase IHC was performed as described by Kwakowsky et al. (27). In brief, sections were washed in PBS with 0.2% Triton X‐100 (PBST) before blocking for endogenous peroxidases (50% methanol and 1% H2O2) for 20 min, followed by three 10‐min washes in PBST and incubated for 72 h in primary antibodies in immunobuffer at 4℃ (Table 3). The sections were then washed in PBST before incubation for 24 h with the biotinylated secondary antibodies (anti‐Mouse IgG‐Biotin antibody produced in goat 1:1000, anti‐rabbit IgG‐Biotin antibody produced in goat 1:1000) in immunobuffer at room temperature (RT). The sections were then washed in PBST before incubation with ExtrAvidin (1:1000, E2886; Sigma, St. Louis, MO, USA) in immunobuffer for 4 h at RT, followed by three 10‐min washes in PBST before development in 0.05% DAB and 0.01% H2O2 and 0.1 M phosphate buffer. Sections were washed in PBST and mounted onto glass slides, dried, dehydrated through a graded series of ethanol, and cleared in xylene. The slides were coverslipped with DPX mountant (1019790500; Merck, Whitehouse Station, NJ, USA). The sections were then imaged on either a Leica DMRB light microscope or a Leica MZ6 dissecting microscope (Wetzlar, Germany).
2.4. Fluorescent immunohistochemistry
A total of 13 cases, 7 control and 6 AD, were used in this experiment. Power calculations (GPower, 3.1.9.6; Heinrich‐Heine‐University Düsseldorf) based on our previous studies determined that a sample size of n = 5 per group would be necessary and sufficient to detect differences with 95% confidence (α = 0.05) and 0.9 power (27). However, additional cases were included to account for potential problems such as tissue damage. Free‐floating fluorescent IHC was performed as described previously by Kwakowsky et al. (27). In brief, sections that did not require antigen retrieval procedures were incubated in PBST overnight at 4℃. Sections that required antigen retrieval (those labeled with the GluA2 antibody) were washed once for 10 min with PBST, then stored 24 h in sodium citrate buffer (pH 6.0) at 4℃. Then the tissue was transferred into fresh citrate buffer in six‐well plates and microwaved in three 10 s blocks, waiting 5 s in between each block and incubated for 30 min at RT. Sections labeled with the GluN1 antibody were incubated in Tris‐EDTA pH9.0 buffer at 4℃ for 24 h followed by the same heat‐induced antigen retrieval method as described above.
These procedures were followed by three 10‐min washes with PBST and incubation for 72 h in the primary antibodies diluted in immunobuffer at 4℃ (Table 3). Sections were washed three times for 10 min in PBST before addition of secondary antibodies goat anti‐rabbit Alexa Fluor 647 (1: 500, A21245, RRID:AB_141775; Invitrogen, Carlsbad, California, USA), goat anti‐mouse Alexa Fluor 488 (1: 500, A11029, RRID: AB_138404; Invitrogen) or goat anti‐mouse Alexa Fluor 647 (1: 500, A21236, RRID:AB_141725; Invitrogen), goat anti‐rabbit Alexa Fluor 488 (1: 500, A11034, RRID:AB_2576217; Invitrogen), goat anti‐chicken Alexa Fluor 488 (1:500, A32931, RRID:AB_2762843; Invitrogen) and incubated for a further 24 h at RT. Sections were then washed for 10 min in PBST before incubation for 35 min at RT with Hoechst nuclei counterstain (1:10,000, 33342, RRID:AB_10626776, Invitrogen) diluted in PBS. After three subsequent 10‐min washes in PBS, sections were mounted onto glass slides, coverslipped with Mowiol mounting medium, and sealed with nail varnish.
2.5. Imaging and analysis
Imaging was conducted using a Zeiss 710 inverted confocal laser‐scanning microscope (Carl Zeiss, Jena, Germany). Brain regions and layers were differentiated based on cell type and relative location, utilizing NeuN and Hoechst staining. An argon laser was used to excite NeuN‐positive neurons at a 488‐nm wavelength, a helium‐neon laser with a 633nm wavelength was used for Alexa 647 immunolabeled antigens of interest, and a blue diode laser with a 405 nm wavelength was used for Alexa 405 for Hoechst counterstained nuclei with a 20x objective. Using ImageJ software (U. S. National Institutes of Health, Bethesda, Maryland, USA), after background subtraction and grayscale threshold determination, the glutamate receptor subunit density measurements were performed from a 31,000 μm2 area in each analyzed layer in the dentate gyrus (str. granulosum, str. moleculare and hilus), CA1, CA2, and CA3 (str. oriens, str. pyramidale, str. radiatum). Density measurements for the subsequent regions were measured using the following parameters: a 432,000 μm2 region in the subiculum, a 605,000 μm2 region in the entorhinal cortex, and a 692,000 μm2 region in the STG through all cortical layers. Both the threshold and the size of the region of interest were constant across all sections for each region in each experiment. The analysis was performed blinded to the experimental groupings to eliminate bias during the experiment, including image acquisition and analysis. Two hippocampal and two STG tissue sections from each case were randomized following standard simple randomization procedures in a blinded fashion. The 26 sections (2 sections/well; one hippocampal and one STG section) were in six‐well plates labeled, new numbers were allocated to each well 1–26 by a person not involved in the study otherwise, these numbers were written on each well with a different color marker pen, a photo was taken and the old/ new numbers recorded on paper. Subsequently, the sections were transferred to new plates in order (1–26) by the same person.
2.6. Western blotting
Western blotting was performed as described by Kwakowsky et al. (27). Lysis buffer containing 50 mM Tris, 2 mM EDTA, 4% sodium dodecyl sulfate, pH 6.8 supplemented with 1 mM phenylmethanesulfonylfluoride and 1% protease inhibitor cocktail (Sigma‐Aldrich Co., Saint Louis, Missouri, USA; P8340) was added to human hippocapal tissue samples (30μg) and protein extracts prepared using 0.5 mm glass beads (Mo‐Bio Laboratories, Solano Beach, California, USA) using a Mini Bullet Blender Tissue Homogeniser (Next Advance, Inc, New York, USA) at speed 8 for 8 min. The homogenates were incubated for 1 h on ice, then centrifuged at 10,621 g for 10 min. The supernatant was collected and stored at 20℃. Protein concentrations were determined by using the Bio‐Rad Detergent Compatible Protein assay (Bio‐Rad, California, USA).
Twenty micrograms of each protein extract was run on a gradient – polyacrylamide electrophoresis gel (NU PAGE 4%–12% BT 1.5, NP0336BOX; Life Technologies, Carlsbad, CA, USA) and then blotted. Proteins were separated in XCell SureLock Mini‐Cell system (Invitrogen, Scoresby, VIC, Australia) and transferred onto nitrocellulose membranes using a Mini Trans‐Blot Electrophoretic Transfer system (Bio‐Rad, California, USA). Two molecular weight ladders, Magic mark (Thermo Fisher, California, USA) and Precision Plus (Bio‐Rad, California, USA), were also loaded in gels as verification of labeled band size. Following transfer, membranes were washed in Tris‐buffered saline pH 7.6, 0.1% Tween (TBST) for 5 min and then blocked with LiCor Odyssey Blocking Buffer (LI‐COR Biosciences, Nebraska, USA) for 30 min at RT on the orbital shaker. Following blocking, another 5‐min wash with TBST was performed prior to overnight incubation at 4℃ with primary antibodies diluted in 4% BSA‐TBST (Table 3). Subsequently, membranes underwent three 5‐min TBST washes prior to the addition of secondary antibody (1:10,000, goat anti‐rabbit IRDye®680RD, 926–68071, RRID:AB_10956166; goat anti‐mouse IRDye®800CW, 926–32210, RRID:AB_621842), which was incubated for 1 h at RT. Following incubation, membranes were washed three times in TBST for 10 min each and subsequently in TBS for 10 min. Imaging was performed on the Odyssey Infrared Imaging System (LI‐COR Biosciences, Nebraska, USA).
2.7. Statistical analysis
To examine differences between groups, an unpaired Mann–Whitney test was used as the data did not meet the assumptions of parametric tests assessed by the D'Agostino–Pearson omnibus and Brown–Forsythe tests. No data points were identified and excluded as outliers using the ROUT method. All statistical analyses were conducted using Graph‐Pad Prism software version 8 (GraphPad software; RRID:SCR_002798) with a value of p ≤ 0.05 considered significant. Adobe Photoshop CC 2018 (Adobe Systems Software, San Jose, CA, USA) was used to prepare the figures. All experimental data are expressed as the mean ± standard error of mean (SEM).
