

Abstract
To investigate the effect of high fructose diet on ultrastructure and content of hepatic mitochondria, we randomized 6–8 weeks old male C57Bl6/J mice to ad lib chow or high-fat-high-fructose (HF2) diet for 32 weeks. HF2-fed mice gained more weight, had higher plasma alanine aminotransferase, and fasting glucose levels and increased hepatic triglyceride content at all time points compared to chow-fed mice. HF2-fed mice had lower mitochondrial to nuclear DNA ratio compared to chow-fed mice. HF2-fed mice had lower average mitochondrial surface area and the number of mitochondria compared to chow-fed mice. HF2-fed mice had higher expression of the hepatic endoplasmic reticulum stress marker Chop, compared to chow-fed mice. A diet high in fat and fructose leads to enhanced hepatic mitochondrial aging, depletion, and dysfunction, which may be important determinants of nonalcoholic steatohepatitis pathogenesis.
Keywords: fatty liver, NAFLD, NASH, obesity, steatohepatitis
Nonalcoholic fatty liver disease (NAFLD) and its more serious manifestation, nonalcoholic steatohepatitis (NASH), are recognized as one of the leading causes of liver transplantation in the United States (1). NAFLD and NASH are chronic, debilitating diseases without current Food and Drug Administration-approved therapy. Although the molecular mechanism behind NASH is not fully understood, studies suggest that mitochondrial dysfunction and endoplasmic reticulum (ER) stress may play an important role in the progression of the disease (2). Although studies have implicated the presence of abnormal hepatocyte mitochondria in the development of NASH, researchers have not conducted controlled, long-term studies that document changes in mitochondrial morphology or mitochondrial copy number in human or animal models of NASH (3). Furthermore, elucidation of such changes has the potential to uncover novel mechanisms in the development of NASH, diagnostic tools that could help differentiate the severity of the disease, and ultimately treatment options.
Mitochondria are dynamic organelles, changing in shape, volume, and length during the aging process (4). Studies have confirmed that mammalian mitochondria swell, decrease in surface area, and decrease in hepatic mitochondrial number during aging and periods of weight gain (5). A high-fat diet has also been shown to diminish mitochondrial copy number and increase mitochondrial triglyceride content in rats (6). Additionally, hepatic ER stress has shown strong associations with fat accumulation, insulin resistance, and apoptosis, all of which increase with age and NASH (7). Murine and human studies have indicated that PRKR-like ER kinase-CCAAT/enhancer-binding protein-homologous protein (PERK–CHOP) plays a role in the transition of NAFLD to NASH (8). PERK and CHOP play a role in the propagation of signals from the ER to the mitochondria and hepatocyte changes during lipotoxic events (9); however, there is a lack of studies determining the effects of a NASH-inducing diet on the crosstalk between mitochondria and ER stress.
We studied the impact of high fat-high carbohydrate (HFHC) diet, that induces a NASH phenotype in mice (10), on changes in hepatic mitochondrial morphology, function, and content.
METHODS
Animal
Six- to eight-week old male C57/bl6 mice (Jackson Laboratories, Bar Harbor, ME) were randomized to a chow (C) diet (LabDiet, St. Louis, MO), or high-fat, high fructose (HF2) diet (Surwit 58% kcal fat plus drinking water enriched with 42 g/L of carbohydrate: 55% fructose [Alfa Aesar, Ward Hill, MA] and 45% sucrose [Fisher-Scientific, Fair Lawn, NJ] by weight). Animals were group-housed in cages in a temperature-controlled (22 ± 2°C) vivarium at Cincinnati Children’s Hospital Research Foundation (CCHRF) with a 12-hour (07:00–19:00 PM) light/dark cycle. Animals were provided ad libitum access to these diets for 8, 16, or 32 weeks. Body weights were measured biweekly. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Cincinnati Children’s Hospital Research Foundation.
