

Abstract
Microglial cells are the main reservoir for HIV-1 within the brain and potential exists for negative immune checkpoint blockade therapies to purge this viral reservoir. Here, we investigated cytolytic responses of CD8+ T lymphocytes against microglia loaded with peptide epitopes. Initially, flow cytometric analysis demonstrated efficient killing of HIV-1 p24 AI9 or YI9 peptide-loaded splenocytes in MHC-matched recipients. Cytolytic killing of microglia was first demonstrated using ovalbumin (OVA) as a model antigen for in vitro cytotoxic T lymphocyte (CTL) assays. Peptide-loaded primary microglia obtained from programmed death ligand (PD-L) 1 knockout (KO) animals showed significantly more killing than cells from wild-type (WT) animals when co-cultured with activated CD8+ T-cells isolated from rAd5-OVA primed animals. Moreover, when peptide loaded-microglial cells from WT animals were treated with neutralizing α-PD-L1 Ab, significantly more killing was observed compared to either untreated or IgG isotype-treated cells. Most importantly, significantly increased in vivo killing of HIV-1 p24 YI9 peptide-loaded microglia from PD-L1 KO animals, as well as AI9 peptide-loaded BALB/c microglial cells treated with α-PD-L1, was observed within brains of rAd5-p24 primed-CNS boosted C57BL/6 or BALB/c mice, respectively. Finally, ex vivo responses of brain CD8+ T-cells in response to AI9 stimulation showed significantly increased IFN-γ and IL-2 production when treated with α-PD-1 Abs. Greater proliferation of CD8+ T-cells from the brain was also observed following blockade. Taken together, these studies demonstrate that PD-L1 induction on microglia restrains CTL responses and indicate that immune checkpoint blockade targeting this pathway may be beneficial in clearing viral brain reservoirs.
Keywords: brain-resident memory T-cells, cytotoxic T lymphocyte, HIV-associated neurocognitive disorders, microglia, programmed death (PD)-1: PD-L1 pathway
Graphical Abstract
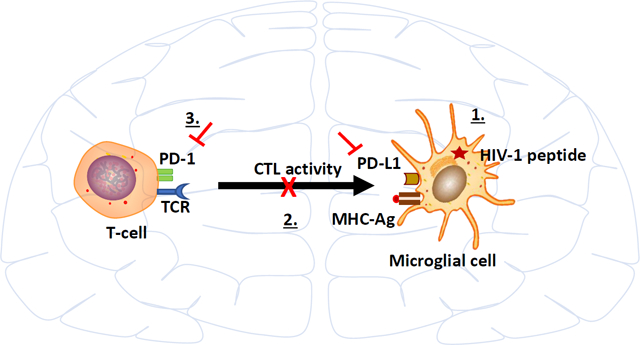
1 |. INTRODUCTION
Negative immune checkpoint blockade has revolutionized contemporary treatment for a variety of cancers with the most widely used and successful therapies targeting the programmed death (PD)-1: PD-L1 pathway (Kerr & Chisholm, 2019; Rosato et al., 2019; Wu et al., 2019). However, individual patient responses are unpredictable and the susceptibility of particular cell types to checkpoint blockade remain highly variable and difficult to predict (Cortese et al., 2019). Therefore, despite major successes, checkpoint blockade immunotherapy is ineffective for the majority of patients. We have previously shown that glial cells govern the activity of brain-infiltrating anti-viral T-cells via upregulation of PD-L1 expression, likely in an effort to mitigate potentially damaging encephalitis (Schachtele, Hu, Sheng, Mutnal, & Lokensgard, 2014). Although these immunosuppressive responses are undeniably beneficial to the host, preventing damage to this vital organ, establishment of a prolonged anti-inflammatory milieu may also result in deficiencies in viral clearance from infected brain cells (Chauhan & Lokensgard, 2019).
Microglia are the main targets of HIV-1 infection in the central nervous system (CNS) and act as a chronic, latent viral reservoir within the brain (Kumar & Herbein, 2014; Wallet et al., 2019). It is well-established that the human brain is an anatomical sanctuary for persistent HIV-1 infection during suppressive antiretroviral therapy (ART), despite reduction of cerebrospinal fluid (CSF) viral loads to undetectable levels. The microglial cell and perivascular macrophage populations within the CNS have been shown to harbor HIV DNA (Ko et al., 2019). Notably, in some virally suppressed aviremic cases, viral RNA can also be detected in the brain indicating spontaneous reactivation or ongoing low-level viral replication despite suppressive ART. Tso et al. demonstrated colocalization of HIV-1 subtype C viral DNA with macrophages and microglia in the brain of virally suppressed cases (Tso et al., 2018). Several other reports also demonstrate the presence of HIV DNA in the brains of ART-treated subjects (Gelman et al., 2013; Lamers et al., 2016). For these reasons, microglial cells are the major potential target cell for anti-HIV therapy using immune checkpoint blockade in attempts to purge the persisting viral reservoir.
Our laboratory has developed an experimental murine model in which the brain becomes populated with long-lived CD8+ T-cells specific for immunodominant HIV-1 Gag epitopes (Prasad, Hu, Sheng, Chauhan & Lokensgard, 2019). High numbers of activated CD8+ T-cells against HIV antigens in the CSF of acutely infected patients have been reported (Kessing et al., 2017; Schrier et al., 2015). It is well-established that CD8+ T-cells responding to infection can differentiate into tissue resident-memory T-cells (TRM), which reside permanently in non-lymphoid tissues including brain (bTRM) in disconnect from the circulation, and provide immunosurveillance for reinfection and reactivation of latently infected cells (Mockus, Ren, Shwetank, & Lukacher, 2019; Smolders et al., 2018; Szabo, Miron, & Farber, 2019). In a number of infection models, high PD-1 expression has been demonstrated to be an inherent property of bTRM (Bhadra, Gigley, & Khan, 2012; Prasad et al., 2017; Shwetank et al., 2017; Wakim, Woodward-Davis, & Bevan, 2010). In addition to mice, analyses of human brain autopsy also describe increased PD-1 expression on bTRM. Moreover, upregulation of PD-L1 was reported on glial cells in demyelinating MS lesions as compared to controls (Smolders et al., 2018). Results from these models indicate that the responses of bTRM may be sensitive to negative checkpoint inhibitors (Prasad, Hu, Sheng, Chauhan, & Lokensgard, 2018; Shwetank et al., 2019).
Using our heterologous prime-CNS boost approach, mice are first immunized via an intravenous (IV) injection of rAd5-p24, a recombinant adenovirus vector expressing the HIV-1 p24 capsid protein. Priming with rAd5-p24 induces a strong peripheral immune response (Hosseini Rouzbahani et al., 2016). This systemic priming via IV administration is followed by a CNS-boost consisting of intracerebral injection of Pr55Gag/Env virus-like particles (HIV-VLPs) into the striatum, which promotes T-cell infiltration into the brain. This heterologous prime-CNS boost establishes a population of brain-resident CD8+ T-cells which are retained long-term, express residency markers of TRM (i.e., CD103, CD69, CD49a, CD127), and also express high levels of PD-1 (Prasad, Hu, Sheng, Chauhan, & Lokensgard, 2019). Interestingly, despite the high level of PD-1 expression, these cells are not exhausted because they produce high levels of cytokines, granzyme B, and proliferate in response to cognate antigen (Prasad et al., 2019). In addition, we have previously shown that both microglial cells and astrocytes upregulate PD-L1 upon activation and this PD-L1 expression negatively regulated cytokine production by activated T-cells (Schachtele et al., 2014). Furthermore, this negative regulation was partially reversed following α-PD-1 or α-PD-L1 (but not α-PD-L2) treatment (Chauhan & Lokensgard, 2019; Schachtele et al., 2014). The current study expands on our previous work by investigating the role of the PD-1: PD-L1 pathway in restraining cytolytic responses of bTRM against HIV-1 peptide-presenting microglia.
2 |. MATERIALS AND METHODS
2.1 |. Ethical statement
This study was carried out according to guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the University of Minnesota’s Institutional Animal Care and Use Committee (Protocol Number: 1701-34539A). All animals were routinely cared for in accordance to RAR (Research Animal Resources) guidelines. All surgery was performed under Ketamine/Xylazine (100 mg/kg/body weight) anesthesia and all efforts were made to ameliorate animal suffering. Animals were euthanized after isoflurane inhalation, whenever required.
2.2 |. Experimental animals
BALB/c and C57BL/6 (6–8 weeks old) mice were primed intravenously with rAd5-p24 (1 × 1010 PFU/mouse). Animals were either used for in vivo cytotoxic T-lymphocyte (CTL) assays in the spleen or boosted via intracranial injection of 300 fluorescent units (FU) in no more than 5 μl volume of HIV virus-like particles (HIV-VLPs; 7 days later) to promote immune cell infiltration and retention within the brain. These animals were then used to carry out in vivo CTL assays in the brain or ex vivo stimulation assays. We also employed OT-1 mice for in vitro CTL assays and PD-L1 KO animals for both in vitro and in vivo CTL assays. PD-L1 KO animals were obtained from Arlene Sharpe (Latchman et al., 2004) (Boston). Pathogen free OT-1 (JAX stock#003831), BALB/c (JAX stock#000651), C57BL/6 (JAX stock#000664), and EGFP-expressing mice [C57BL/6-Tg (CAG-EGFP) 10sb/j), (JAX stock#003291)] mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). Animals were housed in individually ventilated cages and were provided with food and water ad libitum at RAR, University of Minnesota.
2.3 |. Peptides
AMQMLKETI (i.e., AI9; Gag197–205) was purchased from MBL Life Science (Sunnyvale, CA), while YSPTSILDI (i.e., YI9; Gag277–285) and SIINFEKL (i.e., SL8; OVA257–264) were bought from GenScript (Piscataway, NJ).
2.4 |. Virus
Production of adenovirus 5 expressing HIV-1 capsid protein p24 under the control of the CMV IE promoter (i.e., rAd5-p24) was out-sourced to Cyagen Biosciences (Santa Clara, CA). This is a second-generation (i.e., ΔE1 + ΔE3), replication incompetent adenovirus vector. Adenovirus vector expressing ovalbumin (rAd5-OVA) was purchased from Abm (Richmond, BC, Canada). It is also a second-generation vector derived from human adenovirus type 5.
2.5 |. Purification of HIV virus-like particles (HIV-VLPs)
For the production of HIV-VLPs, HEK 293 T-cells were co-transfected with a (a) Gag-expressing codon-optimized plasmid, pEYFP-N3 HIV-1 which encodes 55 kDa Gag precursor protein fused to EYFP (enhanced yellow fluorescent protein) under the control of human CMV IE promoter (obtained from Louis Mansky, University of Minnesota); (b) p3NL(ADA) env plasmid that encodes a full length R5-tropic envelope protein under the control of the HIV-1 LTR promoter (procured with permission from Eric Freed, NCI, Frederick, MD); (c) pCEP4-Tat plasmid. After 72 hr of co-transfection, HIV-VLPs were harvested from supernatants and clarified at 1,000g for 10 min. Clarified supernatants were then purified by layering onto 32% sucrose cushion and centrifugation at 100,000g for 2 hr at 4° using an Optima XPN-100 Ultracentrifuge (Beckman Coulter, Brea, CA). HIV-VLPs were quantified using a Spectramax M2 Fluorescent reader (485ex 538em 530 cutoff) and fluorescent units (FU) were used as an indicator of quantity. 300 FU were used for the CNS boost of the rAd5-p24 primed animals as described previously (Prasad et al., 2019).
