

Abstract
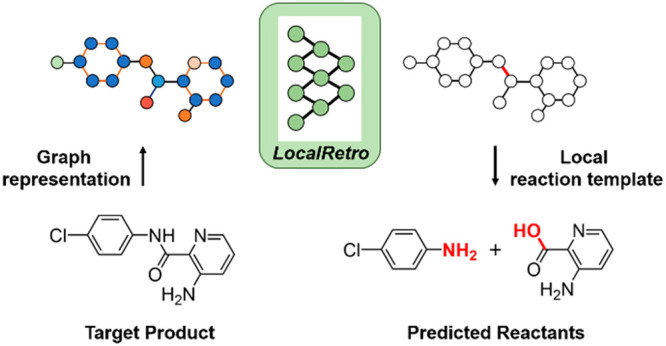
As a fundamental problem in chemistry, retrosynthesis aims at designing reaction pathways and intermediates for a target compound. The goal of artificial intelligence (AI)-aided retrosynthesis is to automate this process by learning from the previous chemical reactions to make new predictions. Although several models have demonstrated their potentials for automated retrosynthesis, there is still a significant need to further enhance the prediction accuracy to a more practical level. Here we propose a local retrosynthesis framework called LocalRetro, motivated by the chemical intuition that the molecular changes occur mostly locally during the chemical reactions. This differs from nearly all existing retrosynthesis methods that suggest reactants based on the global structures of the molecules, often containing fine details not directly relevant to the reactions. This local concept yields local reaction templates involving the atom and bond edits. Because the remote functional groups can also affect the overall reaction path as a secondary aspect, the proposed locally encoded retrosynthesis model is then further refined to account for the nonlocal effects of chemical reaction through a global attention mechanism. Our model shows a promising 89.5 and 99.2% round-trip accuracy at top-1 and top-5 predictions for the USPTO-50K dataset containing 50 016 reactions. We further demonstrate the validity of LocalRetro on a large dataset containing 479 035 reactions (UTPTO-MIT) with comparable round-trip top-1 and top-5 accuracy of 87.0 and 97.4%, respectively. The practical application of the model is also demonstrated by correctly predicting the synthesis pathways of five drug candidate molecules from various literature.
Keywords: retrosynthesis reaction prediction, graph neural networks, local reactivity, global attention mechanism
Introduction
Designing molecules and materials with desired properties is a practical goal of chemistry and materials science. Cheminformatics deals with the use of data to understand the relationships between the structures of the molecules and their properties that can eventually lead to the discovery of novel functional molecules. In fact, there has been a significant surge in using machine learning in chemical science to accelerate the process of new discovery by predicting various molecular properties given their structures or designing novel molecules inversely from the desired functionality as input.1−3 However, the latter machine-enabled molecular designs mainly give the optimized molecular structures that meet the desired property without considering how to synthesize them, which makes the synthesis planning task a critical last step to bring the in silico designed molecules into the practice.
Predicting chemical reactions generally involves two mapping directions, either forward (from the given reactants to predict the product) or backward (from the target product to design the proper reactants), and the term retrosynthesis refers to the synthesis pathway planning of the latter kind. A forward prediction may be generally more straightforward, because the desired task is a one-to-one mapping, that is, for a given set of reactants, the reaction products are usually uniquely defined (within the variations in experimental conditions). Retrosynthesis, on the other hand, is a one-to-many mapping and more challenging in the sense that there might be several different reaction pathways to synthesize a target compound. Thus, the synthesis planning has traditionally been the realm of expert synthetic chemists, and to speed up and scale up the retrosynthesis in a more automated fashion, researchers have sought effective and accurate methods based on the computer-assisted synthesis planning (CASP) for decades.4−6Chematica (now known as Synthia),7 for example, uses 70 000 rules encoded by synthetic experts and uses a decision tree to decide which reaction rule to use and penalize nonselective reactions in an automated and hierarchical fashion in computers. Yet, automated retrosynthesis still remains as a major unsolved problem, and we briefly survey a few latest developments for the data-driven automatic synthesis planning below.