Blood Glucose, Plasma ALT, Liver Triglycerides
Glucose was measured on whole blood collected by tail bleed after a four hour fast at 8, 16, and 26 weeks using an Accu-Check glucose meter (Roche Diagnostics, Indianapolis, IN). Blood was collected at sacrifice and was used to measure alanine aminotransferase (ALT) using a Discret Pak ALT Reagent Kit (Catachem, Bridgeport, CT). To measure liver triglycerides, wet liver tissue was weighed and placed in a relative amount of Tris-Ethylenediaminetetraacetic acid-homogenization buffer solution to make a 100 mg/mL concentration and the tissue was homogenized to extract triglyceride and protein. An enzymatic assay was performed using Triglycerides Reagent Set (Pointe Scientific, Inc., Canton, MI). Photometric absorbance was read at 500 nm.
Histology
Liver sections for histology were obtained at the time of sacrifice at 8, 16, and 32 weeks, fixed in 10% formalin and stained with hematoxylin-eosin and trichrome by the Cincinnati Digestive Health Center Histopathology Core.
Electron Microscopy
Liver sections for electron microscopy were obtained at the time of sacrifice at 8, 16, and 32 weeks and fixed in 3% glutaral-dehyde. Processing was performed by the Cincinnati Digestive Health Center Electron Microscopy Core. Specimens were scanned at 7200× and images of single hepatocytes obtained. Magnification was then increased to 12,000× and images obtained of five areas rich in mitochondria per cell. Images imported to Adobe Photoshop CS 6 (San Jose, CA) and mitochondria magnified at 12,000× in five images per mouse were outlined and hi-lighted. Hi-lighted images were imported to ImageProPlus 7.0 (Media Cybernetics, Bethesda, MD) to count marked mitochondria and measure the surface area.
Relative Gene Expression
Liver tissue was flash frozen and used for RNA extraction using a standard TRIzol reagent protocol (Molecular Research Center, Cincinnati, OH). Quantitative, real-time PCR using Fluorescein amidites-labeled primers specific for CHOP and PERK (Applied Biosystems, Grand Island, NY) was performed using a Stratagene Mx 3000P and/or Mx 3005P (Agilent, Santa Clara, CA) and normalized to the reference gene Rpl18.
Mitochondrial DNA Quantification
Liver tissue was snap-frozen in liquid nitrogen and used for DNA extraction with a QIAamp DNA Mini Kit (Qiagen, Maryland, USA). The same quantity of total DNA was loaded and utilizing Mus16 s Fluorescein amidites-labeled primer, mitochondrial DNA was quantified by quantitative real-time PCR assay. This was normalized to Rpl18 quantified from the same samples.
Statistics
All values are expressed as mean SEM. Statistical significance was evaluated by one- or two-way ANOVA and, where indicated, Student t-test to compare two groups. P values of <0.05 were significant. Statistical analyses were performed using GraphPad Prism Software.
RESULTS
HF2 diet-fed mice gained significantly more weight than chow-fed mice (8 weeks: 42.4 ± 1.2 (HF2) vs 28.8 ± 1.0 (chow), 16 weeks: 47.5 ± 3.5 (HF2) vs 30.6 ± 4.0 (chow), 32 weeks: 56.8 ± 3.1 g (HF2) vs 34.1 ± 2.0 g (chow)) (Fig. 1A). HF2-fed mice had higher fasting glucose level compared to chow-fed mice (8 week: 203.50 ± 1.80 mg/dL (HF2) vs 150.50 ± 1.50 mg/dL (chow), P < 0.05; 16 weeks: 246.500 ± 12.292 mg/dL (HF2) vs 131.833 ± 8.477 mg/dL (chow), P < 0.0001; 32 weeks: 187.750 ± 10.308 mg/dL (HF2) vs 122.167 ± 8.167 mg/dL (chow), P < 0.001) (Fig. 1B). The plasma ALT of HF2-fed mice increased progressively from week 8 to week 32 compared to chow-fed mice (8 weeks: 151.929 ± 10.851 U/L (HF2) vs 69.212 ± 1.5 U/L (chow), P < 0.05; 16 weeks: 162.621 ± 39.976 U/L (HF2) vs 61.344 ± 2.978 U/L (chow), P < 0.05; 32 weeks: 337.058 ± 30.202 U/L (HF2) vs 68.650 ± 6.846 U/L (chow), P < 0.0001) (Fig. 1C).