2.6 |. Intracranial injection of mice
Mice were injected intracranially using a method previously described by Cheeran, Hu, Sheng, Peterson, & Lokensgard (2003). Briefly, mice were anesthetized using a mixture of Ketamine (100 mg/kg body weight; Akorn, Inc. Lake Forest, IL) and Xylazine (10 mg/kg body weight; Bimeda Inc., Le Sueur, MN) and immobilized on a small animal stereotactic apparatus equipped with a Cunningham mouse adapter (Stoelting Co., Wood Dale, IL). Skin was sterilized using Betadine solution (Stamford, CT) and subcutaneous injection of the analgesic bupivacaine (Hospira, Inc. Lake Forest, IL); [1–2 mg/kg (0.4–0.8 ml/kg of a 0.25% solution)] was administered in the head area prior to incision to minimize pain. The skin and the underlying tissues were exposed to reflect the reference structures (sagittal and coronal plane) on the skull. The sagittal plane was attuned so that bregma and lambda were on the same coordinates on the vertical plane. A burr hole was drilled at pre-determined co-ordinates (AP = 0 mm, ML = 2.0 mm from bregma, and DV = 3.0 from skull surface) to access the left striatum. Animals were injected with HIV-VLPs into the striatum using a 10 μl Hamilton syringe fitted to a 27 G needle over a period of 5 min. The needle was retracted slowly with caution and the hole was sealed with sterile bone-wax (Guaynabo, Puerto Rico). The animal was then removed from the stereotactic instrument and the incision was closed with FS-2 needle attached to the 4.0 silk sutures (Ethicon, Somerville, NJ).
2.7 |. Isolation of brain leukocytes
Brain mononuclear cells (BMNCS) were isolated from brain tissue of rAd5-p24 prime-CNS boost animals using a previously described procedure with minor modifications (Cheeran et al., 2007; Marten, Stohlman, Zhou, & Bergmann, 2003; Mutnal, Hu, Little, & Lokensgard, 2011). In brief, whole brain was isolated and minced finely using a scalpel in DMEM/5%FBS and digested in 0.0625% trypsin (in Ca/Mg-free HBSS) at room temperature for 20–30 min. Cells were then washed in HBSS and re-suspended in 30% Percoll (Sigma-Aldrich, St. Louis, MO). This was followed by underlaying 70% Percoll and banding the cells by centrifugation at 900g for 30 min at 15°C. Brain leukocytes were carefully collected from the 30–70% Percoll interface and washed in DMEM/5% FBS.
2.8 |. Primary murine microglial cell cultures
Murine cerebral cortical cells from 1-day-old mice were dissociated after a 30 min trypsinization (0.25%) and plated in 75-cm2 Falcon culture flasks in DMEM containing 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), gentamicin (50 μg/ml), and Fungizone® (250 pg/ml). The medium was replenished after 1 and 4 days of plating. On d 12 of culture, floating microglial cells were harvested and plated onto 6-well tissue culture plates and incubated at 37°C. Purified microglial cells were stained >95% positively with Iba-1 Abs and <2% stained positively with Abs specific to glial fibrillary acidic protein (GFAP) (phenotypic marker of astrocytes).
2.9 |. In vivo CTL assays using peptide-pulsed splenocytes
An in vivo CTL assay was performed similarly to previously described studies (Barber, Wherry, & Ahmed, 2003; Clemente, Dominguez, Vieira, Rodrigues, & Amarante-Mendes, 2013; Fiege, Beura, Burbach, & Shimizu, 2016). Mice (either BALB/c or C57BL/6) were primed with rAd5-p24 intravenously to generate the effector cell population. After 7 days, bulk splenocytes were isolated from naïve animals and were used as targets. These cells were labeled with an intravital dye, CFSE (Invitrogen, Carlsbad, CA). Briefly, cells were suspended in PBS with 5% FBS at 2 × 107 cells/ml. CFSE was diluted in PBS to make 2 concentrations; CFSE-hi (10 μM) and CFSE-lo (1 μM) and then was added to the cells in 1:1 ratio [5 μM (hi) or 0.5 μM (lo) final concentrations]. The cells were incubated at 37°C for 20 min. This was followed by washing the cells twice with PBS/2% FBS. An aliquot of CFSE-lo labeled cells was then pulsed with 1 μg/ml of peptide [either AI9 (AMQMLKETI) or YI9 (YSPTSILDI)] for 30 min at 37°C. Cells were washed twice with PBS/2% FBS before mixing 1:1 with CFSE-hi labeled cells. CFSE mixed populations of peptide-pulsed or unpulsed cells were injected intravenously into rAD5-p24 primed animals. Splenocytes were harvested after 16–18 hr and the CFSE-hi/CFSE-lo ratio was determined by flow cytometry. Cells were acquired on a LSRII H4760 (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (FlowJo, Ashland, OR). Specific killing was calculated by [1 − (control group ratio/experimental group ratio)] × 100.
2.10 |. In vitro microglial cell cytotoxicity assays using OT-1 mice
OT-1 mice were primed with rAd5-OVA (1 × 1010 PFU/mouse) intravenously to generate an immune response against ovalbumin. Seven days later, splenocytes were harvested and CD8+ T-cells were isolated using the MagCellect Mouse CD8+ T-Cell Isolation Kit (R&D Systems, Minneapolis, MN). Purified CD8+ T-cells were treated as effector cells. Microglial cells from C57BL/6 and PD-L1 KO animals were labeled with CFSE-hi and CFSE-lo concentrations as described in previous section. The CFSE-lo labeled microglial cells were either unpulsed or pulsed with 1 μg/ml of SIINFEKL peptide. The cells were washed and mixed with CFSE-hi labeled cells. CFSE-labeled and peptide pulsed microglial cells from C57BL/6 animals were also treated with 10 μg/ml and 30 μg/ml of α-PD-L1 (clone M1H5; eBioscience, San Diego, CA) and rat IgG2a isotype control (eBioscience). This was followed by co-culture of these labeled microglial cells with CD8+ T-cells in different effector to target ratios (10:1, 3:1, 1:1) for 16–18 hr. Cells were then harvested and the CFSE-hi/CFSE-lo ratio as well as PD-L1 expression (using anti-PD-L1-PEcy7, eBioscience) was determined by flow cytometry. Specific killing was calculated as described previously. The co-culture was also stained using rabbit antibody to Iba-1 (2 μg/ml; Wako Chemicals, Richmond, VA) and rat anti-mouse CD8 (10 μg/ml; R&D Systems Inc., Minneapolis, MN). For fluorescent detection, cells were incubated with NL557-conjugated goat anti-rabbit or NL557-conjugated goat anti-rat Ab (R&D Systems) followed by nuclear labeling with Hoechst 33342 (1 μg/ml; Chemicon, Temecula, CA), and viewed under a fluorescent microscope.
2.11 |. In vivo CTL assays in brain using peptide-pulsed microglial cells
C57BL/6 and BALB/c mice were primed with rAd5-p24 and CNS-boosted with HIV-VLPs. After 30 days, C57BL/6 animals were injected intracranially with microglia from C57BL/6 and PD-L1 KO animals-labeled with CFSE and pulsed with YI9 peptide (1 μg/ml), while the BALB/c animals were injected with microglial cells from BALB/c mice labeled with CFSE and pulsed with AI9 (1 μg/ml) ± α-PD-L1 (10 μg/ml; M1H5 clone; eBioscience). The CFSE labeling and peptide pulse was carried out as described in the previous section. BMNCs were isolated after 72 hr and the CFSE-hi/CFSE-lo ratio was determined by flow cytometry. Percent CTL activity was calculated using the same formula as described before.
2.12 |. Immunohistochemistry
Brains were harvested from rAd5-p24 prime-CNS boost C57BL/6 mice injected with microglial cells from EGFP-expressing mice loaded with YI9 peptide. Animals were perfused with 2% sodium nitrate and phosphate-buffered saline (PBS) and prefixed with 4% paraformaldehyde. Brains were then submerged in 4% paraformaldehyde for 24 hr and transferred to 25% sucrose solution for 2 days prior to sectioning. After blocking (PBS with 10% normal goat serum and 0.3% Triton X-100) for 1 hr at RT, brain sections (30 μm) were incubated overnight at 4°C with rat anti-mouse CD8 antibody (10 μg/ml; R&D Systems Inc., Minneapolis, MN). Brain sections were washed four times with PBS. After washing, secondary antibody (Cy3-conjugated donkey anti-goat or donkey anti-rabbit Ab (Jackson Immunoresearch Labs), Alexa Fluor 488 conjugated donkey anti-mouse and/or Alexa Flour 546 conjugated donkey anti-rat antibodies (Molecular Probes) was added for 1 hr at RT followed by nuclear labeling with Hoechst 33342 (1 μg/ml; Chemicon, Temecula, CA) and viewed under a fluorescent microscope.
2.13 |. ELISA
BMNCs were isolated from BALB/c mice primed with rAd5-p24 and CNS-boosted with HIV-VLPs for 30 days. Cells were either left untreated or treated with α-PD-L1 neutralizing Ab (10 μg/ml; M1H5 clone; eBioscience) or rat IgG2a isotype control (10 μg/ml; eBioscience) for 1 hr. This was followed by addition of 100 μM AI9 in all the groups for 1–2 hr prior to the addition of microglial cells. Supernatants were collected after 72 hr and IFN-γ and IL-2 concentrations were measured by employing mouse IFN-γ or IL-2 ELISA kits according to the manufactures instructions (eBioscience).
2.14 |. Ex-vivo antigen stimulation and intracellular staining
BMNCs were isolated from rAd5-p24 prime-CNS boost BALB/c animals as described previously. Cells were stimulated with 100 μM of AI9 and treated with or without α-PD-1 (10 μg/ml; RMP1–14 clone; eBioscience) Ab for 4–6 hr at 37°C in DMEM supplemented with 10% FBS and Brefeldin A (1 μl/ml; eBioscience). Cells were then collected and surface stained for CD8-BV510 (Biolegend, San Diego, CA) and CD103-FITC (eBioscience) prior to permeabilization and fixation using Cytofix/cytoperm kit (eBioscience). Cells were then stained for Ki67-PE and IFN-γ-eFluor450 (eBioscience), according to the manufacturer’s protocol.
2.15 |. Statistical analyses
GraphPad Prism software was used to determine statistical significance (version 5.03; Graphpad Software, La Jolla, CA). For comparing groups, a Student two-tailed unpaired T-test was employed. A p value <.05 was considered significant.