By representing molecules as simplified molecular-input line-entry system (SMILES) string, Liu et al.8 trained a neural sequence-to-sequence (seq2seq) model to convert the SMILES string of the reaction product into the SMILES string of its precursors. Their fully data-driven model showed a comparable performance with a rule-based expert system baseline model. On the other hand, some research groups used Morgan Fingerprint,9 an algorithm that extracts the substructure information on a given molecule to a feature vector, to train their models. Coley et al.10 utilized the chemical structural similarity between the target product and the compounds in the corpus to derive possible synthesis pathways, intuitively conjecturing that similar molecules would have similar reaction pathways, and ranked the predictions by the reaction similarity. Segler and Waller11 proposed a hybrid neural-symbolic approach to predict the retrosynthesis pathway using either hand-coded or automatically extracted rules by a deep neural network. On top of that, Segler et al.12 implemented three different deep neural networks (expansion, in-scope filter, and rollout) with a Monte Carlo tree search (3N-MCTS) to solve a multistep retrosynthesis problem. With a large number of reactions (3.5 million) used to train the model, the model was shown to be able to suggest a professional level of synthesis planning for a benzopyran sulfonamide derivative as an antagonist of the 5-HT6 receptor.13 However, given the nature of Morgan Fingerprint used to represent the molecules in the latter works, which includes only the existence of substructure information on a molecule, the connectivity and relative locations of substructures that could act a critical role in synthesis planning might not be fully captured.
More recently, computer scientists have served molecules as heterogeneous graphs and introduced a graph neural network (GNN) to improve the prediction accuracy of the retrosynthesis.14−17 Dai et al.14 applied RDChiral(18) to extract thousands of reaction templates from the training dataset and predict the probability distribution of these reaction templates to be applied to a given molecule by Conditional Graph Logic Network (GLN). The probability of the template and precursor predicted by GLN is formulated as a function of the probability of the reaction template with a given molecule and the probability of the reactant with a given molecule and template, where the number of predictable templates and reactants varies with different molecule conditions. Shi et al.15 divides a retrosynthesis task into two parts: reaction center identification and variational graph translation. During the reaction center identification, each bond in the molecule is either predicted to be cut or preserved. After the reaction center is identified and edited, the resulting molecule (synthon) is completed by the GNN to generate the corresponding reactants. This identify-and-complete concept is similar to how an experienced chemist designs the synthetic pathway of a given molecule. However, we argue that the identifying step and completing step are highly correlated. In other words, the resulting reactants are highly dependent on the identified reaction center, and thus these two steps should be consolidated and conducted at the same time.
In most elementary chemical reactions, the changes in molecular formula and structures via a bond breaking or bond formation occur mostly locally. However, almost all existing retrosynthesis methods described above employ the global structures of the target molecules to make predictions. For example, in most existing graph-based retrosynthesis methods, the global features are obtained by summing up or averaging over all atomic features and used to predict the reactants. In molecular similarity-based retrosynthesis, the global similarity between the molecules is also used. The use of global features to make synthesis pathway prediction, however, may yield undesired focus on the details not directly relevant to the target reactions.
Here, we design a graph-based retrosynthesis framework by locally deriving the reaction templates and evaluate the suitability of these local templates at all enumerated possible reaction centers for a target molecule. All the chemical transformation information is included in the local reaction template, and therefore, once the correct template is predicted at a chosen reaction center, the reactants can be immediately obtained by simply applying the derived template. In other words, our local approach combines the identify-and-complete two-step processes into a one-pot learning. Additionally, we enable all reaction centers to exchange information by attention mechanism to consider the global scenario. This corresponds to a reflection of nonlocal effects of chemical reactions in which the reactivity can be sometimes altered because of a chemical change in remote locations.
Methods and Datasets
Because the reactant molecules preserve most of their substructures and fragments during and after the reaction, our approach focuses on only the changes in the molecular structure (atoms or bonds) to complete the retrosynthesis. That is, instead of finding the proper reactants from scratch, we focus on inferencing what local change was occurred to form the given product in terms of the formation or breaking of bonds and/or the addition or removal of atoms.
In this work, we used two reaction databases, UTPTO-50K and USTOP-MIT, which contain 50 016 and 479 035 reactions, respectively, with the correct atom-mapping for all reactions. The USPTO-50K dataset, labeled with 10 different reaction classes in the U.S. patent literature curated by Schneider et al.,19 is mainly used for comparison with other methods, because many previous methods were benchmarked on USPTO-50K. The UTPTO-MIT dataset, curated by Jin et al.20 by removing duplicates and erroneous reactions, was used to further generalize our method on a larger dataset for practical applications. We partitioned the USPTO-50K dataset into a 40K/5K/5K train/validation/test split following ref (10). For the USPTO-MIT dataset, we partitioned the dataset into 410K/40K/30K following ref (20).