3. RESULTS
3.1. Expression of AMPAR GluA2 subunit in the human hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus
GluA2 immunoreactivity within the DG was relatively sparse, with substantial neuronal staining within the hilus area (Figure 2A1) and staining surrounding cell bodies within the str. granulosum and most likely astrocytic processes throughout all the layers (Figures 2A1,E1 and 3Da,b). GluA2 within the CA2 subregion showed dense fibrous staining, with labeling concentrated around the pyramidal neurons and staining of neuronal and glial processes within the str. radiatum (Figures 2C1, arrows and 3Ba,b). Immunoreactivity within the CA3 subregion followed similar patterns to the CA2 region but the labeling was weaker in the str. pyramidale (Figures 2D1 and 3Ca,b). The CA1 subregion revealed weaker GluA2 immunoreactivity in comparison to CA2 and CA3, although the str. pyramidale within the CA1 displayed strong localization in some cases to neuronal cell bodies and their adjacent processes (Figure 2B1, arrows) as well as glial processes (Figures 2B1 and 3Aa,b). GluA2 neuronal cell body labeling could be seen within the subiculum (Figures 2H1 and 4Aa,b), whilst labeling within the entorhinal cortex was comparatively weaker and primarily localized to glial and neuronal fibers (Figures 2G1, arrow and 4Ba,b). The STG displayed dense GluA2 immunoreactivity in superficial cortical layers (layer I‐II), with sparse labeling of cell bodies and neuronal and glial processes throughout the lower cortical layers (Figures 2G1, arrow and 4Ca,b).
FIGURE 2.
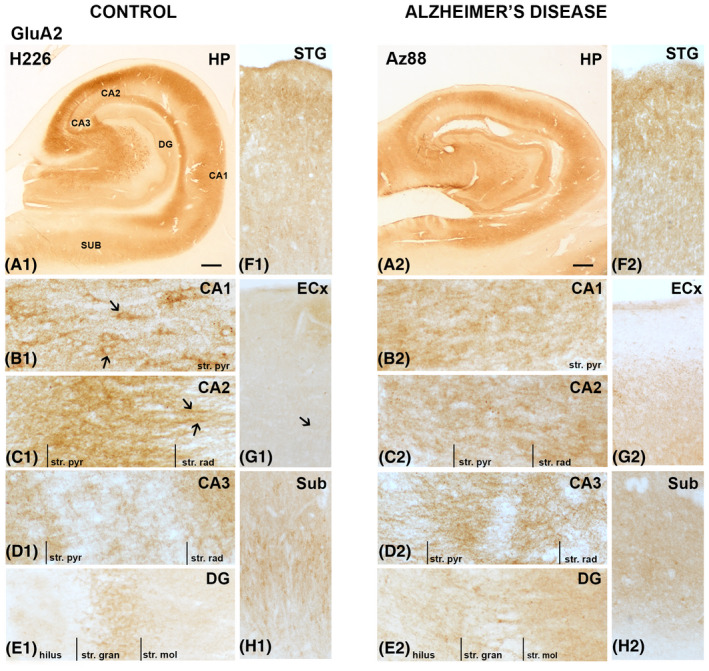
GluA2 expression in the hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus in human control and Alzheimer's disease cases visualized by 3,3′‐diaminobenzidine‐peroxidase immunohistochemistry. Staining appeared relatively strong within the str. pyramidale of the CA regions (A–D), with an increase in expression within the stratum (str.) moleculare of the dentate gyrus in AD (E1,2). Arrows indicating localization to processes and around pyramidal neurons. CA, cornu ammonis; DG, dentate gyrus; ECx, entorhinal cortex; HP, hippocampus; STG, superior temporal gyrus; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. gran, stratum granulosum; Sub, subiculum. Scale bars: A1,2 = 1000 µm; B1–E1, B2‐E2 = 100 µm; F1–H1, F2–H2 = 400 µm
FIGURE 3.

GluA2 expression is altered in the dentate gyrus in Alzheimer's disease. Photomicrographs of representative regions of the CA1(A), CA2 (B), CA3 (C), and dentate gyrus (D) showing GluA2 (red) and GluA2 overlaid with NeuN (green) immunoreactivity for representative Alzheimer's disease and control cases. AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; str. ori, straum oriens; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. mol, stratum moleculare; str. gran, stratum granulosum. Scale bars A–C = 100 µm, D = 50 µm
FIGURE 4.
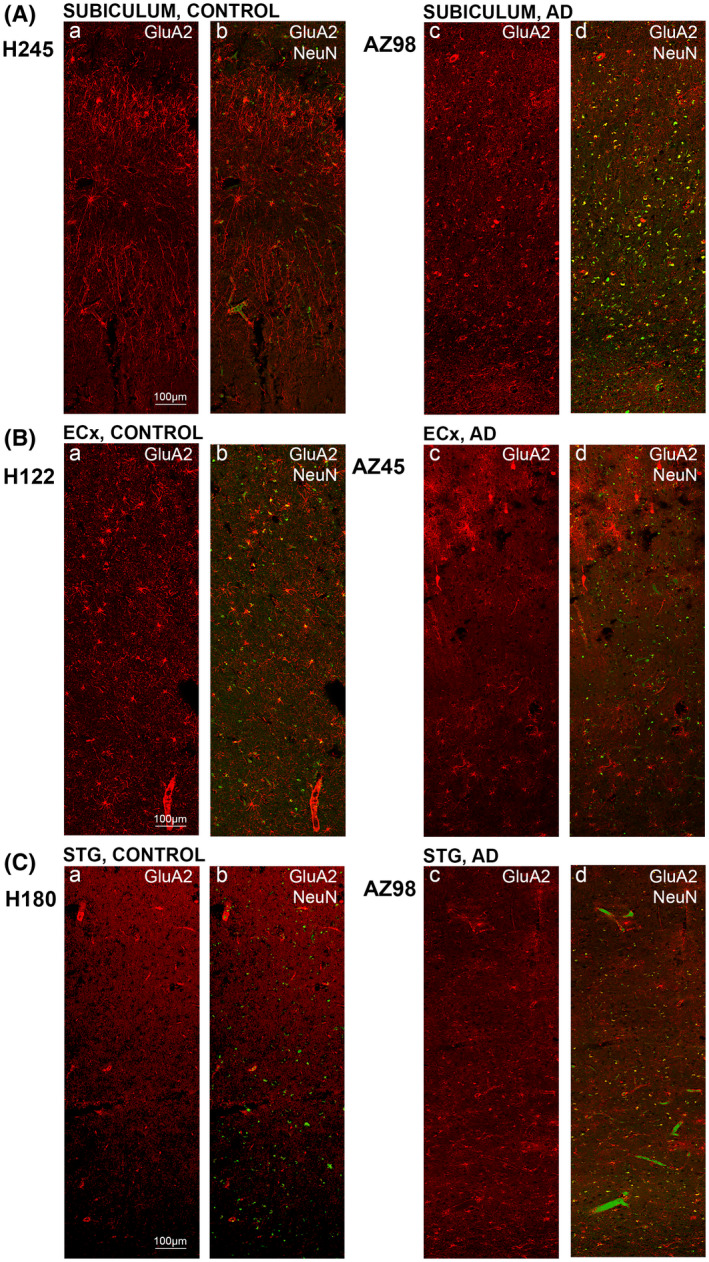
GluA2 expression in the subiculum, entorhinal cortex and superior temporal gyrus in human control and Alzheimer's disease cases. Photomicrographs of representative regions of the subiculum (A), entorhinal cortex (B), and superior temporal gyrus (C) showing GluA2 (red) and GluA2 overlaid with NeuN (green) immunoreactivity for representative Alzheimer's disease and control cases. AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; ECx, entorhinal cortex: STG, superior temporal gyrus. Scale bars A–C, E–G = 100 µm, D = 50 µm
AD cases displayed increased immunoreactivity within the str. granulosum of the DG compared with control (Figures 2E1,2 and 3Da–d). Labeling within the CA2 and CA3 regions was weaker along fibrous structures compared to control cases (Figures 2C1,2;D1,2 and 3Ba–d;Ca–d). In AD, the staining patterns of the CA1 appear to become more homogenous in comparison to control, with an absence of the neuronal localization seen in control cases (Figures 2B1,2 and 3Aa–d). GluA2 immunoreactivity within the subiculum displayed a slight decrease in AD, especially along the neuronal processes (Figures 2H1,2 and 4Aa–d). The entorhinal cortex and STG appeared similar in AD compared to control cases (Figures 2F1,2;G1,2 and 4Ba‐d;Ca‐d).
A significant increase in AMPAR GluA2 subunit expression was detected in AD compared to control cases in the str. moleculare of the DG region (p = 0.0047; Figures 2E1,2, 3Da–d, and 5D). Otherwise, there was no significant change in GluA2 subunit density in AD compared to control cases in any of the brain regions examined (Figures 2, 3, 4, 5; Table 4).
FIGURE 5.