3 |. RESULTS
3.1 |. In vivo CTL-mediated killing of AI9 and YI9 peptide-loaded target cells
To investigate CTL responses generated in rAd5-p24-primed animals, we first tested killing efficacy using AI9 and YI9 peptide-pulsed splenocytes by in vivo CTL as described by Clemente et al. (2013) (Figure 1a). These assays use flow cytometry to measure the amount of CFSE dye that is retained in target cells, which are not killed as shown in Figure S1. Splenocytes obtained from naïve animals were loaded with either AI9 (AMQMLKETI; Gag197–205) or YI9 (YSPTSILDI; Gag277–285) peptides (corresponding to previously identified H-2Kd or H-2Db MHC class I-restricted immunodominant HIV Gag epitopes, respectively), (Chowell et al., 2015; H. X. Tan et al., 2016). In these studies, MHC-matched splenocytes loaded with the AI9 peptide were found to be efficiently killed when injected into rAd5-p24 primed BALB/c (H-2Kd) mice (60.5 ± 6.2% CTL activity). However, as expected, when BALB /c splenocytes were pulsed with the YI9 peptide epitope, no killing of these targets was observed (Figure 1b,c). In contrast, when splenocytes from C57BL/6 (H-2Db) mice were pulsed with AI9, and injected into the tail vein of MHC-matched recipients, no CTL activity was observed. However, when these same cells were loaded with the YI9 peptide, very efficient killing was detected (89.7 ± 1.0% CTL activity), (Figure 1b,c).
FIGURE 1.
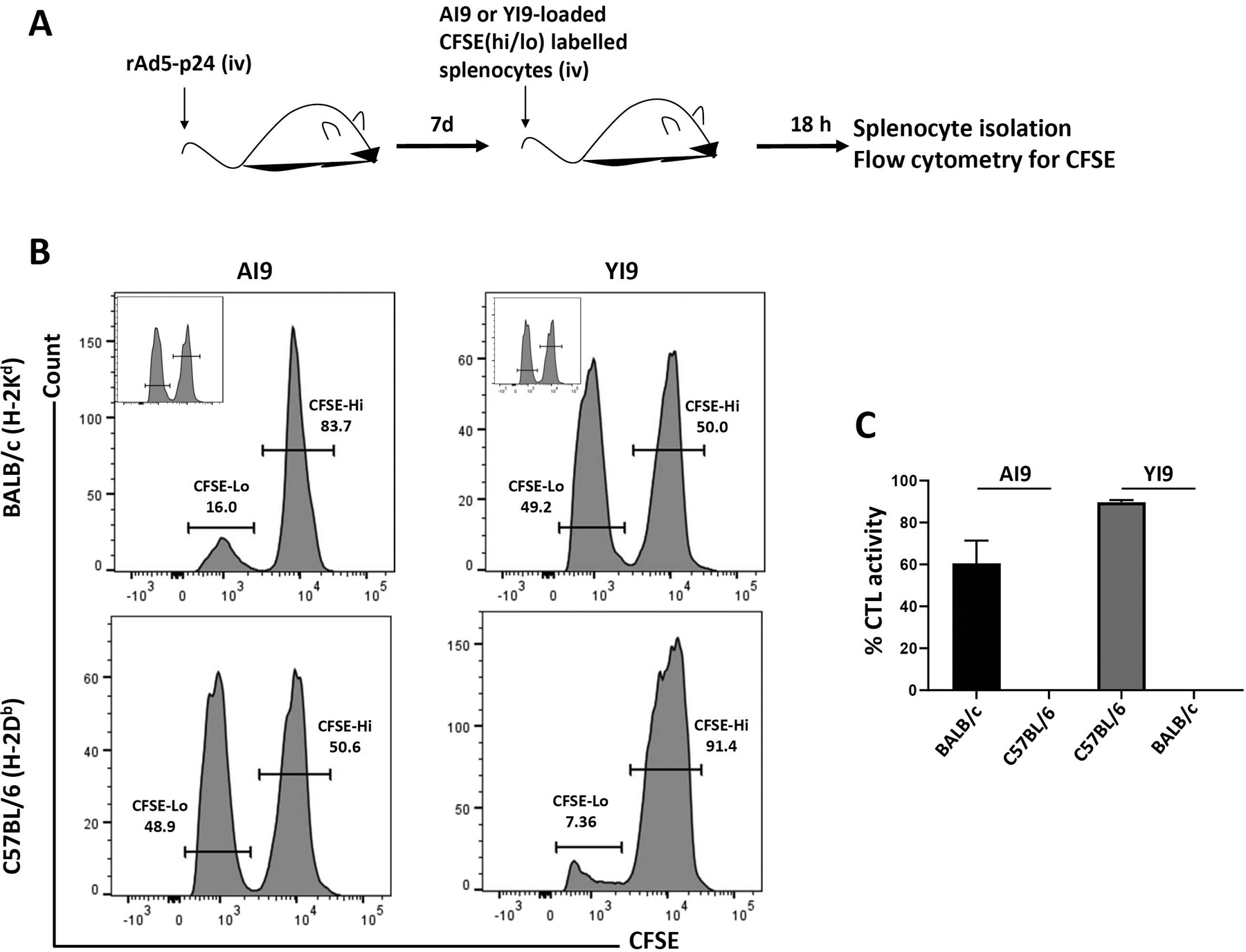
In vivo HIV-specific CTL-mediated killing of peptide-loaded splenocytes. (a) BALB/c and C57BL/6 mice were primed with rAd5-p24. Seven days later, in vivo cytotoxicity was determined 18 hr following intravenous injection of each mouse with syngeneic CFSE-labeled splenocytes pulsed with 1 μg/ml of the HIV-specific AI9 or YI9 peptide. (b) Representative histograms showing CFSE-labeled cells recovered from spleens of rAd5-p24 immunized hosts. Insets are the representative no peptide controls. (c) Graphical representation of % CTL activity [1 − (control group hi/lo ratio: experimental group hi/lo ratio)] × 100. Data shown are representative of three independent experiments
3.2 |. In vitro microglial cell cytotoxicity assays
To investigate killing using primary microglial cells as targets, we first employed in vitro CTL assays using a model antigen, ovalbumin (OVA). In these studies, primary murine microglial cells obtained from either WT or PD-L1 KO mice were first CFSE-labeled and then pulsed with the SIINFEKL (OVA257–264) peptide. CD8+ T-cells were obtained from OT-1 T-cell receptor transgenic mice which had been primed 7 days previously with a recombinant adenovirus vector expressing ovalbumin (rAd5-OVA), (Hogquist et al., 1994). These ovalbumin-specific CD8+ T-cells were then mixed with the SIINFEKL-pulsed primary microglia at 1:1, 3:1, and 10:1 effector: target ratios. The co-cultures were incubated for 18 hr and CTL activity was assessed using flow cytometry. When microglial cells from WT animals were used in these assays, the CD8+ T-cells were found to kill these peptide-loaded brain cells (Figure 2a). We also visually demonstrated aggregation of CD8+ T- cells from OT-1 T-cell receptor transgenic mice with primary microglial cells in the presence of SL8 peptide (Figure S2a). The level of CTL activity observed was associated with induction of PD-L1 (26.3 ± 7.8%) on the microglia following 18 hr of being co-cultured in the presence of activated CD8+ T-cells, which was not seen on PD-L1 KO microglia with or without CD8+ T-cell co-culture (Figure 2b and Figure S3). Interestingly, when microglia from PD-L1 KO animals were used in these assays, significantly more microglial cell killing (34.9 ± 0.7%) than WT cells (25.6 ± 1.8%) was observed after 18 hr at the 10:1 effector: target ratio (Figure 2a,c). CTL-mediated killing of microglial cells either from WT or PD-L1 KO animals was also found to be dependent on the effector to target ratio (Figure 2c).
FIGURE 2.
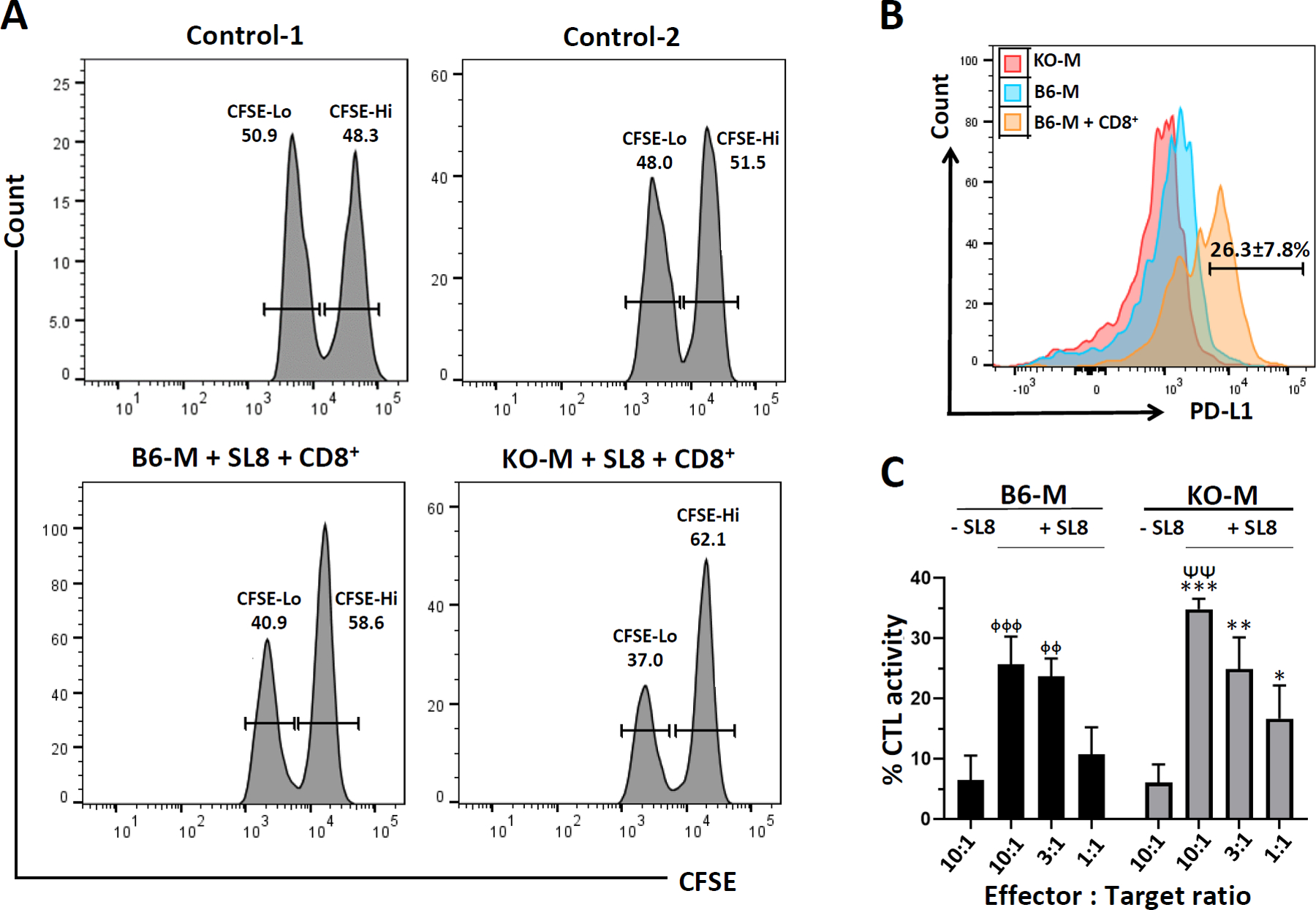
In vitro microglial cell cytotoxicity assays. CD8+ T-cells were isolated from the spleens of rAd5-OVA-primed OT-1 mice using a negative selection kit. CD8+ T-cells were then transferred to microglial cell cultures (10:1, 3:1, and 1:1 effector: target ratios) from WT C57BL/6 (B6-M) or PD-L1 KO (KO-M) loaded with ovalbumin-specific SIINFEKL peptide (SL8) and labeled with CFSE. in vitro cytotoxicity was determined after 18 hr. (a) Representative histograms show CFSE labeled microglia harvested from the plate after 18 hr of co-culture with OT-1 CD8+ T-cells (10:1 effector: target ratio). Control-1 is the CFSE staining control representative of either B6-M or KO-M. Control-2 is the representative histogram of either B6-M or KO-M without peptide. (b) Histogram overlay showing expression of PD-L1 on microglia from PD-L1 KO (KO-M), C57BL/6 (B6-M), and C57BL/6 animals after co-culture with CD8+ T-cells (B6-M + CD8+). The experiment was performed twice in triplicate wells and data are expressed as mean ± SE on the representative histogram. (c) Graphical representation of % CTL activity [1 − (control group hi/lo ratio: experimental group hi/lo ratio)] × 100. p < .01 compared to B6-M + SL8 at 10:1 effector: target ratio. ᶲᶲp < .01 and ᶲᶲᶲp < .001 compared to B6-M − SL8. **p < .01 and ***p < .01 compared to KO-M-SL8. Data shown are representative of three experiments
CFSE-labeled, peptide-loaded microglial cells from WT mice were also treated with neutralizing α-PD-L1 Ab prior to co-culture with CD8+ T-cells for 18 hr. In the untreated and IgG-treated control group, 27.4 ± 1.4% and 26.7 ± 1.4% killing of microglial cells was observed, respectively (Figure 3a). This killing was significantly increased when the PD1: PD-L1 pathway was blocked using 10 μg/ml (33.1 ± 0.8%), or 30 μg/ml (42.7 ± 1.0%), of α-PD-L1 Ab (Figure 3b).