We first derived a set of local reaction templates for the USPTO-50K and USPTO-MIT reaction database. These local reaction templates contain the changes in the atom and bond information before and after the reaction. Because the target product and reactants are atom-mapped, we specified the reaction center by comparing the difference of atoms and bonds between the product and reactants. The atom reaction template refers to a change that occurs on an atom with no bond changes. For example, as one of the atom reaction templates, Figure 1a shows a deprotection reaction in which an ethyl group on oxygen reacts and forms a hydroxide group. The bond reaction template is derived if the reaction involves any bond changes or disconnections. Figure 1b shows an example of the bond reaction template of the Suzuki reaction, which describes a carbon–carbon bond (C–C) formed by a boronic acid (R-OBO) and halocarbon (R-X). In this reaction, a C–C bond is disconnected and replaced by a boronic acid from one reactant and halocarbon by another reactant. As a result, for each reaction in the datasets having a single atom change or bond change, either an atom reaction template or bond reaction template can be derived. For reactions having multiple products or multiple atoms and bonds changes, the derived template includes all the atoms and bonds that are changed during the reactions. If both atoms and bonds are changed in the reaction, the derived reaction template is noted as both atom reaction template and bond reaction template. Figure 1c shows an example reaction in which a primary amine is transferred from an oxygen atom to an aromatic nitrogen atom. Because the reaction occurs on the N–N bond in one molecule and an oxygen atom in another molecule, this template is noted as both an atom reaction template and a bond reaction template. More details on the local reaction template are given in the Supporting Information. If the reaction class is given in the training set, we can further categorize each derived local reaction template to one or more reaction classes depending on the reaction class labeled with the reaction.
Figure 1.

Derivation of (a) atom reaction template, (b) bond reaction template, and (c) multiple change reaction template. An atom reaction template is derived if the reaction does not involve any bond change or disconnection, otherwise a bond reaction template is derived. If both atoms and bonds are changed in the reaction, the derived template is noted as both atom reaction template and bond reaction template. The change of atoms and bonds are highlighted in red color.
Next, we developed a model to predict the correct local reaction template at each atom and bond that leads to the given product by learning their local environments. The overall architecture of LocalRetro and the feature transformation of a target product is shown in Figure 2. We represent the molecule as a heterogeneous graph G = (V,E) with V (vertices) denoting atoms and E (edges) denoting bonds. The features of atoms and bonds are first initialized by the atom and bond properties using the DGL-LifeSci(21) python package. The details of the feature initialization are given in the Supporting Information. To encode the surrounding environmental information for each atom, we applied a message passing neural network (MPNN) described in the literature22 to update each atom feature. We denote the message passing function by MPNN(•), with the atomic features of atom a as va, the atomic features of its neighboring atom b as vb, and the features of their connecting bond as eab. The atomic features of atom a is updated by the MPNN via
| 1 |
The curly bracket {} denotes a set of neighboring atoms around a given atom. After the atomic features are updated, the bond feature is represented by concatenating two atomic features (va′||vb′) and goes through a fully connected layer
| 2 |
Where w is the weights and c is the bias of the fully connected layer, to avoid confusion with the symbol of atom b.
Figure 2.

Model architecture of LocalRetro and the example of feature transformation when predicting the reactants of lenalidomide. First, the molecule graph (G), atom features (va), and bond features (eab) of a given molecule are initialized by the atom and bond properties Next, the atom and bond features are updated by message passing neural network (MPNN), bond feature encoding layer, and global reactivity attention (GRA) layer to encode the local environment and nonlocal reactivity dependency by the atoms and bonds in the molecule. Finally, the score of each local reaction template on each atom and bond is predicted by the atom reaction template classifier and bond reaction template classifier. The predicted reactants are obtained by applying the predicted local reaction template on the predicted atoms and bonds and ranked by their predicted scores.
Because chemical reactions are not always local and some reactions might be affected because of the existence of certain chemical environments remotely within the molecule, to account for such a global dependency and nonlocality of the reaction, we updated the features of all atoms and bonds by applying a global attention mechanism. To capture the different reactivity relationship between atoms and bonds, we applied multihead self-attention, the attention mechanism applied in Transformer(23) to learn the different context by learning the key, query, and value of a given feature. Features of atoms and bonds are updated by all the atoms and bonds in the molecule altogether. We call our nonlocal attention operation as global reactivity attention (GRA) to differentiate our method from the locally encoding graph attention (GAT) commonly used in graph neural network.24 The details of the GRA algorithm are given in the Supporting Information. We denote the atomic features xa and bond features xab updated with all the existing atomic features {va′} and bond features {eab′} by GRA(•)
| 3 |
| 4 |
We then trained an atom reaction template classifier and a bond reaction template classifier to calculate their outputs o using their updated representations xa and xab.
| 5 |
| 6 |
uA and wA are the weights and cA is the bias of the atom reaction template classifier, where uB and wB are the weights and cB is the bias of the bond reaction template classifier. σ stands for the ReLU activation function.
The score of each atom reaction template T applied at atom a is converted from oa by a Softmax function
| 7 |
Similarly, the score of each bond reaction template T applied at bond ab is obtained from oab by a Softmax function
| 8 |
A set of local reaction templates at each chemical center was predicted as the model output. These predicted templates were ranked by the value of the score to be applied to the given product to derive the final reactants. If the reaction class was given as in USPTO-50K, we only applied the local reaction templates, which lie in the template pool belonging to the given reaction class.
