

Abstract
PTEN is a phosphatidylinositol-3,4,5-triphosphate (PIP3) phospholipid phosphatase that is commonly mutated or silenced in cancer. PTEN’s catalytic activity, cellular membrane localization, and stability are orchestrated by a cluster of C-terminal phosphorylation events on Ser380, Thr382, Thr383, and Ser385 but the molecular details for this multifaceted regulation have been uncertain. Here we use a combination of protein semisynthesis, biochemical analysis, NMR, X-ray crystallography, and computational simulations to obtain a detailed picture of how the phospho-C-tail interacts intramolecularly with PTEN’s C2 and phosphatase domains. A key element of this investigation involved characterizing the sea squirt homolog of PTEN, the voltage-sensing phosphatase (VSP), which was amenable to NMR mapping of the interactions of individual tail phospho-sites with particular VSP amino acid residues. What emerged from this multi-disciplinary structural analysis is an extended phosphorylated C-tail peptide that belts around much of VSP/PTEN and coats the phospholipid membrane binding interface and protrudes into the enzyme active site. This structural model provides a framework for how C-tail phosphorylation can comprehensively impact PTEN’s cellular functions.
PTEN catalyzes the conversion of the phospholipid, phosphatidylinositol-3,4,5-triphosphate (PIP3), to PIP2 (Fig. 1a) and is critical to governing cell growth and metabolic pathways in normal and disease states1,2. Loss of function mutations in PTEN are common in many types of cancer and PTEN is a major tumor suppressor gene 3–6. Elevated PIP3 levels are associated with activation of the Akt/mTOR cell proliferation/anti-apoptotic pathway and PTEN provides a brake to this signaling pathway7,8. Therapeutics that can target components in this signaling cascade are at various stages of preclinical and clinical development9,10.
Figure 1. Phosphorylation of PTEN influences PIP3 hydrolysis.
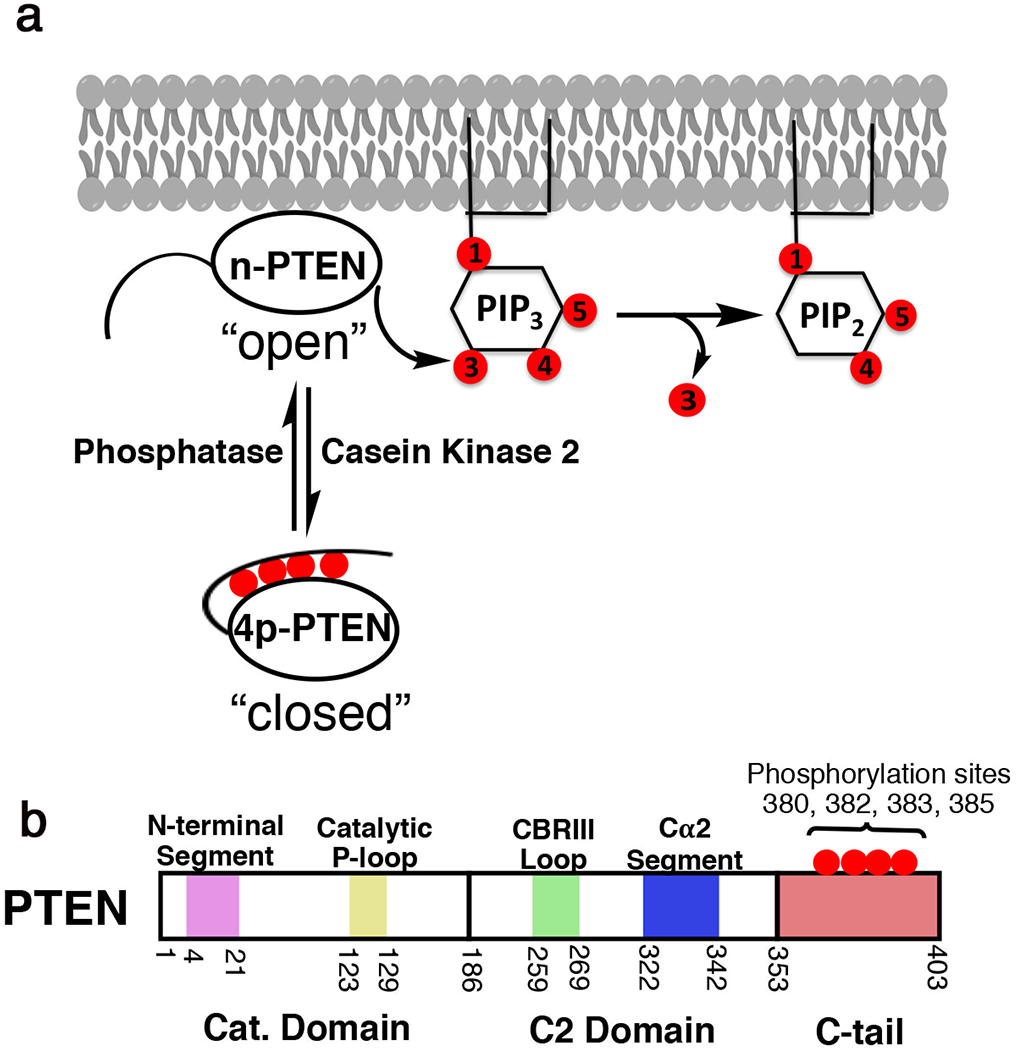
a, Phosphorylation of PTEN on Ser380, Thr383, Thr383, and Ser385 inhibits its enzymatic function towards PIP3 and reduces its plasma membrane binding. b, Schematic of PTEN domains and embedded regions important to function. Purple-N-terminal segment, yellow – catalytic P-loop, green – CBRIII loop*, blue - Cα2 segment*, red – 50 aa disordered C-tail. * represents regions previously identified to bind phosphorylated tail.
PTEN is a 403 amino acid polypeptide that contains an N-terminal catalytic (phosphatase) domain, a membrane-binding C2 domain, and terminates in a 50 amino acid disordered C-terminal tail (Fig. 1b).2 The N-terminal segment of the catalytic domain (aa 6-14) has also been implicated in mediating PTEN membrane association.11–13. PTEN’s physiological substrate PIP3 is embedded in the plasma membrane, and therefore PTEN’s catalytic action is in part dependent on membrane association1,2,14. A prior crystal structure of the core region of PTEN protein has revealed that the catalytic domain exists as a PTP (protein tyrosine phosphatase)-fold that closely abuts the C2 domain14. Although this structure has informed on PTEN biology over the past 20 years, a detailed portrait of the N-terminal segment and C-terminal tail has remained elusive.
In addition to mutation, PTEN function is silenced epigenetically as well as by post-translational modifications (PTMs). The most intensively studied PTEN PTMs are a cluster of four Ser/Thr phosphorylation sites, Ser380, Thr382, Thr383, and Ser385 in the C-terminal tail15. First discovered in 200015, these PTEN phosphorylations are catalyzed by casein kinase 216 and perhaps other kinases, are present in high stoichiometry and are relatively resistant to cellular protein phosphatases17–19. It is now clear that these phosphorylation events drive an intramolecular conformational change in which the PTEN C-tail folds over and interacts with the C2 and/or catalytic domain resulting in a closed state (Fig. 1)18,20–22. Phospho-PTEN dissociates from phospholipid membranes, shows reduced catalytic action, and is resistant to WWP2 E3 ligase-mediated ubiquitination15,17,18,23–28.
In spite of its central importance to PTEN regulation, the structural basis of C-terminal phosphorylation regulation of PTEN remains poorly understood. It has been established using expressed protein ligation29 that each of the four phosphates contributes in an additive fashion to enhance the affinity of the interaction between the phospho-C-tail and the PTEN body21,30. Hydrogen/Deuterium exchange mass spectrometry, site-directed mutagenesis, and photocrosslinking experiments have provided evidence that supports interactions of the phospho-tail with both the C2 and catalytic domains18,20–22,27. The predicted disorder of the phospho-C-tail has limited detailed structural analysis.
The voltage-sensing phosphatase (VSP) is a phospholipid hydrolase expressed in the sea squirt, C. intestinalis, and is a close homolog of PTEN31–33 that is structurally and functionally similar to PTEN34,35. In contrast to PTEN, VSP harbors a large N-terminal transmembrane segment, lacks PTEN’s C-terminal tail (Fig. S1a), and is not known to be regulated by Ser/Thr phosphorylation. In contrast to PTEN, where the recombinant expression can only be reliably produced in its functional state from insect cells, recombinant VSP can be efficiently generated from E. coli. Here we analyze PTEN and VSP using a range of biophysical and biochemical approaches to establish that VSP can faithfully recapitulate the binding interactions between the PTEN C-tail and VSP’s core. Moreover, we employ macromolecular NMR to map the interactions between the individual phosphates of the PTEN C-tail and the body of VSP and develop a detailed model of the tail-core interactions for VSP and PTEN.