FIGURE 3.
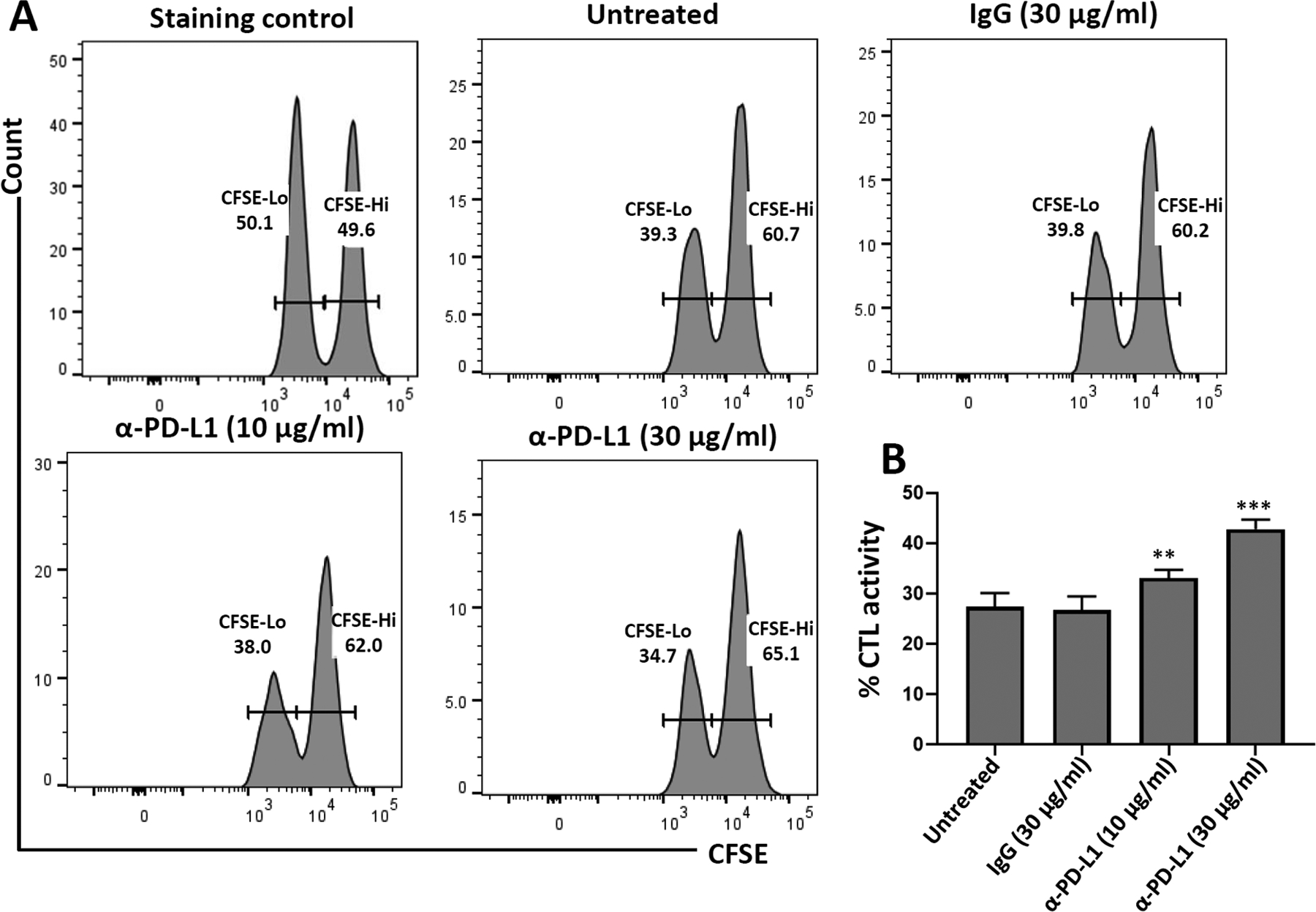
In vitro microglial cell cytotoxicity assays using neutralizing Ab against PD-L1. CD8+ T-cells were isolated from spleens of rAd5-OVA-primed OT-1 mice using a negative selection kit. CD8+ T-cells were then transferred to microglial cells (10:1 effector: target ratio) from C57BL/6 mice loaded with ovalbumin-specific peptide, labeled with CFSE, and either left untreated or treated with IgG control (30 μg/ml) or α-PD-L1 neutralizing Ab (10 μg/ml and 30 μg/ml). in vitro cytotoxicity was determined after 18 hr. (a) Representative histograms show CFSE-labeled microglia harvested from the plate after 18 hr of co-culture with CD8+ T-cells. (b) Graphical representation of % CTL activity [1 − (control group hi/lo ratio: experimental group hi/lo ratio)] × 100. **p < .01; ***p ≤ .001 compared to IgG control. Data shown are representative of three independent experiments
3.3 |. In vivo CTL-mediated killing of HIV-1 YI9 peptide-loaded C57BL/6 and PD-L1 KO microglial cells within the brain
We went on to investigate CTL killing within the unique microenvironment of the brain by combining the in vivo assay presented in Figure 1 with the in vitro assay using microglia presented in Figure 2. For these experiments, primary microglial cells from WT C57BL/6 and PD-L1 KO animals were labeled with CFSE, loaded with the YI9 peptide, and transferred into the brains of MHC-matched animals which had received rAd5-p24 priming along with HIV-VLP CNS-boost 30 days prior to stereotactic injection (Figure 4a). Using this approach, minimal peptide-specific CTL killing of microglial cells was detected within the brains of bTRM-populated, WT C57BL/6 animals after 72 hr (3.2 ± 1.8% vs. 5.1 ± 1.6% without peptide), (Figure 4b). In contrast, CTL killing of microglia increased to 29.6 ± 4.9% when YI9-loaded microglial cells from PD-L1 KO animals were used as targets (Figure 4c).
FIGURE 4.
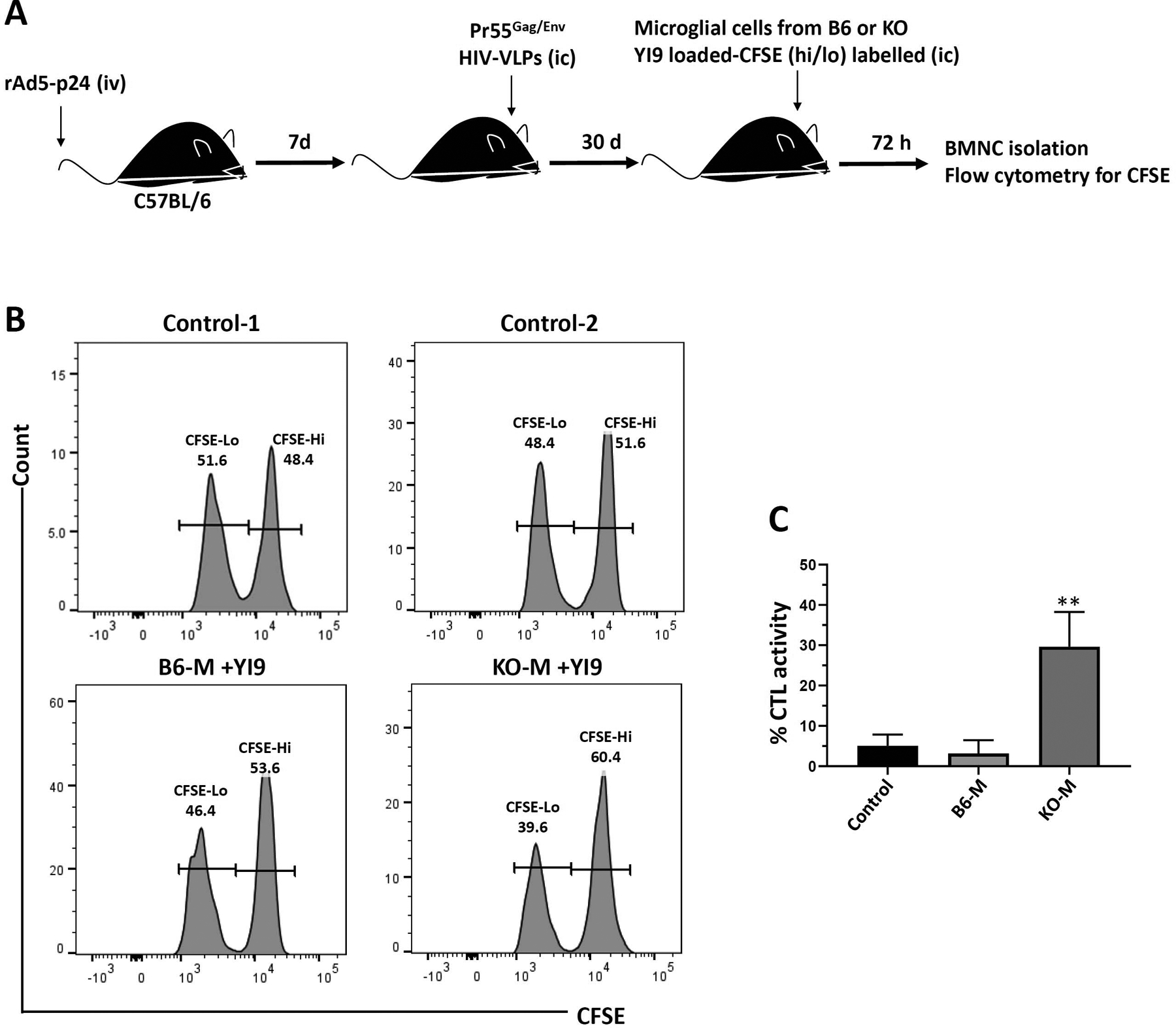
In vivo CTL killing of YI9 peptide-loaded C57BL/6 and PD-L1 KO microglial cells within the brain. (a) C57BL/6 mice were primed via tail-vein injection with rAd5-p24 and boosted in the CNS with HIV-VLPs for 30 days. in vivo brain cytotoxicity was determined 72 hr after intracranial (i.c.) injection of syngenic CFSE-labeled primary microglial cells (0.5 × 106 cells) from either C57BL/6 (B6-M) or PD-L1 KO (KO-M) animals pulsed with or without 1 μg/ml of Gag-specific YI9 peptide. (b) Representative histograms showing CFSE-labeled microglia recovered from the brains of rAd5-p24 primed-CNS boosted hosts. Control-1 is the CFSE staining control representative of either B6-M or KO-M. Control-2 is the representative histogram of either B6-M or KO-M without YI9. (c) Graphical representation of percent specific killing [1 − (control group hi/lo ratio: experimental group hi/lo ratio)] × 100. **p ≤ .01 compared to B6-M as well as the control (B6-M without YI9). Data shown are representative of three experiments using n = 3 animals/group
3.4 |. Effect of α-PD-L1 treatment on in vivo CTL-mediated killing of HIV-1 AI9 peptide-loaded BALB/c microglial cells within the brain
AI9-loaded BALB/c microglia were treated with α-PD-L1 Abs prior to being injected into the brains of bTRM-populated, MHC-matched recipients (Figure 5a). No CTL-mediated killing of microglial cells was observed within the brain when non-Ab-treated target cells or cells treated with isotype control Abs were used (Figure 5b). However, when peptide-loaded microglial cells were treated with α-PD-L1 Abs prior to injection into the brain, CTL activity increased to 17 ± 4.7% (Figure 5c).