As baseline models, we compared our prediction results with five state-of-the-art retrosynthesis models: GLN (conditional graph logic network),14G2G (graph to graph),15GraphRetro,16MEGAN (molecule edit graph attention network),17 and Augmented Transformer.25 We denote our method by LocalRetro to emphasize the core idea, local reactivity prediction. However, as the global structure also plays an important secondary role, unless noted otherwise, LocalRetro refers to the LocalRetro with the GRA attention mechanism included.
Results and Discussion
To present the performance of LocalRetro, we used two types of accuracy, the exact match accuracy and round-trip accuracy. The exact match accuracy is computed by considering whether the set of reactants represented in canonical SMILES are an exact match with the ground truth reactants in the database. We note that some ground-truth reactants with stereocenters do not have the exact stereoinformation specified, and for these cases, the predicted reactants are considered to be correct as long as all the atoms and bond connectivities are correct. The round-trip accuracy was obtained by comparing the desired product with the product predicted by a pretrained forward-synthesis model using the predicted precursors. The consideration of round-trip accuracy reflects the fact that a given chemical product could be synthesized by multiple precursors’ combinations.26 The pretrained Molecular Transformer (MT)27 was used to evaluate the round-trip accuracy. Specifically, we marked the predicted precursors as correct if the predicted precursors were the same as the ground truth or the proposed precursors yielded the target product using MT, and otherwise incorrect. We also evaluated the MaxFrag accuracy for the prediction of the largest fragment introduced by Tetko et al.25 and the results (similar to those in Table 1-3) are given in the Supporting Information.
Table 1. Top-k Exact Match Accuracy on USPTO-50K Dataset and USPTO-MIT Dataset without Given Reaction Classa.
| top-k accuracy (%) |
||||||
|---|---|---|---|---|---|---|
| dataset | model | K = 1 | 3 | 5 | 10 | 50 |
| USPTO-50K | GLN(14) | 52.5 | 69.0 | 75.6 | 83.7 | 92.4 |
| G2G(15) | 48.9 | 67.6 | 72.5 | 75.5 | ||
| GraphRetro(16) | 53.7 | 68.3 | 72.2 | 75.5 | ||
| Augmented Transformer(25) | 53.5 | 69.4 | 81.0 | 85.7 | ||
| MEGAN(17) | 48.1 | 70.7 | 78.4 | 86.1 | 93.2 | |
| LocalRetro wo/GRA (this work) | 49.8 | 75.8 | 84.0 | 91.3 | 97.7 | |
| LocalRetro (this work) | 53.4 | 77.5 | 85.9 | 92.4 | 97.7 | |
| USPTO-MIT | LocalRetro wo/GRA (this work) | 49.9 | 70.7 | 77.0 | 83.1 | 89.8 |
| LocalRetro (this work) | 54.1 | 73.7 | 79.4 | 84.4 | 90.4 | |
The best top-k exact match accuracy is highlighted with bold font.
Table 3. Top-k Round-Trip Accuracy on the USPTO-50K Dataset and USPTO-MIT Dataseta.
| top-k accuracy (%) |
||||
|---|---|---|---|---|
| dataset | model | K = 1 | 3 | 5 |
| USPTO-50K | GLN(14) | 88.4 | 95.0 | 97.1 |
| LocalRetro wo/GRA (this work) | 88.2 | 97.8 | 98.9 | |
| LocalRetro (this work) | 89.5 | 97.9 | 99.2 | |
| USPTO-MIT | LocalRetro wo/GRA (this work) | 85.7 | 95.7 | 97.3 |
| LocalRetro (this work) | 87.0 | 95.9 | 97.4 | |
The best top-k round-trip accuracy is highlighted with bold font.
Total 731 local reaction templates were derived from the USPTO-50K training set. These total 731 local reaction templates cover 98.1% of the reactions in the test set, meaning that 98.1% would be the theoretical upper bound in the exact match accuracy of our approach. For the USPTO-MIT dataset, a total of 21 081 local reaction templates were derived from the 400K reactions in the training set. The latter 21 081 local reaction templates cover 97% of the reactions in the test set, implying the same theoretical limit of the exact match accuracy. We, however, note that the round-trip accuracy is not limited by these theoretical upper bounds of the exact match accuracy due to the possibility of multiple reaction pathways with different templates utilized.
The results of top-k exact match accuracy on the USPTO-50K dataset are shown in Tables 1 and 2. LocalRetro outperforms all the other methods with and without the reaction class given except for the top-1 prediction. For example, at the top-3 predictions, LocalRetro outperforms the current best method with 4.8 and 6.8% margins with and without reaction class given, respectively. The prediction accuracies of LocalRetro are consistently enhanced when applying GRA to the model. For round-trip accuracy, we compared LocalRetro with GLN only because the models trained on the same data split used in other works are not available in public. The round-trip accuracy results are shown in Table 3. Both models with and without GRA outperform GLN(14) by a large margin and reach nearly 99% round-trip accuracy at top-5 predictions.