Quantification of GluA2 immunoreactivity within the CA1, CA2, CA3, dentate gyrus hippocampal subfields, subiculum, entorhinal cortex, and superior temporal gyrus in control and Alzheimer's disease groups. Data are expressed as mean with error bars representing SEM. The figure shows significant increases in GluA2 expression in the stratum moleculare (D) in AD cases (white circles; n = 5–6) compared to controls (black circles; n = 5–7; Unpaired Mann‐Whitney test). AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; str. ori, straum oriens; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. mol, stratum moleculare; str. gran, stratum granulosum
TABLE 4.
Summary of results for fluorescent immunohistochemistry in the human AD hippocampus subiculum, entorhinal cortex and STG
| Receptor subunit | CA1 | CA2 | CA3 | DG | Sub | ECx | STG | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| str. ori | str. pyr | str. rad | str. ori | str. pyr | str. rad | str. ori | str. pyr | str. rad | str. mol | str. gran | hilus | ||||
| GluA2 | – | – | – | – | – | – | – | – | – | ↑↑ | – | – | – | – | – |
| GluN1 | – | – | ↑ | ↑ | ↑↑ | ↑ | ↑↑ | – | ↑↑ | ↑↑ | – | ↑↑ | – | ↑ | – |
| GluN2A | ↑↑ | ↑↑ | ↑↑ | – | – | – | – | – | – | – | – | – | – | – | – |
↑↓ in AD cases compared to control cases; ↑ = p < 0.05, ↑↑ = p < 0.01.
Abbreviations: CA, cornu ammonis; DG, dentate gyrus; ECx, entorhinal cortex; STG, superior temporal gyrus; Sub, subiculum.
3.2. Expression of NMDAR GluN1 subunit in the human hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus
GluN1 immunoreactivity was primarily localized to neurons of the str. pyramidale and adjacent dendritic processes within the CA1 subregion (Figures 6A1 arrows and 6B1). A similar staining pattern was observed in the CA2 (Figures 6C1 and 7Ba,b) and CA3 (Figures 6D1 and 7Ca,b) subregions, but with less localization to neurons and processes. In the DG, GluN1 immunoreactivity was very weak surrounding cell bodies of the str. granulosum and slightly stronger in the str. moleculare (Figures 6A1,E1 and 7Da–d). The subiculum displayed stronger and denser immunoreactivity concentrated around neuronal bodies and associated processes (Figures 6H1 and 8Aa,b). The entorhinal cortex (Figures 6G1 and 8Ba,b) and STG (Figures 6F1 and 8Ca,b) revealed diffuse immunoreactivity with weak labeling along fibers and surrounding some neurons.
FIGURE 6.
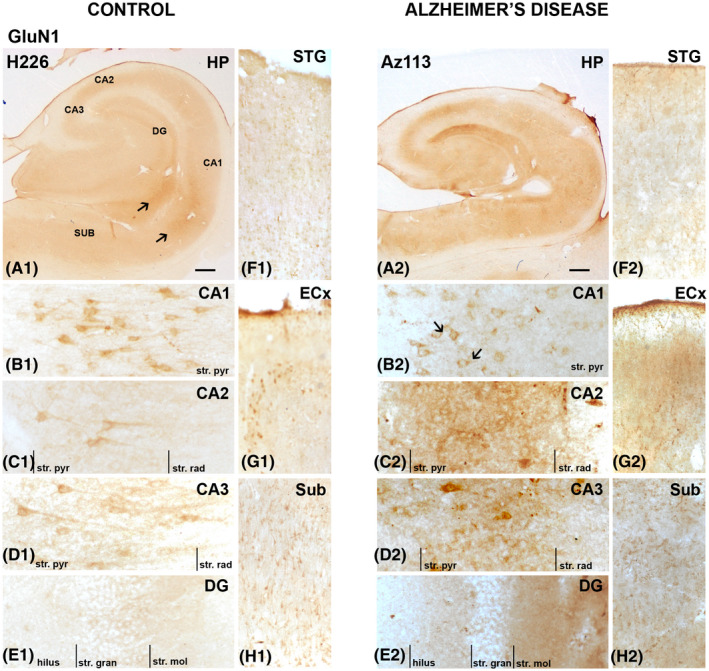
GluN1 expression in the hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus in human control and Alzheimer's disease cases visualized by 3,3′‐diaminobenzidine‐peroxidase immunohistochemistry. Staining appeared moderate within the str. pyramidale of the CA regions (A–C), with an increase in expression within these regions and the stratum (str.) moleculare of the dentate gyrus in AD (E1,2). Arrows indicating localization to processes and around pyramidal neurons. CA, cornu ammonis; DG, dentate gyrus; ECx, entorhinal cortex; HP, hippocampus; STG, superior temporal gyrus; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. gran, stratum granulosum; Sub, subiculum. Scale bars: A1,2 = 1000 µm; B1–E1, B2–E2 = 100 µm; F1–H1, F2–H2 = 400 µm
FIGURE 7.

GluN1 expression is altered in hippocampal subfields in Alzheimer's disease. Photomicrographs of representative regions of the CA1(A), CA2 (B), CA3 (C), and dentate gyrus (D) showing GluN1 (red) and GluN1overlaid with NeuN (green) immunoreactivity for representative Alzheimer's disease and control cases. AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; str. ori, straum oriens; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. mol, stratum moleculare; str. gran, stratum granulosum. Scale bars A–C = 100 µm, D = 50 µm
FIGURE 8.
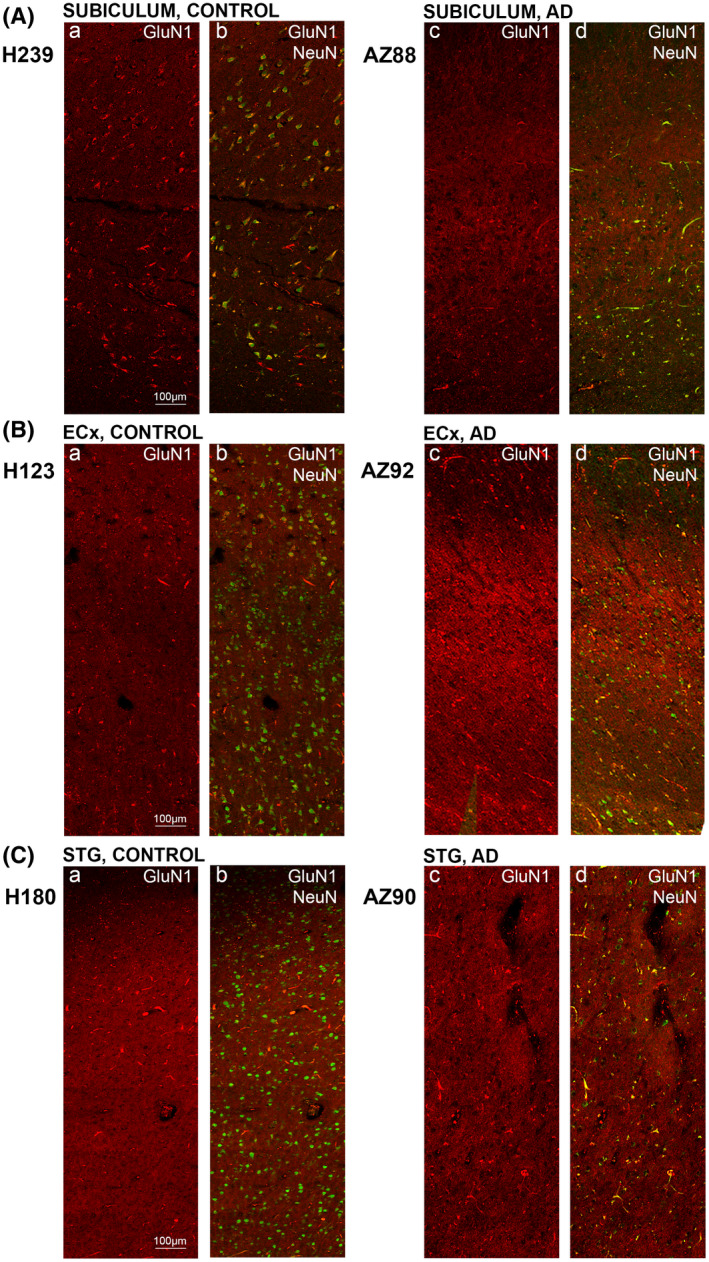
GluN1 expression in the subiculum, entorhinal cortex and superior temporal gyrus in human control and Alzheimer's disease cases. Photomicrographs of representative regions of the subiculum (A), entorhinal cortex (B), and superior temporal gyrus (C) showing GluN1 (red) and GluN1overlaid with NeuN (green) immunoreactivity for representative Alzheimer's disease and control cases. AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; ECx, entorhinal cortex: STG, superior temporal gyrusstr; ori, straum oriens; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. mol, stratum moleculare; str. gran, stratum granulosum. Scale bars A–C, E–G = 100 µm, D = 50 µm
In AD, there is a decrease in GluN1 density surrounding neurons throughout the hippocampus, subiculum, entorhinal cortex, and STG (Figures 6, 7, 8), with a more membrane‐localized labeling in the CA1 region (Figure 6B1,2) and increased staining of neuropil in all regions examined (Figures 6, 7, 8). AD cases displayed increased immunoreactivity within the str. moleculare of the DG compared with control (Figures 6E1,2 and 7Da–d).