FIGURE 1.
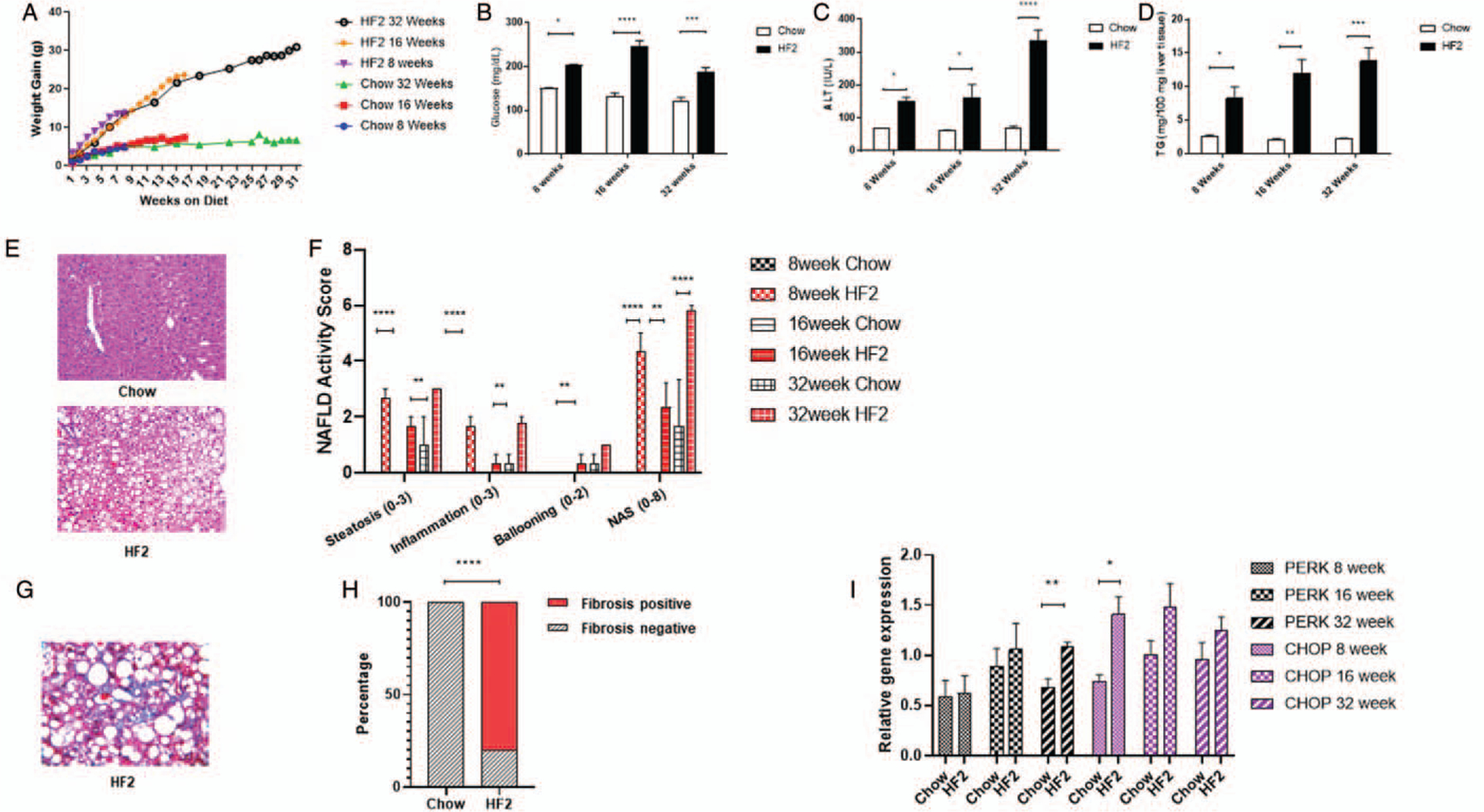
Six to eight weeks old male C57Bl6/J mice fed on high-fat high carbohydrate (HF2) diet for 32 weeks. (A) HF2-fed mice gain more bodyweight compared to chow-fed mice. HF2-fed mice at 8th,16th, and 32nd week on diet have higher (B) fasting blood glucose concentration (mg/dL); (C) plasma alanine aminotransferase (ALT) concentration (IU/L) and (D) hepatic triglyceride concentration (mg/100 mg liver tissues) compared to chow-fed mice. (E) Representative histological image of the liver section showing increased hepatic steatosis HF2-fed mice compared to chow-fed mice. (F) HF2-fed mice have shown a progressive increase in hepatic steatosis, inflammation, and ballooning score at 8th, 16th, and 32nd week compared to chow-fed mice. (G) Representative Masson’s Trichrome stained liver section showing hepatic fibrosis in HF2-fed mice at 32nd week on diet. (H) 80% of mice fed on the HF2 diet for 32 weeks have developed hepatic fibrosis. (I) HF2-fed mice have shown higher expression of hepatic ER stress marker PERK and Chop, a downstream target of PERK mediated ER stress pathway at 8th, 16th, and 32nd week compared to chow-fed mice. Mean ± SEM. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
HF2-fed mice had higher liver triglyceride concentration compared to chow-fed mice at 8, 16, 32 weeks (8 weeks: 10.99 ± 2.50 (HF2) vs 2.78 ± 0.53 (chow), P < 0.05; 16 weeks: 11.96 ± 3.54 vs 2.14 ± 0.27 (chow), P < 0.01; 32 weeks: 13.84 ± 4.39 (HF2) vs 2.21 ± 0.19 (chow) mg/100 mg liver; P < 