FIGURE 5.
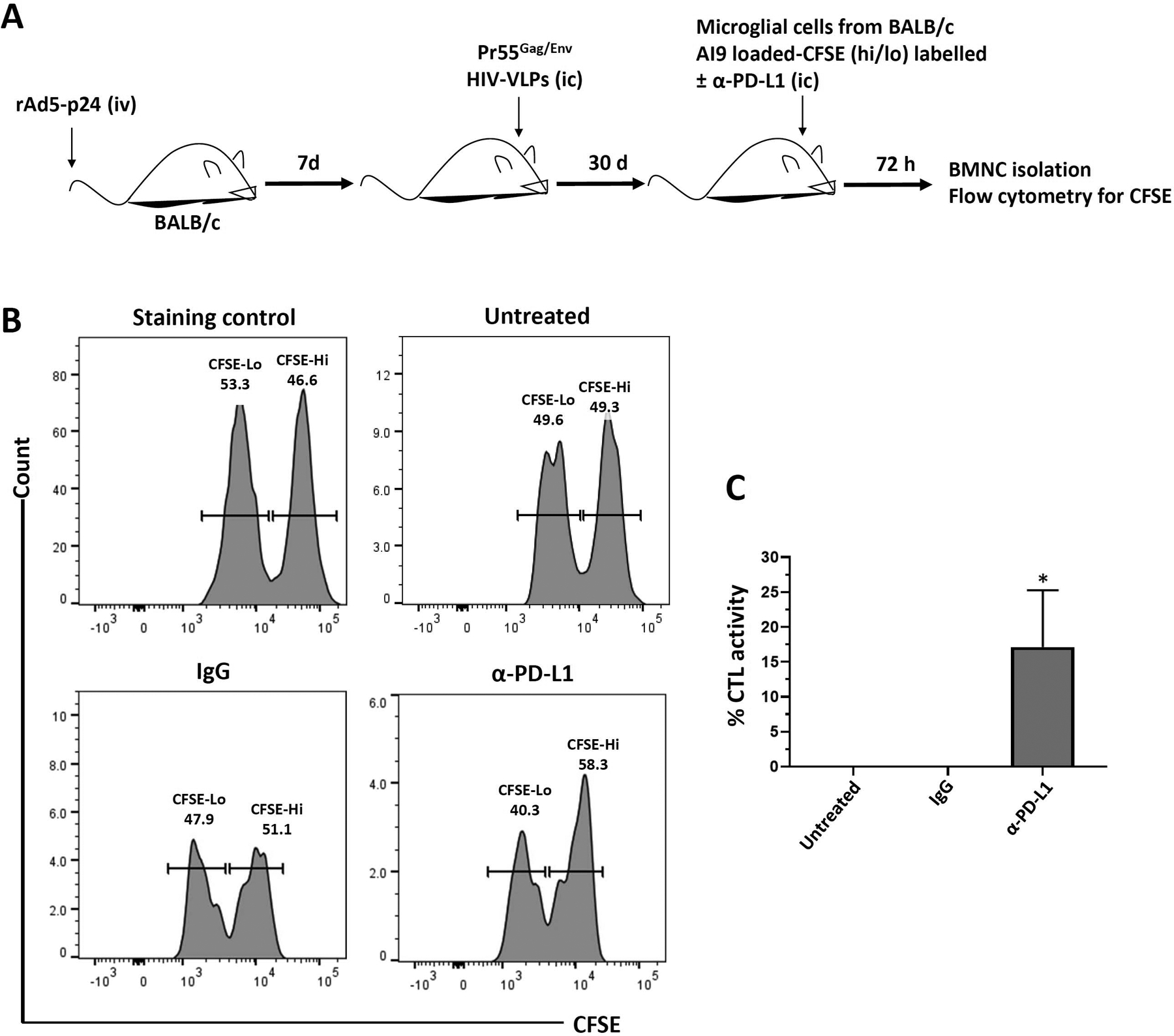
Effect of α-PD-L1 Ab treatment on in vivo CTL killing of AI9 peptide-loaded BALB/c microglia within the brain. (a) BALB/c mice were primed with rAd5-p24 and boosted in the CNS with HIV-VLPs for 30 days. in vivo cytotoxicity was determined after 72 hr of injecting (i.c.) each mouse with syngenic CFSE-labeled primary microglial cells (0.5 × 106 cells) pulsed with or without 1 μg/ml of Gag-specific AI9 peptide and left untreated or treated with either IgG or neutralizing α-PD-L1 Ab. (b) Representative histograms showing CFSE-labeled microglial cells recovered from the brains of rAd5-p24 primed-CNS boosted hosts. (c) Graphical representation of percent specific killing [1 − (control group hi/lo ratio: experimental group hi/lo ratio)] × 100. *p ≤ .05 compared to IgG or untreated control. Data shown are representative of three experiments (n = 3 animals/group)
3.5 |. Blocking the PD-1: PD-L1 Pathway enhances non-cytolytic responses of bTRM to re-stimulation in microglial cell co-cultures
Finally, using BMNCs from rAd5-p24 primed-CNS boost mice, we investigated the effects of blocking the PD-1: PD-L1 pathway on the non-cytolytic responses of bTRM themselves. Brain cells were activated by 100 μM of CD8-specific HIV-p24 AI9 peptide. Prior to the addition of peptide, the cultures were treated with α-PD-L1 neutralizing Ab, control rat IgG isotype, or left untreated. Supernatants were collected after 72 hr and analyzed for IFN-γ and IL-2 levels using ELISA. As expected, α-PD-L1-treated brain cells displayed a significant increase in IFN-γ (p < .01) (Figure 6a) and IL-2 (p < .01) production (Figure 6b).
FIGURE 6.