Table 2. Top-k Exact Match Accuracy on USPTO-50K Dataset with Given Reaction Classa.
| top-k accuracy (%) |
||||||
|---|---|---|---|---|---|---|
| dataset | model | K = 1 | 3 | 5 | 10 | 50 |
| USPTO-50K | GLN(14) | 64.2 | 79.1 | 85.2 | 90.0 | 93.2 |
| G2G(15) | 61.0 | 81.3 | 86.0 | 88.7 | ||
| GraphRetro(16) | 63.9 | 81.5 | 85.2 | 88.1 | ||
| MEGAN(17) | 60.7 | 82.0 | 87.5 | 91.6 | 95.3 | |
| LocalRetro wo/GRA (this work) | 62.3 | 86.1 | 91.8 | 96.0 | 97.9 | |
| LocalRetro (this work) | 63.9 | 86.8 | 92.4 | 96.3 | 97.9 | |
The best top-k exact match accuracy is highlighted with bold font.
The results of the top-k exact match accuracy on the USPTO-MIT dataset are shown in Table 1. Although the size of the dataset and the number of templates for the USPTO-MIT are 10 times and 30 times larger than those for the USPTO-50K, respectively, the top-10 exact match accuracy still reaches 84.4% and the top-50 exact match accuracy reaches 90.4%, comparable to the results of previous methods evaluated on the USPTO-50K dataset. The round-trip accuracy of our model evaluated on the USPTO-MIT dataset is more promising. The top-1, -3, and -5 round-trip accuracy of our LocalRetro trained/benchmarked on the USPTO-MIT are 87.0, 95.9, and 97.4%, respectively, quite comparable to LocalRetro trained/benchmarked on the USPTO-50K, namely, 89.5, 97.9, and 99.2%. These results clearly demonstrate the generalizability of our proposed LocalRetro to a larger database for practical applications.
As shown in Tables 1–3, a 1–2% additional statistical improvement in prediction accuracy was obtained by using global reactivity attention (GRA). Although 1–2% appears to be a small improvement numerically, it is an important improvement. This is because not all the reactions in the dataset require a nonlocal effect to describe the reaction. In fact, the majority of chemical reactions are indeed local, and that is why our baseline local encoding model without nonlocal effects already gives promising results. However, there are still many chemical reactions in which the nonlocal effect is an important factor that chemists consider to explain the selectivity. Therefore, considering a small fraction of reactions in the dataset in which the nonlocal effects can indeed play an important role, we suggest that 1–2% (getting an additional 5000–10 000 reactions correct) is a statistically noticeable and chemically important improvement. In addition, GRA plays an important role when a retrosynthesis task includes more than one product. Because of the awareness of other atoms existing in the other molecules, LocalRetro with GRA shows 12.3% higher top-1 exact match accuracy than the model without GRA for the reactions including multiple products in the test set of the USPTO-MIT dataset (Table S2). We note that there are total 471 reactions (1.2%) with multiple products in the USPTO-MIT dataset.
To understand how GRA helps to increase the prediction accuracy, we compared the models with and without GRA and visualized the nonlocal attention in Figure 3. In both examples, reactants are correctly predicted by the model with GRA, but incorrectly predicted by the model without GRA. Particularly, the areas highlighted with orange in Figure 3 show the nonlocal aspect between the predicted reaction center and the other atoms and bonds located remotely. In example 1, GRA allows the machine to be aware that there is a chloride group in the existing skeleton with a high reactivity, so the stronger electron-donator chloride should be attached, but without GRA the machine proposes oxygen instead and so the reaction incorrectly occurs at the other more reactive chloride center. Similarly, example 2 in Figure 3 shows that GRA is aware of the high reactivity of existing amine so the reaction center is predicted to be close to the amine and the other undesired reaction might not occur. Therefore, GRA allows the algorithm to prioritize the reactive centers and find the correct reaction center, preventing the undesired products that might be formed without the global attention mechanism.
Figure 3.