RESULTS
Defining the spatial constraints for conformational closure of phospho-PTEN
In order to provide a deeper mechanistic insight into the regulation by PTEN’s phosphorylated C-terminal tail, we sought to measure the distance required for the tail to reach its autoinhibitory position in. tetraphosphorylated PTEN (4p-PTEN). In an effort to optimize the distance between the C-terminal phospho-cluster and the core of PTEN to maximize the energetics of its intramolecular interaction, we used expressed protein ligation to prepare a series of semisynthetic 4p-PTEN forms with artificial spacer lengths (Fig. 2a)18. These semisynthetic proteins are comprised of a crystallizable PTEN, aa 7-353, with an internally deleted D-loop aa 286-30914 henceforth referred to as crPTEN, followed by a variable flexible segment (spacer, 13aa to 22aa Gly/Thr/Ser residues) and culminating in a tetra-phosphorylated synthetic C-tail. The spacers are defined as all the residues following Glu353 until (p)Ser380 and will be described as Nsp where N denotes the spacer length and the tail is denoted as T1-T3 (Fig. 2b–c, Table S1) corresponding to its length and phosphorylation status. To prepare these PTEN proteins, the N-Cys synthetic peptide tails were ligated chemoselectively to the recombinant PTEN-spacer thioester fragment expressed in insect cells. Each of these four tetraphosphorylated proteins along with a control non-phosphorylated PTEN (n-crPTEN-13sp-T1) was purified and analyzed by size exclusion chromatography which revealed that they were all monomeric. The catalytic activities of these semisynthetic PTENs were assessed for their degree of autoinhibition associated with each spacer using a soluble PIP3 substrate. The phosphorylated tail with a 13 aa spacer 4p-crPTEN demonstrated nearly identical activity to the non-phosphorylated n-crPTEN form whereas longer spacer 4p-crPTENs showed increasing degrees of autoinhibition as the spacer length increased to 22 aa (Fig. 2b and c). These results suggest that 13 aa is insufficiently long to allow the phospho-peptide tail to engage the PTEN body but that the 16 aa tail and beyond can achieve varying levels of conformational closure (Fig. 2d). Based on the extended length of the spacer and tail residues, we estimate that a minimum of 65 Å is needed for the C-tail phosphates to access the sites on the PTEN core for a stable intramolecular interaction. The varying degrees of autoinhibition with the 16 aa – 22 aa spacers are likely the result of strain imposed on the tail from shorter spacers to reach its closed position. As the spacer length is increased, the phospho-tail may more easily sample its regulatory site leading to a higher occupancy and greater autoinhibition. Furthermore, the spacer in the native PTEN sequence appears to be well configured to occupy the autoinhibited conformation as its natural length (26 aa) resembles that of the most autoinhibited PTEN form (22 aa) tested here.
Figure 2. “Molecular ruler” to measure distance requirements for the tail to autoinhibit PTEN’s enzymatic function.
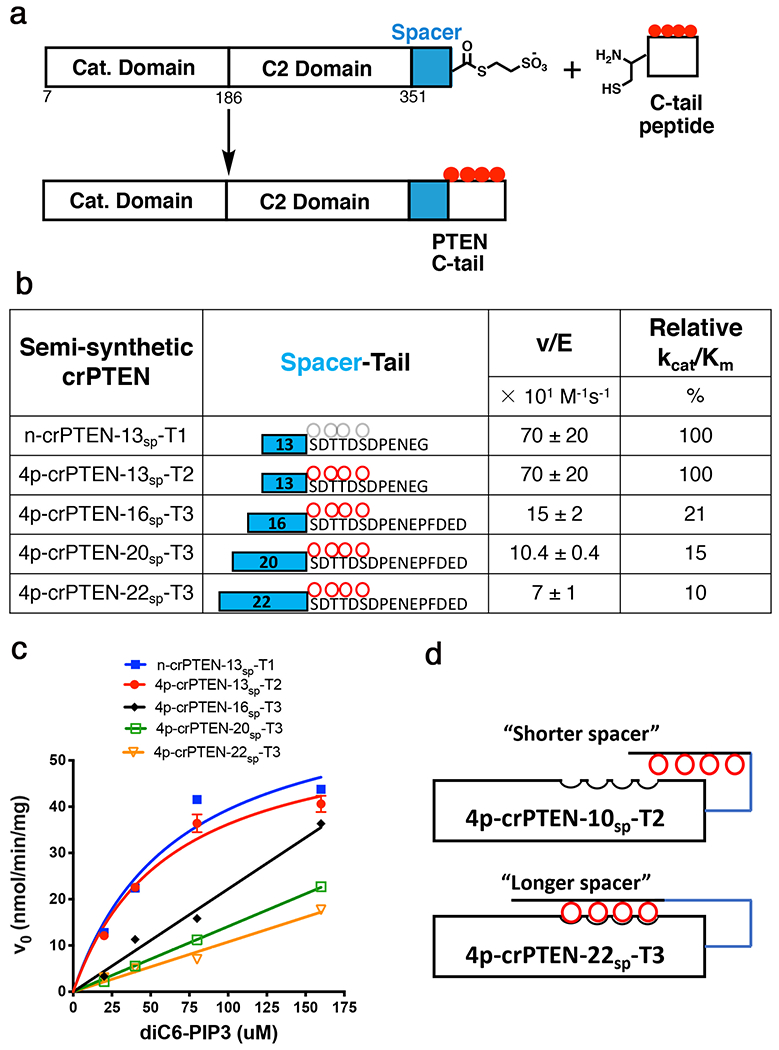
a, EPL scheme to generate semi-synthetic crPTEN proteins. b, Table of kinetic parameters for each crPTEN evaluated in this study. c, Kinetic plot showing change in initial velocity vs [diC6-PIP3] for all crPTEN protein evaluated in this study. d, Cartoon representing how the length of the spacer impacts the ability of the phosphates to reach their autoinhibited position. Blue line represents the “spacer”. Red circles are phosphorylated residues. Uncertainty reported as ± SEM, n=2.
X-ray crystallographic analysis of semisynthetic PTEN forms
We successfully crystallized and determined the X-ray structures of four semisynthetic PTEN forms which are deposited in the PDB database with the following PDB codes, names, and resolutions: 7JUL, n-crPTEN-13sp-T1, 2.25 Å; 7JUK, 4p-crPTEN-13sp-T2, 3.15 Å; 7JVX, 4p-crPTEN-20sp-T3, 3.2 Å, and 7JTX, 4p-crPTEN-22sp-T3, 3.05 Å (Fig. S2–3, Table S2). These structures were similar to the original PTEN structure with a rmsd of 0.49 Å over 303 Cα carbons14. The 13 aa spacer and 20 aa spacer PTENs displayed an additional N-terminal alpha-helix (aa7-14, Fig. 3a) that had not been observed previously but hypothesized to exist36. The helix neighbors the active site and is stabilized by a network of hydrogen bonds. Some key contacts that position this helix include Arg15 bridging of Asp22 to Ser10, Asp24 making a salt bridge with Lys13 and Arg159 that helps position residues in the catalytic P-loop (aa 123-129), and the hydrogen bond between Asn12 and Asp162 (Fig. 3b–c). Most likely, this helix is a dynamic element as suggested by the higher B factors (Fig. S3c–f) and the lack of density for this region in the original (PDB 1D5R) and our 22 aa spacer structures (Fig. S3f). Furthermore, the positioning of the basic residues of this region (Arg11, Lys13, Arg14, Arg15) extends a positively charged surface that surrounds the active site (Fig. S3a–b).
Figure 3. The N-terminal segment of PTEN folds as a helix.
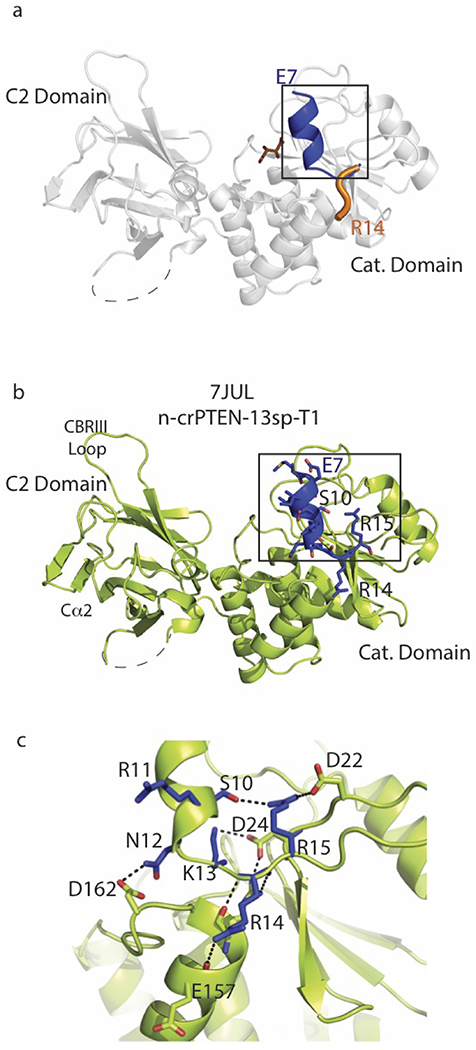
a, PTEN crystal structure (PDB ID 1D5R, aa 14-351) in grey ribbons. Black box displays the overlay of N-terminal PTEN 1D5R R14-Y16 residues folded as a loop in orange with N-terminal 4p-crPTEN-13sp-T1 (PDB ID 7JUL) E7-Y16 folded as a helix-loop in blue. Dashed line represents the D loop (aa 285-309) not present in the construct. b, 4p-crPTEN-13sp-T1 crystal structure (PDB ID 7JUL, aa 7-351) in green ribbons with the new observed N-terminal helix in navy blue. c, Zoom in of b displaying the interactions that position the N-terminal helix (aa 7-15) close to the active site. Residues from N-terminal helix in blue sticks with hydrogen bonding distance as dashed lines.