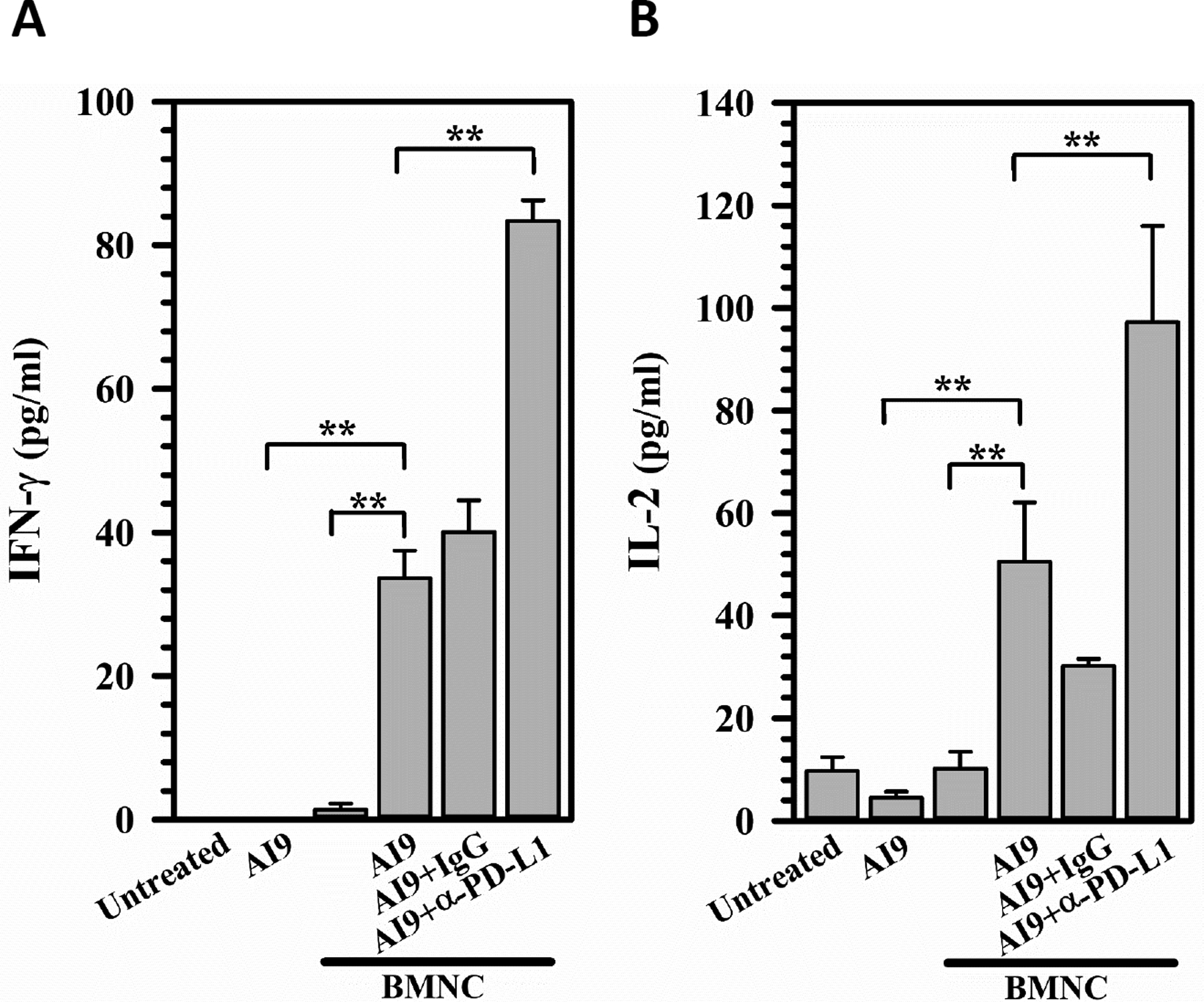
Blocking the PD-1: PD-L1 pathway enhances non-cytolytic responses of bTRM to re-stimulation in microglial cell co-cultures. BMNCs were isolated from the brains of rAd5-p24 primed-CNS boost mice at 30 days post prime-boost and cultured (1.3 × 105 cells/well) in the presence of 100 μM CD8+ T-cells-specific HIV AI9 peptide. Prior to the addition of peptide, the cultures were treated with PD-L1 neutralizing Ab (α-PD-L1), control rat IgG2a isotype (IgG) or left untreated. Supernatants were collected after 72 hr in culture with microglial cells and analyzed for IFN-γ (a) and IL-2 (b) levels using ELISA (pg/ml). Data are representative of two experiments (n = 5 animals/experiment). **p ≤ .01
3.6 |. Enhanced ex vivo bTRM recall responses following α-PD-1 treatment
To investigate the effect of blocking PD-1 on the response of bTRM to restimulation with AI9 peptide, we evaluated IFN-γ as well as Ki67 (a proliferation marker) production. Analysis was carried out using intracellular staining and flow cytometry following ex-vivo treatment with α-PD1 and stimulation with AI9 peptide. bTRM phenotyped as CD8+CD103+ T-cells were found to produce IFN-γ following AI9 stimulation (21.6 ± 1.0%). However, significantly higher cell frequencies of bTRM produced it following α-PD-1 treatment (32.3 ± 1.5%), when compared with untreated control (Figure 7a). Similarly, Ki67-expressing bTRM cells were also detected in a significantly larger proportion in the α-PD-1-treated group (35.7 ± 3.0%) when compared with untreated group (21.5 ± 0.7%) following AI9 restimulation (Figure 7b).
FIGURE 7.
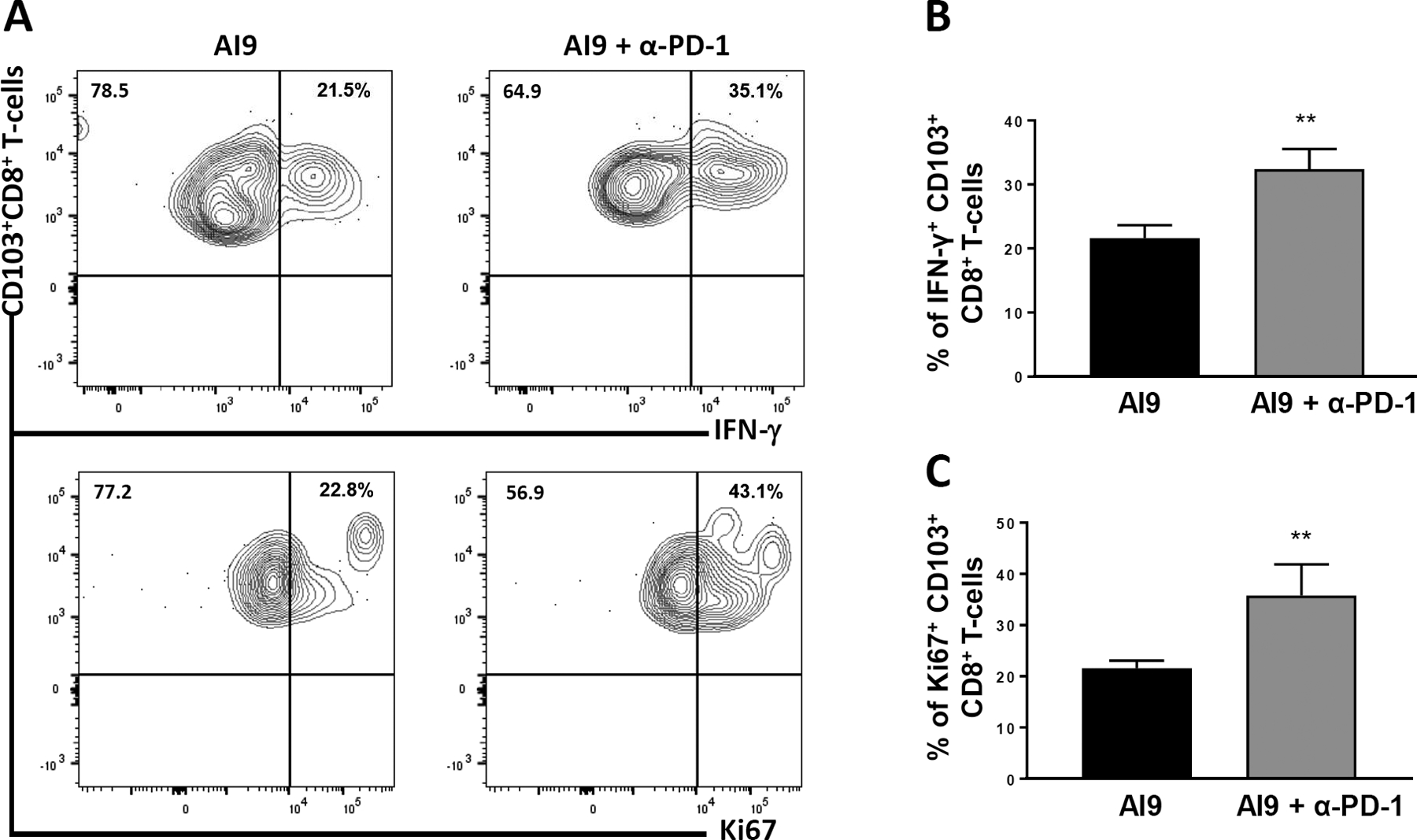
Enhanced ex vivo bTRM recall responses following α-PD-1 Ab treatment. BMNCs were obtained from BALB/c animals at 30 days post prime-CNS boost and were stimulated ex vivo with AI9 peptide. The cells were then left untreated or treated with α-PD-1 for 4–6 hr prior to intracellular staining. (a) Representative contour plots present expression of IFN-γ and Ki67 by CD103+CD8+ T-cells. (b,c) Bar graphs of pooled data show frequencies of IFN-γ expression, as well as Ki67 production, by CD103+CD8+ T-cells. Data shown are representative of three experiments (n = 3 animals/group). **p ≤ .01
4 |. DISCUSSION
A primary goal of contemporary HIV-1 research is to identify approaches to effect a functional cure, where viral load is completely suppressed for extended periods in the absence of antiretroviral therapy (Henderson, Reoma, Kovacs, & Nath, 2020). HIV-specific CD8+ T-cell responses are vital in suppressing acute viral infection within the brain, but eventually fail in their ability to completely eradicate the virus from infected microglia. Ongoing viral transcription, protein production, and replication despite virologic suppression in the CSF likely contribute to the development of HAND (Saylor, 2018). Although patients on successful combination antiretroviral therapy (cART) are viremically suppressed both in CSF and plasma, there exists a persistent HIV-1 reservoir within the CNS, that is believed to contribute to the inability to completely cure the viral infection (Chen, Gill, & Kolson, 2014; Dahl et al., 2014; Stam, Nijhuis, van den Bergh, & Wensing, 2013). Immunotherapeutic approaches using negative checkpoint inhibitors have shown substantial improvements in the treatment of various cancers, but their efficacy against HIV brain infection remains to be determined. It is currently not known whether HIV-infected microglia are viable cellular targets for immune checkpoint blockade. Studies show that targeting the PD-1 pathway in HIV infection has potential to aid in clearance of persisting virus (Henderson et al., 2020; Schwartz et al., 2017; Velu, Shetty, Larsson, & Shankar, 2015). For these reasons, there is great interest in immunotherapeutic approaches to target the CNS reservoir of HIV infection and a phase 1 clinical trial is currently underway to determine whether pembrolizumab is safe and tolerable in this patient population (Nath (17-N-0145), n.d.).
In this study, we used our heterologous prime-CNS boost model in which adaptive CD8+ T-cell-mediated immune responses to HIV-1 p24 protein, including immunodominant peptide epitopes, are generated within murine brains. Mice are first primed with an adenovirus vector expressing the capsid protein followed by a CNS boost using HIV-VLPs, carrying M-tropic CCR5-utilizing Env protein. Recombinant adenovirus type 5 (rAd5) vaccines have long been a vector of choice for generating cellular, as well as humoral immunity (Hosseini Rouzbahani et al., 2016). The systemic immunity generated via adenovirus priming is further augmented by heterologous boosting of these animals by injection of HIV-VLPs into the brain. HIV-VLPs present viral antigens in an authentic configuration and induce strong innate immune responses (Glimcher, Townsend, Sullivan, & Lord, 2004; Russell & Ley, 2002). Following their intracranial injection, there is a production of cytokines and chemokines; which eventually drive peripheral immune cell infiltration into the brain. Using this heterologous prime-boost model, we have previously demonstrated that the murine brain becomes populated by both short-lived effector (SLEC) and memory precursor effector (MPEC) cells; with a population of bTRM persisting long-term within the brain (Prasad et al., 2019).
To investigate CTL responses in our rAd5-p24 primed animal model (both BALB/c and C57BL/6), we first tested killing efficacy using in vivo CTL assays against p24 peptide-loaded splenocytes. Anti-HIV CD8+ T-cells in the primed animals must distinguish peptide-specific from peptide non-specific to respond effectively against Ag. Hence, we determined the CTL efficacy generated in H-2Kd and H-2Db restricted mice against their corresponding immunodominant MHC class-I restricted epitope. AMQMLKETI (i.e., AI9; Gag197–205) has previously been identified as an H-2Kd-restricted peptide (H. X. Tan et al., 2016; H. X. Tan et al., 2018) and YSPTSILDI (i.e., YI9; Gag277–285) as an H-2Db-restricted epitope (Chowell et al., 2015) for the p24 capsid protein. Our results demonstrated efficient killing of AI9-loaded splenocytes in BALB/c (H-2Kd) mice, but not in C57BL/6 (H-2Db) animals. Correspondingly, YI9-loaded splenocytes were very efficiently killed in C57BL/6 mice, but not in BALB/c animals (Figure 1).