Effects of global reactivity attention (GRA). In example 1, the reaction centers predicted with and without GRA are the same, and in example 2, the reaction centers predicted with and without GRA are different. In the cases with GRA, chemical units highlighted (attended) are denoted as shaded circles in red. The expected product using the predicted reactants is obtained by Molecular Transformer (MT).27
The ultimate goal of retrosynthesis is to solve the practical synthesis planning problem, so we further validate our model trained on the USPTO-MIT dataset for five different drug candidates considered in various retrosynthesis and inverse design works in the literature: lenalidomide, salmeterol, a 5-HT6 receptor ligand, and two DDR1 kinase inhibitors. The former two molecules are the retrosynthesis examples demonstrated by Coley et al.,10 the third example is the retrosynthesis example demonstrated by Segler et al.,12 and the last two examples are the compounds proposed by a reinforcement learning-based generative model, GENTRL.28 The full retrosynthetic pathways are obtained by sequentially conducting our retrosynthesis prediction tasks. The comparisons with earlier predictions and actual experimental pathways are summarized in Figure 4.
Figure 4.

Multistep retrosynthesis predictions by LocalRetro for (a) lenalidomide, (b) salmeterol, (c) a 5-HT6 receptor ligand, (d) a DDR1 kinase inhibitor INS015_037, and (e) another DDR1 kinase inhibitor INS015_032. The reaction center and the atom and bond transformations are highlighted in different colors at different reaction steps. Out of total 25 individual predicted synthesis steps considered, 19 steps were predicted correctly within the rank-2 predictions.
In the first example, Ponomaryov et al.29 proposed a process of synthesizing lenalidomide, a cancer drug that can be used to treat multiple myeloma, by three synthesis steps. The synthesis starts with brominating by N-bromosuccinimide (NBS), and then the ring is formed after introducing an ammonia. Lastly, lenalidomide is obtained by condensing the precursor with 3-aminopiperidine-2,6-dione and nitro oxidation. Our retrosynthesis prediction (Figure 4a) reveals the exact same synthesis pathway shown in the literature.
In the second example, Guo et al.30 synthesized salmeterol, a potent β2-adrenoreceptor agonist, through a key step Henry reaction. Our model predicts a reduction and then correctly predicts an amination reaction with rank-1 (Figure 4b). At the next two steps, our model suggests that the product can be synthesized from the Henry reaction, a classic carbon–carbon bond formation reaction. The synthetic pathway predicted by our model is identical to those in the literature, including steps 3 and 4 together, forming the Henry reaction at either rank-1 or rank-2 predictions.
In the third example, Nirogi et al.13 proposed a benzopyran sulfonamide derivative as an antagonist of 5-HT6 receptor. This is a challenging synthesis planning problem that requires a total seven synthesis steps. The synthesis planning suggested by our model is shown in Figure 4c. Except for step 5 predicted at the rank-7 prediction, our model successfully predicts the rest of the steps (steps 1 through 7 except for step 5) with top-3 predictions. This result clearly shows that our model is able to predict the synthesis on the potential drug molecule even in the case that requires a long sequence of synthetic steps.
Another challenging but interesting fourth example is the retrosynthesis task on DDR1 kinase inhibitor INS015_037 proposed by Zhavoronkov et al.28 It is a potential DDR1 kinase inhibitor (Figure 4d) obtained by generative machine learning methods, also experimentally shown to have a favorable pharmacokinetics in mice. Zhavoronkov et al. used the convergent synthesis by separately synthesizing two precursors and synthesized the INS015_37 at the last step. Our model successfully predicts the same convergent synthesis pathway with the reported synthesis pathway in ref (28) with rank-1 predictions. Our retrosynthesis prediction of another DDR1 kinase inhibitor INS015_032 proposed by Zhavoronkov et al.28 is also shown in Figure 4e. Although our model is able to suggest an almost identical synthesis pathway within top-4 predictions compared to the literature, it cannot predict the Suzuki coupling reaction with high confidence at step 2, and the reaction at step 7 is not predicted because of the lack of available reaction template.
Thus, to summarize the results of these more practical multistep retrosynthesis problems, all five demonstrated examples yielded nearly identical retrosynthetic pathways to those in the literature, mostly within the rank-2 predictions. To be more specific, out of all 25 individual steps considered, five steps are predicted at rank-3, -4, -7, -10, and -72, and one step is not solvable because of the lack of reaction template, but the remaining 19 steps were predicted correctly within the rank-2 predictions, among which 17 steps were the rank-1 predictions. These results obtained without considering multiple possible pathways along with the promising round-trip performance on the benchmark dataset described earlier demonstrate the great promises of our approach toward more practical retrosynthesis predictions.
Conclusion
Motivated by chemical intuitions, we present a data-driven retrosynthesis model, LocalRetro, which suggests possible synthesis pathways by locally learning the chemical reactivity together with global reactivity attention to account for the remaining nonlocal effects. The LocalRetro trained and evaluated on the USPTO-MIT dataset containing 479 035 chemical reactions yields a promising 97.4% top-5 round-trip accuracy. We further demonstrated the practical validity of our model by successfully predicting the retrosynthesis pathways of several potential drug molecules. Future work should aim to involve an even larger reaction database to further generalize our model. These future reaction databases could also potentially include the reaction conditions such as reagents, temperature, and pH values, which are all critical yet absent information in the current dataset we used in this research. With the advance of reaction mapping methods,31 we expect to be able to train our model using a larger dataset with high quality in the future. We also expect that the local/nonlocal reactivity concept proposed here can be utilized in the forward reaction product prediction models, which is currently under development in our group. The source code of LocalRetro is released at https://github.com/kaist-amsg/LocalRetro.
