

Abstract
Painful diabetic neuropathy occurs in approximately 20% of diabetic patients with underlying pathomechanisms not fully understood. We evaluated the contribution of the CaV3.2 isoform of T-type calcium channel to hyperglycemia-induced changes in cutaneous sensory C-fiber functions and neuropeptide release employing the streptozotocin (STZ) diabetes model in congenic mouse strains including global knockouts (KOs). Hyperglycemia established for 3 – 5 weeks in male C57BL/6J mice led to major reorganizations in peripheral C-fiber functions. Unbiased electrophysiological screening of mechanosensitive single-fibers in isolated hairy hindpaw skin revealed a relative loss of (polymodal) heat sensing in favor of cold sensing. In healthy CaV3.2 KO mice both heat and cold sensitivity among the C-fibers seemed underrepresented in favor of exclusive mechanosensitivity, low-threshold in particular, which deficit became significant in the diabetic KOs. Diabetes also led to a marked increase in the incidence of spontaneous discharge activity among the C-fibers of wildtype mice, which was reduced by the specific CaV3 blocker TTA-P2 and largely absent in the KOs. Evaluation restricted to the peptidergic class of nerve fibers - measuring KCl-stimulated CGRP release - revealed a marked reduction in the sciatic nerve by TTA-P2 in healthy but not diabetic wildtypes, the latter showing CGRP release that was as much reduced as in healthy and, to the same extent, in diabetic CaV3.2 KOs. These data suggest that diabetes abrogates all CaV3.2 functionality in the peripheral nerve axons. In striking contrast, diabetes markedly increased the KCl-stimulated CGRP release from isolated hairy skin of wildtypes but not KO mice, and TTA-P2 reversed this increase, strongly suggesting a de novo expression of CaV3.2 in peptidergic cutaneous nerve endings which may contribute to the enhanced spontaneous activity. De-glycosylation by neuraminidase showed clear desensitizing effects, both in regard to spontaneous activity and stimulated CGRP release, but included actions independent of CaV3.2. However, as diabetes-enhanced glycosylation is decisive for intra-axonal trafficking, it may account for the substantial reorganizations of the CaV3.2 distribution. The results may strengthen the validation of CaV3.2 channel as a therapeutic target of treating painful diabetic neuropathy.
Keywords: T-type calcium channel, neuropathic pain, excitability, Calcitonin Gene-related Peptide, TTA-P2, sciatic nerve, neuropeptide release
INTRODUCTION
One of the most distressing sequelae in diabetic patients is peripheral neuropathy with symptoms of tingling, burning feet, hyperalgesia to heat and touch and, in some patients, spontaneous attacks of sharp deep pain. The prevalence of these “positive” symptoms varies between articles. Recent literature suggests about 20% of diabetic patients suffer from neuropathy (DN) which is accompanied by pain (pDN) in about half of the cases (Calcutt, 2020; Gylfadottir et al., 2019). Underlying mechanisms leading to pDN are still poorly understood. In diabetic patients and rodent models hyperexcitability and spontaneous firing of unmyelinated C fibers were shown and suggested to be linked to the symptomatology (Garcia-Perez et al., 2018; Orstavik et al., 2006,Garcia-Perez, 2018 #109). On a cellular level, an increased expression of the voltage-gated sodium channels NaV1.7 and NaV1.8, responsible for triggering and the upstroke of the action potentials (APs), has been reported in axons of primary nociceptive neurons, causing a reduction of the use-dependent conduction failure in these fibers (Sun et al., 2012). The reactive metabolite methylglyoxal, which is a substrate for non-enzymatic glycation of proteins, is elevated in plasma of pDN patients (Bierhaus et al., 2012). This in turn results in posttranslational modifications of NaV1.8 and the transient receptor potential channel ankyrin 1 (TRPA1), enhancing their activity and the neuronal excitability (Andersson et al., 2013; Eberhardt et al., 2012). However, since pDN is a multifactorial disease, other cellular targets must be considered.
Another ion channel that in recent years has been linked to hyperexcitability of peripheral afferents is the CaV3.2 isoform of low voltage-activated (T-type) Ca++ channel. With its relatively hyperpolarized activation threshold (−50 mV), rapid inactivation and slow deactivation kinetics it augments sub-threshold depolarizations, bringing the membrane closer to action potential threshold (Chemin et al., 2002; Williams et al., 1999). It thus plays an important role in facilitating spontaneous firing and pacemaker activities and is widely studied with regard to neuronal excitability changes for instance in epilepsy and cardiovascular diseases (Cain and Snutch, 2012; David et al., 2010; Emanuel et al., 1998; Todorovic and Lingle, 1998a). In the periphery the CaV3.2 channel is found in small diameter isolectin B4 (IB4)-positive and capsaicin sensitive nociceptive DRG neurons, along their axons as well as their cutaneous endings, and in specific sensory neuronal subtypes such as low threshold Aδ mechanoreceptors, i.e. rapidly adapting D-hair receptors (Bernal Sierra et al., 2017; François et al., 2015; Rose et al., 2013). A function of CaV3.2 in peripheral nerve electrophysiology is suggested by the effect of mibefradil, an unspecific blocker of T-type channels; the drug reduces the propagated compound action potential of the C-fibers (Gomez et al., 2020). CaV3.2 current was further shown to facilitate the synaptic transmission to lamina II dorsal horn neurons, supporting its contribution to peripheral input and spinal sensitization (Candelas et al., 2019; Jacus et al., 2012). Importantly, overexpression of functional CaV3.2 in sensory neurons was found in several neuropathic pain models in rodents such as L5 spinal nerve ligation and spared nerve injury (Feng et al., 2019; Gomez et al., 2020; Kang et al., 2018; Liu et al., 2019). This overexpression was linked to hyperexcitability of specific neurons and concomitant behavioral pain (Duzhyy et al., 2015; Jagodic et al., 2007). Pharmacological inhibition of CaV3.2 alleviated diabetes-induced hyperalgesia and allodynia (Ayoola et al., 2014; Latham et al., 2009; Messinger et al., 2009; Obradovic et al., 2014; Todorovic et al., 2002). Furthermore, post-translational modification via N-glycosylation of CaV3.2 exerts major effects, enhancing channel density in the neuronal membrane, permeability, open probability, and glucose-dependent potentiation (Lazniewska and Weiss, 2017; Orestes et al., 2013; Weiss et al., 2013). Diabetic hyperglycemia increases the N-glycosylation of CaV3.2 (Fukami et al., 2017; Shathili et al., 2018). Importantly, blocking or reversing the glycosylation of CaV3.2 attenuated the channel’s activity and also alleviated diabetic neuropathic pain symptoms in mice (Orestes et al., 2013).
Here, we set out to fill a certain gap in the literature on diabetic neuropathy between cellular/molecular and behavioral data by exploring how the cellular changes reported translate to functional changes in whole nerves and cutaneous nerve endings of mice, both wildtypes and CaV3.2 null mutants. We used methods of quantifying stimulated calcitonin gene-related peptide (CGRP) release and electrophysiological recordings from identified cutaneous single-fibers to evaluate the effect sustained diabetes exerts on sensory neurons in vitro. Stimulated neuropeptide release from peptidergic sensory nerves is commonly used to study mass activation of primary afferents (Levine et al., 1993). Several parts of these neurons, including the peripheral nerve axons, exhibit calcium influx-dependent vesicular exocytosis, and innervated tissues release vasoactive neuropeptides in a physiological manner (Lundberg et al., 1992; Sauer et al., 1999). Previous studies show T-type Ca2+ channels play an important role in controlling CGRP release from trigeminal ganglia (Amrutkar et al., 2011) but the channel’s contribution to release from cutaneous nerve endings and along the axon or its role in adjusting the excitability of specific fiber populations have not yet been explored. Recording from single-fibers enabled to study excitability alterations translating into spontaneous firing and reduced sensory thresholds (parts of the data have been presented at 7th International Congress on Neuropathic Pain in London/UK 2019 and at SfN in Chicago/USA 2020).
METHODS
Animals
All animal experiments were approved by the responsible Animal Protection Authority (Regierung von Unterfranken, Würzburg, Germany). Inbred congenic C57BL/6/J and CaV3.2 knock-out (KO) mice (B6;129-Cacna1htm1Kcam/J; Jackson Laboratories) of both sexes and ranging in weight between 20–25g were housed in group cages in a temperature-controlled environment with a 12h light-dark cycle and were supplied with food and water ad libitum. On the day of the final experiment accounted animals were killed in a rising CO2 atmosphere in accord with German and European laws.
Diabetes model
We used multiple low-dosed streptozotocin (STZ) injections for inducing diabetes in C57BL/6J control and CaV3.2 KO mice (Furman, 2015). On the day of STZ injections food was removed from cages 4 hours prior to injections, water supply remained ad libitum. Prior to STZ injections 6 ml sodium citrate buffer was made by mixing 3 ml 0.1 M Na-citrate 1:1 with 3ml 0.1 M citric acid and pH was adjusted to 4.5. Citrate buffer and STZ powder were kept in darkness and cooled in ice. Immediately prior to injections STZ was dissolved in the citrate buffer to a concentration of 4 mg/ml. Mice were weighted and immediately injected with STZ solution intraperitoneally (i.p.) at 40 mg/kg. Mice were returned to their home cages and provided with food and 10% sucrose water for the rest of the day. STZ injections were repeated for 5 consecutive days; on the last day mice were provided with regular tap water. Glucose tail vein blood levels were measured in mice using a dipstick glucose monitoring device (Accu-Check, ROCHE, Germany) on days 14 and 21 (counting from the first injection day). Animals with blood levels above 250 mg/dL at day 21 were considered diabetic and were sacrificed for further experiments within a maximum of 14 days.
CGRP release
Stimulated neuropeptide release was measured ex vivo in hindpaw hairy skin or desheathed sciatic nerve preparations of healthy and diabetic mice as previously described (Babes et al., 2010; Sauer et al., 1999). Samples were placed in carbogen-gassed SIF solution (see below; 5 mM glucose) or high-glucose SIF (25 mM) in case of preparations from diabetic mice and positioned in a shaking bath set to 32 °C (skin) or 38 °C (nerve) for a washout period of 30 min. The preparations then passed a series of four consecutive elution steps (S1-S4) each lasting 5 minutes The first two incubations were to determine basal CGRP release. In the third incubation step high potassium SIF solution at 40 mM (KCl) concentration was used for stimulation, the fourth incubation step assessed the recovery of the response. CGRP contents were determined using commercial enzyme immunoassays (EIAs; Bertin Pharma, Montigny, France), as described previously (Babes et al., 2010). Samples were photometrically analyzed using a microplate reader (Opsys MR™, Dynex Technologies, Chantilly, VA, USA). To investigate dependence of stimulated CGRP release on CaV3.2 or N- and L-type channels we used TTA-P2 (10 μM) or ω-conotoxin (1 μM) combined with nifedipin (10 μM) to block these calcium channels in the skin of C57BL/6J mice, respectively. These blockers were added from the second to the fourth incubation period. In case of deglycosylation, neuramindase (2 units/ml) was dissolved in freshly bubbled SIF and the tissue samples were incubated as a pretreatment for 2 hours, after 1 hour the incubation solution was renewed to maintain oxygen supply and buffer capacity. Skin preparations were incubated at 32°C and isolated sciatic nerves at 38°C, respectively.
Determination of skin and nerve CGRP content
Skin and sciatic nerve CGRP contents were determined as described previously (Eberhardt et al., 2008). In brief, hairy hindpaw skin and sciatic nerve preparations of wildtype and CaV3.2 knockout mice were heated in 1 ml acetic acid (2 M) for a 10 min period at 95 °C. Thereafter the preparations were homogenized by an Ultra-Turrax T8 (IKA Works, Staufen, Germany) and heated again for 10 min at 95 °C - CGRP is heat-stable. The suspension was then centrifuged for 30 min at 10,000g. The clear supernatant was diluted 1:50 with EIA buffer to adjust the pH value to 7.4 and to cope with the high concentrations of CGRP. The CGRP content of this solution was determined using the commercial assay as already described and the amounts of CGRP content are given in reference to 1g tissue weight in pmol/g (mean ± sem). The mean tissue weights of hairy skin and sciatic nerve preparations were comparable for wildtype and KO mice (wildtype 6.5 ± 0.7 mg and 5.3 ± 0.8 mg in KO, wildtype 2.8 ± 0.2 mg and 2.7 ± 0.6 mg in KO, respectively).
Skin-nerve recordings
The isolated preparation of the mouse hind limb skin and innervating saphenous nerve was used to record extracellular action potentials from functionally singled cutaneous C-fibers using a compact all-in-one rig (Avere Solutions, Erlangen, Germany). A skin flap of the hairy hindpaw skin together with the saphenous nerve was excised and bond with vaseline to the bottom of an organ bath, corium side up. The chamber containing the skin was continuously perfused with SIF solution (for control mice) or high-glucose SIF (for tissue from diabetic mice) warmed to 32°C. The cut end of the nerve trunk was desheathed and thin free filaments were gently sucked into a recording glass pipette to obtain a single unit recording from a fiber with a delimited receptive field (RF). Voltage signals of the selected fiber were amplified (Avere Solutions, Erlangen, Germany), digitized (CED Micro1401, Cambridge Electronic Design, UK) and stored on disk for post-hoc analysis using custom written routines in Spike2 software (Cambridge Electronic Design, UK). Mechanical stimulation was used as search stimulus and receptive fields were mapped using a blunt glass rod. Once a mechanical RF was identified, a metal microelectrode was placed on the skin inside the RF and served as the cathode for electrical stimulation. Axonal conduction velocity was determined on the basis of distance and action potential latency in response to electrical stimulation. Units conducting at < 1.4 m/s were classified as unmyelinated C-fibers (Bessou and Perl, 1969). The marking technique, in which shifts in latency to electrical stimulation identify mechanically evoked action potentials (Schmelz et al., 1996), was used to confirm that mechanical and electrical stimuli affected one and the same C-fiber. Mechanical thresholds were determined using gravity-driven hand-held von Frey probes with calibrated buckling loads scaled from 1 to 128 mN in a geometric sequence. Responses to cooling or heating stimuli were determined by placing a metal ring (ca. 9 mm diameter) on the skin encircling the RF which was perfused using a feedback-controlled flow-through thermal stimulator (Avere Solutions, Erlangen, Germany). A fiber was considered cold/heat-sensitive, if more than two action potentials occurred during the cooling/heating ramp of 30 s duration to 47° C, respectively. The activation threshold was defined as the temperature at which the second action potential of the response occurred. Individual C-fibers were classified functionally as mechano-heat sensitive (C-MH), high threshold mechanosensitive (> 5.6 mN, C-HTM), low threshold mechanosensitive (< 2.8 mN, C-LTM), mechano-cold (C-MC) or mechano-heat-cold sensitive (C-MHC). During characterization fibers were given at least 2 min rest between stimuli to prevent adaptation. After sensory characterization, in particular after heat stimulation, a proportion of C-fibers develops low-frequent irregular ongoing (“spontaneous”) activity (Bessou and Perl, 1969). This ongoing firing was recorded for at least 10 minutes in SIF (control mice) or ‘hyperglycemic’ SIF (diabetic mice) followed by 10 minutes in hypoxic SIF solutions. To probe the CaV3.2 involvement, a specific blocker TTA-P2 (10μM) in hypoxic normal or ‘hyperglycemic’ SIF solution was subsequently washed-in as a third step while continuously recording spontaneous firing in these fibers. The spontaneous discharge activity was analyzed over the last 8 minutes of each experimental period to exclude eventual after-discharge from previously applied mechanical/thermal stimulation. Spontaneous firing was defined as at least two spikes per minute, i.e. 16 spikes per 8 minutes recording. Bursting discharge pattern was defined as at least five consecutive spikes at a rate beyond the mean frequency of the individual fiber during the 8 min of recording. For instance, for a fiber with a mean firing frequency of 0.2 spikes/sec a burst would be defined as a cluster of at least 5 spikes during a coherent period of 25 seconds.
For evaluating the effects of de-glycosylation skin-flaps were immersed in neuraminidase-containing SIF (NEU, 2 units/ml) for at least 2 hours prior to experiment onset, and spontaneous firing was recorded while the RF was superfused with neuraminidase.