Splenocytes were found to be efficiently killed using our in vivo assays, but primary microglia are intrinsically more resistant to CTL-mediated killing. Twenty years ago, Bergmann et al. demonstrated that the relative insensitivity of these cells to CTL lysis results in viral CNS persistence (Bergmann, Yao, & Stohlman, 1999). Although very little work has been done investigating direct CTL activity against microglia, there have been a number of studies which demonstrate that HIV-infected macrophages are inherently resistant to CD8+ T cell-mediated killing, resulting in inefficient viral suppression (Clayton et al., 2018; Kelleher & Xu, 2018; Kumar & Herbein, 2014). Impaired killing of infected macrophages has been associated with their dependence on caspase-3, granzyme B activity, and their prolonged formation of synapses with the CTLs (Clayton et al., 2018). Moreover, infectious viral particles can assemble in the “virus containing compartment” within the macrophages where they are resistant to degradation by lysosome-associated reactive oxygen species and proteases (J. Tan & Sattentau, 2013). Further, in vivo HIV infection and the persistence of infected macrophages during cART suggest that macrophages contribute to viral pathogenesis (Honeycutt et al., 2017). Infected macrophages have been observed in the lungs, gut, lymph tissues of HIV-infected patients as well as in the brain, and the inefficient killing of these infected macrophages likely contributes to development of chronic inflammation associated with HAND and dementia (DiNapoli, Hirsch, & Brenchley, 2016). Similar observations have been made when macrophages present other viral antigens (e.g., cytomegalovirus, Epstein–Barr virus, and influenza virus) suggesting that they are less susceptible to CTL-mediated killing, even independent of HIV infection (Clayton et al., 2018).
To investigate immune-mediated killing of primary microglial target cells, we first employed in vitro CTL assays using a model antigen, ovalbumin, along with OT-1 mice. The transgenic T-cell receptor in these mice is designed to recognize an immunodominant ovalbumin peptide of residues 257–264 (SIINFEKL) in the context of H2Kb (Hogquist et al., 1994). In these experiments, we loaded SIINFEKL peptide onto CFSE-labeled primary microglial cells obtained from either C57BL/6 mice or PD-L1 KO animals, and co-cultured them along with CD8+ T-cells obtained from rAd5-OVA primed animals. It was evident from these studies that microglia from WT animals were somewhat resistant to killing, which was associated with induced PD-L1 expression following 18 hr in co-culture with T-cells (Figure 2). We have previously shown by qRT-PCR and flow cytometry that upon stimulation with IFN-γ, there is a significantly increased PD-L1 expression (Schachtele et al., 2014). However, this partial resistance to CTL killing was ablated if we used microglial cells from PD-L1 KO animals, in which co-culture did not induce PD-L1 (Figure 2). A similar observation was made when we used neutralizing Ab against PD-L1 to treat microglial cells from WT animals (Figure 3). Hence, these studies demonstrated that KO of PD-L1 or treatment with α-PD-L1 Abs increased CTL activity against microglia, indicating that these cells may be viable targets for negative checkpoint blockade therapy.
In addition to the microglial cells themselves, the unique microenvironment of the brain dictates the efficacy of CTL activity. Hence, we adapted the in vivo CTL assay to the brain using our prime-CNS boost model (in both BALB/c and C57BL/6 animals). As expected, when HIV-1 peptide (YI9) loaded microglial cells from C57BL/6 animals were injected into the prime-CNS boost animals, negligible killing by CTLs was detected (Figure 4). This resistance to killing can be attributed to PD-L1 expression as we have previously shown that upon in vivo AI9 stimulation of prime-CNS boost animals, PD-L1 is induced on approximately 75% of microglial cells (Prasad et al., 2019). Furthermore, when microglia from PD-L1 KO animals were loaded with HIV-1 peptide, we observed significantly increased killing (Figure 4). These data indicate that induced PD-L1 expression on microglial cells restrains their killing by CTLs. The same observation was confirmed in BALB/c animals, where microglial cells loaded with AI9 peptide were treated with neutralizing α-PD-L1 Ab; these cells exhibited significantly more killing compared to the control group (Figure 5). The increased CTL activity against microglia following blockade of the PD-1: PD-L1 pathway indicates that this approach may be effective in clearance of viral brain reservoirs. Further experiments demonstrated the presence of CD8+ T-cells within brain sections of rAd5-p24 prime-CNS boost C57BL/6 animals by immunohistochemistry (Figure S2b). When these animals were injected with microglial cells from EGFP-expressing mice (loaded with YI9 peptide), we observed CD8+ T-cells along with the green microglia (Figure S2b). It is possible that the target microglia, which have been cultured in vitro, may be somewhat different than infected endogenous microglia in terms of their MHC class I expression. We have previously reported that microglial cells in culture express constitutive MHC class I (44.1%), (Schachtele et al., 2014, GLIA). In this study, we carried out experiments to assess the expression levels of MHC I on microglia within the brains of rAd5-p24 primed-HIV-VLP boosted mice, and observed similar expression levels in rAd5-p24 primed-CNS boosted animals (42.23 ± 1.92% vs. 10.6 ± 0.60% in naive BALB/c), (Figure S4).
Finally, we investigated whether blocking the PD-1: PD-L1 pathway can enhance non-cytolytic responses of bTRM to re-stimulation using microglial cell: BMNC co-cultures. Our previous studies have demonstrated an immunoregulatory role for microglia in both in vivo and in vitro murine models (Chauhan, Hu, Sheng, Prasad, & Lokensgard, 2017; Schachtele et al., 2014). Expression of PD-L1 on glial cells is regulated by CD8+ T-cell activation. Blockade of PD-L1 on microglia or PD-1 on CD8+ T-cells using α-PD-L1 or α-PD-1 neutralizing Abs, respectively enhanced T-cell IFN-γ and IL-2 production (Schachtele et al., 2014). Similarly, in this study, we have demonstrated increased IFN-γ and IL-2 production following blockade of PD-L1 in peptide-stimulated microglia: BMNC co-cultures, as compared to IgG-treated or unstimulated cells (Figure 6). In addition, treatment of bTRM obtained from prime-CNS boost mice with α-PD-1 Abs enhanced their proliferation (47%) and IFN-γ production (40%) during ex vivo recall responses (Figure 7).
Data presented here demonstrate how the immunosuppressive, neuroprotective brain microenvironment intervenes with complete clearance of virus from microglial cell reservoirs. Correspondingly, we found that negative immune checkpoint blockade stimulates CTL activity against peptide-presenting microglia. Results from this study support the approach of using immune checkpoint inhibitors, along with other anti-retroviral therapies, to advance towards the functional cure of HIV infection. Besides HIV-1, microglial cells are also the primary target population for Zika virus infection, as demonstrated using fetal brain cells (Lum et al., 2017). So, it seems likely that negative checkpoint inhibition may also be relevant to clear Zika virus from brain reservoirs and impede virus-mediated pathogenesis.
However, the applicability of this approach within the CNS still remains questionable as the brain cannot endure prolonged neuroinflammation without bystander damage resulting in permanent neuronal injury. Off-target CNS effects such as recall responses, reactive gliosis, tissue-wide innate responses, and cumulative neurotoxicity may limit its use and will certainly need to be managed.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by grants from the National Institute of Mental Health (MH-066703) and the National Institute of Neurological Disorders and Stroke (NS-038836). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Jessica Fiege, Department of Microbiology, University of Minnesota, for valuable input regarding CTL assays.
Funding information
National Institute of Mental Health, Grant/Award Number: MH-066703; National Institute of Neurological Disorders and Stroke, Grant/Award Number: NS-038836
DATA AVAILABILITY STATEMENT
All of the supporting data are included in the manuscript.
REFERENCES
- Barber DL, Wherry EJ, & Ahmed R (2003). Cutting edge: Rapid in vivo killing by memory CD8 T cells. Journal of Immunology, 171(1), 27–31. 10.4049/jimmunol.171.1.27 [DOI] [PubMed] [Google Scholar]
- Bergmann CC, Yao Q, & Stohlman SA (1999). Microglia exhibit clonal variability in eliciting cytotoxic T lymphocyte responses independent of class I expression. Cellular Immunology, 198(1), 44–53. 10.1006/cimm.1999.1581 [DOI] [PubMed] [Google Scholar]
- Bhadra R, Gigley JP, & Khan IA (2012). PD-1-mediated attrition of polyfunctional memory CD8+ T cells in chronic toxoplasma infection. Journal of Infectious Diseases, 206(1), 125–134. 10.1093/infdis/jis304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan P, Hu S, Sheng WS, Prasad S, & Lokensgard JR (2017). Modulation of microglial cell Fcgamma receptor expression following viral brain infection. Scientific Reports, 7, 41889. 10.1038/srep41889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan P, & Lokensgard JR (2019). Glial cell expression of PD-L1. International Journal of Molecular Sciences, 20(7), 1677. 10.3390/ijms20071677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeran MC, Hu S, Palmquist JM, Bakken T, Gekker G, & Lokensgard JR (2007). Dysregulated interferon-gamma responses during lethal cytomegalovirus brain infection of IL-10-deficient mice. Virus Research, 130(1–2), 96–102. 10.1016/j.virusres.2007.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeran MC, Hu S, Sheng WS, Peterson PK, & Lokensgard JR (2003). CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10. Journal of Virology, 77(8), 4502–4515. 10.1128/jvi.77.8.4502-4515.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MF, Gill AJ, & Kolson DL (2014). Neuropathogenesis of HIV-associated neurocognitive disorders: Roles for immune activation, HIV blipping and viral tropism. Current Opinion in HIV and AIDS, 9(6), 559–564. 10.1097/COH.0000000000000105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowell D, Krishna S, Becker PD, Cocita C, Shu J, Tan X, … Anderson KS (2015). TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proceedings of the National Academy of Sciences of the United States America, 112(14), E1754–E1762. 10.1073/pnas.1500973112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton KL, Collins DR, Lengieza J, Ghebremichael M, Dotiwala F, Lieberman J, & Walker BD (2018). Resistance of HIV-infected macrophages to CD8(+) T lymphocyte-mediated killing drives activation of the immune system. Nat Immunology, 19(5), 475–486. 10.1038/s41590-018-0085-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente T, Dominguez MR, Vieira NJ, Rodrigues MM, & Amarante-Mendes GP (2013). In vivo assessment of specific cytotoxic T lymphocyte killing. Methods, 61(2), 105–109. 10.1016/j.ymeth.2013.02.007 [DOI] [PubMed] [Google Scholar]