Acknowledgments
We acknowledge the financial support from NRF Korea (NRF- 2017R1A2B3010176). A generous supercomputing time from KISTI is acknowledged. We are grateful to Juhwan Noh and Geun Ho Gu for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00246.
Details about local reaction template, MaxFrag accuracy, results of multiproduct reactions, model implementation, and computational cost analysis (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4 (2), 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W.; Barzilay R.; Jaakkola T.. Junction Tree Variational Autoencoder for Molecular Graph Generation. In International Conference on Machine Learning; PMLR, 2018; pp 2323–2332.
- Segler M. H. S.; Kogej T.; Tyrchan C.; Waller M. P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4 (1), 120–131. 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J.; Wipke W. T. Computer-Assisted Design of Complex Organic Syntheses. Science 1969, 166 (3902), 178–192. 10.1126/science.166.3902.178. [DOI] [PubMed] [Google Scholar]
- de Almeida A. F.; Moreira R.; Rodrigues T. Synthetic Organic Chemistry Driven by Artificial Intelligence. Nature Reviews Chemistry 2019, 3 (10), 589–604. 10.1038/s41570-019-0124-0. [DOI] [Google Scholar]
- Strieth-Kalthoff F.; Sandfort F. S.; Segler M. H.; Glorius F. Machine Learning the Ropes: Principles, Applications and Directions in Synthetic Chemistry. Chem. Soc. Rev. 2020, 49 (17), 6154–6168. 10.1039/C9CS00786E. [DOI] [PubMed] [Google Scholar]
- Mikulak-Klucznik B.; Gołȩbiowska P.; Bayly A. A.; Popik O.; Klucznik T.; Szymkuć S.; Gajewska E. P.; Dittwald P.; Staszewska-Krajewska O.; Beker W.; Badowski T.; Scheidt K. A.; Molga K.; Mlynarski J.; Mrksich M.; Grzybowski B. A. Computational Planning of the Synthesis of Complex Natural Products. Nature 2020, 588 (7836), 83–88. 10.1038/s41586-020-2855-y. [DOI] [PubMed] [Google Scholar]
- Liu B.; Ramsundar B.; Kawthekar P.; Shi J.; Gomes J.; Luu Nguyen Q.; Ho S.; Sloane J.; Wender P.; Pande V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3 (10), 1103–1113. 10.1021/acscentsci.7b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Rogers L.; Green W. H.; Jensen K. F. Computer-Assisted Retrosynthesis Based on Molecular Similarity. ACS Cent. Sci. 2017, 3 (12), 1237–1245. 10.1021/acscentsci.7b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M. H. S.; Waller M. P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. - Eur. J. 2017, 23 (25), 5966–5971. 10.1002/chem.201605499. [DOI] [PubMed] [Google Scholar]
- Segler M. H. S.; Preuss M.; Waller M. P. Planning Chemical Syntheses with Deep Neural Networks and Symbolic AI. Nature 2018, 555 (7698), 604–610. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Nirogi R. V. S.; Badange R.; Reballi V.; Khagga M. Design, Synthesis and Biological Evaluation of Novel Benzopyran Sulfonamide Derivatives as 5-HT6 Receptor Ligands. Asian J. Chem. 2015, 27 (6), 2117–2124. 10.14233/ajchem.2015.17783. [DOI] [Google Scholar]
- Dai H.; Li C.; Coley C. W.; Dai B.; Song L.. Retrosynthesis Prediction with Conditional Graph Logic Network. arXiv (Machine Learning) Jan 6, 2020, 2001.01408, ver. 1. https://arxiv.org/abs/2001.01408 (accessed 2021-03-01).
- Shi C.; Xu M.; Guo H.; Zhang M.; Tang J.. A Graph to Graphs Framework for Retrosynthesis Prediction. arXiv (Machine Learning) Mar 28, 2020, 2003.12725, ver. 1. http://arxiv.org/abs/2003.12725 (accessed 2021-03-01).
- Somnath V. R.; Bunne C.; Coley C. W.; Krause A.; Barzilay R.. Learning Graph Models for Retrosynthesis Prediction. arXiv (Machine Learning) Jun 12 2020, 2006.07038, ver. 2. http://arxiv.org/abs/2006.07038 (accessed 2021-07-30).