Notably, the diffraction of 20-spacer and 22-spacer PTEN forms show no electron density for the phospho-tail (Fig. S3f), even though the enzymatic data suggest that they exist principally in closed conformations. This is not surprising because the conditions needed to crystallize the PTEN forms under study invariably included very high ionic strengths (>1 M salt) in contrast to the PIP3 hydrolysis enzymatic assays which are performed in comparatively low salt concentration (<100 mM salt). Prior studies have shown that the phospho-tail PTEN interactions are abolished at high salt concentration17,18, so we surmise that this elevated ionic strength in the crystallization conditions accounts for the lack of conformational closure seen in the crystal structures of the 20- and 22-spacer forms.
Characterization of the VSP-phospho-PTEN tail interactions to ensure that VSP is a bona fide surrogate of PTEN
Since we were unable to capture phospho-tail-PTEN body interactions by crystallography, we turned our investigation to VSP as a PTEN homolog to gain further insights into phospho-dependent regulation. VSP is structurally similar35 to PTEN (rmsd 1.321 Å) and it notably harbors a comparable N-terminal helix to the one that we have now identified in PTEN as described above (Fig. 4a–b). Although VSP lacks a natural phospho-C-tail, we generated a chimeric protein (4p-VSP) that contains the body of VSP coupled with the 50 aa phospho-C-tail of PTEN using expressed protein ligation (Fig. 4c–d, S1b–c). The propensity of semisynthetic 4p-VSP to adopt a closed conformation was analyzed by determining the rate of dephosphorylation of the C-tail by the non-specific phosphatase, alkaline phosphatase (Fig. 4e). We have previously reported that the phosphates in the C-tail of 4p-PTEN are protected from alkaline phosphatase removal, presumably because of shielding due to phospho-tail/core PTEN interactions18,20. The dephosphorylation kinetics of 4p-VSP catalyzed by alkaline phosphatase closely matched those of 4p-PTEN (Fig. 4e), suggesting a similar conformationally closed state, where the phosphate interactions with the body of VSP protect them from hydrolysis.
Figure 4. VSP as a surrogate to understand the structural basis for phospho-tail interactions with PTEN.
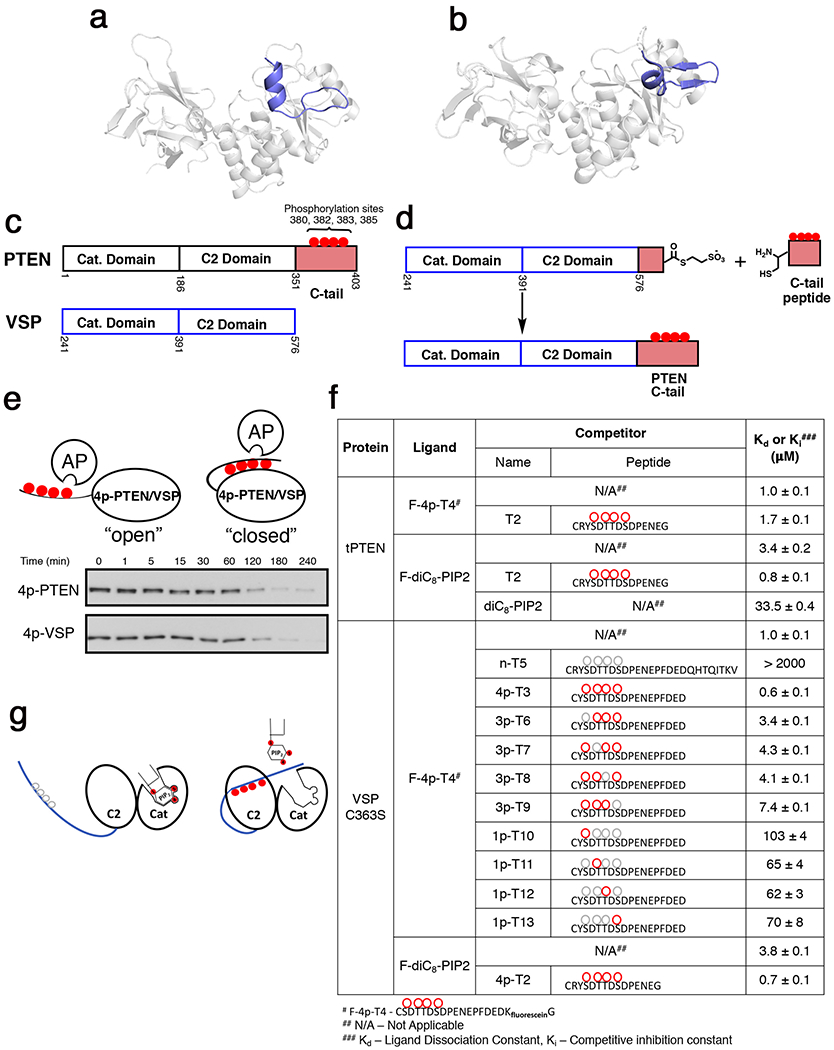
a, New PTEN crystal structure of n-crPTEN-13sp-T2 showing N-terminal helix (blue) that ranges from aa7-14 in the N-terminal segment. b, VSP crystal structure (PDB:3V0D, chain B) showing similar N-terminal helix in blue as our new PTEN crystal structure. c, Domain architecture overlay of PTEN and VSP showing VSP lacking the 50 amino acid C-tail of PTEN. d, EPL of the chimeric VSP that is fused to the C-tail of PTEN. e, Phosphatase sensitivity assay of 4p-PTEN and 4p-VSP using 1 U of alkaline phosphatase to evaluate VSP’s ability to mimic PTEN’s conformational closed state. Half-life for 4p-PTEN = 80 ± 10 min and 4p-VSP = 60 ± 10 min. f, Binding constants determined by fluorescence anisotropy for various ligands with either tPTEN (aa1-379,Y379C) or VSP-C363S. g, Cartoon showing competition of PIP2 with tetraphosphorylated C-tail for binding to VSP/PTEN. Uncertainty reported as ± SEM, n=2.
Prior experiments on PTEN showed that synthetic phosphopeptide PTEN tails can efficiently block PTEN catalytic activity in an intermolecular fashion (i.e., in trans).18 To probe the PTEN phospho-tail/VSP core interactions in a modular fashion, we developed an intermolecular binding assay based on fluorescence anisotropy. Fluorescein-labeled PTEN phospho-peptide (4p-T4) was shown to bind to VSP with a Kd of 1.0 μM (Fig. 4f), the same as 4p-T4’s binding affinity to tPTEN (aa 1-379y379c), and comparable to the IC50 of similar phospho-peptides ability to inhibit PTEN catalysis (Fig. S1d–f)18. Through a series of competition assays of the VSP/FAM-4p-peptide complex treated with varying concentrations of 4p-, 3p-, and 1p-peptides, we showed that the unlabeled phospho-peptides could readily displace the F-4p-T4 peptide from VSP. The extrapolated Kd values from these competition assays were approximately 1 μM for 4p-peptide (4p-T3), about 5 μM for peptides with one phosphate removed at each position (3p-peptides), and they further increased to ~70 μM for the 1p-peptides with the non-phosphorylated peptide showing a Kd >2 mM (Fig. 4f). This data is consistent with previous studies on PTEN that showed that each phosphorylation site partially contributes to a composite effect towards conformational closure 21,30. We also observed that soluble PIP2 (diC8) competed with the binding of phospho-peptides to VSP, indicative of overlapping binding surfaces on the enzyme (Fig. 4g). These results are consistent with prior PTEN biochemical assays that showed competition between the phospho-tail and PIP217,18,21,30. Taken together, these results indicate that the core VSP structure is a faithful surrogate of the core of PTEN with respect to PTEN phospho-tail interactions.