A significant increase in NMDA GluN1 subunit density was detected in AD compared to control cases in the str. radiatum of the CA1 region (p = 0.0140; Figures 6A1,2;B1,2, 7Aa–d, and 9A), the str. moleculare (p = 0.0082), and hilus (p = 0.0012) of the DG (Figures 6A1,2;E1,2, 7Da–d, and 9D), and the entorhinal cortex (p = 0.0140; Figures 6G1,2, 8Ba–d, and 9F). GluN1 expression showed a significant increase in AD in all layers (str. oriens, p = 0.0242; str. pyramidale, p = 0.0051; str. radiatum, p = 0.0480) of the CA2 subregion (Figures 6A1,2;C1,2, 7Ba–d and 9B), and in the str. oriens (p = 0.0012) and str. radiatum (p = 0.0023) of the CA3 subregion (Figures 6A1,2;D1,2, 7Ca–d, and 9C). No significant differences in GluN1 subunit labeling density were seen in AD compared with control in the subiculum (Figures 6A1,2;H1,2, 8Aa–d, and 9E), and STG (Figures 6F1,2, 7Ca–d, and 9G; Table 4).
FIGURE 9.
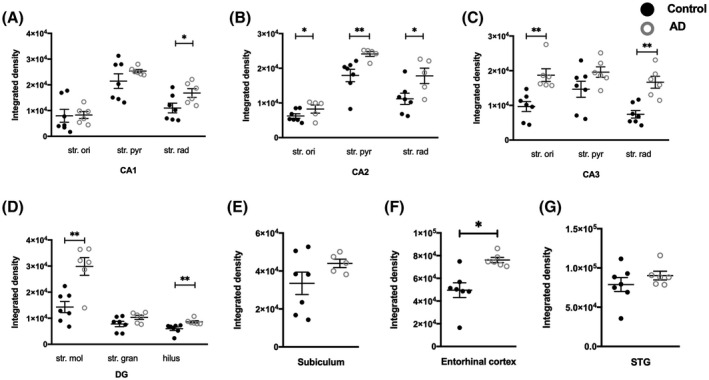
Quantification of GluN1 immunoreactivity within the CA1, CA2, CA3, dentate gyrus hippocampal subfields, subiculum, entorhinal cortex, and superior temporal gyrus in control and Alzheimer's disease groups. Data are expressed as mean with error bars representing SEM. The figure shows significant increases in GluN1 expression in specific layers of the hippocampal subfields (A–D) and the entorhinal cortex (F) in AD cases (white circles; n = 5–6) compared to controls (black circles; n = 6–7; unpaired Mann–Whitney test). AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; str. ori, straum oriens; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. mol, stratum moleculare; str. gran, stratum granulosum
3.3. Expression of NMDAR GluN2A subunit in the human hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus
GluN2A immunohistochemistry revealed glia‐like staining throughout the hippocampus, subiculum, entorhinal cortex, and STG, with increased reactivity in the most superficial layers, particularly of the entorhinal cortex and STG (Figures 10, 11, 12). Co‐labeling of GluN2A and GFAP, an astrocytic marker, revealed GluN2A expression on astrocytes (Figure 13). Lipofuscin was observed in some sections (Figure 11Db, asterisked arrows).
FIGURE 10.

GluN2A expression in the hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus in human control and Alzheimer's disease cases visualized by 3,3′‐diaminobenzidine‐peroxidase immunohistochemistry. Staining appeared relatively strong on glial cells and their processes throughout the hippocampus with an increased labeling in the CA1 subfield in AD (B1,2). Arrows indicating localization to astrocytic cell bodies and processes. CA, cornu ammonis; DG, dentate gyrus; ECx, entorhinal cortex; HP, hippocampus; STG, superior temporal gyrus; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. gran, stratum granulosum; Sub, subiculum. Scale bars: A1,2 = 1000 µm; B1–E1, B2–E2 = 100 µm; F1–H1, F2–H2 = 400 µm
FIGURE 11.
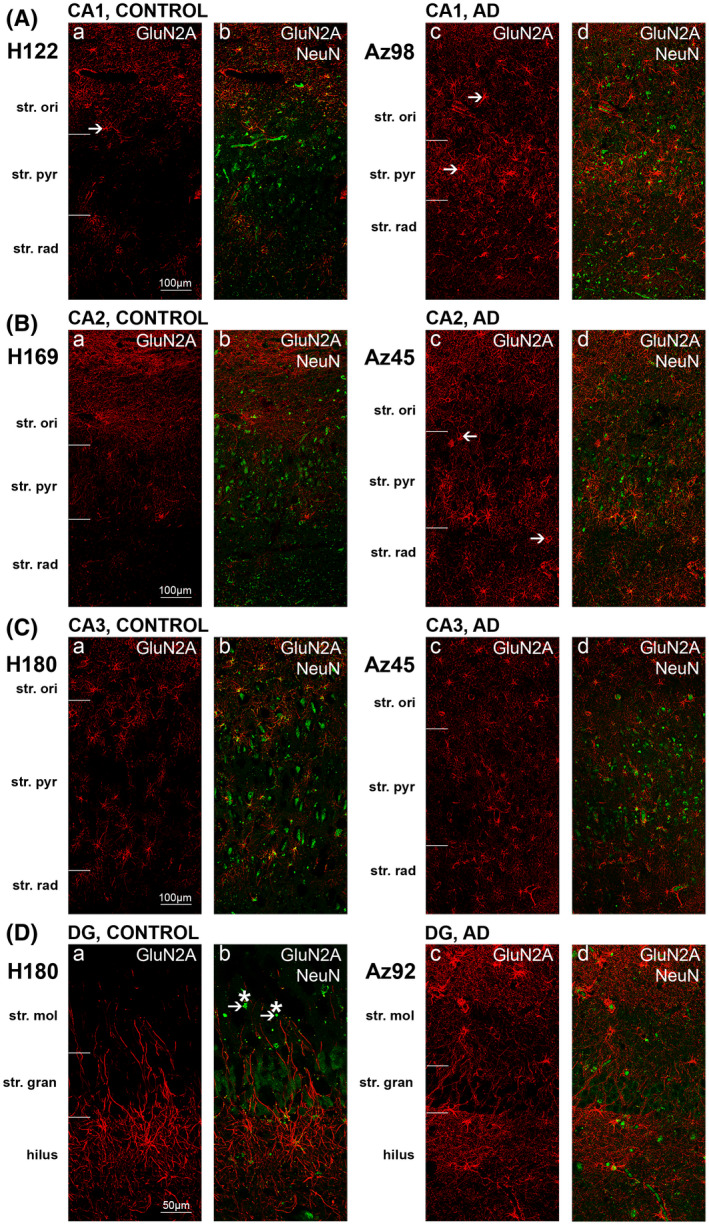
GluN2A expression is altered in hippocampal CA1 subfield in Alzheimer's disease. Photomicrographs of representative regions of the CA1(A), CA2 (B), CA3 (C), and dentate gyrus (D) showing GluN2A (red) and GluN2Aoverlaid with NeuN (green) immunoreactivity for representative Alzheimer's disease and control cases. AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; str. ori, straum oriens; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. mol, stratum moleculare; str. gran, stratum granulosum. Scale bars A–C = 100 µm, D = 50 µm
FIGURE 12.

GluN2A expression in the subiculum, entorhinal cortex and superior temporal gyrus in human control and Alzheimer's disease cases. Photomicrographs of representative regions of the subiculum (A), entorhinal cortex (B), and superior temporal gyrus (C) showing GluN2A (red) and GluN2Aoverlaid with NeuN (green) immunoreactivity for representative Alzheimer's disease and control cases. AD, Alzheimer's disease; ECx, entorhinal cortex: STG, superior temporal gyrus. Scale bars A–C, E–G = 100 µm, D = 50 µm
FIGURE 13.