Chemicals and solutions
Synthetic interstitial fluid (SIF; (Bretag, 1969)) used for all in vitro experiments consisted (in mM) of: 107.8 NaCl, 26.2 NaCO3, 9.64 Na-gluconate, 7.6 sucrose, 5.05 glucose, 3.48 KCl, 1.67 NaH2PO4, 1.53 CaCl2 and 0.69 MgSO4. PH was buffered to 7.4 using carbogen bubbling (95% oxygen and 5% carbon dioxide). To induce unspecific neuronal depolarization, SIF containing 40 mM KCl was generated by equimolar substitution of sodium chloride with potassium chloride. Hyperglycemia was mimicked by increasing the glucose concentration from 2.5 to 25 mM in SIF; tissue preparations from diabetic animals were always bathed in this solution. Hyperglycemic hypoxia: Hypoxia was induced by nitrogen gassing of normal or hyperglycemic HEPES-buffered SIF consisting of (in mM): 95.3 NaCl, 25 Na-gluconate, 7.6 sucrose, 25 glucose, 3.48 KCl, 1.67 NaH2PO4, 1.53 CaCl2, 0.69 MgSO4 and 10 HEPES bubbled with 97.5% N2 and 2.5% CO2 instead of carbogen at pH 7.4. The hypoxic condition was verified in pilot experiments using an oxygen sensor (FireStingO2; Pyro Science, Germany); oxygen levels during continuous nitrogen gassing dropped to 7.5%. ω-conotoxin (Alamone Labs, Israel) combined with nifedipin (Sigma-Aldrich, Germany) and a selective Ttype channel blocker 3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide (TTA-P2; Alamone Labs, Israel) was diluted in normal or hyperglycemic SIF to a final concentration of 10μM. Neuraminidase (Sigma-Aldrich, Germany) was used to deglycosylate CaV3.2, the enzyme was diluted in normal or hyperglycemic SIF to a final concentration of 2 units/ml. Streptozotocin (STZ, Enzo Life Science GmbH, Germany), to induce diabetes, was dissolved in sodium citrate buffer (0.1 M Na-citrate mixed 1:1 with 0.1 M citric acid) – see diabetes model.
Statistics
For iCGRP release experiments data is displayed in pg / ml (mean ± SEM). The column diagrams show the overall stimulated release (AUC, in pg / ml iCGRP). For this, values of the stimulated and the successive sample (S3 + S4) were added up and the sum of the baseline values (S1 + S2) was subtracted to gain quasi an area-under-the-curve (AUC, in pg / ml iCGRP). Within one experimental group data of iCGRP release were analyzed by Wilcoxon matched pairs test and to compare the time course of different groups ANOVA analysis together with Fisher’s LSD post-hoc test were applied. AUC data from multiple groups were compared by one-way ANOVA again followed by Fisher’s LSD post-hoc test.
Group variance of single-fiber recordings and of the ‘success’ rate of STZ treatment (i.e., development of hyperglycemia after STZ-treatment) were analyzed using χ2 test. Excitability changes in fibers were tested using either Wilcoxon matched pairs (for comparison of pre- vs. post-treatment changes within a group) or Mann-Whitney U-test (for comparison between two fiber groups). All statistical tests were performed with Statistica 7 software (StatSoft, Tulsa, USA); differences were considered significant at p < 0.05 and marked with *.
RESULTS
In order to evaluate the possible role that peripheral CaV3.2 may play in pDN we measured stimulated neuropeptide release from isolated skin flaps and desheathed sciatic nerves and recorded sensory and conductive capacities, as well as hypoxia-evoked activation of characterized cutaneous C-nociceptors. Diabetic mice of congenic C57BL/6J controls and CaV3.2 KOs were compared to healthy mice of the same genotypes. Whenever preparations from diabetic animals were used, diabetic tissue conditions were mimicked by elevating the glucose concentration to 25 mM of applied extracellular fluids.
Streptozotocin induction of diabetes
Streptozotocin (STZ) injections (low dosage model as described in Methods) were able to increase blood glucose levels to a diabetic range (>250 mg/dL). Both C57BL/6J and KO groups reached comparable blood sugar levels (307 ± 39 mg/dL, n=42 in C57BL/6J vs. 316 ± 18 mg/dL, n=14 in CaV3.2 KO at day 21). In contrast to the similarity in glucose levels of diabetic mice in both genotypes, the number of mice becoming diabetic through STZ injections differed. More than a third of the C57BL/6J control group had glucose levels above 250 mg/dL following the STZ treatment (42 of 97 mice; 43%) in comparison to below a quarter of the KO mice (14 of 73; 19%, χ2 (df = 1) = 10.9, p = 0.0009). In line with previously published information (Furman, 2015), there was a sex-specific difference in diabetes development of both genotypes. More males than females became diabetic following STZ injections: 49 % of C57BL/6 (42 of 86 males) and 33 % of KO mice (13 of 39 males) vs. 0 % (0 of 11 females) in C57BL/6J and 3 % (1 of 34 females) of KO mice. Thus, males were more susceptible to develop diabetes than female mice (χ2 (df = 1) = 28.3, p = 1e−7). The sex difference applies only to the multiple low-dose, not to the single high-dose, STZ protocol and results from estrogens protecting the pancreatic β-cells from glucose-derived oxidative stress and subsequent apoptosis (Le May et al., 2006; Rossini et al., 1978; Witcher et al., 2010). This led us to employ more males than females in the course of our study.
Diabetes increased stimulated CGRP release from skin in contrast to peripheral nerve
We used stimulus-induced CGRP release as a measure of mass activation of primary peptidergic nociceptors to evaluate diabetes-induced functional changes. Fig. 1 summarizes the iCGRP release from isolated hairy hindpaw skin and desheathed sciatic nerves from healthy (-contr.) and STZ-diabetic (-diab.) C57BL/6J mice. In skin from both healthy and diabetic mice depolarizing stimulation (20 or 40 mM potassium) induced a significant increase in CGRP release over baseline (Wilcoxon matched pairs test, p < 0.001), which was significantly augmented in diabetic animals (Fig. 1A, F(3.69) = 6.07, p < 0.01; LSD p < 0.01). This was true for both 20 mM potassium stimulation (Fig. 1B, F(3.37) = 12.15, p < 0.01, LSD p = 0.04), as well as 40 mM potassium stimulation (LSD p = 0.001). In contrast, neuropeptide release from isolated sciatic nerves revealed an opposite effect; we found a significant reduction of 40 mM KCl-induced iCGRP release in diabetic versus healthy C57BL/6J mice (F(3.54) = 4.14, p < 0.01; LSD p < 0.01, Fig. 1C); also the AUC was significantly reduced (F(1.18) = 4.69, p = 0.04, Fig 1D). We summarize that diabetes affected neuropeptide release differentially: in the skin stimulated iCGRP release was increased while the sciatic release was diminished.
Fig 1: Diabetes-induced changes in stimulated CGRP release from C57BL/6J skin and sciatic nerve.
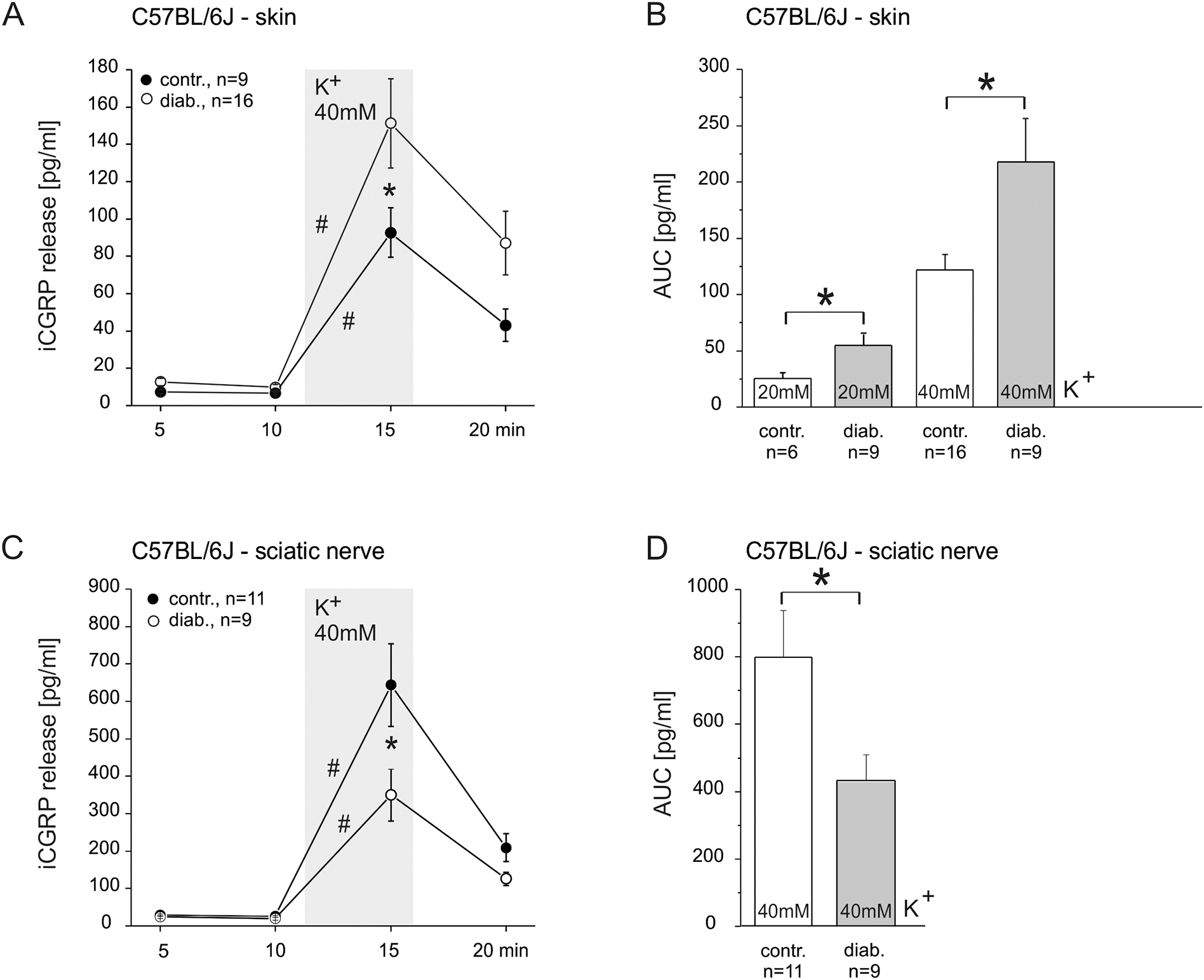
A. KCl-evoked CGRP release from isolated hairy skin of healthy and diabetic mice. B. AUC display of the data in A plus KCl concentration dependency. C. KCl-evoked CGRP release from isolated sciatic nerve of healthy and diabetic mice. D. AUC display of the data in C. Significant increases in CGRP release over baseline and significant differences between healthy and diabetic preparations are marked with a hashtag or an asterisk based on Wilcoxon matched pairs test and ANOVA repeated measurements, respectively.
Diabetes changed the prevalence of sensory fiber subpopulations and of spontaneous activity in skin nociceptors
In addition to neuropeptide release we studied diabetes-induced changes of the distribution of sensory properties and hypoxia-induced activation among C-fiber populations through single-fiber recordings from cutaneous nociceptors. Skin-nerve preparations from diabetic animals were examined in SIF with 25 mM glucose while preparations from healthy mice were bathed in normal SIF (5 mM glucose). After sensory testing we also assessed the eventual development of spontaneous discharge activity, because it has been shown that diabetes increases the propensity of spontaneous discharge among rat nociceptors, in particular under the condition of tissue hypoxia (Fuchs et al., 2010; Suzuki et al., 2002b).
We recorded from 18 Aδ fibers of healthy and diabetic mice but found no obvious diabetes-related in sensory properties or spontaneous activity at least 3 weeks after induction of hyperglycemia. However, among the C-fibers conspicuous differences became quickly apparent. We therefore restricted the search to a total of 44 C-units from healthy and diabetic WT mice. The fibers were assigned to sensory sub-classes according to their responses to mechanical and thermal stimulation. In healthy C57BL/6J preparations the 24 mechanosensitive C-fibers were subclassified as 14 (58%) C-MH, 3 (12.5%) C-MHC, 4 (17%) C-HTM and 3 (12.5%) C-LTM, making C-MH the dominant fiber population in the hindpaw innervation of the saphenous nerve (Fig. 2A, upper graph). These data are consistent with comparable data showing C-MHs to be the most common type of unmyelinated mechanosensitive fibers in the healthy mouse (Hoffmann et al., 2008). Twenty C-fibers from diabetic mice were characterized as 6 (30%) C-MH, 6 (30%) C-MHC, 3 (15%) C-MC, 2 (10%) C-HTM, and 3 (15%) C-LTM (Fig. 2A, lower graph). When comparing the fiber compositions in healthy and diabetic WT mice, diabetes increased the incidence of cold sensitivity from 12.5% to 45% (C-MHC + C-MC) partly at the expense of heat sensitivity (C-MH + C-MHC) which difference appears significant (χ2 (df = 1) = 6.64, p = 0.0099). However, whether this relative shift in prevalence may translate into a perceptual difference would require an analysis of threshold and stimulus-response relationship, which was beyond the scope of the study. The sensory and conductive properties of the different fiber populations are listed in Table 1. There was no diabetes-related change in the conduction velocity, electrical, mechanical or heat threshold of these C-fibers.
Fig. 2: Diabetes-induced changes in C-fiber sensitivities and spontaneous activity - single-fiber recordings from hairy hindpaw skin.
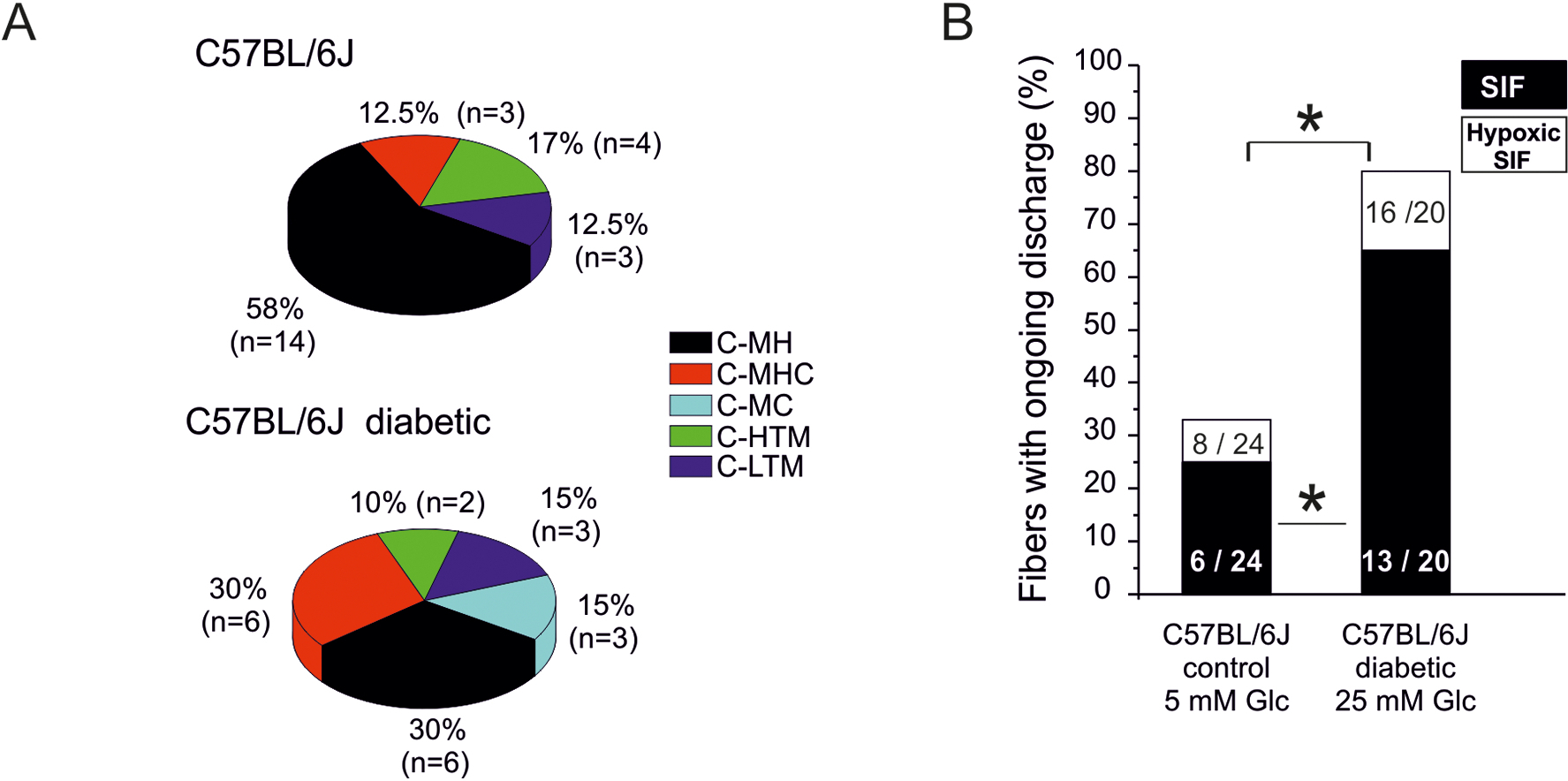
A. Distribution of sensory C-fiber subclasses in healthy versus diabetic C57BL/6J mice: MH = mechano-heat, MHC = mechanoheat-cold, MC = mechano-cold, HTM = high-threshold mechano-, LTM = low-threshold mechano-sensitive. B. Incidence (% of fibers) of spontaneous discharge activity (after sensory testing) in healthy versus diabetic C57BL/6J mice and its increment during 10 min hypoxia. Significant difference between healthy and diabetic preparations is marked with an asterisk based on ANOVA repeated measurements.