0.001) (Fig. 1D). We observed higher hepatic steatosis and hepatic inflammation in the histological analysis of the liver section of mice fed the HF2 diet compared to chow-fed mice (Fig. 1E and F). Eighty percent of mice fed on HF2 diet for 32 weeks had hepatic fibrosis (Fig. 1G and H) whereas HF2 diet-fed mice for 8 and 16 weeks did not have hepatic fibrosis.
HF2 diet-induced hepatic ER stress in mice with a progressive increase of Protein Kinase R-like ER kinase (PERK) expression in the liver from week 8 to week 32 (Fig. 1I). At 8th week on HF2 diet, we observed higher hepatic expression of C/EBP homologous protein (Chop), a downstream target of PERK-mediated ER stress pathway, compared to chow-fed mice (Fig. 1I). Further, at 8 weeks mice fed HF2 diet had lower mitochondrial DNA to nuclear DNA ratio compared to chow-fed mice (Fig. 2D). At 16 weeks, HF2-fed mice had a lower surface area of hepatic mitochondria compared to that of chow-fed mice (16 weeks: 0.45 ± 0.02μ2 (HF2) vs 0.70 ± 0.04 μ2 (chow), P < 0.001). Possibly due to aging effect, chow-fed mice at 32 weeks show decreased mitochondrial surface area; however, HF2-fed mice showed no decrease in mitochondrial surface area at 32 weeks compared to 16 weeks (32 weeks: 0.53 ± 0.03 μ2 (HF2) vs 0.36 ± 0.01 μ2 (chow), P < 0.001) (Fig. 2C). Along with the morphological abnormalities, HF2-fed mice had a lower number of hepatic mitochondria compared to that of chow-fed mice at 32 weeks (32 weeks: 26.48 ± 1.87 (HF2) vs 51.53 ± 2.03 (chow) per field; P < 0.0001).
FIGURE 2.