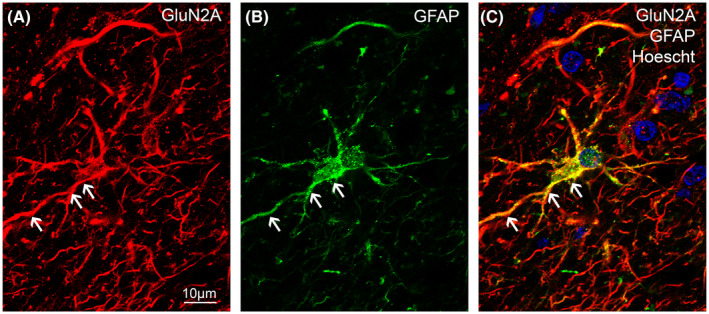
GluN2A immunoreactivity on astrocytic cell bodies and processes in the human hippocampus. GluN2A subunit (red) and GFAP (green) immunoreactivity are co‐localized (yellow) within the CA1 subregion of the human hippocampus. Nuclei were counterstained with Hoechst dye (blue). Scale bars A–C, E–G = 10 µm
In AD, immunoreactivity appeared much stronger, particularly within the CA1 subregion (Figures 10A1,2;B1,2 and 11Aa–d). There is an increase in both glial cell body staining, mainly astrocytic (Figures 10B2 and 11Aa–d, arrows), and increased immunolabeling along the processes (Figures 10B2 and 11Aa–d). The same trend was observed within the CA2 (Figures 10C1,2 and 11Ba–d, arrows), CA3 (Figures 10D1,2 and 11Ca–d), DG (Figures 10E1,2, arrows and 11Da–d), subiculum (Figures 10H1,2 and 12Aa–d), entorhinal cortex (Figures 10G1,2 and 12Ba–d) and STG (Figures 10F1,2 and 12Ca–d). GluN2A immunofluorescence also appeared more variable within AD brains compared to control brains.
Quantification of NMDAR GluN2A immunolabeling revealed statistically significant increases in intensity in AD cases compared to control in all three layers of the CA1 subregion, the str. oriens (p = 0.0095), str. pyramidale (p = 0.0095) and str. radiatum (p = 0.0381; Figure 14A). A similar trend was observed within the CA2 (Figure 14B) and CA3 (Figure 14C) subregions, str. moleculare and str. granulosum of the DG (Figure 14D), subiculum (Figure 14E), and entorhinal cortex (Figure 14F) although this did not reach significance. In the STG, GluN2A density showed no statistically significant change in AD compared to control cases (Figure 14G; Table 4).
FIGURE 14.
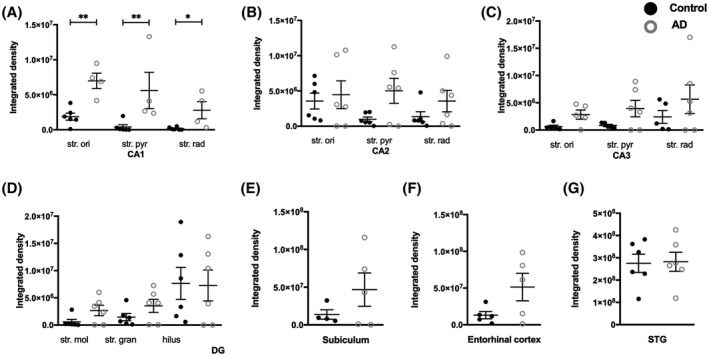
Quantification of GluN2A immunoreactivity within the CA1, CA2, CA3, dentate gyrus hippocampal subfields, subiculum, entorhinal cortex, and superior temporal gyrus in control and Alzheimer's disease groups. Data are expressed as mean with error bars representing SEM. The figure shows significant increases in GluN2A expression in specific layers of the hippocampal subfields (A–D) in AD cases (white circles; n = 4–6) compared to controls (black circles; n = 4–6) (Unpaired Mann‐Whitney test). AD, Alzheimer's disease; CA, cornu ammonis; DG, dentate gyrus; STG, superior temporal gyrus; str. ori, straum oriens; str. pyr, stratum pyramidale; str. rad, stratum radiatum; str. mol, stratum moleculare; str. gran, stratum granulosum
4. DISCUSSION
The present study is the first to provide a comprehensive examination of expression levels and patterns of glutamate receptor subunits GluA2, GluN1, and GluN2A in the human hippocampus, subiculum, entorhinal cortex, and STG, and how this expression is altered in AD. Our findings indicate differential region‐ and layer‐specific changes of these glutamatergic receptor subunits in the AD human hippocampus, subiculum, and entorhinal cortex in comparison to healthy controls. Increased GluA2 receptor expression was found within the str. moleculare of the DG in AD compared to control cases. There was a significant increase in GluN1 receptor subunit expression within the str. moleculare and hilus of the DG, str. radiatum of the CA1, str. oriens, str. pyramidale, and str. radiatum of the CA2, the str. oriens and str. radiatum of the CA3, and the entorhinal cortex. We also observed increased GluN2A receptor subunit expression throughout the CA1 subregion in AD.
4.1. GluA2 subunit expression in the AD human hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus
The GluA2 subunit holds significant importance in regulating the function of the AMPARs, as it is responsible for maintaining the receptor's impermeability to calcium. 40 As such, a loss of GluA2 may result in an increase in calcium‐permeable AMPARs, leading to increased cytosolic calcium accumulation and neuronal death. Alterations in the regulation of GluA2 expression have been observed in a variety of disease states, and can occur through many mechanisms, including and not limited to: reduced or defective GluA2 mRNA editing and transcription, altered post‐translational modifications, and internalization of GluA2 containing AMPARs. 41 Aside from an increase in GluA2 subunit expression within the str. moleculare of the DG, we observed no significant expression changes within other layers and regions of the hippocampus, subiculum, entorhinal cortex, and STG. There is a lack of quantitative studies examining the expression of GluA2 in these brain regions in the AD brain.
Interestingly, Armstrong and Ikonomovic (20) demonstrated variable subunit expression alterations within different regions of the brain, with vulnerable sectors of the hippocampus (e.g., CA1, subiculum) demonstrating loss in GluA2/3 immunolabeling, while less vulnerable regions, for example, CA2/3 and DG demonstrated a marked increase in expression. The authors postulated the heterogenous changes are reflective of extensive cell loss and neurofibrillary pathology within vulnerable regions of the hippocampus (20). These studies offer an interesting insight into potential mechanisms at play, including upregulation of GluA2 within less vulnerable regions of the hippocampus, a decrease in expression within more vulnerable regions secondary to cell loss, and downregulation of GluA2 subunit expression in regions outside of the hippocampus, which is independent of cell loss. It is important to note that these observations were qualitative. Another qualitative immunohistochemical study also found a substantial loss in GluA2 labeled neurons within the entorhinal cortex (42). Later, the Armstrong group demonstrated that GluA2 expression within the CA1, CA2, CA3, and DG were similar across different Braak stages in AD (22). This is in agreement with our findings in the CA subregions. This study involved homogenization of tissue from these hippocampal subregions. Therefore, while it was able to quantify regional differences in receptor subunit expression, possible layer‐specific changes in the DG have been missed. Interestingly, while a significant decrease in expression was observed within the subiculum between mild (Braak stage I and II) and moderate (Braak stage III and IV), this difference was lost when compared to severe cases (Braak stage V and VI), suggesting more complex mechanisms that evolve with the progression of AD. We observed a trend towards decreased GluA2 subunit expression within the subiculum that did not reach significance. The AD cases used in our study are Braak stage IV and VI, but we found no Braak stage‐specific expression of the GluA2 subunit, some of the stage IV cases showed as high receptor subunit density as the stage VI cases. Importantly, this study did not include a healthy control group. Therefore, based on the findings of Carter and colleagues and our results we can conclude that GluA2 subunit expression is robust against AD pathology in all the brain regions examined except the DG. In our earlier mouse study, we also report no significant changes in GluA2 expression between control and Aβ1–42‐injected mice in all layers of the CA1, CA3, and DG (31). However, in the str. moleculare of the DG we detected a trend towards increased GluA2 receptor density suggesting the involvement of Aβ1‐42 in the regulation of GluA2 subunit levels (31).
There are a variety of mechanisms described in the literature which may result in increased GluA2 subunit expression. Protein interacting with C kinase 1 (PICK1) is an integral protein in the scaffolding of GluA2 to the synapse and is necessary for the endocytosis of GluA2 and AMPAR, with a direct role in the subunit composition and expression of GluA2‐containing AMPARs (43, 44). This is exemplified in a study by Citri et al. (45) who demonstrated the possibility of AMPAR internalization being controlled by calcium‐induced modification of PICK1. Therefore, it is possible that NMDAR‐dependent release of calcium, which is dysfunctional in AD, can result in alterations in proteins involved in the maintenance of GluA2‐containing AMPARs at the synaptic membrane. Alfonso et al. (43), through knockout PICK1 mice, has demonstrated a failure of Aβ to induce synaptic dysfunction and GluA2 subunit reduction in the absence of the PICK1 protein. PICK1 also requires tau‐phosphorylation at Ser396 to initiate this process, alongside CaMKII, both of which may lose functionality in AD (44, 46). Tau‐phosphorylation has also been shown to induce neurodegenerative gain‐of‐function at the synapse, preventing endocytosis of synaptic components, including AMPARs (47, 48). As both Aβ and phosphorylated tau are predominant hallmarks in AD and are considered the forefront causative molecules of AD pathophysiology, its potential dual interactions with PICK1 and opposing effects on GluA2 expression warrants further investigations.