Table 1:
Table of fibre populations and sensory properties of healthy control and diabetic mice
| Healthy control mice | diabetic mice | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| strain | fiber type | n | cv (m/s) | median v. Frey (mN) | electrical threshold | heat threshold (°C) | n | cv (m/s) | median v. Frey (mN) | electrical threshold | heat threshold (°C) |
| C57BL/6 | C-MH | 14 | 0.42 ± 0.02 | 4 | 3.3 ± 1.4 | 37.9 ± 0.7 | 6 | 0.52 ± 0.06 | 5.7 | 4.6 ± 2.6 | 39.4 ± 1.3 |
| C-MHC | 3 | 6 | |||||||||
| C-MC | -- | 3 | |||||||||
| C-HTM | 4 | 0.24 ± 0.04 | 8 | 5 ± 3.2 | -- | 2 | 0.28 ± 0.06 | 5.7 | 0.09 ± 0.07 | -- | |
| C-LTM | 3 | 0.3 ± 0.01 | 2 | 0.5 ± 0.9 | -- | 3 | 0.5 ± 0.1 | 2 | 8.1 ± 6.9 | -- | |
| Cav3.2 −/− | C-MH | 6 | 0.49 ± 0.06 | 4.8 | 5.7 ± 1.7 | 37.9 ± 1 | 5 | 0.6 ± 0.2 | 2.4 | 4.2 ± 2.4 | 43.7 ± 1.2 |
| C-MHC | 3 | 2 | |||||||||
| C-MC | 1 | 3 | |||||||||
| C-HTM | 7 | 0.45 ± 0.06 | 5.7 | 10.9 ± 3.8 | -- | 7 | 0.49 ± 0.1 | 5.7 | 1.5 ± 1.1 | ||
| C-LTM | 2 | 0.5 ± 0.14 | 1 | 2 ± 0.7 | -- | 7 | 0.23 ± 0.04 | 4 | 3.3 ± 2.3 | -- | |
C-MH, mechano, heat-sensitive; C-MHC, mechano, heat, cooling-sensitive, C-MC, mechano-cold sensitive; C-LTM, low threshold mechano-sensitive; HTM, high threshold mechano-sensitive
Diabetes significantly increased the incidence of spontaneously firing fibers about 2.5-fold (Fig. 2B, black columns) from 6 of 24 fibers (= 25%) in control C57BL/6 mice to 13 of 20 (= 65%,) in diabetic C57BL/6J, (χ2 (df = 1) = 7.11, p = 0.007). The mean spontaneous discharge rate was higher, but not significantly, under diabetic versus healthy conditions (203 ± 147 spikes/8 min = 0.4/s versus 113 ± 58 spikes/8min = 0.2/s, respectively). The spike rates of fibers did not correlate with blood sugar levels of the mice (Pearson correlation). Firing patterns in the ongoing discharge activity were also evaluated, but none of the 6 spontaneously discharging fibers from healthy and only 2 of the 13 fibers from diabetic C57BL/6J mice fired in bursts (6 and 8 bursts per 8 min recording, data not shown). Thus, STZ diabetes seems to induce an increased incidence of spontaneous activity in the form of irregular low-frequency spiking, which is consistent with previously reported findings (Fuchs et al., 2010; Suzuki et al., 2002b).
Endoneural hypoxic conditions have previously been reported to develop in diabetic peripheral nerves and spinal dorsal roots due to microangiopathy (Yorek, 2015). In order to mimic this diabetic tissue condition, we applied SIF-based hypoxic solution for ten minutes to the receptive fields of the recorded C-fibers: hypoxic SIF in healthy C57BL/6J and hypoxic - high glucose-SIF in diabetic mice. In both groups hypoxia elevated the number of fibers with spontaneous discharge: from 25 % to 33 % in healthy and from 65 % to 80% in diabetic mice so that the significance of the difference between control and diabetic groups was preserved under hypoxia (χ2 (df = 1) =11.99, p = 0.0005). The hypoxia-induced gain in mean discharge rate again tended to be greater under diabetic versus healthy conditions (204 ± 57 spikes/8min versus 65 ± 14 spikes/8min), though this was not statistically significant (Wilcoxon matched pairs, p>0.05). The emergence of spontaneous discharge activity after sensory characterization is a normal finding in a proportion of C-fibers but is clearly exacerbated in diabetic mice, while the hypoxia-induced increase in spontaneous discharge rate remains to be substantiated.
Differential contribution of CaV3.2 channels to neuropeptide release from cutaneous terminals versus peripheral nerve in healthy animals
It has previously been demonstrated that CaV3.2 channels contribute to spinal synaptic glutamate release (Bao et al., 1998; Bellamy et al., 2006,Jacus et al., 2012) and to CGRP release from cultured DRG neurons upon high potassium-induced depolarization (Quallo et al., 2015). In line with this, we have shown that T-type channels contribute to axonal CGRP release in rat sciatic nerves upon membrane depolarization (Spitzer et al., 2008). In skin and nerve preparations from healthy mice high-potassium stimulation induced a significant increase in CGRP release over baseline (Fig. 3, Wilcoxon test p < 0.001). This stimulated CGRP release from isolated mouse sciatic nerves was significantly reduced to about half in healthy CaV3.2 KO mice in comparison to the C57BL/6J mice (Fig. 3C, rep. ANOVA F(3,51) = 4.23, p < 0.001; LSD post hoc p < 0.001). Accordingly, the T-type blocker TTA-P2 caused a significant reduction of the CGRP release in C57BL/6J (ANOVA F(1.17) = 10.22, LSD post-hoc p < 0.01) (Fig 3D). In contrast, no dependency on T-type channels could be detected in depolarization-induced CGRP release from skin of control animals. When TTA-P2 at 10μM was used prior and during exposure to KCl 20 or 40 mM, it caused no change of stimulated release (Fig. 3B). In CaV3.2 KO mice a significantly increased cutaneous release was found (ANOVA F(3,69) = 10.99, p < 0.001; LSD post hoc p < 0.001, Fig 3A). We propose that this could be mediated by functionally overexpressed N- and L-type channels which mediate cutaneous KCl-induced CGRP release (Kress et al., 2001). Hence, we used a combination of ω-conotoxin and nifedipine to block these calcium channels in the skin of CaV3.2 KO mice, which abolished the KCl response (Fig. 3A). In skin preparations from WT mice this combination of N- and L-type channel blockers nearly abolished KCl-induced release. Also, an increase of CGRP contents could theoretically be responsible for augmented release from skin of CaV3.2 KO mice, so we determined CGRP contents of skin flaps and isolated sciatic nerves from control and knockout mice. But neither CGRP content of skin preparations (n = 6; C57BL/6J 209.9 ± 9 pmol/g and KO 216.3 ± 16 pmol/g) nor of sciatic nerves (n = 6, 958.3 ± 67 pmol/g and KO 923 ± 88 pmol/g) differed. Our results thus indicate that CaV3.2 channel is expressed and highly involved in K+-induced CGRP release from the axonal membranes of peripheral nerve. However, a contribution of CaV3.2 to CGRP release from skin nerve endings could not be detected, at least not in response to unspecific depolarizing stimulation.
Fig. 3: Contributions of CaV3.2 to stimulated CGRP release in healthy skin and sciatic nerve.

A. KCl-evoked CGRP release from hairy skin of C57BL/6J and CaV3.2 KO mice and inhibitory effect of combined HVA calcium channel blockers nifedipine (10 μM) and ω–conotoxin (1μM). B. AUC display of data in A and no effect of the CaV3.2 blocker TTAP2 (10μM). C. KCl-evoked CGRP release from sciatic nerve of C57BL/6J and CaV3.2 KO mice. D. AUC display of the data in A and effect of the CaV3.2 blocker TTA-P2 (10μM). Significant increases in CGRP release over baseline and significant differences between experimental groups are marked with a hashtag or an asterisk based on Wilcoxon matched pairs test and ANOVA repeated measurements, respectively.
The differential contribution of CaV3.2 channels to diabetes-induced changes of CGRP release
The involvement of T-type calcium channels in diabetes-associated hyperexcitablilty of DRG neurons with nociceptive signature has previously been studied (Bourinet et al., 2016; Todorovic, 2016). We aimed to investigate whether T-type channels are also involved in the diabetes-induced alterations of neuropeptide release and therefore compared K+-stimulated CGRP release from skin and nerve preparations of healthy and diabetic CaV3.2 KO mice.
Our above results indicated that the axonal release in healthy mice is strongly reduced by diabetes in wildtypes and as much decreased in healthy CaV3.2 KOs (Figs. 1 and 3). However, the reduced axonal release in CaV3.2 KO mice showed no further reduction in diabetic KOs (Fig. 4B). Correspondingly, the treatment with a T-type blocker TTA-P2 could not further reduce the lowered axonal release in diabetic wildtypes (Fig. 4D). Thus, diabetes seems to act like deletion of CaV3.2 in the axolemma of peripheral nerve, reducing the K+-evoked axonal CGRP exocytosis.
Fig. 4: Contribution of CaV3.2 to diabetes-induced changes in stimulated CGRP release.
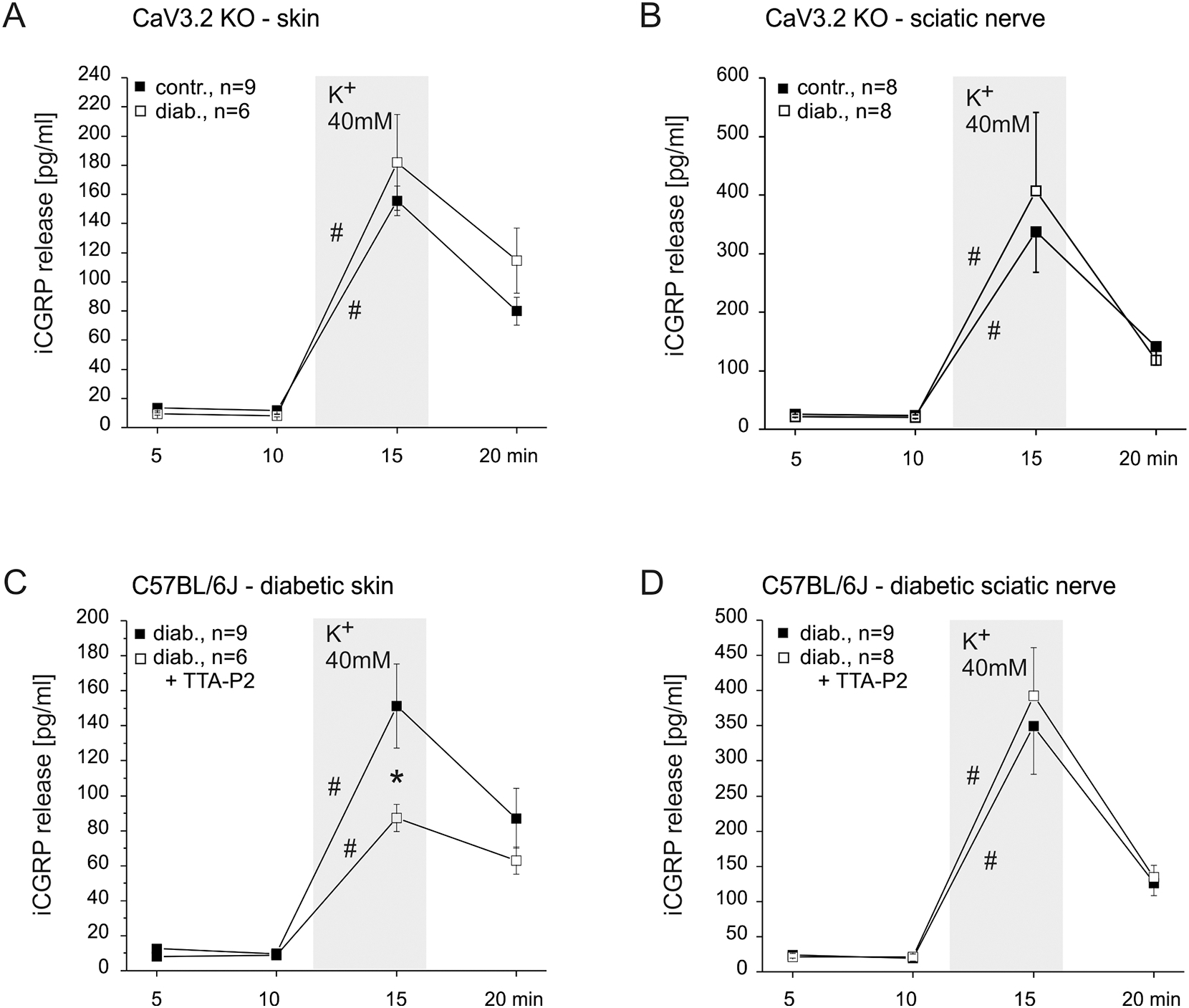
A. KCl-evoked CGRP release from skin of healthy and diabeticCaV3.2 KO mice. B. KCl-evoked CGRP release from sciatic nerve of healthy and diabetic CaV3.2 KO mice. C. KCl-evoked CGRP release from skin of diabetic C57BL/6J and effect of the CaV3.2 blocker TTA-P2 . D. KCl-evoked CGRP release from sciatic nerve of diabetic C57BL/6J and no effect of the CaV3.2 blocker TTA-P2. Significant increases in CGRP release over baseline and significant differences between experimental groups are marked with a hashtag an asterisk based on Wilcoxon matched pairs test and ANOVA repeated measurements, respectively.
Also in the skin, the diabetes-induced changes in wildtypes were about the same in direction and extent as resulting from CaV3.2 depletion in healthy mice, but here the K+-evoked CGRP release was enhanced, not reduced as in the nerve (Figs. 1 and 3). Like in the peripheral nerve, diabetes in CaV3.2 KOs did not further change - enhance - the cutaneous release (Fig. 4A), but in contrast to the nerve TTA-P2 showed a pronounced and significant reducing effect in diabetic wildtypes (ANOVA. F(3.39) = 3.00, p = 0.04, Fig. 4C) which was not seen in healthy WTs (Fig. 4B). This may mean that cutaneous peptidergic nerve endings gain in CaV3.2 functionality during diabetes development while their peripheral nerve axons loose it, both perhaps due to a redirection in axonal trafficking which privileges the terminals to the detriment of the axons.
Deletion of CaV3.2 changes fiber class composition and prevents diabetes-associated spontaneous activity
Single-fiber recording from healthy CaV3.2 KO animals indicated a cutaneous C-fiber class composition that appeared not statistically different from that seen in control C57BL/6 mice (χ2 (df = 4) = 4.7, p > 0.05). In healthy KO mice 19 mechanically sensitive fibers were determined as 6 (31.5%) C-MH, 3 (16%) C-MHC, 7 (37%) C-HTM, 2 (10.5%) C-LTM) and 1 (5%) C-MC (Fig. 6A, upper graph). In KO diabetic mice a total 24 mechanically sensitive C-units was tested and characterized as: 5 (21%) C-MH, 2 (8%) CMHC, 7 (29%) C-HTM, 7 (29%) C-LTM and 3 (13%) C-MC (Fig. 5A, lower graph) demonstrating no significant differences in the incidence of thermally and mechanosensitive fibers in comparison to healthy KO mice (χ2 (df = 1) = 0.51, p = 0.4706). These results suggest that the lack of CaV3.2 channels prevents the diabetes-induced trend towards more cold sensitivity among the polymodal C-fibers that was indicated by the comparison of healthy and diabetic C57BL/6J mice. Further analysis of the single-fiber data showed that there were no significant differences in the conduction velocities, heat and electrical thresholds between healthy and diabetic KO mice and in comparison to their C57BL/6J counterparts (Table 1). However, when comparing the prevalence of thermosensitive versus thermoinsensitive, merely mechanoresponsive, C-fibers it is conspicuous, though not significant, that the CaV3.2 KO mice in contrast to the WT mice yielded more units responding only to mechanical stimuli (47.5% vs. 29.5%; χ2 (d.f. 1) = 1.50, p = 0.2201). This difference became significant when comparing the diabetic genotypes (58% vs. 25%; χ2(d.f. 1 = 5,859761, p = 0.015491), indicating a relative loss in thermosensitivity in CaV3.2 KO mice that was aggravated by diabetes.