NMR analysis of VSP interactions with PTEN phospho-tail peptides
As VSP can be made efficiently in E. coli, we investigated its potential for structural analysis by NMR. 2H, 13C, 15N-labeled VSP was used to obtain backbone chemical shift assignments via a series of triple resonance experiments37. As the core of VSP is relatively large (38 kDa) and shows limited stability and solubility, this proved challenging using conventional approaches. In addition to routine approaches, we recorded additional triple-resonance spectra using the recently developed NMR pulse sequence method beta/alpha decoupling pulse (BADCOP)38. BADCOP employs a set of optimized decoupling schemes to modulate NMR peak shapes. The observed peak shapes of the Cα carbons depend on the Cβ chemical shift frequencies, thus providing additional information on the amino acid type. The observed peak shapes encode Cβ chemical shift frequency. This provided additional information for sequential matching and assignment of resonances in cases where we have degeneracy in the Cα chemical shifts. To aid and validate the assignment process we recorded 15N-1H TROSY-HSQC spectra on samples that were labeled with selective amino acids39 and specifically engineered mutants. This combination of procedures, as fully described in the Methods section, allowed us to confidently assign 28% of the 325 non-proline residues (Fig. 5 and S4). These assigned residues were spread throughout the catalytic and C2 domains of the protein and were sufficient to provide detailed information of phospho-peptide interactions through chemical shift perturbation (CSP) experiments.
Figure 5. Mode of interaction of tetraphosphorylated PTEN C-tail with VSP.
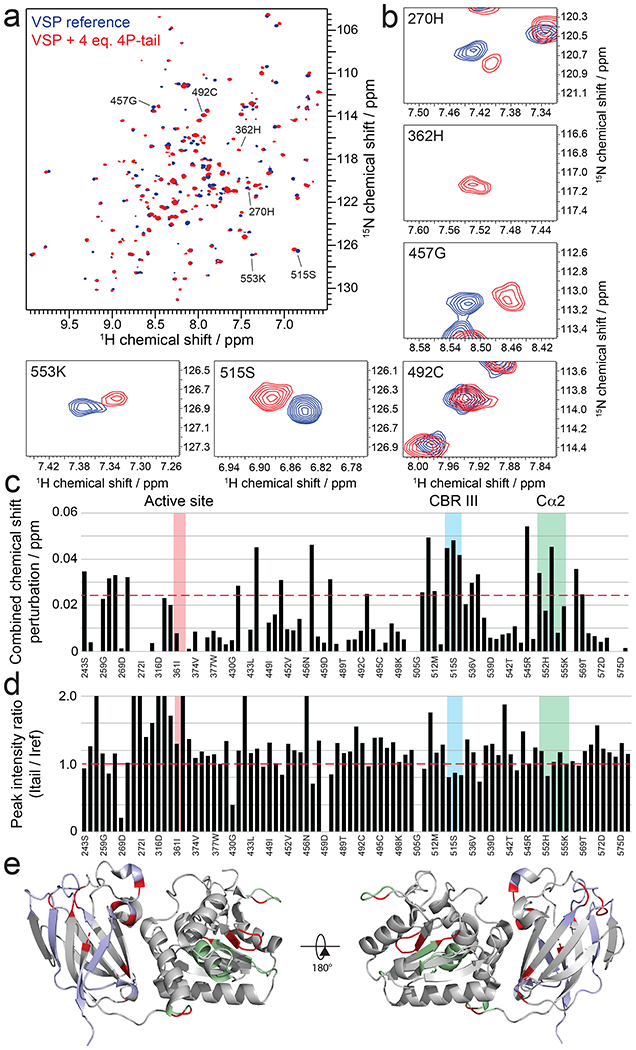
a, Overlay of 15N-1H-HSQC spectra of VSP alone (blue) and VSP in the presence of 4 molar equivalent of PTEN C-tail (red). A bigger figure including all obtained peak assignments can be found in Figure S4. b, Expanded views of spectra in (a) around select assigned peaks. c, Combined chemical shift perturbations corresponding to the spectra in (a-b) and plotted against VSP primary sequence. Dashed red line corresponds to the standard deviation to the mean, excluding outliers (higher than 3xSDM). Notable regions of VSP for their strong interaction or biological relevance are shown in colored areas. d, Peak intensity ratios corresponding to the spectra in (a) and plotted against VSP primary sequence. Notable regions of VSP for their strong interaction or biological relevance are shown in colored areas. e, Structure of VSP (PDB:3V0G) colored according to NMR results. Unassigned residues are in grey. Assigned residues that do not experience significant chemical shift perturbations or peak intensity changes are shown in light blue for the C2 domain and light green for the catalytic domain. Residues that show perturbations are shown in red.
The VSP backbone assignments were made in the presence of excess 4p-tail PTEN peptide (4p-T3) to maximize the stability of the protein during the multi-day time frames needed for data acquisition. To assess the stoichiometry of the 4p-tail peptide/VSP interactions, we recorded 15N-1H TROSY-HSQC spectra on a sample of VSP with different molar equivalents of peptide. Dramatic changes in chemical shifts occurred between 0 and 1 equivalents of 4p-tail peptide vs. VSP and further addition of 4p-tail peptide beyond 1 equivalent up to 10 equivalents of 4p-tail peptide resulted in minimal additional changes. These results suggest that there is one primary binding site of the 4p-tail PTEN peptide on VSP. The primary CSPs caused by the addition of the 4p-tail peptide was localized to the CBRIII loop (aa514-525 in VSP), Cα2 segment (aa545-558 in VSP), as well as patches in the catalytic domain (Fig. 5a–e).
To attempt to map the position of the individual phosphates of the 4p-tail onto VSP, we performed a series of NMR titrations with the four variations of the 3p-phospho-PTEN peptide (Figs. 6a and S5). The rationale was that the CSP experienced by VSP amide groups with a particular 3p-tail peptide would resemble the apo vs the 4p-tail for the deleted phosphate that make specific contacts with VSP. Analysis of these results showed that the four 3p-peptide NMR titration CSP results readily separated into two distinct groups. pSer380 and pThr382 displayed similar behaviors and impacted CBRIII loop residues the most (Fig. 6b–c). In contrast, pSer383 and pSer385 impacted the chemical shifts of residues in the Cα2 segment more significantly (Fig. 6d–e). In general, CSP data from the single phosphate deletions from 4p-tail did not show major differences on residues in the catalytic domain suggesting that the phospho-cluster makes its closest contacts to the C2 domain. Furthermore, we performed an additional NMR titration on VSP with p380 mono-phosphorylated tail and this showed CBRIII loop interactions that were consistent with the 4p- and 3p-tail data (Fig. S6).
Figure 6. Mode of interaction of triphosphorylated PTEN C-tail with VSP.
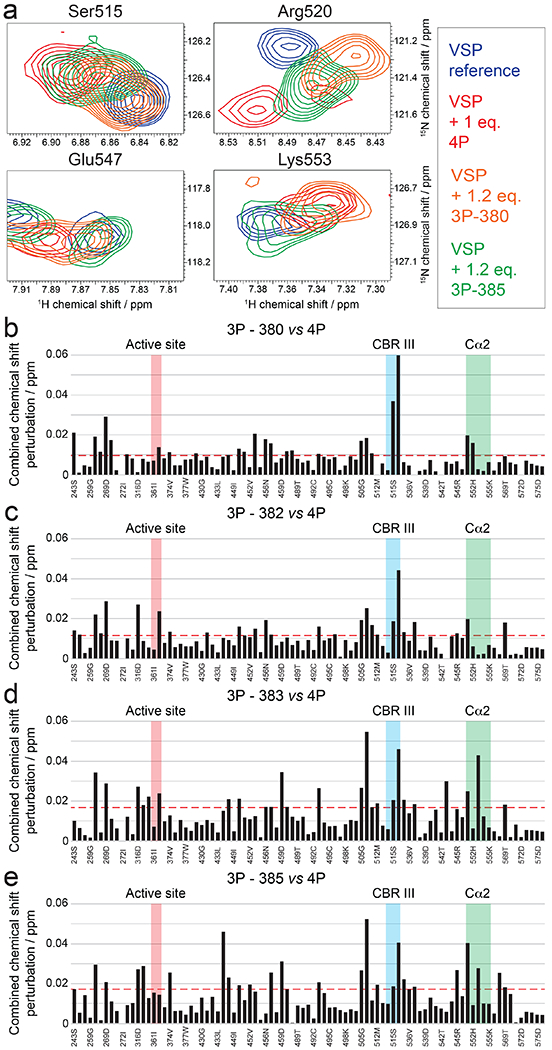
a, Overlay of 15N-1H-HSQC spectra of VSP alone (blue) and VSP in the presence of 1 molar equivalent tetra-phosphorylated (red), 1.2 molar equivalent tri-phosphorylated lacking pSer380 (orange) and 1.2 molar equivalent tri-phosphorylated lacking pSer385 (green) PTEN C-tail. Spectra are shown as expanded views around the peaks assigned to selected residues of the CBRIII and Cα2 loops. Corresponding full spectra can be found in Figure S5. b-e, Combined chemical shift perturbations for 1.2 molar equivalent tri-phosphorylated lacking pSer380 b, 1.2 molar equivalent tri-phosphorylated lacking pSer382 c, 1.2 molar equivalent tri-phosphorylated lacking pSer383 d, and 1.2 molar equivalent tri-phosphorylated lacking pSer385 e, referenced to a spectrum of VSP alone and plotted against VSP primary sequence. Dashed red line corresponds to the standard deviation to the mean, excluding outliers (higher than 3xSDM). Notable regions of VSP for their strong interaction or biological relevance are shown in colored areas.