4.2. GluN1 subunit expression in the AD human hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus
GluN1 functions as a ubiquitous subunit within the NMDARs, with all NMDARs requiring the presence of GluN1 to be functional (49). The aberration of NMDARs has long been associated with Aβ toxicity (10, 11). Despite its central role in NMDAR function, there is little literature documenting GluN1 subunit concerning its functional significance and how it is altered in AD.
The GluN1 subunit displayed increased expression in AD brains compared to controls in many areas measured, including the CA1, CA2, CA3, DG, and entorhinal cortex. In agreement with our findings, no significant difference was reported in GluN1 mRNA expression within different regions of the brain, including the frontal lobe, parahippocampal gyrus, subiculum, and the STG in AD compared to controls (50). Also, in line with our observations, a significantly increased expression of GluN1 receptor subunit within the entorhinal and frontal cortex of AD brains was found compared to controls (51). However, there is scarce literature surrounding the expression of the GluN1 subunit in the AD hippocampus.
An early study utilizing immunohistochemistry demonstrated a qualitative increase in GluN1‐positive pyramidal neurons within the CA regions, particularly in severe AD cases compared to mild AD and controls (21). However, Sze et al. (23) demonstrated a marked decrease in GluN1 subunit expression in the whole hippocampus in AD brains compared to controls, using Western blot. Sze et al. (23) also observed no changes in GluN1 levels in early AD compared to control brains, suggesting the expression decrease in AD occurs at a “later” stage. This “later” stage is represented by two stage III and V and two stage VI cases while our AD cohort has three stage IV and three stage VI cases with more severe deficits based on neuropsychological scores. This indicates our AD group is representing an even later stage of AD. Interestingly, in agreement with our findings we also reported increased expression of the GluN1 subunit in the CA1 str. radiatum, CA3 str. radiatum and str. oriens, DG hilus and ventral str. granulosum in response to acute injection of Aβ1–42 in mice (39). Thirty days after injection of Aβ1–42 there were no significant alterations in GluN1 subunit expression but we observed a trend towards increased GluN1 density in the CA3 and DG subregions (31).
4.3. GluN2A subunit expression in the AD human hippocampus, subiculum, entorhinal cortex, and superior temporal gyrus
The expression of NMDA receptors has been well characterized within the rat and monkey neocortex (52), but its expression in the hippocampus has remained relatively unexplored. Our recent comprehensive studies provided a detailed examination of GluN1 and GluN2A subunit expression in the mouse hippocampus, with region and layer‐specific glial and neuronal labeling (31, 39). Previous rodent studies also indicate predominant localization to neuronal dendritic spines and astrocytes (19, 53, 54, 55, 56). GluN2A subunit localization to synapses has been linked to neuroprotection in mice, inhibiting apoptotic mechanisms by regulating synaptic calcium influx (57). Only one human study reported neuronal GluN2A/B expression (58). However, the antibody used in this study does not distinguish between the GluN2A and GluN2B subunits. Because the GluN2B subunit has been reported to localize to neurons in humans and rodents, it is difficult to extrapolate GluN2A expression patterns based on this study (59, 60). Interestingly, using the same antibody as in our previous mouse studies, our results indicated strong astrocytic staining, which is in line with previous animal studies. However, there is no evidence of neuronal labeling in the human hippocampus, subiculum, entorhinal cortex, and STG.
Our results indicated an upregulation of GluN2A subunit levels within all three layers of the CA1 subregion in the AD hippocampus compared to controls, with GluN2A typically expressed along glial processes surrounding cell bodies. The altered expression levels of the GluN2A subunit in AD is a contentious topic. Reductions in mRNA levels of GluN2A have been reported within the entorhinal cortex and hippocampus (61). In the following study, Sze et al. (23) also demonstrated a decrease in GluN2A protein levels within the entorhinal cortex, although no change was seen within the hippocampus. However, Western blotting experiments conducted by Sze et al. (23) did not examine region‐specific changes, as the preparation of the tissue involved homogenization of the whole hippocampus as opposed to specific regions. As a result, changes we observed specifically in the CA1 subregion were lost in their study. A study involving an Aβ1‐42‐ and AlCl3‐injected AD rat model indicated no significant change in GluN2A expression within the hippocampus (59). However, again, a potential reason for this study observing no expression change may be because of homogenization of the entire hippocampus for Western blot that removes the ability to discern any layer‐specific changes. We also report no acute Aβ1‐42‐induced alteration in GluN2A subunit density within the mouse hippocampus (39), but chronic exposure resulted in downregulated subunit levels secondary to reduced neuronal labeling (31). In the mouse CA1 subregion 3‐days post‐Aβ1‐42 injection astrogliosis is high and extended throughout the region, but at 3‐days post‐injection, increased GFAP density and astrocytes with activated morphology are localized to the injection site only (62). In the human CA1 subfield we did not detect GluN2A on neurons and the upregulation of GluN2A is associated with increased astrogliosis, the characteristic feature of AD and other neurodegenerative disorders (63, 64, 65), and increased expression of the subunit is localized to GFAP positive cells and processes. Recent studies confirmed that astrocytes can respond to neuronal stimuli mediated by a variety of neurotransmitters, including glutamate, and modulate neuronal activity (66, 67, 68). Glutamate or selective NMDAR agonists mediate astrocytic ATP release, ion currents, and intracellular calcium waves (67, 69). NMDAR‐dependent calcium signaling mediates astrocytic modulation of presynaptic strengths in the CA1 hippocampal subfield (70). Furthermore, persistent stimulation of NMDARs in astrocytes has been suggested to occur in AD and pathologies associated with excitotoxicity (67). The upregulated astrocytic GluN2A subunit expression observed in our study can therefore directly be linked to neuronal loss related to excitotoxic damage mediated by these receptors.
The limitation of our approach is that dynamic expression changes cannot be examined using post‐mortem tissue. Glutamate receptor expression is regulated at different levels transcriptionally and post‐transcriptionally (6, 71, 72, 73). Expression within the cell and even targeting specific domains of the plasma membrane is regulated and can be affected by AD (72, 74). In addition, some glutamate receptor subunits can act as binding sites for Aβ. This binding has a direct effect on glutamate receptor expression, with interactions inducing the endocytosis of both AMPA and NMDA receptors (75, 76, 77). AMPAR surface expression can be regulated through a series of Aβ‐driven mechanisms, including and not limited to ubiquitination, dephosphorylation, and endocytosis (78, 79, 80, 81). Additional experiments are required to clarify the effect of AD on tightly controlled expression and post‐transcriptional modification of glutamate receptors and the link between these processes and the modulation of neuronal activity.
In conclusion, subunit‐specific glutamate receptor expression alterations provide evidence of spatial receptor composition‐dependent changes in AD in the hippocampus and entorhinal cortex. The glutamatergic system has a critical role in regulating synaptic activity, and as such any disruption to the composition of glutamatergic receptors could have significant consequences for normal neuronal function and contribute to AD pathology (11, 17, 18, 82). The increased expression of the GluN1 and GluN2A receptor subunits indicates functional changes associated with calcium dysregulation and excitotoxicity leading to neuronal death and/or cognitive deficits. A better understanding of glutamatergic receptor changes in AD may lead to new therapeutic approaches to target specific components of the glutamatergic signaling pathway. However, selective targeting of these specific receptor subunits will require novel effective pharmacological compounds (76, 82, 83). Our findings warrant further investigation into the potential of these glutamate receptor subunits as novel therapeutic targets. Glutamate receptor subunit‐, cell type‐, and brain region‐specific therapies may provide a fine‐tune targeted approach to the management and treatment of AD, offering an interesting avenue for future drug development.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHORS CONTRIBUTIONS
Jason H. Y. Yeung, Joshua L. Walby, Thulani H. Palpagama, and Andrea Kwakowsky performed the research; Clinton Turner carried out the pathology for all human brain tissue; Andrea Kwakowsky, Richard L. M. Faull, and Henry J. Waldvogel designed the research; Richard L. M. Faull and Andrea Kwakowsky were responsible for the funding acquisition; Andrea Kwakowsky was responsible for project administration; Henry J. Waldvogel, Richard L. M. Faull, and Andrea Kwakowsky supervised the study; Jason H. Y. Yeung, Joshua Walby, Thulani H. Palpagama, Henry J. Waldvogel, Richard L. M. Faull, and Andrea Kwakowsky wrote the manuscript.
ETHICS APPROVAL
All procedures were approved by the University of Auckland Human Participants' Ethics Committee (Approval number: 001654). All experiments were conducted in compliance with the ARRIVE guidelines.