Fig. 6: Pharmacological effects on spontaneous C-fiber activity of healthy versus diabetic C57BL/6J.
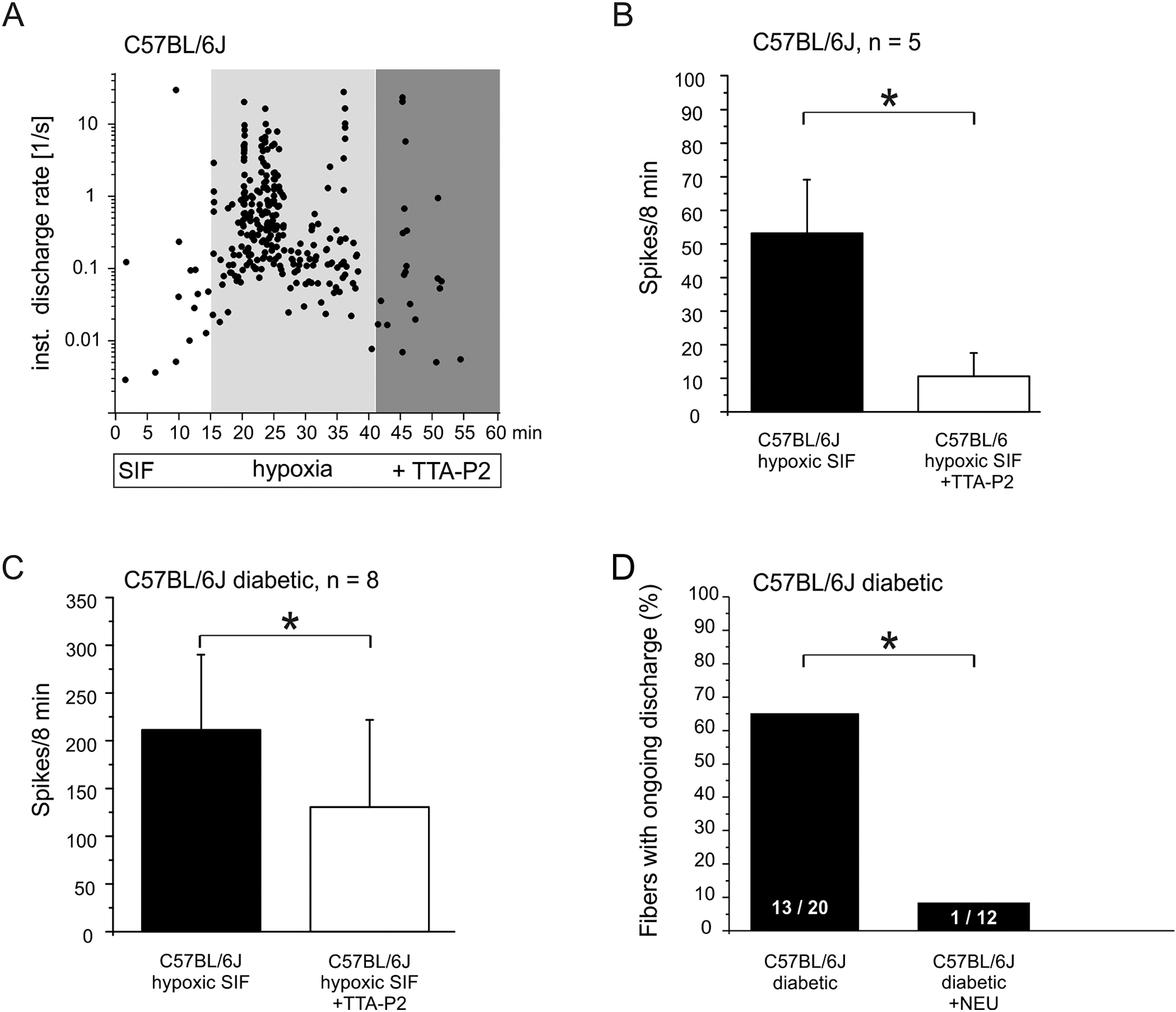
A. Single-fiber example of ongoing discharge intensified by hypoxia and inhibitory effect of CaV3.2 blocker TTA-P2. B. Mean ongoing discharge in final 8 min of hypoxia and subsequent inhibitory effect of TTA-P2. C. As in B but diabetic animals. D. Incidence (% of fibers) of spontaneous activity among C-fibers of diabetic skin (from Fig. 3B) versus diabetic skin after de-glycosylation pretreatment with neuraminidase. Significant differences between experimental groups are marked with an asterisk based on ANOVA repeated measurements.
Fig. 5: Contribution of CaV3.2 to C-fiber sensitivities, spontaneous discharge, and diabetic changes.
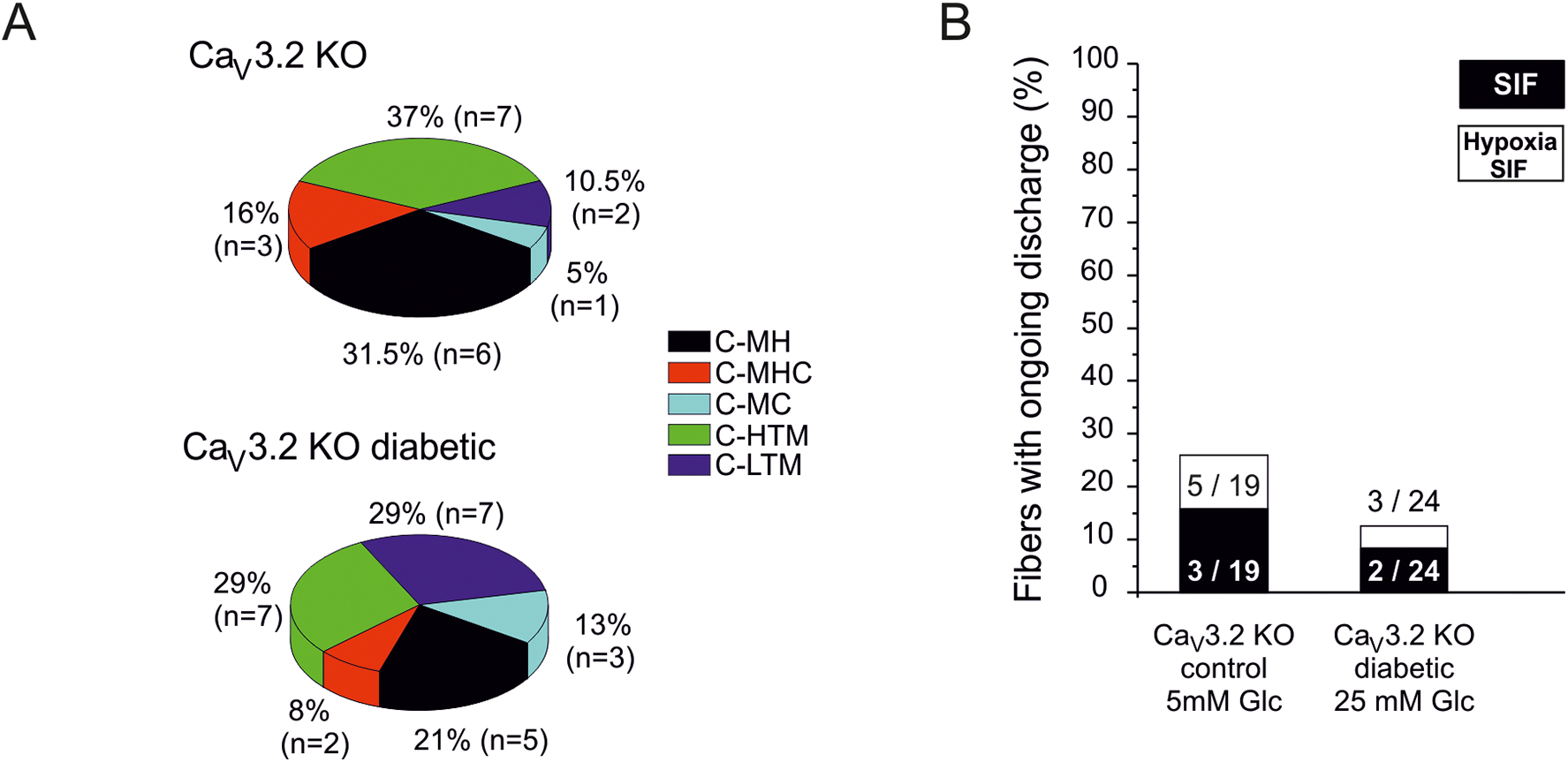
A. Distribution of sensory C-fiber subclasses in in healthy versus diabetic CaV3.2 KO mice. B. Incidence (% of fibers) of spontaneous discharge activity (after sensory testing) in healthy versus diabetic CaV3.2 KO mice and its increment during 10 min hypoxia.
The number of fibers spontaneously discharging in healthy KO mice (3 of 19 fibers = 16%) did not significantly differ from that seen in C57BL/6J healthy controls (6 of 24 fibers = 23%; χ2 (d.f. 1) = 0,57897023, p = 0.44, Fig. 5B). Diabetes in KO mice did not increase the number of spontaneously active fibers like it did in diabetic C57BL/6J: only 2 of 24 (= 8.33%) fibers in diabetic KOs exhibited spontaneous activity (Fig. 5B). In the KO animals we again investigated whether hyperglycemic hypoxia elevates the number of spontaneously discharging fibers similar to the increase seen in control animals (Fig. 2B). A 10% increase of spontaneously firing fibers in healthy KOs (16 % to 26 %) and 4% in diabetic KO mice (8 % to 12 %, Fig. 5B) was observed. Thus, the number of diabetes-induced spontaneous fibers under hypoxia (12 %) was significantly lower in the CaV3.2 KO mice than in diabetic C57BL/6J controls (80 %) under hypoxia (χ2 (df = 1) = 20.158, p = 0.00000067), suggesting CaV3.2 contributes essentially to diabetes-induced hyperexcitability among C-fibers.
There is an evidence that ongoing peripheral nerve activity contributes to spontaneous pain and hyperalgesia in patients with pDN (Bennett, 2012; Haroutounian et al., 2014). Whether the CaV3.2 channels contribute to spontaneous activity was also pharmacologically tested applying TTA-P2 to samples of fibers from diabetic C57BL/6J. Indeed, Figure 6A depicts the TTA-P2 diminishing effect on hypoxia-induced ongoing discharge of one diabetic C-MHC fiber (cv 0.49 m/s, von Frey threshold 5.7 mN, elect. threshold 4.8 μA). We also examined whether TTA-P2 pretreatment reduces hypoxia-induced activation of nociceptors recorded in healthy control mice: 5 C-nociceptors exhibited a mean discharge activity of 53.2 ± 16 spikes/8 min which was significantly reduced to 10.6 ± 7 spikes/8 min by 10 μM TTA-P2 application (Fig. 6B left panel, p = 0.04, Wilcoxon matched pairs test). Also in hyperglycemic hypoxia-activated fibers from diabetic C57BL/6J mice that were tested with TTA-P2 (8 out of the 16 spontaneous fibers under hypoxic conditions) the mean discharge activity was significantly reduced from 211.4 ± 77 spikes/8min to 131.5 ± 91 spikes/8min (Fig. 6C right panel, Wilcoxon matched pairs test, p = 0.04). These findings support the notion that CaV3.2 plays a role in diabetes-related increased neuronal excitability and in hypoxia-induced facilitation of spontaneous discharge. Interestingly, the fact that TTA-P2 reduced spontaneous firing also in healthy control animals suggests that CaV3.2 channels are involved in neuronal hyperexcitability under both physiological and diabetic conditions.
De-glycosylation by neuraminidase treatment reduces stimulated neuropeptide release in diabetic animals
It has previously been shown that posttranslational modification by asparagine-linked glycosylation of CaV3.2 channels controls the channels’ expression pattern and is involved in the development of diabetic neuropathic pain (Joksimovic et al., 2020; Orestes et al., 2013; Weiss et al., 2013). We investigated whether de-glycosylation by neuraminidase alters stimulated neuropeptide release under healthy and diabetic conditions. This enzyme cleaves glycosidic bonds in many transmembrane proteins including CaV3.2 which shows enhanced glycosylation under diabetic conditions (Lazniewska and Weiss, 2017; Weiss et al., 2013) Neuraminidase (NEU, 2u/ml dissolved in SIF) was applied for 2 hours prior to and throughout the release experiment at 32°C and 38°C for skin and nerve preparations, respectively. In skin and sciatic nerve preparations from healthy and diabetic C57BL/6J and KO mice high-potassium stimulation induced significant increases in CGRP release over baseline (Figs. 7A and B, Wilcoxon test p < 0.001). As depicted in figure 7A, NEU treatment did not change the stimulated cutaneous CGRP release in healthy control C57BL/6J mice. In contrast, in preparations from diabetic C57BL/6J mice the augmentation of cutaneous release was reversed by NEU treatment and the release was reduced to below control level (one-way ANOVA comparing AUC in pg/ml F(3.39) = 8.03, p = 0.002; p < 0.001 for both effects indicated; the control data already appeared in Fig. 2B). Also in skin preparations of healthy KO mice, NEU treatment did not change KCl-induced CGRP release, but in skin from diabetic KO mice NEU treatment still caused a reduction of stimulated release although less so than in diabetic WT mice (Fig. 7B, one-way ANOVA comparing AUC in pg/ml F(3.31) = 3.57, p = 0.02; p < 0.01).
Fig. 7: Effects of de-glycosylation on stimulated CGRP release from healthy and diabetic skin and sciatic nerve.
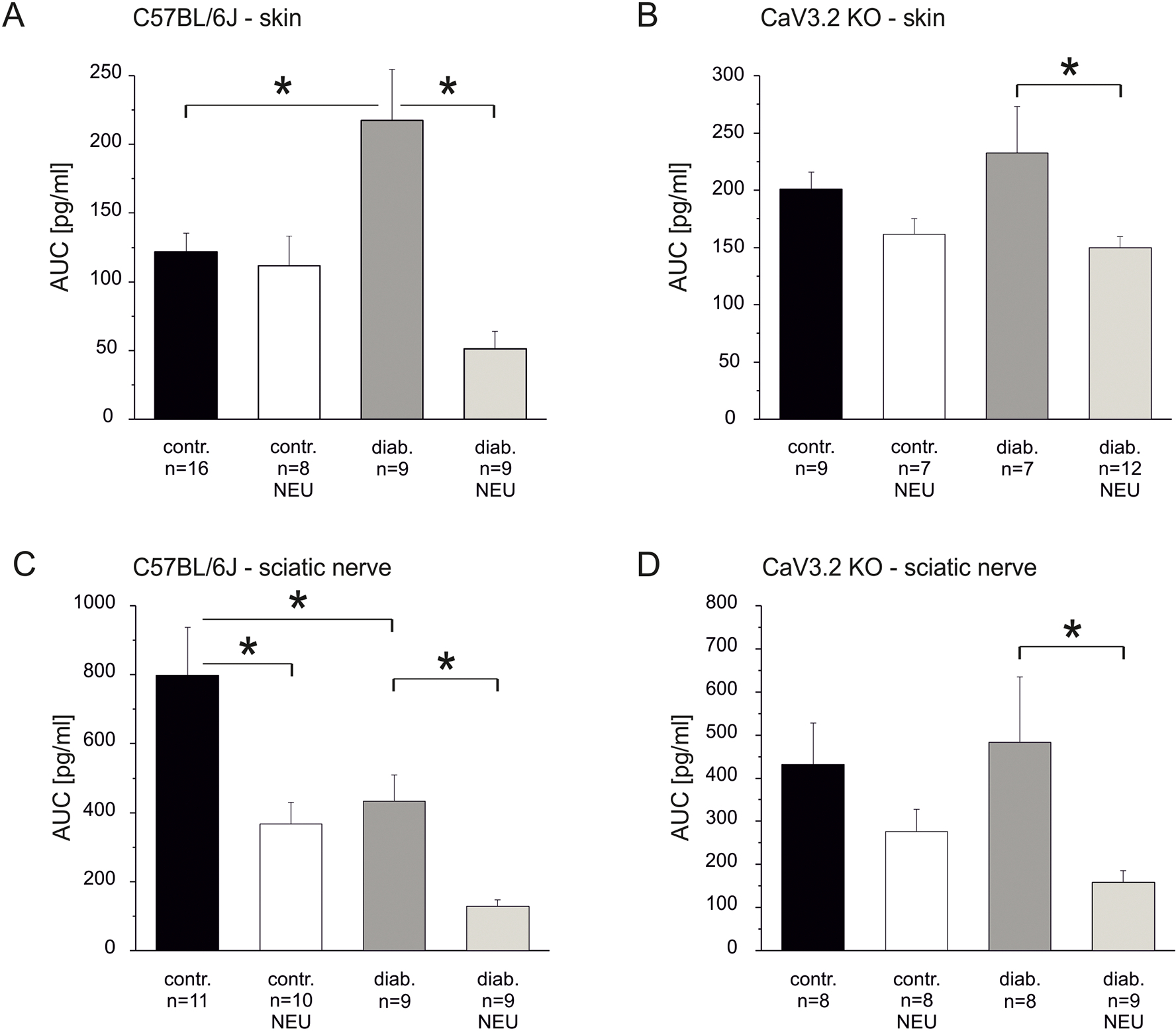
A. KCl-evoked CGRP release from skin of healthy versus diabetic C57BL/6J mice with and without pretreatment by the de-glycosilating enzyme neuraminidase. B. KCl-evoked CGRP release from skin of healthy versus diabetic CaV3.2 KO mice with and without pretreatment by neuraminidase. C. KCl-evoked CGRP release from sciatic nerve of healthy versus diabetic C57BL/6J mice with and without pretreatment by neuraminidase. D. KCl-evoked CGRP release from sciatic nerve of healthy versus diabetic CaV3.2 KO mice with and without pretreatment by neuraminidase. Significant differences between experimental groups are marked with an asterisk based on ANOVA repeated measurements.