We also carried out a related NMR analysis of soluble PIP2 interaction with VSP. In contrast to the results with 4p-peptide, VSP CSPs from PIP2 were smaller in the C2 domain. However, the prominent CSPs induced by PIP2 were localized to the catalytic domain, in and around the active site (Fig. 7a–d). This indicates that, despite the 4p-peptide and PIP2 both being multi-phosphorylated entities, PIP2 most closely associates with the VSP/PTEN catalytic domain whereas the PTEN tail phosphates largely engage with the VSP/PTEN C2 domain.
Figure 7. Mode of interaction of PIP2 with VSP.
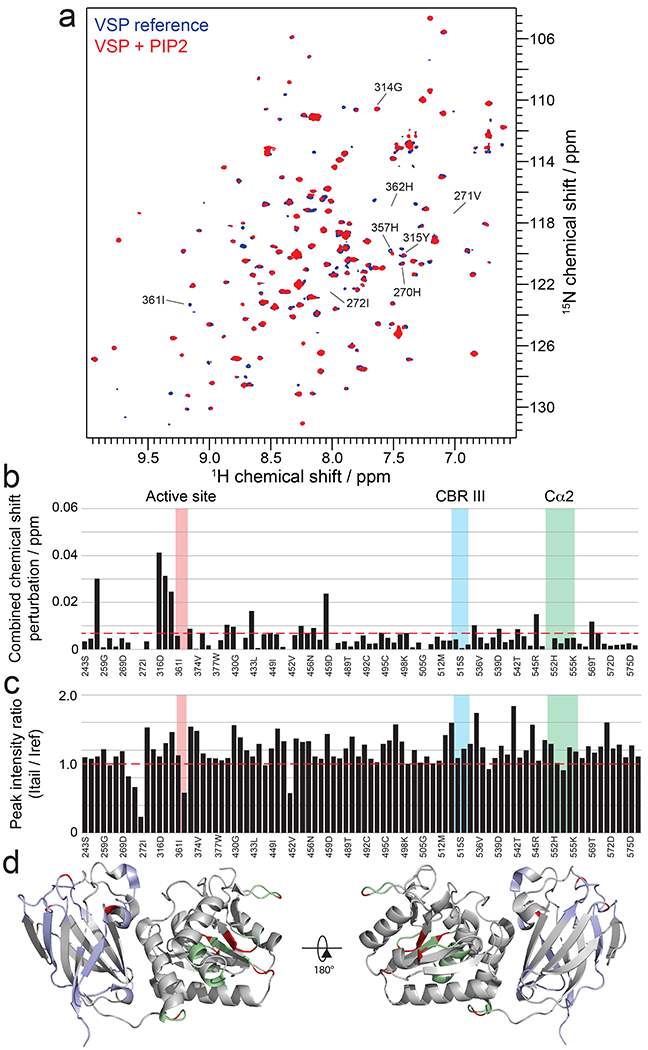
a, Overlay of 15N-1H-HSQC spectra of VSP alone (blue) and VSP in the presence of 25 μM PIP2 (red). b, Combined chemical shift perturbations corresponding to the spectra in (a) and plotted against VSP primary sequence. Dashed red line corresponds to the standard deviation to the mean, excluding outliers (higher than 3xStDev). Notable regions of VSP for their strong interaction or biological relevance are shown in colored areas. c, Peak intensity ratios corresponding to the spectra in (a) and plotted against VSP primary sequence. Notable regions of VSP for their strong interaction or biological relevance are shown in colored areas. d, Structure of VSP (PDB:3V0G) colored according to NMR results. Unassigned residues are in grey. Assigned residues that do not experience significant chemical shift perturbations or peak intensity changes are shown in light blue for the C2 domain and light green for the catalytic domain. Residues that show perturbations due to PIP2 are shown in red.
NMR driven modelling of the phospho-tail peptide on VSP
Our next goal was to build a model representing a biologically relevant mode of interaction between VSP and the phospho-C-tail using the NMR data. We employed Rosetta FloppyTail40,41 to generate possible conformations of the C-tail, introduced the phosphorylations and subsequently refined these models (Fig. S7a–b). We then selected a low energy model and performed computational docking using the CSP information observed on VSP in the presence of 4p-tail PTEN peptide. These constraints were used in the software HADDOCK42,43 to determine an energetically stable pose between the peptide and protein. The modeling constraints also position the N-terminus of the C-tail peptide next to the C-terminus of the core of PTEN in addition to including a previous photocrosslinking experiment which placed PTEN 4p-tail peptide residue Phe392 in close proximity to a catalytic domain segment (aa45-50 in PTEN, aa285-290 in VSP)21. Two possible poses were modeled by HADDOCK (Fig. S7c–d). The best scoring one populated more than 90% of all calculated structures, thus we considered this pose as relevant. The results of this modeling are depicted in Fig. 8a which shows a detailed portrait of the closed conformation that reinforce the current and previous biochemical and structural experimental data available for PTEN and VSP. The model positions the phosphorylated C-tail cradled by a basic groove on VSP (Fig. 8b–d) with the phosphorylated residues of the C-tail positioned close to the CBRIII loop and Cα2 segment, that are consistent with the surfaces suggested from our FloppyTail simulations (Fig. S7a–b). The acidic residues (PTEN aa 386-395) of the tail that are C-terminal to the phosphorylated residues extend into the catalytic domain (Fig. 8a). The major driving force behind tail binding comes from the phospho-cluster as evidenced by the more than 2000-fold weaker affinity of the nonphosphorylated peptide relative to the tetraphosphorylated peptide for VSP (Fig. 4f). However, the contacts of the C-terminal half of the PTEN peptide with VSP also appear to contributet to the overall binding affinity. This is evident from analysis of binding affinities to VSP of various phosphorylated C-terminal truncated PTEN peptides (Fig. 8e), where a moderate drop-off in binding affinity from 18aa (4p-T3) to 15aa (4p-T2) was observed and a more dramatic weakening with a 7-mer (3p-T14) was measured. Moreover, PTEN PIP3 phosphatase inhibition assays using peptides of similar lengths, added in trans to PTEN (Fig. S1d–f), afforded results that correlated well with the binding data obtained for VSP.
Figure 8. NMR-driven HADDOCK model for how the phosphorylated C-tail interacts with VSP.
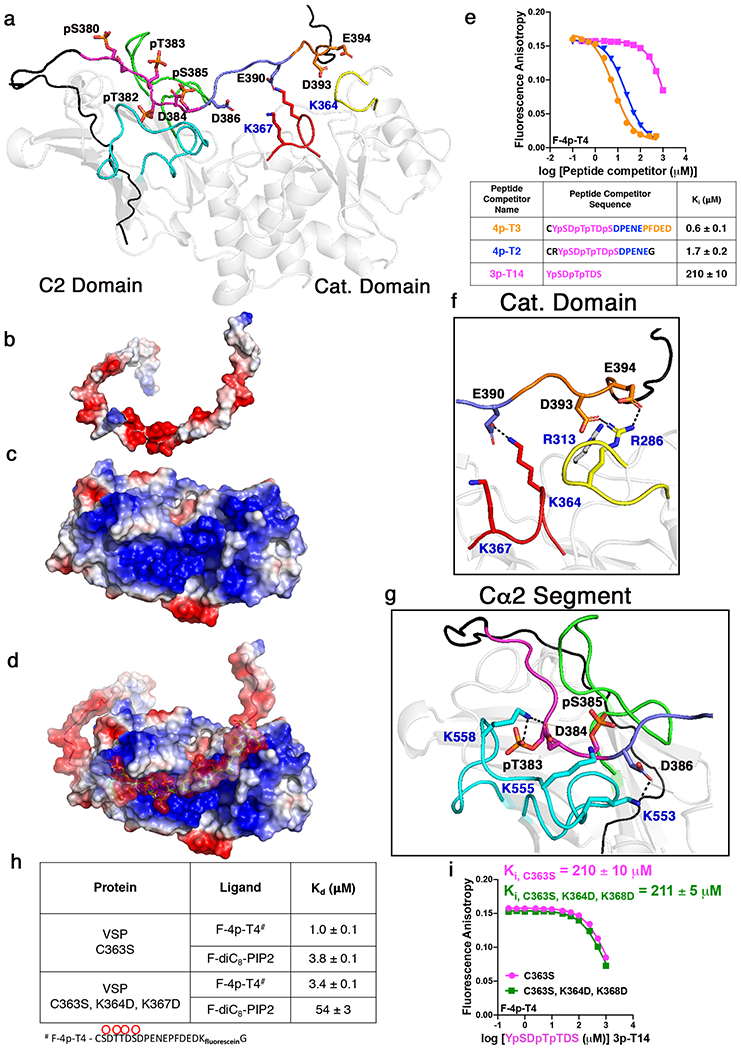
a, Molecular docking of PTEN phosphorylated C-tail (aa 353-403) with VSP (PDB:3V0G) showing lowest energy model. For VSP structure: Cα2 segment in cyan, CBRIII loop in green, active site P-loop in red, and site of previous determined crosslinking in yellow. For phosphorylated C-tail, black region represents segment of tail not used in NMR experiments whereas magenta is the phospho-cluster, dark blue is the second acidic patch spanning residues 386-390, and orange is the third acidic patch spanning residues 391-394. b-d, Surface electrostatic map of HADDOCK model showing VSP and phospho-PTEN C-tail (b), VSP (c) and overlay (d). Basic surfaces in blue, neural in white, and acidic in red. e, Fluorescence anisotropy competition assays to measure commitment to binding for different length peptides with VSP-C363S. Coloring of peptide lengths corresponds to colors used in panel a and in table below. f, Residues in the catalytic domain including Arg286 and Arg313 which correspond to Arg47 and Arg74 in PTEN that are important for tail binding, respectively. g, Residues in the Cα2 segment including Lys553, Lys555, and Lys558 which correspond to Lys330, Lys332, and Arg335 in PTEN that are important for tail binding, respectively. h, Binding constants for VSP-C363S and VSP-C363S, K364D, K367D with fluorescein-labeled tetraphosphorylated C-tail or PIP2 determined by measuring the change in fluorescence anisotropy. i, Fluorescence anisotropy competition assay comparing binding affinities of VSP-C363S and VSP-C363S, K364D, K367D with phospho-cluster limited peptide (3p-T14). Uncertainty reported as ± SEM, n=2.