- Sacha M.ła.; Błaz M.ła.; Byrski P.; Dabrowski-Tumanski P.ł; Chrominski M.ła.; Loska R.ł; Włodarczyk-Pruszynski P.ł; Jastrzebski S.ła. Molecule Edit Graph Attention Network: Modeling Chemical Reactions as Sequences of Graph Edits. J. Chem. Inf. Model. 2021, 61, 3273. 10.1021/acs.jcim.1c00537. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Green W. H.; Jensen K. F. RDChiral: An RDKit Wrapper for Handling Stereochemistry in Retrosynthetic Template Extraction and Application. J. Chem. Inf. Model. 2019, 59 (6), 2529–2537. 10.1021/acs.jcim.9b00286. [DOI] [PubMed] [Google Scholar]
- Schneider N.; Stiefl N.; Landrum G. A. What’s What: The (Nearly) Definitive Guide to Reaction Role Assignment. J. Chem. Inf. Model. 2016, 56 (12), 2336–2346. 10.1021/acs.jcim.6b00564. [DOI] [PubMed] [Google Scholar]
- Jin W.; Coley C.; Barzilay R.; Jaakkola T. Predicting Organic Reaction Outcomes with Weisfeiler-Lehman Network. Advances in Neural Information Processing Systems 2017, 30. [Google Scholar]
- awslabs/dgl-lifesci https://github.com/awslabs/dgl-lifesci (accessed 2021-02-10).
- Gilmer J.; Schoenholz S. S.; Riley P. F.; Vinyals O.; Dahl G. E.. Neural Message Passing for Quantum Chemistry. arXiv (Machine Learning) Jun 12, 2017, 1704.01212, ver. 2. http://arxiv.org/abs/1704.01212 (accessed 2021-03-01).
- Vaswani A.; Shazeer N.; Parmar N.; Uszkoreit J.; Jones L.; Gomez A. N.; Kaiser L.; Polosukhin I.. Attention Is All You Need. arXiv (Machine Learning) Dec 5, 2017, 1706.03762, ver. 2. http://arxiv.org/abs/1706.03762 (accessed 2021-03-01).
- Veličković P.; Cucurull G.; Casanova A.; Romero A.; Liò P.; Bengio Y.. Graph Attention Networks. arXiv (Machine Learning) Feb 4, 2018, 1710.10903, ver. 3. https://arxiv.org/abs/1710.10903 (accessed 2021-03-01).
- Tetko I. V.; Karpov P.; Van Deursen R.; Godin G. State-of-the-Art Augmented NLP Transformer Models for Direct and Single-Step Retrosynthesis. Nat. Commun. 2020, 11 (1), 5575. 10.1038/s41467-020-19266-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller P.; Petraglia R.; Zullo V.; Nair V. H.; Haeuselmann R. A.; Pisoni R.; Bekas C.; Iuliano A.; Laino T. Predicting Retrosynthetic Pathways Using Transformer-Based Models and a Hyper-Graph Exploration Strategy. Chemical Science 2020, 11 (12), 3316–3325. 10.1039/C9SC05704H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller P.; Laino T.; Gaudin T.; Bolgar P.; Hunter C. A.; Bekas C.; Lee A. A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5 (9), 1572–1583. 10.1021/acscentsci.9b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhavoronkov A.; Ivanenkov Y. A.; Aliper A.; Veselov M. S.; Aladinskiy V. A.; Aladinskaya A. V.; Terentiev V. A.; Polykovskiy D. A.; Kuznetsov M. D.; Asadulaev A.; Volkov Y.; Zholus A.; Shayakhmetov R. R.; Zhebrak A.; Minaeva L. I.; Zagribelnyy B. A.; Lee L. H.; Soll R.; Madge D.; Xing L.; Guo T.; Aspuru-Guzik A. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019, 37 (9), 1038–1040. 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- Ponomaryov Y.; Krasikova V.; Lebedev A.; Chernyak D.; Varacheva L.; Chernobroviy A. Scalable and Green Process for the Synthesis of Anticancer Drug Lenalidomide. Chem. Heterocycl. Compd. 2015, 51 (2), 133–138. 10.1007/s10593-015-1670-0. [DOI] [Google Scholar]
- Guo Z.-L.; Deng Y.-Q.; Zhong S.; Lu G. Enantioselective Synthesis of (R)-Salmeterol Employing an Asymmetric Henry Reaction as the Key Step. Tetrahedron: Asymmetry 2011, 22 (13), 1395–1399. 10.1016/j.tetasy.2011.08.008. [DOI] [Google Scholar]
- Schwaller P.; Hoover B.; Reymond J.-L.; Strobelt H.; Laino T. Extraction of Organic Chemistry Grammar from Unsupervised Learning of Chemical Reactions. Science Advances 2021, 7 (15), eabe4166 10.1126/sciadv.abe4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.