Next we evaluated contribution of CaV3.2 channel glycosylation to stimulated axonal CGRP release from sciatic nerve. We had already shown that this strongly depends on depolarization-induced activation of CaV3.2 channels (Figs. 3C and D). However, in contrast to skin, in sciatic preparations from healthy WT mice NEU treatment caused a significant reduction of stimulated CGRP release, and in preparations from diabetic WT mice the already reduced release was further diminished (Fig. 7C, one-way ANOVA comparing AUC in pg/ml F(3.35) = 9.32, p < 0.001 for all effects indicated). Interestingly, in nerve preparations of healthy mice lacking CaV3.2 channels de-glycosylation treatment caused only a non-significant reduction, whereas in diabetic CaV3.2 KO mice NEU treatment strongly reduced the KCl-induced CGRP release (Fig. 7D, one-way ANOVA comparing AUC in pg/ml F(3.29) = 2.71, p = 0.06; p = 0.04). Comparing these figures (7C and D) shows again that diabetes does not reduce stimulated axonal CGRP release if CaV3.2 is missing.
Taken together, cutaneous CGRP release in healthy animals was not changed by de-glycoslation treatment, neither in C57BL/6J control mice nor in CaV3.2 KO mice, whereas in preparations from diabetic animals NEU treatment strongly reduced stimulated CGRP release. In diabetic skin the NEU effect was much more pronounced in C57BL/6J (76% reduction) than in CaV3.2 KO mice (40% reduction). In contrast to healthy skin, stimulated axonal release was reduced by NEU treatment in sciatic nerve preparations from healthy C57BL/6J mice. This is consistent with the TTA-P2 effects, signifying the high functional expression and therefore contribution of CaV3.2 channels to axonal release (Figs. 3C, D). In healthy KO mice the axonal release was hardly reduced by NEU treatment, however, in diabetic KO mice de-glycosylation reduced the stimulated CGRP release in the nerve to the same extent as in control C57BL/6J (about 70% reduction in both genotypes). This result and the NEU effect in skin of diabetic KOs indicates that not only de-glycosylation of CaV3.2 channel but also of other not yet identified ion channels contributes to neuraminidase-induced reductions of neuropeptide release from nociceptors, both cutaneous terminals and along their sciatic axons.
De-glycosylation reduces diabetes-induced spontaneous activity of sensory fibers
Single-fibre recordings also showed that NEU (2 units/ml) treatment dramatically reduced the increased neuronal activity induced by diabetes. Skin from diabetic C57BL/6J mice was incubated for 2 hours prior to experiment and superfused during recordings with hyperglycemic SIF containing NEU; 12 C-fibers were tested for spontaneous discharge and characterized as 8 C-LTM and 4 C-HTM (Fig. 6D). Given the high percentage of thermosensitivity among cutaneous C-fibers (70 %, Fig. 2A), the complete absence of heat and/or cold sensitivity among 12 C-fibers harvested from diabetic mouse skin under NEU treatment appears significant and suggests loss of thermosensitivity in consequence of de-glycosylation (χ2 (df = 4) = 26.65, p = 0.000023). Conduction velocity and von Frey threshold of diabetic C- fibers treated with NEU (mean cv = 0.5 ± 0.04 m/s, median of v. Frey threshold of C-LTMs 4 mM and C-HTM 5.7 mN) were comparable with untreated diabetic C-fibers (see table 1), only the electrical threshold was significantly lowered (4.8 ± 2.1 μA in diabetic C57BL/6J vs. 1.1 ± 0.6 μA in diabetic C57BL/6J with NEU, MW U-Test, p<0.05, data not shown). In striking contrast to this increased electrical excitability, NEU treatment significantly reduced the number of spontaneously firing neurons in diabetic C57BL/6J mice from 13 of 20 tested fibers (= 65 %, data from Fig. 2B) to only 1 of 12 units (= 8.3%; Fig. 9, χ2 (df = 1) =10.12, p = 0.0014). The one spontaneously active fiber under NEU treatment fired 28 sporadic spikes in the 8 min recording period (data not shown), which is markedly less in comparison to the 203 ± 147 spikes fired by spontaneous fibers in diabetic C57/BL6J (Fig. 2C).
DISCUSSION
Our studies on early mechanisms of painful diabetic neuropathy (pDN) pursued two main objectives, first to reconfirm and extend previous pathophysiological findings on cutaneous sensory C-fiber functions and, second, to determine if and to which extent the CaV3.2 isoform of T-type calcium channel and its glycosylation are involved in their aberrant properties. The research strategy was to compare WT mice with congenic global CaV3.2 null mutant mice, both healthy and diabetic, four to five weeks after disease induction by repeated low-dose STZ.
Altered sensory spectrum of diabetic C-fibers and in CaV3.2 KO mice
Previous electrophysiological works in diabetic rats were not intended to investigate the incidence of sensory subtypes among the C-fibers (Fuchs et al., 2010; Suzuki et al., 2002a). Our bias-free search ex vivo for mechanosensitive C-fibers yielded two conspicuous features with regard to incidence, first in diabetic WTs, a relative increase of cold responsiveness at the expense of heat sensitivity and, second in CaV3.2 KO mice, a marked deficit in thermosensitivity, both heat and cold sensing, with concomitant increase of exclusive mechanosensitivity, which imbalance appeared further enhanced in diabetic KO mice. The numerical reduction of heat sensors in diabetic WTs may be a discrete portent of the developing loss of warm and heat pain sensing that is reported as an early symptom of pDN (Kramer et al., 2004). This paradox is taken to a peak by another clinical finding in pDN patients: The fewer intraepidermal nerve endings (including the heat sensors) are found in skin biopsies, the more intense pain symptoms are reported (Sorensen et al., 2006). We speculate that the numerical increase of cold sensing in our data may be related to the frequent complaint of “cold feet” feelings in diabetic patients. From various rodent models of diabetes it is reported that an early developing hyperalgesia to heat turns into a later and final stage of hypoalgesia, and both anomalies are strongly correlated with an up- and down-regulation, respectively, in expression and function of the heat-activated cation channel TRPV1 (Pabbidi et al., 2008). On the other hand, the deficit in heat sensing of CaV3.2 KO mice was not unexpected, as the KO mice had previously shown a relative behavioral insensitivity to noxious heat (Choi et al., 2007). An interesting functional link between TRPV1 and CaV3.2 has been discovered when both ion channels were co-expressed in HEK cells: Activation of TRPV1 (by capsaicin) induced a profound and very sustained inhibition of the calcium channel, reminiscent of a similar finding in co-expressing rat DRG neurons (Comunanza et al., 2011; Kerckhove et al., 2014). The phenomenon was explained by the massive calcium influx through TRPV1 that activates various protein kinases which synergistically may inhibit CaV3.2 currents (Zhang et al., 2013). Heat activation of TRPV1 would, thus, prevent the support by CaV3.2 of the depolarizing, excitatory generator potential. Such a support is theoretically required when temperature increase reduces the membrane resistance which attenuates generator potentials (Touska et al., 2018). Either way, loss of sensitivity corresponds to the established role of CaV3.2 channel which is to contribute to the control of neuronal excitability, a function that is enhanced in DRG neurons of diabetic mice (Chemin et al., 2002; Hall et al., 1995; Jagodic et al., 2007; Latham et al., 2009; Todorovic and Jevtovic-Todorovic, 2011; Williams et al., 1999).
Spontaneous discharge activity in diabetic C-fibers of WT and much less in CaV3.2 KO mice
Enhanced excitability is also a possible cause of spontaneous neuronal discharge activity. Cutaneous C-fiber nociceptors are normally silent in the absence of overt stimulation, however, after probing their receptive fields with thermal and mechanical stimuli they often develop sustained ongoing activity with low-frequent irregular discharge, a behavior that was already described as spontaneous activity in the very first original article on “polymodal” chemosensory C-nociceptors (Bessou and Perl, 1969). This kind of ‘secondary’ spontaneous activity among the C-fibers was assessed in our single-fiber recordings and found markedly enhanced in diabetic WTs in terms of incidence and, in tendency, of discharge rate. In our previous work on diabetic rat skin it was hypoxia during high glucose (25 mM) superfusion that provoked ongoing ‘spontaneous’ discharge activity in all C-fibers (n = 16) of a sample, while in non-diabetic skin only a fraction of units showed marginal activity (Fuchs et al., 2010). In an earlier study high glucose (20 mM) superfusion of receptive fields was sufficient to induce spontaneous activity and increased mechanical responsiveness in C-fibers of STZ-diabetic rat skin, which anomalies were absent if receptive fields were superfused with normal glucose concentration (Suzuki et al., 2002b). From the same lab a firm assignment of spontaneous activity to the polymodal, mechano-heat sensitive, C-fibers is available for diabetic rat skin (Suzuki et al., 2002a). However, some provocation by sensory or chemical stimulation appears to be necessary, in vivo at least, as recently only low-threshold mechanoreceptive Aδ-fibers but no C-fibers were found to become spontaneously active in STZ-diabetic rat skin (Djouhri et al., 2020). The same (first) author, however, found spontaneous C-fiber activity after CFA-induced cutaneous inflammation and in a (modified) spinal nerve transection model of neuropathic pain. Notably, this study reported a close association in the treated rats of rate and incidence of spontaneous discharge activity with spontaneous foot liftings which were interpreted as behavioral expressions of spontaneous pain attacks (Djouhri et al., 2006). Spontaneous peripheral C-fiber activity has also been reported from spontaneously diabetic BB/Wistar rats and from STZ-diabetic rats (Burchiel et al., 1985; Serra et al., 2012). The latter study, using human microneurography in addition, also documented significant C-fiber spontaneous activity in neuropathic, including diabetic, pain patients.
An obvious concern with spontaneous activity in neuropathic C-fibers is whether its low frequency can be considered responsible for the allodynia and hyperalgesia symptoms in pDN. Indeed, the mean discharge rate we found in diabetic WTs was 203 spikes in 8 min corresponding to 0.4 spikes/s, but this was averaged across as much as 57% (13/23 units) spontaneously active ones of the recorded C-fibers. These 13 fibers had receptive fields distributed over the small area of about half the mouse hindpaw dorsum, which suggests the possibility of spatial summation and convergence onto spinal dorsal horn relay neurons. It is well known that spatial, as well as temporal summation together with facilitating neuropeptide release from C-fibers firing at low frequency play an essential role in ‘central sensitization’ of synaptic transmission which is responsible for mechanical allodynia and (secondary) hyperalgesia (Sandkühler, 2009). In our mouse model, as in the previous rat model, it was the increased incidence of spontaneous activity among the C-fibers rather than their rate of discharge that made the significant difference between healthy and diabetic skin (Suzuki et al., 2002a): Also in a partial nerve injury rat model of neuropathic hyperalgesia the increased incidence but not discharge rate of C-fiber spontaneous activity correlated with the mechanical hypersensitivity of the animals. In the same study healthy rats were treated at the heel for 10 min with electrocutaneous field stimulation at 0.2/s pulse rate and C-fiber strength which induced a marked and sustained mechanical hyperalgesia (Wu et al., 2002). With regard to translation from animal to human conditions, a clinical study on ultrasound-guided peripheral nerve blocks appears particularly relevant: pDN patients were injected with lidocaine which temporarily abolished all spontaneous pain, hyperalgesia, and allodynia symptoms, signifying the essential role of ongoing peripheral input to the spinal cord and the short life span of the much-invoked “pain memory” (Haroutounian et al., 2014).
‘Hyperglycemic’ hypoxia and role of ‘threshold’ channels
Our method of enhancing spontaneous activity by ‘hyperglycemic’ hypoxia is derived from fundamental work in Peter Grafe’s lab; they achieved a marked increase in axonal (electrical) excitability by superfusing healthy rat peripheral nerve with hypoxic high-glucose solution and found intraaxonal acidosis responsible for the effect which blocked voltage-gated potassium channels (Grafe et al., 1994; Schneider et al., 1993). The model is thought to mimic a pathomechanism that may lead to the lancinating deep pain attacks that dDN patients are reporting. An early diabetic impairment of endoneural and epineurial arterioles (microangiopathy) in peripheral nerves and spinal roots leads to episodes of inadequate blood supply and reduced buffer capacity eventually combined with hyperglycemia; this coincidence may overstrain the axonal lactic acid extrusion and clearance which may account for the acidosis (Catrina et al., 2004; Yorek, 2015; Zochodne et al., 1994). In our hands, the 10 min hypoxia period enhanced the incidence and rate of spontaneous discharge activity among the C-fibers, less in healthy and more in diabetic skin of WTs but the difference of the increments was not significant. An ion channel that takes relevant influence on the threshold to firing of nociceptive C-fibers is the voltage-gated sodium channel NaV1.9, activating close to resting membrane potential, TTX resistant, and enormously accelerated in kinetics by heating to and beyond body temperature (Touska et al., 2018). This NaV1.9 was included in a study on healthy rat DRG neurons exposed overnight to either ‘hyperglycemic’ (25 mM) or hypoxic (4% O2) nutrient solution, both of which increased the voltage sensitivity of the TTX resistant sodium current in a protein kinase A and C dependent way (Bich-Hoai et al., 2010). Phosphorylation by these and other protein kinases can also play a, however desensitizing, role in case of CaV3.2 channel but has not yet been studied in the context of pDN (Blesneac et al., 2015; Chemin et al., 2019; Gomez et al., 2020). Worth considering is also the hyperpolarization-activated cAMP-gated cation channel HCN2 which also contributes to nociceptive sensitization in models of inflammation and neuropathy, not only through direct gating by cAMP but, even more, through phosphorylation by PKA which increases the voltage sensitivity of the channel (Herrmann et al., 2017). In rodent models of pDN, the HCN2-dependent mechanical allodynia was associated with a drastic increase of cAMP in DRG neurons, and pharmacological block of cAMP formation was as effective against the allodynia as HCN2 block (Ma et al., 2021; Tsantoulas et al., 2017). Although different channels may work in concert to contribute to sensory neuron sensitization in pDN, our present study found that deletion of CaV3.2 completely prevented the marked gain in spontaneous activity among C-fibers in diabetic mice that had been observed in WTs. This strongly suggests a predominant role of CaV3.2 in controlling the excitability of cutaneous C-fibers, as was previously reported by studies on DRG neurons from diabetic animals showing also enhanced excitability, increased T-current densities, and a threefold upregulation of CaV3.2 mRNA (Jagodic et al., 2007; Latham et al., 2009).