Six to eight weeks old male C57Bl6/J mice fed on high-fat high carbohydrate (HF2) diet for 32 weeks. (A) Graphical illustration of mitochondria surface area measurement using ImagePro Plus 7.0 after digital electron microscopy. (B) Representative digital electron microscopy image of liver section from chow-fed and HF2-fed mice at 16th week and 32nd week on diet. (C) HF2-fed mice have a lower average mitochondrial surface area in the liver compared to chow-fed mice at 16th week of the diet. (D) HF2-fed mice have lower mitochondrial DNA to nuclear DNA ratio compared to chow-fed mice. Mean ± SEM. ***P < 0.001; *P < 0.05.
DISCUSSION
In the present study, we explored the progressive changes that occur to mitochondrial morphology and ER transcription factors in a diet-induced murine model of a NASH. Consistent with that model liver triglycerides and ALT concentration increased progressively in the HF2 diet from 8 weeks to 32 weeks. By 32 weeks, 80% of the HF2 mice had hepatic fibrosis. Additionally, mice fed the HF2 diet had higher expression of hepatic PERK and CHOP compared to that of chow-fed mice. Our data show steep decreases of hepatic mitochondrial DNA content at 8 weeks for mice fed HF2 diet compared to that of chow-fed mice. The surface area of hepatic mitochondria decreased rapidly and inconsistently for mice fed HF2 diet compared to chow-fed mice. Collectively, our data demonstrate that HF2 diet induces mitochondrial dysfunction with the onset of NAFLD and accelerates NASH progression.
High-calorie diets increase hepatic triglyceride concentration (11). Studies have reported that increased hepatic triglyceride concentration causes a decrease in hepatic mitochondrial DNA content and may induce hepatic ER stress (12). From our data, we observed that hepatic triglyceride concentration increased at 8 weeks with a steep decrease in hepatic mitochondrial DNA content compared to chow-fed mice. Previous studies have reported that high-calorie diet may induce PERK mediated hepatic ER stress with increase CHOP expression (13). In our study, we have observed that HF2 diet causes a decrease in hepatic mitochondrial DNA content at 8 weeks and 16 weeks increases in CHOP expression, downstream of the PERK mediated ER stress pathway. Our data support findings from studies that have reported decreased abundance of and dysfunction in hepatic mitochondria induced by hepatic ER stress (14). The present study demonstrates a steep decrease in hepatic mitochondrial DNA content as early as 8 weeks on the HF2 diet. Our data demonstrate that HF2 diet may induce cellular aging in hepatocytes that contributes to the progression of NAFLD to NASH.
Liver diseases other than NASH/NAFLD, such as viral hepatitis and alcoholic liver disease, have been shown to have hepatic mitochondrial dysfunction. Even extrahepatic diseases such as myopathies (Duchenne muscular dystrophy), idiopathic pulmonary fibrosis, hypertensive cardiomyopathy (15), and radiation-induced skin injury are known to affect mitochondrial content, function, and physiology. Similar to the liver fibrosis in NASH, mitochondrial dysfunction leads to increased fibrosis in the solid organs and tissues (16). Treatment with agents that rescue mitochondrial function, such as Epicatechin, has been shown to increase mitochondrial biogenesis and reverse the mitochondrial dysfunction in preclinical disease models of Duchenne muscular dystrophy (17). Preclinical studies have reported that enhanced mitochondrial function via inhibition of α-amino-β-carboxymuconate-ε-semial-dehyde decarboxylase (ACMSD) induces de novo NAD+, SIRT1 activity reversing NAFLD (18).
The present study provides experimental evidence of the effect that a diet high in fat and carbohydrate/sugar may have on the ultrastructure of hepatic mitochondria in mice similar to that long observed in adult patients with NASH (3). Similar defects in mitochondrial ultrastructure have also been borne out anecdotally in children with NASH (19). Thus, through our study, we hope to advocate for an improvement in the diet of our children to help prevent ongoing mitochondrial damage that may result in them having NASH as adults.