The PTEN phosphopeptide/VSP conformational model is consistent with previous mutagenesis efforts that showed a weakening of the phospho-tail interaction with the core of PTEN. In particular, mutations in the catalytic domain (Arg47 and Arg74), Cα2 segment (Lys330, Lys332, Arg335), and CBRIII loop all decreased the binding affinity for the phosphorylated C-tail18,21. This NMR-based model for tail-VSP involves the corresponding residues in the catalytic domain and Cα2 segments in VSP (Fig. 8f–g). Although the specific amino acids that are modeled to interact with the phospho-tail peptide in the CBRIII loop are not highly conserved between PTEN and VSP, the role of this loop generally in promoting the intramolecular association in PTEN has been clearly documented.18
Interaction of the PTEN phospho-tail peptide with the active site of VSP
The model of the PTEN phospho-tail peptide engagement with VSP showed that the C-terminal acidic region of the tail extends to the VSP catalytic domain. In particular, the sidechain of Glu390 of the PTEN C-tail peptide is proximal to the sidechains of Lys364 and Lys367, which neighbor the active site Cys363 (replaced here with a Ser to prevent phospho-peptide hydrolysis during binding and structural analysis). To understand the potential significance of these Lys residues located in the active site of VSP, on the tail interactions with the C-tail, we generated the VSP mutant K364D/K367D and measured its affinity with the phospho-tail peptide. The double mutant displayed a Kd of 3.4 μM compared with a Kd of 1 μM for the VSP form that harbored Lys residues at these positions (Fig. 8h). This 3.4-fold weakened affinity conferred by Lys mutations, a moderate effect, is consistent with the proposed extended and multifaceted nature of the PTEN peptide-VSP protein interaction. Moreover, binding of the 7-mer triphosphorylated peptide to VSP was unaffected by the K364D/K367D replacements (Fig. 8i), supporting the computational model that places the C-terminal segment of the PTEN phosphopeptide close to the catalytic domain of VSP/PTEN whereas the phosphate-rich N-terminal segment of the tail primarily targets the C2 domain. Interestingly, the K364D/K367D mutations in VSP showed a much sharper, 14-fold effect on soluble PIP2 binding, with the Kd increasing to 54 μM compared with 3.8 μM for the non-mutant form (Fig. 8h). These results highlight that the active site of VSP is much more central to PIP2 interaction relative to phospho-peptide tail engagement.
The role for the N-terminal region in promoting phospho-tail interactions
Our model and biochemical data establish that the phospho-tail in VSP/PTEN extends to the catalytic domain, engaging multiple surfaces including the active site. Our discovery in this study that PTEN possesses an N-terminal α-helix. led us to assess the role that this region may play in phospho-tail binding. We generated N-terminally truncated proteins of VSP (VSP-NΔ16, aa 256-576) and semi-synthetic PTEN (4p-PTEN-NΔ16, aa 17-403Y379C) that delete these helical regions and analyzed phospho-tail-VSP/PTEN interactions. The affinity of 4p-F-T4 for VSP-NΔ16 in trans is weakened by 9-fold relative to the non-deleted VSP protein (Fig. S8a). With semisynthetic 4p-PTENs we observed that dephosphorylation of the C-tail by alkaline phosphatase was accelerated by 13-fold with 4p-PTEN-NΔ16 relative to that of full-length 4p-PTEN (Fig. S8b). Taken together, these results suggest that the N-terminal α-helix containing segment in both VSP and PTEN contributes about 1.4 kcal/mol to the binding energy between the phospho-tail and core of both proteins.
DISCUSSION
Here we have provided a new structural understanding of how phosphorylation regulates the tumor suppressor PTEN. Although phosphorylation of proteins is generally accepted to be the most ubiquitous and best characterized class of post-translational modifications44, the structural details of their functional consequences are poorly understood in a plethora of cases. This is particularly true with poly-phosphorylation of proteins in which phosphoprotein sample preparation and biophysical analysis can be extremely difficult. The structural challenges posed by phospho-PTEN are especially daunting, likely accounting for why it has been recalcitrant to a detailed understanding for two decades. In addition to PTEN’s requirement of insect cell expression to obtain well-behaved protein18, the phosphorylated tail is long, disordered, and highly acidic. This acidic phospho-tail is reminiscent of polyanionic polymers like heparin which are notoriously flexible45.
In prior studies, we were able to generate site-specifically modified phospho-PTEN forms using various semisynthetic strategies17,18,21,30. We extended that work here, but we showed that crystallization of these phospho-proteins does not capture the physiological closed state because of the need for high salt concentrations for crystal growth. Unexpectedly, three of the new PTEN crystal structures showed the appearance of an N-terminal alpha-helix which has been proposed previously36 but not observed. The positioning of this helix, its close proximity to the phosphatase domain’s active site, and its high B-factor suggest a dynamic role that could function in catalytic turnover and regulation. This possibility is consistent with this region’s enzymatic mutational sensitivity11,18,46–50 and number of reported cancer mutations (COSMIC database). An important role for the N-terminal helix is also supported by our deletion experiments where removing this region weakens the phospho-tail interaction with the core of the protein. Among the four crystal structures reported here three, except PTEN with the 22 aa spacer, have the N-terminal helix. Furthermore, the loss of this helix in the PTEN crystal structure that possessed the greatest capability to associate with the phospho-C-tail (22 aa spacer) may be due to dynamic intramolecular interactions that are partially formed even at high salt concentrations. The presumed dynamic nature of this N-terminal helix may explain why we were unable to assign these residues in the NMR analysis.
The helical character of the N-terminal residues in both PTEN and VSP and the previously known conservation in core structural features prompted us to investigate this PTEN homolog as a surrogate for analyzing PTEN phospho-tail interactions35. Validation of VSP as a reliable PTEN mimic in this regard was achieved through both intra- and intermolecular experiments and this encouraged a detailed NMR analysis since VSP, unlike PTEN, is readily expressed in E. coli. There have been only a few PTP-family members characterized in detail by NMR51–53. Assigning the VSP backbone chemical shifts by triple resonance experiments proved challenging due to limiting concentrations and the long-term stability of the protein, but we were ultimately able to obtain sequence information for a third of the non-proline residues distributed throughout the protein. With these assignments, through a series of 1H-15N HSQC peptide titration experiments we were able to map the four specific phosphates onto interaction sites within the CBRIII loop and Cα2 segments on the body of VSP.
Our NMR derived model shows that phospho-tail engages the body of VSP through a multivalent interaction, with an electrostatic contribution in which the acidic residues of the C-tail interact with the basic patch in the body of VSP. Each phosphorylation event increases the negative charge of the tail and concomitantly the strength of the interaction of the phospho-tail with the body. Since there are multiple phosphorylation sites in the C-tail, it is conceivable that nature uses phosphorylation as molecular rheostat to fine tune the activity of the PTEN. Previous mutagenesis18,21,27 and HDX experiments22 have highlighted the importance of these regions in PTEN but heretofore whether these were direct or indirect phospho-peptide contacts was inconclusive. The individual 3p-peptide NMR CSPs provides strong evidence of specific and direct interactions. HADDOCK42,43computational docking based on these CSPs as well as the spatial boundaries defined by semisynthetic linker length analysis performed here and prior photocrosslinking add evidence to the proposed model. Particularly informative binding experiments that corroborated this structural model were the differential affinity effects of the 4p-18mer vs. the 3p-7mer with the K364D/K367D and the wt VSP. The lack of an influence of these VSP Lys residues on the shorter phospho-peptide binding affinity strongly suggests that the peptide phosphates are localized to the C2 domain as indicated by the NMR CSPs.