CGRP release and CaV3.2 in healthy mice
Single-fiber recording and measurements of stimulated CGRP release are in principle dealing with overlapping populations of sensory C-fibers, however, the latter technique provides lump data from an incomparably greater number of fibers, if only from peptidergic units. In addition, although CGRP release can be evoked by propagated action potentials, it does not depend on the spikes but is entirely determined by depolarization, including subthreshold generator potentials, which increase the intracellular calcium ion concentration mostly by evoking calcium influx through voltage-gated calcium channels (Gebhardt et al., 2020; Huang and Neher, 1996; Kress et al., 1999). Block of action potentials by TTX or lidocaine takes no influence on KCl- or capsaicin-induced CGRP release from innervated tissues (Nemeth et al., 2003; Spitzer et al., 2008). Thus, CGRP release studies are potentially more sensitive than single-fiber recordings to chemical or genetic influences on stimulated membrane potential changes, although the informative value is limited to CGRP-expressing nerve fibers. Convergent evidence from Calca (CGRPα) gene reporter mice, DRG neuron recording and labeling, and skin immunohistochemistry indicates that CGRP+ neurons are large numbers of polymodal and high-threshold mechanosensitive (HTM) C-nociceptors and, smaller numbers, of HTM Aβ and Aδ-nociceptors (Kestell et al., 2015; Lawson et al., 2002; McCoy et al., 2012). Optogenetic inhibition of these CGRP-expressing neurons reversed mechanical, cold, and heat hyperalgesia as well as spontaneous pain-related behavior in mice subjected to ‘spared nerve injury’, an established model of neuropathic pain (Cowie et al., 2018).
In case of depolarization, e.g. resulting from a sensory transduction process, high-voltage activated (HVA) N-and L-type calcium channels are gated open and induce neuropeptide release; this has been verified for spinal terminals as well as for peripheral nerve endings (Del Bianco et al., 1991; Kress et al., 2001; Nemeth et al., 2003). Also the involvement of low-voltage activated (LVA) T-type calcium channels such as CaV3.2 in “low-threshold exocytosis” has been established (Weiss et al., 2012). DRG neurons and isolated sciatic nerve preparations release neuropeptides with participation of T-type channels (Amrutkar et al., 2011; Quallo et al., 2015; Spitzer et al., 2008). Our present results confirm the previous data from isolated rat sciatic nerves that show the involvement of L as well as T-type calcium channels in mediating calcium influx, triggering vesicular exocytosis and axonal CGRP release (Spitzer et al., 2008). It was previously shown in isolated skin that pharmacological blocks of either L-type or N-type calcium channels result in around 50% inhibition of high-potassium induced CGRP release, but specific T-type blockers were not tested (Kress et al., 2001). Now, our results reveal a differential contribution to CGRP release of CaV3.2 channels in peripheral nerve axons versus cutaneous terminals of peptidergic nociceptors. In the isolated sciatic nerve preparation about 50% of the stimulated release was clearly based on T-type activation. In contrast, in skin preparations of CaV3.2 knockouts we surprisingly found no reduced but rather an enhanced KCl-induced CGRP release as compared to wildtypes, which effect was for the most part dependent on HVA, L and N-type calcium channels (Fig. 4A). Our determinations of CGRP content in skin and nerve exclude an upregulation of CGRP expression in the CaV3.2 knockout animals. A possible compensatory upregulation of HVA calcium channels has not been reported in CaV3.2 knockouts, nor did it occur after silencing of CaV3.2 by antisense oligonucleotides in nociceptive DRG neurons (Bourinet et al., 2005; Chen et al., 2003). Further below we speculate about a relocation of HVA channels from axons to nerve endings to account for the unexpected finding in the KOs.
The expression pattern of T-type calcium channels has primarily been studied in DRG neurons and it was established that they are expressed predominantly in medium and small size DRG neurons (Nelson and Todorovic, 2006; Schroeder et al., 1990; Scroggs and Fox, 1992). Other studies have supported the idea that at least 20 to 30 % of all DRG neurons of these sizes are positive for T-type calcium channels (François et al., 2015; Watanabe et al., 2015). At least one study reported large CaV3.2 current densities in a subset of DRG neurons termed “T-rich” (Nelson et al., 2005). However, it has been reported that in most small DRG neurons T-type channels have a relatively low expression density (Blair and Bean, 2002; Todorovic and Lingle, 1998b). But for our investigation the actual co-localization of T-type channels and CGRP together with a possible allocation to specific tissues is more important. About 20% of CGRP positive (and IB4 negative) DRG neurons co-express CaV3.2 immunoreactivity (Rose et al., 2013) and vice versa about 40% of all CaV3.2 positive DRG neurons express CGRP (Watanabe et al., 2015). These numbers indicate that T-type calcium channels overlap with CGRP expression in sensory neurons and therefore can contribute to depolarization-induced stimulation of CGRP release. Beside expression in neuronal cell bodies, T-type calcium channels have been localized by immunostaining along the axons of sciatic nerve sections and, using an immuno-gold labeling technique, CaV3.2 was found preferentially along unmyelinated axons (Rose et al., 2013). Our data confirm a robust expression of functional T-type channels along the axons that contributes to high-potassium stimulated CGRP release (Fig.4 C and D). In peripheral nerves this vasodilatory neuropeptide may play a role in activity-dependent blood flow increase and as a paracrine trophic factor, for Schwann cells are well equipped with the CGRP receptor complex (Lennerz et al., 2008).
In glabrous skin CaV3.2 was first discovered in nociceptive nerve endings, here CaV3.2 is co-localized with MrgprD, a marker for IB4-positive (non-peptidergic) fibers (Rose et al., 2013). In hairy skin it is established that CaV3.2 is a marker for D(down)-hair mechanosensors (low-threshold rapidly adapting Adelta fibers) and expressed in tactile C-fibers that contribute to cold allodynia in humans (Dubreuil et al., 2004; Samour et al., 2015; Shin et al., 2003; Wang and Lewin, 2011). Recently, genetic tracing revealed that sensory neurons in the periphery expressing CaV3.2 are deep tissue nociceptors but do not innervate the hairy skin (Bernal Sierra et al., 2017). This is in full agreement with our functional results that KCl-induced cutaneous CGRP release was neither altered in CaV3.2 KO mice, nor by TTA-P2 block in WT mice.
CGRP release and CaV3.2 in diabetic mice
When we studied high-potassium induced CGRP release in diabetic wildtype mice, we again observed differential effects in preparations of hairy skin and isolated sciatic nerves. In the skin of diabetic mice CGRP release was augmented compared to healthy WTs, whereas in the diabetic sciatic nerve we found a massive reduction of stimulated CGRP release (see Fig. 2). In both opposite diabetic changes CaV3.2 must play a decisive role, since diabetes in CaV3.2 KO mice showed no influence on stimulated CGRP release; the release remained augmented in the skin and reduced in the sciatic nerve, just as in healthy KOs versus WTs. Supporting the role of CaV3.2 in diabetic WTs, we found that TTA-P2 normalized the augmented release in the skin, while it was ineffective in healthy WTs. Thus, the lack of CaV3.2 function in healthy skin was replaced by a gain of its function in diabetic skin, accounting for the augmentation of KCl-evoked CGRP release. The exact opposite occurred in the sciatic nerve, TTA-P2 in healthy WTs and CaV3.2 deletion both led to a similarly profound reduction of stimulated release, whereas both interventions were completely ineffective in diabetic animals of both genotypes. The striking inverse proportionality of diabetic changes in skin and nerve invites the speculation that the CaV3.2 channels may be relocated within the same neurons from peripheral nerve to nerve endings. A diabetic alteration of the axonal transport could be responsible for the redistribution of the CaV3.2 cargo, ending the en passant translocation of the protein into the axolemma but maintaining the traffic towards the terminals. The diabetic changes were astonishingly similar to the results from CaV3.2 deletion: KCl-evoked CGRP release was enhanced in the skin and to a similar extent decreased in the nerve, inversely proportional. As in CaV3.2 KO mice the vast majority of depolarization-evoked release was mediated by L and N-type subtypes of HVA calcium channels, applying the above logic could mean that these proteins may also be redistributed from peripheral nerve to nerve endings by an alteration of the axonal trafficking.
Stimulated neuropeptide release has been found reduced in trachea preparations from diabetic animals (Calcutt et al., 1998), but also an increase of stimulated neuropeptide release from the skin was reported (Ellington et al., 2002; Fuchs et al., 2010). Accounting for enhanced cutaneous release, most recently, in human skin biopsies taken from patients with pDN, the proportion of peptidergic, SP and CGRP expressing, intraepidermal nerve endings was found much increased (> 40%) and in good correlation with the pain ratings, in contrast to diabetic patients without pain and healthy controls (Karlsson et al., 2021). The conflictive reports from animal studies are probably due to different diabetes models, time points of observation after diabetes induction, and different stimuli used, e.g. activation of TRPV1 or G-protein coupled receptors that undergo differential regulation during diabetic neuropathy (Hong and Wiley, 2005; Pabbidi et al., 2008; Talbot and Couture, 2012). We used high-potassium solution to circumvent these complex activation mechanisms and to depolarize the peptidergic fibers. In both the peripheral endings and axons of peptidergic nociceptors an increase in intracellular calcium is indispensable for increasing the exocytosis rate. Upregulation and increase of HVA calcium currents together with a reduced capacity to eliminate calcium from the cytosol has been described in cultured DRG neurons from diabetic animals (Huang et al., 2002; Kostyuk et al., 1995; Yusaf et al., 2001). Likewise, there are studies that report an up-regulation of CaV3.2 in different models of diabetic neuropathy (Todorovic and Jevtovic-Todorovic, 2014). In STZ-diabetic rats an up-regulation of T-type currents in IB4-positive DRG neurons by about twofold is described (Cao et al., 2011; Jagodic et al., 2007; Khomula et al., 2013). This up-regulation is accompanied by altered biophysical properties of the T-type current in diabetic animals that render CaV3.2 expressing neurons more excitable. In addition, this upregulation of T-currents correlates with the hyperglycemia that develops after the STZ treatment of the rats (Messinger et al., 2009).
However, the above findings were largely achieved from cultured and acutely dissociated DRG neurons. Investigating the peripheral compartments of these sensory neurons we found that diabetes induced a de novo functionality of T-type channels in peptidergic cutaneous nerve terminals accompanied by a loss of T-type function in the peripheral nerve axons with reduction of stimulated CGRP release. In peripheral nerve, diabetic neuropathy in patients primarily affects the axons of the sensory neurons: axonal dysfunction, degeneration and abortive regeneration occur (Sima and Zhang, 2014). Already four weeks after diabetes induction in rodents substance P and CGRP contents of sciatic nerves are decreased due to reduced synthesis in the somata (Brewster et al., 1994; Diemel et al., 1992) and to reduced axonal transport (Fernyhough et al., 1994; Prior et al., 2017). However, reduced availability of CGRP cannot be the cause of diminished axonal release in our experiments three weeks after diabetes induction, since TTA-P2 block of CaV3.2 channels acutely achieved the same reduction in healthy WTs and completely lost its effect in diabetic WTs (Figs. 4D versus 5D). Thus, loss of CaV3.2 expression in the axolemma most likely accounts for the diabetic nerve impairment, an interpretation supported by the ineffectiveness of diabetes on nerves of CaV3.2 KO mice (Fig. 5B). The impaired calcium homeostasis in axons and Schwann cells in diabetes has previously been reviewed (Verkhratsky and Fernyhough, 2014). By the same token, the diabetic enhancement of cutaneous CGRP release in our and other’s experiments is likely not due to an excess supply of the neuropeptide but rather to an acquired overexpression of CaV3.2 in the nerve endings: TTA-P2 was ineffective in healthy WTs but normalized the release in diabetic WTs, and diabetes was ineffective on the skin of CaV3.2 KO mice (Figs. 4B vs. 5C and 5A).
CGRP release and de-glycosylation of CaV3.2
Neuraminidase, a member of an enzyme family expressed in bacteria and mammalian lysosomes, is specific in removing all sorts of glycosylation but it is entirely indifferent to the lipids and proteins to be de-glycosylated. Nonetheless, neuraminidase was significantly effective reducing KCl-induced CGRP release from the healthy sciatic nerve of WTs, while it was ineffective in CaV3.2 KO mice and in healthy WT skin where CaV3.2 is not expressed in the nerve endings (see above). This would argue for an important role of CaV3.2 in axonal “low-threshold exocytosis” of the neuropeptide (Weiss et al., 2012). Diabetic hyperglycemia, including STZ-diabetes, enhances N-glycosylation of proteins in general and of CaV3.2 in particular which increases the channel’s expression and density in neuronal plasma membranes (Ficelova et al., 2020; Fukami et al., 2017; Lazniewska and Weiss, 2017; Orestes et al., 2013; Shathili et al., 2018). CaV3.2 glycosylation has little effect on the voltage sensitivity of the channel but increases its conductance and open probability as well as its potentiation by increased glucose levels (Ondacova et al., 2016; Orestes et al., 2013; Weiss et al., 2013). This all fits with our result that stimulated CGRP release was almost doubled in skin of diabetic WTs and drastically reduced by neuraminidase treatment (as well as normalized by TTA-P2 block of CaV3.2), corroborating the de novo expression of CaV3.2 in diabetic nerve endings of hairy skin. However, neuraminidase was also effectively reducing the CGRP release from the diabetic sciatic nerve in WTs, where CaV3.2 is no longer expressed (and TTA-P2 ineffective), and it was effective in CaV3.2 KO mice. Thus, the de-glycosylation must have affected other voltage-gated calcium channels such as the L-types (CaV1.x) which, for example, take over the role of T-type channels in KCl-evoked CGRP release in CaV3.2 KO mice, according to our result (Fig. 4A). Indeed, CaV1.x HVA channels present with four N-glycosylation sites (asparagine residues), mutation of which causes substantial reductions of voltage sensitivity, current density, and surface expression of the channels (Lazniewska and Weiss, 2017). In addition, the off-target effects of neuraminidase may also affect other glycosylated ion channels, e.g. potassium channels, that are involved in the depolarizing action of high extracellular KCl concentration (Baycin-Hizal et al., 2014). Our neuraminidase experiments indicate that glycosylation of CaV3.2, enhanced by diabetes, is jointly responsible for the augmented neuropeptide release in diabetic skin, although the effect size could not exactly be determined. Given the decisive role of glycosylation in coding, routing, directional trafficking, and finally surface expression of proteins (Abad-Rodríguez and Díez-Revuelta, 2015), it may not be far-fetched to assume that the substantial reorganization of functional CaV3.2 expression in diabetes - loss of T-types in nerve, gain in skin nerve endings - also results from enhanced posttranslational modifications by glycosylation of CaV3.2 channel.
In conclusion, a few weeks of STZ-induced hyperglycemia in male C57BL/6J mice led to major reorganisations in peripheral C-fiber functions. An unbiased screening of mechanosensitive single-fibers in isolated skin revealed a relative loss of (polymodal) heat sensing in favor of cold sensing; a downregulation of TRPV1 may be responsible (Pabbidi et al., 2008). In healthy CaV3.2 KO mice both heat and cold sensitivity among the C-fibers seemed underrepresented in favor of exclusive mechanosensitivity, low-threshold in particular, which deficit became significant in the diabetic KO mice. Diabetes also led to a marked increase in the incidence of spontaneous discharge activity among the C-fibers of wildtype mice, which was reduced by blocking CaV3.2 channels with TTA-P2 and it was largely absent in the KOs. An evaluation restricted to the peptidergic class of nerve fibers - measuring KCl-stimulated CGRP release - revealed a marked reduction in the sciatic nerve by TTA-P2 in healthy but not diabetic wildtypes, the latter showing CGRP release that was as much reduced as in healthy and, equally, in diabetic CaV3.2 KO mice. These data suggest that diabetes abrogates all CaV3.2 functionality in the peripheral nerve axons. In striking contrast, diabetes markedly increased the KCl-stimulated CGRP release from isolated hairy skin in wildtypes but not KOs, and TTA-P2 reversed this increase, suggesting a de novo expression of CaV3.2 in peptidergic cutaneous nerve endings which may contribute to the enhanced spontaneous activity. De-glycosylation by neuraminidase showed clear desensitizing effects, both in regard to spontaneous activity and stimulated CGRP release, but included actions independent of CaV3.2. However, as diabetes-enhanced glycosylation is decisive for intraaxonal trafficking, it may account for the substantial reorganisations of the CaV3.2 distribution – leading to enhanced excitability of nociceptors during diabetic neuropathy. The results may strengthen the validation of CaV3.2 as a therapeutic target of treating painful diabetic neuropathy.
Highlights.
Diabetic glycosylation of CaV3.2 increases spontaneous activity in nociceptors.
Diabetic spontaneous activity reduced by CaV3.2 block and absent in knockouts.
Diabetes depletes CaV3.2 in peripheral nerve and accumulates it in the skin terminals.
CGRP release enhanced in diabetic skin and reduced by CaV 3.2 block and in knockouts.
Acknowledgements:
The authors thank Annette Kuhn and Birgit Vogler for excellent technical assistance.
Funding information:
This study was funded by the grant from the National Institutes of Health (1R01NS091353-01A1) to SMT and to SKS/PWR.