Our HF2 diet-fed mice demonstrate quick, persistent decreases in mitochondrial DNA while developing histologic findings consistent with NASH. In our model, we observed an early decrease in mitochondria size. We simultaneously observed increased expression of ER stress markers in the liver of HF2-fed mice. Our data demonstrate that mitochondrial dysfunction plays a key role in promoting aging of hepatic mitochondria. Deciphering the molecular mechanism triggered by HF2 diet that causes decreases in hepatic mitochondria content and mitochondrial dysfunction would provide a therapeutic option to treat NASH. In future studies, we will investigate the extrahepatic effect of HF2 diet in peripheral blood mononuclear cells (PBMCs). A correlation of hepatic mitochondrial content and dysfunction to PBMCs could be used as a biomarker to follow NASH pathogenesis.
Acknowledgments
This work was supported by NIH R01DK100314 (R.K.), T32 DK007727 18 (K.S.B.), Cincinnati Digestive Health Center PHS Grant P30 DK078392, NASPGHAN Foundation Mentored Summer Student Research Program (R.G.).
Footnotes
The authors have no conflict of interest to declare.
REFERENCES
- 1.Golabi P, Bush H, Stepanova M, et al. Liver transplantation (LT) for cryptogenic cirrhosis (CC) and nonalcoholic steatohepatitis (NASH) cirrhosis data from the Scientific Registry of Transplant Recipients (SRTR): 1994 to 2016. Med (United States) 2018;97:e11518. doi: 10.1097/MD.0000000000011518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Léveillé M, Estall JL. Mitochondrial dysfunction in the transition from NASH to HCC. Metabolites 2019;9:223. doi: 10.3390/metabo9100233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001;120:1183–92. [DOI] [PubMed] [Google Scholar]
- 4.Cedikova M, Pitule P, Kripnerova M, et al. Multiple roles of mitochondria in aging processes. Physiol Res 2016;65:S519–31. [DOI] [PubMed] [Google Scholar]
- 5.Herbener GH. A morphometric study of age dependent changes in mitochondrial populations of mouse liver and heart. J Gerontol 1976;31:8–12. [DOI] [PubMed] [Google Scholar]
- 6.Mansouri A, Gattolliat CH, Asselah T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology 2018;155:629–47. [DOI] [PubMed] [Google Scholar]
- 7.Bozaykut P, Sahin A, Karademir B, et al. Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic steatohepatitis. Mech Ageing Dev 2016;157:17–29. [DOI] [PubMed] [Google Scholar]
- 8.Kim JY, Garcia-Carbonell R, Yamachika S, et al. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell 2018;175:133.e15–45.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verfaillie T, Rubio N, Garg AD, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012;19:1880–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohli R, Kirby M, Xanthakos SA, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010;52:934–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisinger K, Krautbauer S, Hebel T, et al. Lipidomic analysis of the liver from high-fat diet induced obese mice identifies changes in multiple lipid classes. Exp Mol Pathol 2014;97:37–43. [DOI] [PubMed] [Google Scholar]
- 12.Jo H, Choe SS, Shin KC, et al. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013;57:1366–77. [DOI] [PubMed] [Google Scholar]
- 13.Rutkowski DT, Wu J, Back SH, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell 2008;15:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim JH, Lee HJ, Ho Jung M, et al. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cell Signal 2009;21:169–77. [DOI] [PubMed] [Google Scholar]
- 15.Dai DF, Johnson SC, Villarin JJ, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 2011;108:837–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guimarães EL, Best J, Dollé L, et al. Mitochondrial uncouplers inhibit hepatic stellate cell activation. BMC Gastroenterol 2012;12:68. doi: 10.1186/1471-230X-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald CM, Ramirez-Sanchez I, Oskarsson B, et al. (−)-Epicatechin induces mitochondrial biogenesis and markers of muscle regeneration in adults with Becker muscular dystrophy. Muscle Nerve 2020;63:239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katsyuba E, Mottis A, Zietak M, et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018;563:354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lotowska JM, Sobaniec-Lotowska ME, Bockowska SB, et al. Pediatric non-alcoholic steatohepatitis: the first report on the ultrastructure of hepatocyte mitochondria. World J Gastroenterol 2014;20:4335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]