- Cortese I, Muranski P, Enose-Akahata Y, Ha SK, Smith B, Monaco M, … Nath A (2019). Pembrolizumab treatment for progressive multifocal Leukoencephalopathy. New England Journal of Medicine, 380(17), 1597–1605. 10.1056/NEJMoa1815039 [DOI] [PubMed] [Google Scholar]
- Dahl V, Peterson J, Fuchs D, Gisslen M, Palmer S, & Price RW (2014). Low levels of HIV-1 RNA detected in the cerebrospinal fluid after up to 10 years of suppressive therapy are associated with local immune activation. Aids, 28(15), 2251–2258. 10.1097/QAD.0000000000000400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNapoli SR, Hirsch VM, & Brenchley JM (2016). Macrophages in progressive human immunodeficiency virus/simian immunodeficiency virus infections. Journal of Virology, 90(17), 7596–7606. 10.1128/JVI.00672-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiege JK, Beura LK, Burbach BJ, & Shimizu Y (2016). Adhesion- and degranulation-promoting adapter protein promotes CD8 T cell differentiation and resident memory formation and function during an acute infection. Journal of Immunology, 197(6), 2079–2089. 10.4049/jimmunol.1501805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman BB, Lisinicchia JG, Morgello S, Masliah E, Commins D, Achim CL, … Soukup VM (2013). Neurovirological correlation with HIV-associated neurocognitive disorders and encephalitis in a HAART-era cohort. Journal of Acquired Immune Deficiency Syndromes, 62(5), 487–495. 10.1097/QAI.0b013e31827f1bdb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH, Townsend MJ, Sullivan BM, & Lord GM (2004). Recent developments in the transcriptional regulation of cytolytic effector cells. Nature Reviews Immunology, 4(11), 900–911. 10.1038/nri1490 [DOI] [PubMed] [Google Scholar]
- Henderson LJ, Reoma LB, Kovacs JA, & Nath A (2020). Advances toward curing HIV-1 infection in tissue reservoirs. Journal of Virology, 94(3), e00375. 10.1128/JVI.00375-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, & Carbone FR (1994). T cell receptor antagonist peptides induce positive selection. Cell, 76(1), 17–27. 10.1016/0092-8674(94)90169-4 [DOI] [PubMed] [Google Scholar]
- Honeycutt JB, Thayer WO, Baker CE, Ribeiro RM, Lada SM, Cao Y, … Garcia JV (2017). HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nature Medicine, 23(5), 638–643. 10.1038/nm.4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini Rouzbahani N, Bayanolhagh S, Gholami M, Esmaeilzadeh A, Bayat Jozani Z, Mohraz M, & Pourfathollah AA (2016). Enhanced immune responses against HIV-1 with Adenovector (gag and tat) prime/protein boost regimen and GM-CSF injection. Iran Journal of Allergy, Asthma and Immunology, 15(5), 403–412. [PubMed] [Google Scholar]
- Kelleher P, & Xu XN (2018). Hard-to-kill macrophages lead to chronic inflammation in HIV. Nature Immunology, 19(5), 433–434. 10.1038/s41590-018-0089-z [DOI] [PubMed] [Google Scholar]
- Kerr WG, & Chisholm JD (2019). The next generation of immunotherapy for cancer: Small molecules could make big waves. Journal of Immunology, 202(1), 11–19. 10.4049/jimmunol.1800991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessing CF, Spudich S, Valcour V, Cartwright P, Chalermchai T, Fletcher JL, … Trautmann L (2017). High number of activated CD8+ T cells targeting HIV antigens are present in cerebrospinal fluid in acute HIV infection. Journal of Acquired Immune Deficiency Syndromes, 75(1), 108–117. 10.1097/QAI.0000000000001301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, & Kim WK (2019). Macrophages but not Astrocytes Harbor HIV DNA in the brains of HIV-1-infected Aviremic individuals on suppressive antiretroviral therapy. Journal of Neuroimmune Pharmacology, 14(1), 110–119. 10.1007/s11481-018-9809-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, & Herbein G (2014). The macrophage: A therapeutic target in HIV-1 infection. Molecular and Cellular Therapies, 2, 10. 10.1186/2052-8426-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers SL, Rose R, Maidji E, Agsalda-Garcia M, Nolan DJ, Fogel GB, … Singer EJ (2016). HIV DNA is frequently present within pathologic tissues evaluated at autopsy from combined antiretroviral therapy-treated patients with undetectable viral loads. Journal of Virology, 90(20), 8968–8983. 10.1128/JVI.00674-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, … Sharpe AH (2004). PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proceedings of the National Academy of Sciences of the United States of America, 101(29), 10691–10696. 10.1073/pnas.0307252101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum FM, Low DK, Fan Y, Tan JJ, Lee B, Chan JK, … Ng LF (2017). Zika virus infects human fetal brain microglia and induces inflammation. Clinical Infectious Diseases, 64(7), 914–920. 10.1093/cid/ciw878 [DOI] [PubMed] [Google Scholar]
- Marten NW, Stohlman SA, Zhou J, & Bergmann CC (2003). Kinetics of virus-specific CD8+ −T-cell expansion and trafficking following central nervous system infection. Journal of Virology, 77(4), 2775–2778. 10.1128/jvi.77.4.2775-2778.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockus TE, Ren HM, Shwetank, & Lukacher AE (2019). To go or stay: The development, benefit, and detriment of tissue-resident memory CD8 T cells during central nervous system viral infections. Viruses, 11(9), 842. 10.3390/v11090842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutnal MB, Hu S, Little MR, & Lokensgard JR (2011). Memory T cells persisting in the brain following MCMV infection induce long-term microglial activation via interferon-gamma. Journal of NeuroVirology, 17(5), 424–437. 10.1007/s13365-011-0042-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath, A. (17-N-0145). (n.d.). PD-1 Inhibition to Determine CNS Reservoir of HIV-Infection. National Institute of Neurological Disorders and Stroke (NINDS), Clinical Trials Number-NCT03239899. Retrieved from https://clinicalstudies.info.nih.gov/ProtocolDetails.aspx?A_2017-N-0145.html.
- Prasad S, Hu S, Sheng WS, Chauhan P, & Lokensgard JR (2018). Reactive glia promote development of CD103(+) CD69(+) CD8(+) T-cells through programmed cell death-ligand 1 (PD-L1). Immunity, Inflammation and Disease, 6(2), 332–344. 10.1002/iid3.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S, Hu S, Sheng WS, Chauhan P, & Lokensgard JR (2019). Recall responses from brain-resident memory CD8(+) T cells (bTRM) induce reactive gliosis. iScience, 20, 512–526. 10.1016/j.isci.2019.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S, Hu S, Sheng WS, Chauhan P, Singh A, & Lokensgard JR (2017). The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. Journal of Neuroinflammation, 14(1), 82. 10.1186/s12974-017-0860-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, … Masopust D (2019). Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nature Communications, 10(1), 567. 10.1038/s41467-019-08534-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JH, & Ley TJ (2002). Lymphocyte-mediated cytotoxicity. Annual Review of Immunology, 20, 323–370. 10.1146/annurev.immunol.20.100201.131730 [DOI] [PubMed] [Google Scholar]
- Saylor D (2018). Neurologic complications of human immunodeficiency virus infection. Continuum (Minneap Minn), 24(5, Neuro-infectious Disease), 1397–1421. 10.1212/CON.0000000000000647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtele SJ, Hu S, Sheng WS, Mutnal MB, & Lokensgard JR (2014). Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia, 62(10), 1582–1594. 10.1002/glia.22701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier RD, Hong S, Crescini M, Ellis R, Perez-Santiago J, Spina C, … Group H (2015). Cerebrospinal fluid (CSF) CD8+ T-cells that express interferon-gamma contribute to HIV associated neurocognitive disorders (HAND). PLoS One, 10(2), e0116526. 10.1371/journal.pone.0116526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz C, Bouchat S, Marban C, Gautier V, Van Lint C, Rohr O, & Le Douce V (2017). On the way to find a cure: Purging latent HIV-1 reservoirs. Biochemical Pharmacology, 146, 10–22. 10.1016/j.bcp.2017.07.001 [DOI] [PubMed] [Google Scholar]
- Shwetank Abdelsamed, H. A, Frost EL, Schmitz HM, Mockus TE, Youngblood BA, & Lukacher AE (2017). Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunology & Cell Biology, 95(10), 953–959. 10.1038/icb.2017.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shwetank Frost, E. L, Mockus TE, Ren HM, Toprak M, Lauver MD, … Lukacher AE (2019). PD-1 dynamically regulates inflammation and development of brain-resident memory CD8 T cells during persistent viral encephalitis. Frontiers in Immunology, 10, 783. 10.3389/fimmu.2019.00783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders J, Heutinck KM, Fransen NL, Remmerswaal EBM, Hombrink P, Ten Berge IJM, … Hamann J (2018). Tissue-resident memory T cells populate the human brain. Nature Communications, 9 (1), 4593. 10.1038/s41467-018-07053-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam AJ, Nijhuis M, van den Bergh WM, & Wensing AM (2013). Differential genotypic evolution of HIV-1 quasispecies in cerebrospinal fluid and plasma: A systematic review. AIDS Reviews, 15(3), 152–161. [PubMed] [Google Scholar]
- Szabo PA, Miron M, & Farber DL (2019). Location, location, location: Tissue resident memory T cells in mice and humans. Science Immunology, 4(34), eaas9673. 10.1126/sciimmunol.aas9673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HX, Gilbertson BP, Jegaskanda S, Alcantara S, Amarasena T, Stambas J, … De Rose R (2016). Recombinant influenza virus expressing HIV-1 p24 capsid protein induces mucosal HIV-specific CD8 T-cell responses. Vaccine, 34(9), 1172–1179. 10.1016/j.vaccine.2016.01.030 [DOI] [PubMed] [Google Scholar]
- Tan HX, Wheatley AK, Esterbauer R, Jegaskanda S, Glass JJ, Masopust D, … Kent SJ (2018). Induction of vaginal-resident HIV-specific CD8 T cells with mucosal prime-boost immunization. Mucosal Immunology, 11(3), 994–1007. 10.1038/mi.2017.89 [DOI] [PubMed] [Google Scholar]
- Tan J, & Sattentau QJ (2013). The HIV-1-containing macrophage compartment: A perfect cellular niche? Trends in Microbiology, 21(8), 405–412. 10.1016/j.tim.2013.05.001 [DOI] [PubMed] [Google Scholar]
- Tso FY, Kang G, Kwon EH, Julius P, Li Q, West JT, & Wood C (2018). Brain is a potential sanctuary for subtype C HIV-1 irrespective of ART treatment outcome. PLoS One, 13(7), e0201325. 10.1371/journal.pone.0201325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velu V, Shetty RD, Larsson M, & Shankar EM (2015). Role of PD-1 co-inhibitory pathway in HIV infection and potential therapeutic options. Retrovirology, 12, 14. 10.1186/s12977-015-0144-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim LM, Woodward-Davis A, & Bevan MJ (2010). Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proceedings of the National Academy of Sciences of the United States of America, 107(42), 17872–17879. 10.1073/pnas.1010201107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallet C, De Rovere M, Van Assche J, Daouad F, De Wit S, Gautier V, … Schwartz C (2019). Microglial cells: The Main HIV-1 reservoir in the brain. Frontiers in Cellular and Infection Microbiology, 9, 362. 10.3389/fcimb.2019.00362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Maxwell R, Xia Y, Cardarelli P, Oyasu M, Belcaid Z, … Lim M (2019). Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. Journal of Neuro-Oncology, 143(2), 241–249. 10.1007/s11060-019-03172-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the supporting data are included in the manuscript.