Abbreviation list:
- AP
Action potential
- AMC
A mechano-cold fiber
- AMH
A mechano-heat fiber
- AMHC
A mechano-heat-cold fiber
- Aδ-LTM
Aδ low threshold mechanosensitive fiber
- Aδ-HTM
Aδ high threshold mechanosensitive fiber
- CGRP
Calcitonin gene-related peptide
- CAP
Compound action potentials
- CV
Conduction velocity
- C-MC
C mechano-cold fiber
- C-MH
C mechano-heat fiber
- C-MHC
C mechano-heat-cold fiber
- C-LTM
C low threshold mechanosensitive fiber
- C-HTM
C high threshold mechanosensitive fiber
- DN
Diabetic neuropathy
- DRG
Dorsal root ganglia
- i.p.
Intraperitoneally
- KO
Knock – out mice
- NEU
Neuraminidase
- PDN
Painful diabetic neuropathy
- RF
Receptive field
- STZ
Streptozotocin
- SIF
Synthetic interstitial fluid
- TRPA1
Transient receptor potential channel Ankyrin 1
- TRPV1
Transient receptor potential vanilloid 1
- TTXs
Tetrodotoxin sensitive
- TTA-P2
3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abad-Rodríguez J, Díez-Revuelta N, 2015. Axon glycoprotein routing in nerve polarity, function, and repair. Trends in biochemical sciences 40, 385–396. [DOI] [PubMed] [Google Scholar]
- Amrutkar DV, Ploug KB, Olesen J, Jansen-Olesen I, 2011. Role for voltage gated calcium channels in calcitonin gene-related peptide release in the rat trigeminovascular system. Neuroscience 172, 510–517. [DOI] [PubMed] [Google Scholar]
- Andersson DA, Gentry C, Light E, Vastani N, Vallortigara J, Bierhaus A, Fleming T, Bevan S, 2013. Methylglyoxal evokes pain by stimulating TRPA1. PloS one 8, e77986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoola C, Hwang SM, Hong SJ, Rose KE, Boyd C, Bozic N, Park JY, Osuru HP, DiGruccio MR, Covey DF, Jevtovic-Todorovic V, Todorovic SM, 2014. Inhibition of CaV3.2 T-type calcium channels in peripheral sensory neurons contributes to analgesic properties of epipregnanolone. Psychopharmacology 231, 3503–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babes A, Fischer MJ, Reid G, Sauer SK, Zimmermann K, Reeh PW, 2010. Electrophysiological and neurochemical techniques to investigate sensory neurons in analgesia research. Methods Mol. Biol 617:237–59., 237–259. [DOI] [PubMed] [Google Scholar]
- Bao J, Li JJ, Perl ER, 1998. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J. Neurosci 18, 8740–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baycin-Hizal D, Gottschalk A, Jacobson E, Mai S, Wolozny D, Zhang H, Krag SS, Betenbaugh MJ, 2014. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochemical and biophysical research communications 453, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy J, Bowen EJ, Russo AF, Durham PL, 2006. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur. J. Neurosci 23, 2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, 2012. What is spontaneous pain and who has it? The journal of pain : official journal of the American Pain Society 13, 921–929. [DOI] [PubMed] [Google Scholar]
- Bernal Sierra YA, Haseleu J, Kozlenkov A, Bégay V, Lewin GR, 2017. Genetic Tracing of Ca(v)3.2 T-Type Calcium Channel Expression in the Peripheral Nervous System. Frontiers in molecular neuroscience 10, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessou P, Perl ER, 1969. Response of cutaneous sensory units with unmyelinated fibers to noxious stimuli. Journal of neurophysiology 32, 1025–1943. [DOI] [PubMed] [Google Scholar]
- Bich-Hoai TT, Marin A, Dinu C, Banciu D, Maria-Luiza F, Ristoiu V, 2010. Hypoxia and high glucose activate tetrodotoxin-resistant Na(+) currents through PKA and PKC. Acta neurobiologiae experimentalis 70, 351–361. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnolzer M, Lasitschka F, Neuhuber WL, Kichko TI, Konrade I, Elvert R, Mier W, Pirags V, Lukic IK, Morcos M, Dehmer T, Rabbani N, Thornalley PJ, Edelstein D, Nau C, Forbes J, Humpert PM, Schwaninger M, Ziegler D, Stern DM, Cooper ME, Haberkorn U, Brownlee M, Reeh PW, Nawroth PP, 2012. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nature medicine 18, 926–933. [DOI] [PubMed] [Google Scholar]
- Blair NT, Bean BP, 2002. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 10277–10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesneac I, Chemin J, Bidaud I, Huc-Brandt S, Vandermoere F, Lory P, 2015. Phosphorylation of the Cav3.2 T-type calcium channel directly regulates its gating properties. Proceedings of the National Academy of Sciences of the United States of America 112, 13705–13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrère C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J, 2005. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. The EMBO journal 24, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Francois A, Laffray S, 2016. T-type calcium channels in neuropathic pain. Pain 157 Suppl 1, S15–22. [DOI] [PubMed] [Google Scholar]
- Bretag AH, 1969. Synthetic interstitial fluid for isolated mammalian tissue. Life science 8, 319–329. [DOI] [PubMed] [Google Scholar]
- Brewster WJ, Diemel LT, Leach RM, Tomlinson DR, 1994. Reduced sciatic nerve substance P and calcitonin gene-related peptide in rats with short-term diabetes or central hypoxaemia co-exist with normal messenger RNA levels in the lumbar dorsal root ganglia. Neuroscience 58, 323–330. [DOI] [PubMed] [Google Scholar]
- Burchiel KJ, Russell LC, Lee RP, Sima AA, 1985. Spontaneous activity of primary afferent neurons in diabetic BB/Wistar rats. A possible mechanism of chronic diabetic neuropathic pain. Diabetes 34, 1210–1213. [DOI] [PubMed] [Google Scholar]
- Cain SM, Snutch TP, 2012. Voltage-Gated Calcium Channels in Epilepsy, in: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (Eds.), Jasper’s Basic Mechanisms of the Epilepsies. National Center for Biotechnology Information (US) [PubMed] [Google Scholar]
- Copyright © 2012, Michael A Rogawski, Antonio V Delgado-Escueta, Jeffrey L Noebels, Massimo Avoli and Richard W Olsen., Bethesda (MD). [Google Scholar]
- Calcutt NA, 2020. Diabetic neuropathy and neuropathic pain: a (con)fusion of pathogenic mechanisms? Pain 161, S65–s86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcutt NA, Chen P, Hua XY, 1998. Effects of diabetes on tissue content and evoked release of calcitonin gene-related peptide-like immunoreactivity from rat sensory nerves. Neurosci. Lett 254, 129–132. [DOI] [PubMed] [Google Scholar]
- Candelas M, Reynders A, Arango-Lievano M, Neumayer C, Fruquière A, Demes E, Hamid J, Lemmers C, Bernat C, Monteil A, Compan V, Laffray S, Inquimbert P, Le Feuvre Y, Zamponi GW, Moqrich A, Bourinet E, Méry PF, 2019. Cav3.2 T-type calcium channels shape electrical firing in mouse Lamina II neurons. Scientific reports 9, 3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XH, Byun HS, Chen SR, Pan HL, 2011. Diabetic neuropathy enhances voltage-activated Ca2+ channel activity and its control by M4 muscarinic receptors in primary sensory neurons. Journal of neurochemistry 119, 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L, 2004. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes 53, 32263232. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P, 2002. Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. The Journal of physiology 540, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Stamenic TT, Cazade M, Llinares J, Blesneac I, Todorovic SM, Lory P, 2019. A novel phospho-modulatory mechanism contributes to the calcium-dependent regulation of T-type Ca(2+) channels. Scientific reports 9, 15642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, Cribbs LL, England SK, Sigmund CD, Weiss RM, Williamson RA, Hill JA, Campbell KP, 2003. Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science (New York, N.Y.) 302, 1416–1418. [DOI] [PubMed] [Google Scholar]
- Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic-Todorovic V, Todorovic SM, 2011. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol 80, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Na HS, Kim J, Lee J, Lee S, Kim D, Park J, Chen CC, Campbell KP, Shin HS, 2007. Attenuated pain responses in mice lacking Ca(V)3.2 T-type channels. Genes, brain, and behavior 6, 425–431. [DOI] [PubMed] [Google Scholar]
- Comunanza V, Carbone E, Marcantoni A, Sher E, Ursu D, 2011. Calcium-dependent inhibition of T-type calcium channels by TRPV1 activation in rat sensory neurons. Pflugers Archiv : European journal of physiology 462, 709–722. [DOI] [PubMed] [Google Scholar]
- Cowie AM, Moehring F, O’Hara C, Stucky CL, 2018. Optogenetic Inhibition of CGRPα Sensory Neurons Reveals Their Distinct Roles in Neuropathic and Incisional Pain. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 5807–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LS, Garcia E, Cain SM, Thau E, Tyson JR, Snutch TP, 2010. Splice-variant changes of the Ca(V)3.2 T-type calcium channel mediate voltage-dependent facilitation and associate with cardiac hypertrophy and development. Channels (Austin, Tex.) 4, 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bianco E, Santicioli P, Tramontana M, Maggi CA, Cecconi RG,P, 1991. Different pathways by which extracellular Ca2+ promotes calcitonin gene-related peptide release from central terminals of capsaicin-sensitive afferents of guinea pigs: effect of capsaicin, high K+ and low pH media. Brain Research 566, 46–53. [DOI] [PubMed] [Google Scholar]
- Diemel LT, Sevens EJ, Willars GB, Tomlinson DR, 1992. Depletion of substance P and calcitonin gene-related peptide in sciatic nerve of rats with experimental diabetes; effects of insulin and aldose reductase inhibition. Neuroscience Letters 137, 253–256. [DOI] [PubMed] [Google Scholar]
- Djouhri L, Koutsikou S, Fang X, McMullan S, Lawson SN, 2006. Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri L, Zeidan A, Abd El-Aleem SA, Smith T, 2020. Cutaneous Aβ-Non-nociceptive, but Not C-Nociceptive, Dorsal Root Ganglion Neurons Exhibit Spontaneous Activity in the Streptozotocin Rat Model of Painful Diabetic Neuropathy in vivo. Frontiers in neuroscience 14, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil AS, Boukhaddaoui H, Desmadryl G, Martinez-Salgado C, Moshourab R, Lewin GR, Carroll P, Valmier J, Scamps F, 2004. Role of T-type calcium current in identified D-hair mechanoreceptor neurons studied in vitro. J. Neurosci 24, 8480–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duzhyy DE, Viatchenko-Karpinski VY, Khomula EV, Voitenko NV, Belan PV, 2015. Upregulation of T-type Ca2+ channels in long-term diabetes determines increased excitability of a specific type of capsaicin-insensitive DRG neurons. Molecular pain 11, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt M, Hoffmann T, Sauer SK, Messlinger K, Reeh PW, Fischer MJ, 2008. Calcitonin gene-related peptide release from intact isolated dorsal root and trigeminal ganglia. Neuropeptides 42, 311–317. [DOI] [PubMed] [Google Scholar]
- Eberhardt MJ, Filipovic MR, Leffler A, de la Roche J, Kistner K, Fischer MJ, Fleming T, Zimmermann K, Ivanovic-Burmazovic I, Nawroth PP, Bierhaus A, Reeh PW, Sauer SK, 2012. Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. J. Biol. Chem 287, 28291–28306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington HC, Cotter MA, Cameron NE, Ross RA, 2002. The effect of cannabinoids on capsaicin-evoked calcitonin gene-related peptide (CGRP) release from the isolated paw skin of diabetic and non-diabetic rats. Neuropharmacology 42, 966–975. [DOI] [PubMed] [Google Scholar]
- Emanuel K, Mackiewicz U, Pytkowski B, Lewartowski B, 1998. Effects of mibefradil, a blocker of T-type Ca2+ channels, in single myocytes and intact muscle of guinea-pig heart. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society 49, 577–590. [PubMed] [Google Scholar]
- Feng XJ, Ma LX, Jiao C, Kuang HX, Zeng F, Zhou XY, Cheng XE, Zhu MY, Zhang DY, Jiang CY, Liu T, 2019. Nerve injury elevates functional Cav3.2 channels in superficial spinal dorsal horn. Molecular pain 15, 1744806919836569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernyhough P, Diemel LT, Brewster WJ, Tomlinson DR, 1994. Deficits in sciatic nerve neuropeptide content coincide with a reduction in target tissue nerve growth factor messenger RNA in streptozotocin-diabetic rats: effects of insulin treatment. Neuroscience 62, 337–344. [DOI] [PubMed] [Google Scholar]
- Ficelova V, Souza IA, Cmarko L, Gandini MA, Stringer RN, Zamponi GW, Weiss N, 2020. Functional identification of potential non-canonical N-glycosylation sites within Ca(v)3.2 T-type calcium channels. Molecular brain 13, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François A, Schüetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, Mantilleri A, Nargeot J, Noël J, Wood JN, Moqrich A, Pongs O, Bourinet E, 2015. The Low-Threshold Calcium Channel Cav3.2 Determines Low-Threshold Mechanoreceptor Function. Cell reports 10, 370–382. [DOI] [PubMed] [Google Scholar]
- Fuchs D, Birklein F, Reeh PW, Sauer SK, 2010. Sensitized peripheral nociception in experimental diabetes of the rat. Pain 151, 496–505. [DOI] [PubMed] [Google Scholar]
- Fukami K, Asano E, Ueda M, Sekiguchi F, Yoshida S, Kawabata A, 2017. High glucose induces N-linked glycosylation-mediated functional upregulation and overexpression of Ca(v)3.2 T-type calcium channels in neuroendocrine-like differentiated human prostate cancer cells. Journal of pharmacological sciences 133, 57–60. [DOI] [PubMed] [Google Scholar]
- Furman BL, 2015. Streptozotocin-Induced Diabetic Models in Mice and Rats. Current protocols in pharmacology 70, 5.47.41–45.47.20. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez E, Schönberger T, Sumalla M, Stierstorfer B, Solà R, Doods H, Serra J, Gorodetskaya N, 2018. Behavioural, morphological and electrophysiological assessment of the effects of type 2 diabetes mellitus on large and small nerve fibres in Zucker diabetic fatty, Zucker lean and Wistar rats. European journal of pain (London, England) 22, 1457–1472. [DOI] [PubMed] [Google Scholar]
- Gebhardt LA, Kichko TI, Fischer MJM, Reeh PW, 2020. TRPA1-dependent calcium transients and CGRP release in DRG neurons require extracellular calcium. The Journal of cell biology 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez K, Calderón-Rivera A, Sandoval A, González-Ramírez R, Vargas-Parada A, Ojeda-Alonso J, Granados-Soto V, Delgado-Lezama R, Felix R, 2020. Cdk5-Dependent Phosphorylation of Ca(V)3.2 T-Type Channels: Possible Role in Nerve Ligation-Induced Neuropathic Allodynia and the Compound Action Potential in Primary Afferent C Fibers. The Journal of neuroscience : the official journal of the Society for Neuroscience 40, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe P, Bostock H, Schneider U, 1994. The effects of hyperglycaemic hypoxia on rectification in rat dorsal root axons. J Physiol Lond 480, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylfadottir SS, Weeracharoenkul D, Andersen ST, Niruthisard S, Suwanwalaikorn S, Jensen TS, 2019. Painful and non-painful diabetic polyneuropathy: Clinical characteristics and diagnostic issues. Journal of diabetes investigation 10, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KE, Sima AA, Wiley JW, 1995. Voltage-dependent calcium currents are enhanced in dorsal root ganglion neurones from the Bio Bred/Worchester diabetic rat. The Journal of physiology 486 ( Pt 2), 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrøm JB, Jensen TS, 2014. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155, 1272–1279. [DOI] [PubMed] [Google Scholar]
- Herrmann S, Rajab H, Christ I, Schirdewahn C, Höfler D, Fischer MJM, Bruno A, Fenske S, Gruner C, Kramer F, Wachsmann T, Wahl-Schott C, Stieber J, Biel M, Ludwig A, 2017. Protein kinase A regulates inflammatory pain sensitization by modulating HCN2 channel activity in nociceptive sensory neurons. Pain 158, 2012–2024. [DOI] [PubMed] [Google Scholar]
- Hong S, Wiley JW, 2005. Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J. Biol. Chem 280, 618–627. [DOI] [PubMed] [Google Scholar]
- Huang LY, Neher E, 1996. Ca(2+)-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron 17, 135–145. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Sayers NM, Fernyhough P, Verkhratsky A, 2002. Diabetes-induced alterations in calcium homeostasis in sensory neurones of streptozotocin-diabetic rats are restricted to lumbar ganglia and are prevented by neurotrophin-3. Diabetologia 45, 560–570. [DOI] [PubMed] [Google Scholar]