In contrast to the tail-peptide phosphates, the NMR and mutagenesis binding experiments with PIP2 revealed that the PIP2 phosphates are closely associated with the catalytic rather than the C2 domain of VSP. This result was unexpected given the phosphate clusters present in both molecules. Nevertheless, due to the extended surface of the interaction of VSP with the phospho-peptide tail, that is rich in Asp and Glu residues in its C-terminal segment, the phospho-peptide competes with PIP2 for binding to VSP. Prior studies have indicated a role for the C2 domain binding to the phospholipid bilayer of the plasma membrane and our overall structural model is fully consistent with this possibility14,22,54. Therefore, the extended interactions between the phospho-peptide tail across the C2 and the catalytic domains of PTEN described here prevent plasma membrane interaction and block phosphatase catalysis even with soluble PIP3 substrates.
In summary, these studies have integrated protein semisynthesis, crystallographic, NMR, biochemical, and computational approaches to address a longstanding structural challenge with the key tumor suppressor PTEN. This multifaceted attack was needed because of the unusual challenge of capturing interactions between the phosphate-rich disordered and highly dynamic C-terminal tail and the PTEN core. We propose that marshaling this range of methods may help solve other complex phospho-protein structural interactions that have often proven resistant to classical X-ray crystallographic analysis.
METHODS
Peptide synthesis
All peptides were synthesized using standard Fmoc solid phase peptide synthesis strategies using the methods previously described in Dempsey et al 20. In brief, Wang resin coupled with the corresponding C-terminal residue was extended using 6 molar equivalents (eq) of the Fmoc residue with 5.8 eq of HATU, and 10 eq of diisopropylethylamine in organic solvent consisting of 1:1 mixture of dichloromethane: dimethylformamide (DMF) until complete coupling was observed by the semi-quantitative ninhydrin test. Deprotection of Fmoc was carried out by the addition of 20% piperidine in DMF two times for 15 min per addition. For the F-4p-T4 peptide, Fmoc-Lys(Alloc)-OH was used in the peptide synthesis where the Alloc protective group was selectively removed by incubating the reaction mixture with 4.3 eq of tetrakis(triphenylphosphine)palladium(0) in a 20:1:0.5 (v/v) of chloroform:acetic acidN-methylmorpholine (NMM) for three hours. Following deprotection, the peptide-resin was washed with a solution of 0.5% sodium diethyldithiocarbamate in DMF, 5% NMM in DMF, and then with DMF. Fluorescein was then coupled to the ε-amine of the lysine by incubating with 1.1 eq of fluorescein-NHS (Thermo Fisher Scientific), 5% NMM in DMF overnight. All coupling and deprotection steps were performed at room temperature with gentle mixing. The final peptides were cleaved from the Wang resin and deblocked by the addition of Reagent K [82.5% TFA, 5% phenol, 5% H2O, 5% thioanisole, 2.5% 1,2-ethanedithiol (v/v)] for four hours. After precipitation with ice-cold ether, the precipitate was dissolved in water and purified by reversed phase HPLC using a Varian Dynamax Microsorb 100-5 C18 column (250 x 21.4 mm, 5 μm). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-MS) with α-cyano-4-hydroxycinnamic acid as the matrix in 50% acetonitrile was used to analyze the final purified peptides, confirming the correct structures and high purities. Unphosphorylated peptides were evaluated in positive ion mode whereas phosphorylated peptides were evaluated in negative ion mode with the addition of 5 mM ammonium citrate to the matrix. MALDI-MS data were collected on an Applied Biosystems Voyager-DE STR BioSpectrometry Workstation or 4800 MALDI TOF/TOF Analyzer.
n-T1: [M+H]+ calculated as m/z 1688 Da, observed as m/z 1689 Da
4p-T2: [M−H]− calculated as m/z 2008 Da, observed as m/z 2006 Da
4p-T3: [M−H]− calculated as m/z 2398 Da, observed as m/z 2395 Da
F-4p-T4: [M−H]− calculated as m/z 2778 Da, observed as m/z 2776 Da
n-T5: [M+H]+ calculated as m/z 3169 Da, observed as m/z 3170 Da
3p-T6: [M−H]− calculated as m/z 2318 Da, observed as m/z 2316 Da
3p-T7: [M−H]− calculated as m/z 2318 Da, observed as m/z 2316 Da
3p-T8: [M−H]− calculated as m/z 2318 Da, observed as m/z 2316 Da
3p-T9: [M−H]− calculated as m/z 2318 Da, observed as m/z 2316 Da
1p-T10: [M−H]− calculated as m/z 2158 Da, observed as m/z 2156 Da
1p-T11: [M−H]− calculated as m/z 2158 Da, observed as m/z 2156 Da
1p-T12: [M−H]− calculated as m/z 2158 Da, observed as m/z 2156 Da
1p-T13: [M−H]− calculated as m/z 2158 Da, observed as m/z 2156 Da
3p-T14: [M−H]− calculated as m/z 997 Da, observed as m/z 994 Da
4p-T15: [M−H]− calculated as m/z 1749 Da, observed as m/z 1746 Da
4p-T16: [M−H]− calculated as m/z 2235 Da, observed as m/z 2232 Da
4p-T17: [M−H]− calculated as m/z 3170 Da, observed as m/z 3168 Da
PTEN expression
All PTEN forms expressed in this study were generated as reported in Bolduc et al 18. In brief, PTEN fused with the GyrA intein and chitin binding domain (CBD) was sub-cloned into a pFastBac1 vector and transformed into DH10Bac E. coli cells for bacmid production. The purified bacmid was then transfected into Sf21 insect cells to generate the P1 baculovirus. The viral titer was then increased by infecting more Sf21 insect cells with the P1 virus to generate P2 virus. The P2 virus was then used to infect HighFive insect cells at cell density of 1 million/mL in liquid suspension with a multiplicity of infection (MOI) of 1. All insect cells were maintained at 27°C. After 48 hours, the HighFive insect cells that expressed corresponding PTEN was harvested by centrifugation at 2,000 rpm for 20 min at 4°C. The resulting cell pellet was flash frozen with liquid nitrogen and stored at −80°C until purification.
PTEN Expressed protein ligation (EPL)
Semi-synthetic PTENs and VSP were made using the same standard method 18. Semi-synthetic tPTEN is comprised of amino acids 1-379Y379C, whereas 4p-PTEN has the full-length sequence of amino acids 1-403Y379C. Semi-synthetic PTENs prepared to determine the spacer-length dependence for autoinhibition used a crystallizable form of PTEN (crPTEN) which is truncated at its N-terminus, contains an internal D-loop, and possesses a C-terminal tail thereby consisting of aa residues 7-285, and aa 310-353. The “spacer” segment is located between residues Glu353 until pSer380 (or Ser380). The composition of this spacer begins with an artificial polypeptide chain installed during the expression of PTEN followed by amino acids from the EPL-ligated peptide up until pSer380. Four spacers were used which have the following sequences: 13sp - TGGGSGGTGGCRY, 16sp - TGGGSGGTGGGSGGCY, 20sp - TGGGSGGTGGGSGGTGGGCY, and 22sp - TGGGSGGTGGGSGGTGGGSGCY. All residues in the “spacer” that include and follow the Cys come from the synthetic peptide. We made five combinations of crPTEN proteins with varying spacers and C-terminal peptides which are 13sp-T1, 13sp-T2, 16sp-T3, 20sp-T3, 22sp-T3. The C-terminal peptide tail (n-T1 – CRYSDTTDSDPENEG, 4p-T2 - CRYpSDpTpTDpSDPENEG, or 4p-T3 - CYpSDpTpTDpSDPENEPFDED) was inserted by EPL using the same procedure as previously reported in Bolduc et al 18. Briefly, the cell pellet from the insect cell expression was resuspended in 50 mM HEPES pH 7.5, 250 mM NaCl, 1 mM EDTA, 10% glycerol, and one tablet of complete EDTA-free protease inhibitor cocktail (Pierce) and lysed using 40 strokes with a Dounce homogenizer. After centrifugation of the mixture at 27,000g for 40 min, the supernatant was loaded onto chitin resin. The mixture was then washed with 20 column volumes of an aqueous solution containing 25 mM HEPES pH 7.5, 250 mM NaCl, 1 mM EDTA, 0.1% Triton and the 10 column volumes of a solution containing 50 mM HEPES pH 7.5, 250 mM NaCl, 1 mM EDTA. Semi-synthetic tPTENs were generated by incubating the immobilized protein with 50 mM HEPES pH 7.5, 250 mM NaCl, 1 mM EDTA, 400 mM MESNA, and 50 mM cysteine for 24 hours at room temperature. The peptide-ligated proteins were all generated by incubating the immobilized protein in a buffered solution containing 50 mM HEPES pH 7.5, 250 mM NaCl, 1 mM EDTA, 400 mM MESNA, and 2 mM of the corresponding peptide for 48-72 hours at room temperature. Following EPL, all semi-synthetic proteins were dialyzed into Buffer A (50 mM Tris pH 8.0, 5 mM NaCl, 10 mM DTT) and then loaded onto a MonoQ anion exchange column (GE Healthcare) and further purified with a 240 mL gradient with 0-50% Buffer B (50 mM Tris pH 8.0, 1 M NaCl, 10 mM DTT). Purified fractions (>90% by Coomassie-stained SDSPAGE) were collected, concentrated, and then loaded onto either a Superdex 200 or Superdex 75 column with a mobile phase of either 25 mM Tris pH 8.0, 200 mM NaCl, 10 mM DTT for crPTENs or 50 mM Tris pH 8.0, 200 mM NaCl, 1 mM TCEP for tPTEN and 4p-PTEN. Purified fractions were collected, concentrated and stored at −80°C.