ACKNOWLEDGMENTS
We acknowledge the excellent work and assistance of Kristina Hubbard, Marika Eszes, and Jacqueline Ross. This work was supported by Alzheimers New Zealand Charitable Trust (AK; 370836), Alzheimers New Zealand (AK; 3718869), Freemasons New Zealand (AK; 3719321), Aotearoa Foundation, Centre for Brain Research and University of Auckland (AK; 3705579), Neurological Foundation of New Zealand (AK, TP; 848010), Brain Research New Zealand (AK, HJW, RLF), Health Research Council of New Zealand (RLF and HJW; 3627373).
Yeung JHY, Walby JL, Palpagama TH, Turner C, Waldvogel HJ, Faull RLM, et al. Glutamatergic receptor expression changes in the Alzheimer's disease hippocampus and entorhinal cortex. Brain Pathology. 2021;31:e13005. 10.1111/bpa.13005
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. [DOI] [PubMed] [Google Scholar]
- 2. Perry G, Cash AD, Smith MA. Alzheimer disease and oxidative stress. J Biomed Biotechnol. 2002;2:120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sims NR, Bowen DM, Allen SJ, Smith CC, Neary D, Thomas DJ, et al. Presynaptic cholinergic dysfunction in patients with dementia. J Neurochem. 1983;40:503–9. [DOI] [PubMed] [Google Scholar]
- 4. Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer's disease. J Alzheimers Dis. 2018;62:1403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steinhäuser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–45. [DOI] [PubMed] [Google Scholar]
- 6. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 7. Pin J‐P, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. [DOI] [PubMed] [Google Scholar]
- 8. Cull‐Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–35. [DOI] [PubMed] [Google Scholar]
- 9. Butterfield DA, Pocernich CB. The glutamatergic system and Alzheimer's disease: therapeutic implications. CNS Drugs. 2003;17:641–52. [DOI] [PubMed] [Google Scholar]
- 10. Kwakowsky A, Waldvogel HJ, Faull RL. The effects of amyloid‐beta on hippocampal glutamatergic receptor and transporter expression. Neural Regen Res. 2021;16:1399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bukke VN, Archana M, Villani R, Romano AD, Wawrzyniak A, Balawender K, et al. The dual role of glutamatergic neurotransmission in Alzheimer's disease: from pathophysiology to pharmacotherapy. Int J Mol Sci. 2020;21(20):7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cowburn RF, Wiehager B, Trief E, Li‐Li M, Sundstrom E. Effects of beta‐amyloid‐(25–35) peptides on radioligand binding to excitatory amino acid receptors and voltage‐dependent calcium channels: evidence for a selective affinity for the glutamate and glycine recognition sites of the NMDA receptor. Neurochem Res. 1997;22:1437–42. [DOI] [PubMed] [Google Scholar]
- 13. Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, et al. Soluble β‐amyloid1‐40 induces NMDA‐dependent degradation of postsynaptic density‐95 at glutamatergic synapses. J Neurosci. 2005;25:11061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fernandez‐Tome P, Brera B, Arevalo MA, de Ceballos ML. Beta‐amyloid25‐35 inhibits glutamate uptake in cultured neurons and astrocytes: modulation of uptake as a survival mechanism. Neurobiol Dis. 2004;15:580–9. [DOI] [PubMed] [Google Scholar]
- 15. Zhao D, Watson JB, Xie CW. Amyloid beta prevents activation of calcium/calmodulin‐dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long‐term potentiation. J Neurophysiol. 2004;92:2853–8. [DOI] [PubMed] [Google Scholar]
- 16. Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–50. [DOI] [PubMed] [Google Scholar]
- 17. Greenamyre JT, Maragos WF, Albin RL, Penney JB, Young AB. Glutamate transmission and toxicity in alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12(4):421–30. [DOI] [PubMed] [Google Scholar]
- 18. Maragos WF, Greenamyre JT, Penney JB Jr, Young AB. Glutamate dysfunction in Alzheimer's disease: an hypothesis. Trends Neurosci. 1987;10(2):65–8. [Google Scholar]
- 19. Aoki C, Venkatesan C, Go C, Mong JA, Dawson TM. Cellular and subcellular localization of NMDA‐R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci. 1994;14:5202–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Armstrong DM, Ikonomovic MD. AMPA‐selective glutamate receptor subtype immunoreactivity in the hippocampal dentate gyrus of patients with Alzheimer disease. Evidence for hippocampal plasticity. Mol Chem Neuropathol. 1996;28:59–64. [DOI] [PubMed] [Google Scholar]
- 21. Ikonomovic MD, Mizukami K, Warde D, Sheffield R, Hamilton R, Wenthold RJ, et al. Distribution of glutamate receptor subunit NMDAR1 in the hippocampus of normal elderly and patients with Alzheimer's disease. Exp Neurol. 1999;160:194–204. [DOI] [PubMed] [Google Scholar]
- 22. Carter TL, Rissman RA, Mishizen‐Eberz AJ, Wolfe BB, Hamilton RL, Gandy S, et al. Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer's disease patients according to Braak stage. Exp Neurol. 2004;187:299–309. [DOI] [PubMed] [Google Scholar]
- 23. Sze C, Bi H, Kleinschmidt‐DeMasters BK, Filley CM, Martin LJ. N‐Methyl‐D‐aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J Neurol Sci. 2001;182:151–9. [DOI] [PubMed] [Google Scholar]
- 24. Waldvogel HJ, Curtis MA, Baer K, Rees MI, Faull RL. Immunohistochemical staining of post‐mortem adult human brain sections. Nat Protoc. 2006;1:2719–32. [DOI] [PubMed] [Google Scholar]
- 25. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer's disease; Neurology. 1991;41(4):479. [DOI] [PubMed] [Google Scholar]
- 26. Mai J, Paxinos A, Voss T. Atlas of the human brain. 3rd ed. San Diego: Elsevier Inc.; 2008. [Google Scholar]
- 27. Kwakowsky A, Calvo‐Flores Guzman B, Pandya M, Turner C, Waldvogel HJ, Faull RL. GABAA receptor subunit expression changes in the human Alzheimer's disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J Neurochem. 2018;145:374–92. [DOI] [PubMed] [Google Scholar]
- 28. Erlenhardt N, Yu H, Abiraman K, Yamasaki T, Wadiche JI, Tomita S, et al. Porcupine controls hippocampal AMPAR levels, composition, and synaptic transmission. Cell Rep. 2016;14:782–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morimura N, Yasuda H, Yamaguchi K, et al. Autism‐like behaviours and enhanced memory formation and synaptic plasticity in Lrfn2/SALM1‐deficient mice. Nat Commun. 2017;8:15800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seigneur E, Südhof TC. Genetic ablation of all cerebellins reveals synapse organizer functions in multiple regions throughout the brain. J Neurosci. 2018;38:4774–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yeung JHY, Calvo‐Flores Guzman B, Palpagama TH, et al. Amyloid‐beta1‐42 induced glutamatergic receptor and transporter expression changes in the mouse hippocampus. J Neurochem. 2020;155:62–80. [DOI] [PubMed] [Google Scholar]
- 32. Yeung JHY, Palpagama TH, Tate WP, Peppercorn K, Waldvogel HJ, Faull RLM, et al. The acute effects of amyloid‐beta1‐42 on glutamatergic receptor and transporter expression in the mouse hippocampus. Front Neurosci. 2020;13:1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Atkin G, Moore S, Lu Y, et al. Loss of F‐box only protein 2 (Fbxo2) disrupts levels and localization of select NMDA receptor subunits, and promotes aberrant synaptic connectivity. J Neurosci. 2015;35:6165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kahlfuß S, Simma N, Mankiewicz J, et al. Immunosuppression by N‐methyl‐D‐aspartate receptor antagonists is mediated through inhibition of Kv1.3 and KCa3.1 channels in T cells. Mol Cell Biol. 2014;34:820–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Konstantoudaki X, Chalkiadaki K, Tivodar S, Karagogeos D, Sidiropoulou K. Impaired synaptic plasticity in the prefrontal cortex of mice with developmentally decreased number of interneurons. Neuroscience. 2016;322:333–45. [DOI] [PubMed] [Google Scholar]
- 36. Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–46. [DOI] [PubMed] [Google Scholar]
- 37. Roshanravan H, Kim EY, Dryer SE. NMDA receptors as potential therapeutic targets in diabetic nephropathy: increased renal NMDA receptor subunit expression in Akita mice and reduced nephropathy following sustained treatment with memantine or MK‐801. Diabetes. 2016;65:3139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tindi JO, Chavez AE, Cvejic S, Calvo‐Ochoa E, Castillo PE, Jordan BA. ANKS1B Gene Product AIDA‐1 Controls Hippocampal Synaptic Transmission by Regulating GluN2B Subunit Localization. J Neurosci. 2015;35:8986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yeung JHY, Palpagama TH, Tate WP, Peppercorn K, Waldvogel HJ, Faull RLM, et al. The acute effects of amyloid‐beta1–42 on glutamatergic receptor and transporter expression in the mouse hippocampus. Front Neurosci. 2020;13:1–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hollmann M, Hartley M, Heinemann S (1991) Ca2+ permeability of KA‐AMPA–gated glutamate receptor channels depends on subunit composition. Science 252(5007):851–853. [DOI] [PubMed] [Google Scholar]