- Jacus MO, Uebele VN, Renger JJ, Todorovic SM, 2012. Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 9374–9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, Bayliss DA, Jevtovic-Todorovic V, Todorovic SM, 2007. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J. Neurosci 27, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksimovic SL, Evans JG, McIntire WE, Orestes P, Barrett PQ, Jevtovic-Todorovic V, Todorovic SM, 2020. Glycosylation of Ca(V)3.2 Channels Contributes to the Hyperalgesia in Peripheral Neuropathy of Type 1 Diabetes. Frontiers in cellular neuroscience 14, 605312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang XJ, Chi YN, Chen W, Liu FY, Cui S, Liao FF, Cai J, Wan Y, 2018. Increased expression of Ca(V)3.2 T-type calcium channels in damaged DRG neurons contributes to neuropathic pain in rats with spared nerve injury. Molecular pain 14, 1744806918765808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson P, Provitera V, Caporaso G, Stancanelli A, Saltalamacchia AM, Borreca I, Manganelli F, Santoro L, Jensen TS, Nolano M, 2021. Increased peptidergic fibers as a potential cutaneous marker of pain in diabetic small fiber neuropathy. Pain 162, 778–786. [DOI] [PubMed] [Google Scholar]
- Kerckhove N, Mallet C, François A, Boudes M, Chemin J, Voets T, Bourinet E, Alloui A, Eschalier A, 2014. Ca(v)3.2 calcium channels: the key protagonist in the supraspinal effect of paracetamol. Pain 155, 764–772. [DOI] [PubMed] [Google Scholar]
- Kestell GR, Anderson RL, Clarke JN, Haberberger RV, Gibbins IL, 2015. Primary afferent neurons containing calcitonin gene-related peptide but not substance P in forepaw skin, dorsal root ganglia, and spinal cord of mice. The Journal of comparative neurology 523, 2555–2569. [DOI] [PubMed] [Google Scholar]
- Khomula EV, Viatchenko-Karpinski VY, Borisyuk AL, Duzhyy DE, Belan PV, Voitenko NV, 2013. Specific functioning of Cav3.2 T-type calcium and TRPV1 channels under different types of STZ-diabetic neuropathy. Biochimica et biophysica acta 1832, 636–649. [DOI] [PubMed] [Google Scholar]
- Kostyuk E, Pronchuk N, Shmigol A, 1995. Calcium signal prolongation in sensory neurones of mice with experimental diabetes. Neuroreport 6, 1010–1012. [DOI] [PubMed] [Google Scholar]
- Kramer HH, Rolke R, Bickel A, Birklein F, 2004. Thermal thresholds predict painfulness of diabetic neuropathies. Diabetes Care 27, 2386–2391. [DOI] [PubMed] [Google Scholar]
- Kress M, Guthmann C, Averbeck B, Reeh PW, 1999. Calcitonin gene-related peptide and prostaglandin E2 but not substance P release induced by antidromic nerve stimulation from rat skin, in vitro. Neuroscience 89, 303–310. [DOI] [PubMed] [Google Scholar]
- Kress M, Izydorczyk I, Kuhn A, 2001. N- and L- but not P/Q-type calcium channels contribute to neuropeptide release from rat skin in vitro. Neuroreport 12, 867–870. [DOI] [PubMed] [Google Scholar]
- Latham JR, Pathirathna S, Jagodic MM, Choe WJ, Levin ME, Nelson MT, Lee WY, Krishnan K, Covey DF, Todorovic SM, Jevtovic-Todorovic V, 2009. Selective T-type calcium channel blockade alleviates hyperalgesia in ob/ob mice. Diabetes 58, 2656–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson SN, Crepps B, Perl ER, 2002. Calcitonin gene-related peptide immunoreactivity and afferent receptive properties of dorsal root ganglion neurones in guinea-pigs. The Journal of physiology 540, 989–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazniewska J, Weiss N, 2017. Glycosylation of voltage-gated calcium channels in health and disease. Biochimica et biophysica acta. Biomembranes 1859, 662–668. [DOI] [PubMed] [Google Scholar]
- Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F, 2006. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proceedings of the National Academy of Sciences of the United States of America 103, 9232–9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennerz JK, Rühle V, Ceppa EP, Neuhuber WL, Bunnett NW, Grady EF, Messlinger K, 2008. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. The Journal of comparative neurology 507, 1277–1299. [DOI] [PubMed] [Google Scholar]
- Levine JD, Fields HL, Basbaum AI, 1993. Peptides and the primary afferent nociceptor. The Journal of neuroscience : the official journal of the Society for Neuroscience 13, 2273–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen W, Fan X, Wang J, Fu S, Cui S, Liao F, Cai J, Wang X, Huang Y, Su L, Zhong L, Yi M, Liu F, Wan Y, 2019. Upregulation of interleukin-6 on Ca(v)3.2 T-type calcium channels in dorsal root ganglion neurons contributes to neuropathic pain in rats with spinal nerve ligation. Experimental neurology 317, 226–243. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Franco-Cereceda A, Alving K, Delay-Goyet P, Lou YP, 1992. Release of calcitonin gene-related peptide from sensory neurons. Annals of the New York Academy of Sciences 657, 187–193. [DOI] [PubMed] [Google Scholar]
- Ma Y, Chen J, Yu D, Wei B, Jin H, Zeng J, Liu X, 2021. cAMP-PKA signaling is involved in regulation of spinal HCN channels function in diabetic neuropathic pain. Neurosci Lett 750, 135763. [DOI] [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Zylka MJ, 2012. CGRPalpha-expressing sensory neurons respond to stimuli that evoke sensations of pain and itch. PLoS. One 7, e36355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, Orestes P, Latham JR, Todorovic SM, Jevtovic-Todorovic V, 2009. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 145, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Perez-Reyes E, Todorovic SM, 2005. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J. Neurosci 25, 8766–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Todorovic SM, 2006. Is there a role for T-type calcium channels in peripheral and central pain sensitization? Mol. Neurobiol 34, 243–248. [DOI] [PubMed] [Google Scholar]
- Nemeth J, Helyes Z, Oroszi G, Jakab B, Pinter E, Szilvassy Z, Szolcsanyi J, 2003. Role of voltage-gated cation channels and axon reflexes in the release of sensory neuropeptides by capsaicin from isolated rat trachea. Eur. J. Pharmacol 458, 313–318. [DOI] [PubMed] [Google Scholar]
- Obradovic A, Hwang SM, Scarpa J, Hong SJ, Todorovic SM, Jevtovic-Todorovic V, 2014. CaV3.2 T-type calcium channels in peripheral sensory neurons are important for mibefradil-induced reversal of hyperalgesia and allodynia in rats with painful diabetic neuropathy. PloS one 9, e91467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondacova K, Karmazinova M, Lazniewska J, Weiss N, Lacinova L, 2016. Modulation of Cav3.2 T-type calcium channel permeability by asparagine-linked glycosylation. Channels (Austin, Tex.) 10, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orestes P, Osuru HP, McIntire WE, Jacus MO, Salajegheh R, Jagodic MM, Choe W, Lee J, Lee SS, Rose KE, Poiro N, Digruccio MR, Krishnan K, Covey DF, Lee JH, Barrett PQ, Jevtovic-Todorovic V, Todorovic SM, 2013. Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T-type calcium channels. Diabetes 62, 3828–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orstavik K, Namer B, Schmidt R, Schmelz M, Hilliges M, Weidner C, Carr RW, Handwerker H, Jorum E, Torebjork HE, 2006. Abnormal function of C-fibers in patients with diabetic neuropathy. J. Neurosci 26, 11287–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabbidi RM, Yu SQ, Peng S, Khardori R, Pauza ME, Premkumar LS, 2008. Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol. Pain 4:9., 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior R, Van Helleputte L, Benoy V, Van Den Bosch L, 2017. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiology of disease 105, 300–320. [DOI] [PubMed] [Google Scholar]
- Quallo T, Gentry C, Bevan S, Broad LM, Mogg AJ, 2015. Activation of transient receptor potential ankyrin 1 induces CGRP release from spinal cord synaptosomes. Pharmacol. Res. Perspect 3, e00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic-Todorovic V, Todorovic SM, 2013. Immunohistological demonstration of CaV3.2 T-type voltage-gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 250, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossini AA, Williams RM, Appel MC, Like AA, 1978. Complete protection from low-dose streptozotocin-induced diabetes in mice. Nature 276, 182–184. [DOI] [PubMed] [Google Scholar]
- Samour MS, Nagi SS, Mahns DA, 2015. Cav3.2-expressing low-threshold C fibres in human hairy skin contribute to cold allodynia--a non-TRPV1- and non-TRPM8-dependent phenomenon. Pain 156, 1566–1575. [DOI] [PubMed] [Google Scholar]
- Sandkühler J, 2009. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev 89, 707–758. [DOI] [PubMed] [Google Scholar]
- Sauer SK, Bove GM, Averbeck B, Reeh PW, 1999. Rat Peripheral Nerve Components Release Calcitonin Gene-Related Peptide and Prostaglandin E2 In Response to Noxious Stimuli: Evidence That Nervi Nervorum are Nociceptors. Neuroscience 92, 319–325. [DOI] [PubMed] [Google Scholar]
- Schmelz M, Schmidt R, Ringkamp M, Forster C, Handwerker HO, Torebjork HE, 1996. Limitation of sensitization to injured parts of receptive fields in human skin C-nociceptors. Exp Brain Res 109, 141–147. [DOI] [PubMed] [Google Scholar]
- Schneider U, Quasthoff S, Mitrovic N, Grafe P, 1993. Hyperglycaemic hypoxia alters after-potential and fast K+ conductance of rat axons by cytoplasmic acidification. J Physiol Lond 465, 679–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JE, Fischbach PS, McCleskey EW, 1990. T-type calcium channels: heterogeneous expression in rat sensory neurons and selective modulation by phorbol esters. The Journal of neuroscience : the official journal of the Society for Neuroscience 10, 947–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS, Fox AP, 1992. Multiple Ca2+ currents elicited by action potential waveforms in acutely isolated adult rat dorsal root ganglion neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 12, 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra J, Bostock H, Solà R, Aleu J, García E, Cokic B, Navarro X, Quiles C, 2012. Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain 153, 42–55. [DOI] [PubMed] [Google Scholar]
- Shathili AM, Brown HM, Everest-Dass AV, Tan TCY, Parker LM, Thompson JG, Packer NH, 2018. The effect of streptozotocin-induced hyperglycemia on N-and O-linked protein glycosylation in mouse ovary. Glycobiology 28, 832–840. [DOI] [PubMed] [Google Scholar]
- Shin NS, Lee IS, Yoon YS, Lee HS, 2003. Immunohistochemical localization of substance P, calcitonin gene-related peptide, galanin and calcium-binding proteins in trigeminal ganglia of goat (Capra hircus). Anatomia, histologia, embryologia 32, 310–315. [DOI] [PubMed] [Google Scholar]
- Sima AA, Zhang W, 2014. Mechanisms of diabetic neuropathy: axon dysfunction. Handbook of clinical neurology 126, 429–442. [DOI] [PubMed] [Google Scholar]
- Sorensen L, Molyneaux L, Yue DK, 2006. The relationship among pain, sensory loss, and small nerve fibers in diabetes. Diabetes Care 29, 883–887. [DOI] [PubMed] [Google Scholar]
- Spitzer MJ, Reeh PW, Sauer SK, 2008. Mechanisms of potassium- and capsaicin-induced axonal calcitonin gene-related peptide release: involvement of L- and T-type calcium channels and TRPV1 but not sodium channels. Neuroscience 151, 836–842. [DOI] [PubMed] [Google Scholar]
- Sun W, Miao B, Wang XC, Duan JH, Wang WT, Kuang F, Xie RG, Xing JL, Xu H, Song XJ, Luo C, Hu SJ, 2012. Reduced conduction failure of the main axon of polymodal nociceptive C-fibres contributes to painful diabetic neuropathy in rats. Brain 135, 359–375. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Sato J, Kawanishi M, Mizumura K, 2002a. Lowered response threshold and increased responsiveness to mechanical stimulation of cutaneous nociceptive fibers in streptozotocin-diabetic rat skin in vitro--correlates of mechanical allodynia and hyperalgesia observed in the early stage of diabetes. Neurosci. Res 43, 171–178. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Sato J, Kawanishi M, Mizumura K, 2002b. Tissue glucose level modulates the mechanical responses of cutaneous nociceptors in streptozotocin-diabetic rats but not normal rats in vitro. Pain 99, 475–484. [DOI] [PubMed] [Google Scholar]
- Talbot S, Couture R, 2012. Emerging role of microglial kinin B1 receptor in diabetic pain neuropathy. Experimental neurology 234, 373–381. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, 2016. Painful Diabetic Neuropathy: Prevention or Suppression? International review of neurobiology 127, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V, 2011. T-type voltage-gated calcium channels as targets for the development of novel pain therapies. British journal of pharmacology 163, 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V, 2014. Targeting of CaV3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflugers Archiv : European journal of physiology 466, 701–706. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Lingle CJ, 1998a. Pharmacological properties of T-type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. J. Neurophysiol 79, 240–252. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Lingle CJ, 1998b. Pharmacological properties of T-type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. Journal of neurophysiology 79, 240–252. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Meyenburg A, Jevtovic-Todorovic V, 2002. Mechanical and thermal antinociception in rats following systemic administration of mibefradil, a T-type calcium channel blocker. Brain Res 951, 336–340. [DOI] [PubMed] [Google Scholar]
- Touska F, Turnquist B, Vlachova V, Reeh PW, Leffler A, Zimmermann K, 2018. Heat-resistant action potentials require TTX-resistant sodium channels Na(V)1.8 and Na(V)1.9. The Journal of general physiology 150, 1125–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsantoulas C, Laínez S, Wong S, Mehta I, Vilar B, McNaughton PA, 2017. Hyperpolarization-activated cyclic nucleotide-gated 2 (HCN2) ion channels drive pain in mouse models of diabetic neuropathy. Science translational medicine 9, eaam6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Fernyhough P, 2014. Calcium signalling in sensory neurones and peripheral glia in the context of diabetic neuropathies. Cell Calcium 56, 362–371. [DOI] [PubMed] [Google Scholar]
- Wang R, Lewin GR, 2011. The Cav3.2 T-type calcium channel regulates temporal coding in mouse mechanoreceptors. J. Physiol 589, 2229–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Ueda T, Shibata Y, Kumamoto N, Shimada S, Ugawa S, 2015. Expression and Regulation of Cav3.2 T-Type Calcium Channels during Inflammatory Hyperalgesia in Mouse Dorsal Root Ganglion Neurons. PloS one 10, e0127572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss N, Black SA, Bladen C, Chen L, Zamponi GW, 2013. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Archiv : European journal of physiology 465, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Weiss N, Zamponi GW, De Waard M, 2012. How do T-type calcium channels control low-threshold exocytosis? Communicative & integrative biology 5, 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Washburn MS, Hans M, Urrutia A, Brust PF, Prodanovich P, Harpold MM, Stauderman KA, 1999. Structure and functional characterization of a novel human low-voltage activated calcium channel. Journal of neurochemistry 72, 791–799. [DOI] [PubMed] [Google Scholar]
- Witcher D, Sakai N, Williams B, Rahimian R, Anderson L, 2010. Gender differences in the effects of streptozotocin-induced diabetes on parasympathetic vasodilatation in the rat submandibular gland. Archives of oral biology 55, 745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Ringkamp M, Murinson BB, Pogatzki EM, Hartke TV, Weerahandi HM, Campbell JN, Griffin JW, Meyer RA, 2002. Degeneration of myelinated efferent fibers induces spontaneous activity in uninjured C-fiber afferents. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 7746–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorek MA, 2015. Vascular Impairment of Epineurial Arterioles of the Sciatic Nerve: Implications for Diabetic Peripheral Neuropathy. The review of diabetic studies : RDS 12, 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusaf SP, Goodman J, Pinnock RD, Dixon AK, Lee K, 2001. Expression of voltagegated calcium channel subunits in rat dorsal root ganglion neurons. Neurosci. Lett 311, 137–141. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jiang X, Snutch TP, Tao J, 2013. Modulation of low-voltage-activated T-type Ca2⁺ channels. Biochimica et biophysica acta 1828, 1550–1559. [DOI] [PubMed] [Google Scholar]
- Zochodne DW, Ho LT, Allison JA, 1994. Dorsal root ganglia microenvironment of female BB Wistar diabetic rats with mild neuropathy. Journal of the neurological sciences 127, 36–42. [DOI] [PubMed] [Google Scholar]