- 41. Weiss JH (2011) Ca2+ permeable AMPA channels in diseases of the nervous system. Front Mol Neurosci 4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Armstrong DM, Ikonomovic MD, Sheffield R, Wenthold RJ. AMPA‐selective glutamate receptor subtype immunoreactivity in the entorhinal cortex of non‐demented elderly and patients with Alzheimer's disease. Brain Res. 1994;639:207–16. [DOI] [PubMed] [Google Scholar]
- 43. Alfonso S, Kessels HW, Banos CC, Chan TR, Lin ET, Kumaravel G, et al. Synapto‐depressive effects of amyloid beta require PICK1. Eur J Neurosci. 2014;39:1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anggono V, Clem RL, Huganir RL. PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci. 2011;31:2188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Citri A, Bhattacharyya S, Ma C, Morishita W, Fang S, Rizo J, et al. Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long‐term depression. J Neurosci. 2010;30:16437–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu W, Khatri L, Ziff EB. Trafficking of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPA) receptor subunit GluA2 from the endoplasmic reticulum is stimulated by a complex containing Ca2+/calmodulin‐activated kinase II (CaMKII) and PICK1 protein and by release of Ca2+ from internal stores. J Biol Chem. 2014;289:19218–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ittner A, Ittner LM. Dendritic tau in Alzheimer's disease. Neuron. 2018;99:13–27. [DOI] [PubMed] [Google Scholar]
- 48. Regan P, Piers T, Yi JH, Kim DH, Huh S, Park SJ, et al. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci. 2015;35:4804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cercato MC, Vázquez CA, Kornisiuk E, Aguirre AI, Colettis N, Snitcofsky M, et al. GluN1 and GluN2A NMDA receptor subunits increase in the hippocampus during memory consolidation in the rat. Front Behav Neurosci. 2017;10:242. 10.3389/fnbeh.2016.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Panegyres P, Zafiris‐Toufexis K, Kakulas B. The mRNA of the NR1 subtype of glutamate receptor in Alzheimer's disease. J Neural Transm. 2002;109:77–89. [DOI] [PubMed] [Google Scholar]
- 51. Leuba G, Vernay A, Kraftsik R, Tardif E, Riederer BM, Savioz A. Pathological reorganization of NMDA receptors subunits and postsynaptic protein PSD‐95 distribution in Alzheimer's disease. Curr Alzheimer Res. 2014;11:86–96. [DOI] [PubMed] [Google Scholar]
- 52. Conti F, Hicks TP. Excitatory amino acids and the cerebral cortex. Cambridge, MA: MIT Press; 1996. [Google Scholar]
- 53. Conti F, DeBiasi S, Minelli A, Melone M. Expression of NR1 and NR2A/B subunits of the NMDA receptor in cortical astrocytes. Glia. 1996;17:254–8. [DOI] [PubMed] [Google Scholar]
- 54. Ngo‐Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca 2+‐mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642. [DOI] [PubMed] [Google Scholar]
- 55. Oshima S, Fukaya M, Masabumi N, Shirakawa T, Oguchi H, Watanabe M. Early onset of NMDA receptor GluRε1 (NR2A) expression and its abundant postsynaptic localization in developing motoneurons of the mouse hypoglossal nucleus. Neurosci Res. 2002;43:239–50. [DOI] [PubMed] [Google Scholar]
- 56. Van de Ven TJ, VanDongen HM, VanDongen AM. The nonkinase phorbol ester receptor α1‐chimerin binds the NMDA receptor NR2A subunit and regulates dendritic spine density. J Neurosci. 2005;25:9488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang X‐M, Luo J‐H. GluN2A versus GluN2B: twins, but quite different. Neurosci Bull. 2013;29:761–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Conti F, Barbaresi P, Melone M, Ducati A. Neuronal and glial localization of NR1 and NR2A/B subunits of the NMDA receptor in the human cerebral cortex. Cereb Cortex. 1999;9:110–20. [DOI] [PubMed] [Google Scholar]
- 59. Liu ZA, Lv CE, Zhao W, Song Y, Pei D, Xu T. NR2B‐containing NMDA receptors expression and their relationship to apoptosis in hippocampus of Alzheimer's disease‐like rats. Neurochem Res. 2012;37:1420–7. [DOI] [PubMed] [Google Scholar]
- 60. Loftis JM, Janowsky A. The N‐methyl‐D‐aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther. 2003;97:55–85. [DOI] [PubMed] [Google Scholar]
- 61. Bi H, Sze CI. N‐methyl‐D‐aspartate receptor subunit NR2A and NR2B messenger RNA levels are altered in the hippocampus and entorhinal cortex in Alzheimer's disease. J Neurol Sci. 2002;200:11–8. [DOI] [PubMed] [Google Scholar]
- 62. Calvo‐Flores Guzman B, Elizabeth Chaffey T, Hansika Palpagama T, et al. The interplay between beta‐amyloid 1–42 (Abeta1‐42)‐induced hippocampal inflammatory response, p‐tau, vascular pathology, and their synergistic contributions to neuronal death and behavioral deficits. Front Mol Neurosci. 2020;13:522073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: an integral player in the pathogenesis of Alzheimer's disease. Prog Neurogibol. 2016;144:121–41. [DOI] [PubMed] [Google Scholar]
- 64. Palombo F, Tamagnini F, Jeynes JCG, et al. Detection of Abeta plaque‐associated astrogliosis in Alzheimer's disease brain by spectroscopic imaging and immunohistochemistry. Analyst. 2018;143:850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Palpagama TH, Waldvogel HJ, Faull RLM, Kwakowsky A. The role of microglia and astrocytes in Huntington's disease. Front Mol Neurosci. 2019;12:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227–38. [DOI] [PubMed] [Google Scholar]
- 67. Skowronska K, Obara‐Michlewska M, Zielinska M, Albrecht J. NMDA receptors in astrocytes: in search for roles in neurotransmission and astrocytic homeostasis. Int J Mol Sci. 2019;20(2):309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. 2018;98:239–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–31. [DOI] [PubMed] [Google Scholar]
- 70. Letellier M, Park YK, Chater TE, Chipman PH, Gautam SG, Oshima‐Takago T, et al. Astrocytes regulate heterogeneity of presynaptic strengths in hippocampal networks. Proc Natl Acad Sci U S A. 2016;113:E2685–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bai G, Hoffman PW. Transcriptional regulation of NMDA receptor expression. In: Van Dongen AM, editor. Biology of the NMDA receptor. Boca Raton, FL: CRC Press; 2009. p. 80–95. [PubMed] [Google Scholar]
- 72. Orlandi C, La Via L, Bonini D, Mora C, Russo I, Barbon A, et al. AMPA receptor regulation at the mRNA and protein level in rat primary cortical cultures. PLoS One. 2011;6:e25350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vazhappilly R, Sucher NJ. Translational regulation of the N‐methyl‐D‐aspartate receptor subunit NR1. Neurosignals. 2004;13:190–3. [DOI] [PubMed] [Google Scholar]
- 74. Perez‐Otano I, Ehlers MD. Learning from NMDA receptor trafficking: clues to the development and maturation of glutamatergic synapses. Neurosignals. 2004;13:175–89. [DOI] [PubMed] [Google Scholar]
- 75. Hamilton A, Zamponi GW, Ferguson SS. Glutamate receptors function as scaffolds for the regulation of beta‐amyloid and cellular prion protein signaling complexes. Mol Brain. 2015;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu J, Chang L, Song Y, Li H, Wu Y. The role of NMDA receptors in Alzheimer's disease. Front Neurosci. 2019;13:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Malinow R. New developments on the role of NMDA receptors in Alzheimer's disease. Curr Opin Neurobiol. 2012;22:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guntupalli S, Widagdo J, Anggono V. Amyloid‐β‐induced dysregulation of AMPA receptor trafficking. Neural Plast. 2016;2016:3204519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR removal underlies Aβ‐induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jurado S. AMPA receptor trafficking in natural and pathological aging. Front Mol Neurosci. 2018;10:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang Y, Guo O, Huo Y, Wang G, Man H‐Y. Amyloid‐β induces AMPA receptor ubiquitination and degradation in primary neurons and human brains of Alzheimer's disease. J Alzheimers Dis. 2018;62:1789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. [DOI] [PubMed] [Google Scholar]
- 83. Zarate CA Jr, Manji HK. The role of AMPA receptor modulation in the treatment of neuropsychiatric diseases. Exp Neurol. 2008;211 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.