Enzyme assay
PTEN’s phosphatase activity was measured using the soluble PIP3 substrate, diC6PIP3 (dihexanoyl-phosphatidyl inositol 3,4,5-triphosphate, Avanti Polar Lipids), and monitoring phosphate release by the Malachite Green assay (R&D Biosystems) at 620 nm. Initial velocities were measured in a 25 μL buffered solution containing 50 mM Tris pH 8.0, 10 mM BME, 0.05 mg/mL ovalbumin, 0.5-20 μg PTEN, and varying concentrations of substrate (20-160 μM) 18.
Crystallization of PTEN
n-crPTEN-13sp-T1, 4p-crPTEN-13sp-T2, 4p-crPTEN-20sp-T3, and 4p-crPTEN-22sp-T3 were all crystallized in this study. n-crPTEN-13sp-T1 and 4p-crPTEN-13sp-T2, were concentrated to 14 mg/mL and crystals were grown by hanging drop in 24-well plates by adding a 1:1 volume of well solution, 100 mM Bis-Tris propane pH 7.0, 1.2 M DL-malic acid (Hampton Research), to protein and incubated at 20°C for about one week. 4p-crPTEN-20sp-T3 and 4p-crPTEN-22sp-T3 were crystallized using the same procedure except that the well solution was 300 mM sodium citrate tribasic, 20% (v/v) PEG 3350 (Molecular Dimensions). n-crPTEN-13sp-T1, 4p-crPTEN-13sp-T2, and 4p-crPTEN-20sp-T3, crystals were flash frozen and stored in liquid nitrogen until data collection. 4p-crPTEN-22sp-T3 crystals were cryoprotected using 300 mM sodium citrate tribasic, 20% (v/v) PEG 3350, 25% glycerol, flash frozen, and stored in liquid nitrogen until data collection.
Data collection and structural determination
n-crPTEN-13sp-T1, 4p-crPTEN-13sp-T2 and 4p-crPTEN-20sp-T3 data were collected at National Synchrotron Light Source-II at beamlines 17-ID-1(AMX) on a DECTRIS Eiger X 9M detector and 17-ID-2 (FMX) on a DECTRIS Eiger X 16M detector. n-crPTEN-13sp-T1, 4p-crPTEN-13sp-T2 were collected using a vector over the length of the crystal. 4p-crPTEN-20sp-T3 data were collected using crystals ranging in sizes from 10-13 μm using multiple crystals mounted in a single loop55. Three data sets were scaled using XSCALE with a resolution cut-off criterion was 〈I/σ(I)〉 of 2.9 Å. Datasets were indexed, integrated and scaled using fastdp56, XDS57, and aimless58 (Table S1). The structure for the n-crPTEN-13sp-T1, 4p-crPTEN-13sp-T2 and 4p-crPTEN-20sp-T3 were determined by direct refinement using PDB ID 1D5R 59 as the starting model. The data were refined using iterative rounds of refinement with REFMAC560,61 and manual rebuilding in Coot62. Structures were validated using Coot62 and PDB Deposition tools with more than 95 % of residues in preferred and allowed regions according to Ramachandran statistics (Table S1). Figures were rendered in PyMOL (v2.2.3, Schrödinger, LLC, New York, NY). Buried areas were calculated with PDBePISA63.
VSP EPL
A chimeric version of VSP that was fused with 25 C-terminal residues of PTEN (spanning from Pro354 to Arg378), followed by the GyrA intein and chitin binding domain was sub-cloned into a pET28b(+) vector and expressed in BL21(DE3) Rosetta pLysS E. coli cells with 2×YT media supplemented with 40 μg/mL kanamycin. 1 L of culture was inoculated with 10 mL of an overnight cell culture and grown at 37°C until an OD600 of 0.6 was reached. The cell culture was then induced by the addition of 0.5 mM IPTG and further incubated for 16 hours at 22°C. Immediately after harvesting the culture, the cell pellet was resuspended in buffer containing 50 mM HEPES pH 7.5, 250 mM NaCl, 1 mM EDTA, 10% glycerol, and one tablet of complete EDTA-free protease inhibitor cocktail (Pierce) and lysed using a french press. The remaining purification steps for the chimeric semi-synthetic ciVSP were performed as described in the “PTEN expressed protein ligation (EPL)” section. The final semi-synthetic protein 4p-VSP was generated using EPL with the same peptide (CSpDTpTpDSpDPENEPFDEDQHTQITKV) employed for preparing 4p-PTEN.
Phosphate protection assay
Both 4p-VSP and 4p-PTEN-WT were analyzed using a previously described phosphate protection assay 18. This assay monitors the dephosphorylation of pSer380 by Western blot. These assays are performed in a solution of 50 mM Tris pH 8.0, 10 mM BME with 1 μM of 4p-PTEN or 4p-VSP and initiated with 1 or 2.7 U of alkaline phosphatase (CIP, New England Biolabs, catalog number M0290) at room temperature. 1 U is defined as the amount of alkaline phosphatase needed to hydrolyze the C-terminal phosphates of 4p-PTEN with a half-life of 80 min. All data was normalized to 1 U of alkaline phosphatase. Aliquots of the reaction were quenched in SDS-loading dye and the fraction of pSer380 remaining is quantified by Western blot using a phospho-C-tail specific antibody from Cell Signaling Technology (catalog number 9549).
Expression and purification of VSP
Codon optimized ciVSP (Genscript) was sub-cloned into pET-28b(+) vector fused to an N-terminal 6x-His-tag-SUMO for easy removal of the tag by ULP1 (SUMO protease). The VSP-pET28b vector was transformed into BL21(DE3) Rosetta pLysS E. coli cells and expressed in 2×YT media supplemented with 40 μg/mL kanamycin. 1 L of cell culture was inoculated with 10 mL of an overnight culture and incubated at 37°C until an OD600 of 0.6 was reached. The cell culture was then induced by the addition of 0.5 mM IPTG and further incubated for 16 hours at 22°C35. The cell culture was harvested by centrifugation with 4,000 rpm at 4°C and the cell pellet was immediately resuspended in a buffer containing 100 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole, 1 mM TCEP, 1 mM PMSF, and one tablet of complete EDTA-free protease inhibitor cocktail (Pierce). The cells were lysed by french press and the lysate, after centrifugation with 15,000 rpm at 4°C, was loaded onto Ni2+ Sepharose 6 Fast Flow resin (GE Healthcare). The resin was then washed with five column volumes of a solution containing100 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole followed by 100 mM HEPES pH 7.5, 500 mM NaCl, 60 mM imidazole. VSP was then eluted with 20 mL of a buffered solution containing 100 mM HEPES pH 7.5, 500 mM NaCl, 250 mM imidazole, and 1 mM TCEP. Eluted protein was then concentrated using a 10 kDa Amicon filter and then ULP1 (SUMO protease) was added to a final molar ratio of 1:10 (ULP1:VSP). Cleavage by ULP1 proceeded during a stepwise dialysis to slowly remove the imidazole with the same buffer composition as the elution buffer but with decreasing amounts of imidazole (in order: 100 mM, 30 mM, 10 mM imidazole) over a 40 h period at 4°C. Following the stepwise dialysis, any uncleaved VSP and ULP1 were removed by passing the protein sample back over the Ni2+ Sepharose 6 Fast Flow resin followed by a 10 mL chase of 100 mM HEPES pH 7.5, 500 mM NaCl, 75 mM imidazole, 1 mM TCEP. The recombinant VSP protein was then concentrated with a 10 kDa Amicon filter and dialyzed three consecutive times against 2 L of a buffer containing 50 mM Tris pH 8.0, 5 mM NaCl, and 10 mM DTT for one hour per time at 4°C. The freshly dialyzed protein sample was then immediately loaded onto a MonoQ anion exchange column (GE Healthcare) for further purification with a 240 mL gradient of 0-50% Buffer B (Buffer A: 50 mM Tris pH 8.0, 5 mM NaCl, and 10 mM DTT; Buffer B: 50 mM Tris pH 8.0, 1 M NaCl, and 10 mM DTT). Purified fractions (>90% by Coomassie-stained SDSPAGE) were collected, concentrated, and dialyzed against a buffer containing 50 mM Tris pH 8.0, 200 mM NaCl, 1 mM TCEP, and 10% glycerol overnight at 4°C. The mixture was then flash frozen and stored at −80°C. Prior to any biochemical or structural study, the frozen VSP was thawed and further purified by a Superdex 200 gel filtration column with a mobile phase of a buffered solution containing 50 mM Tris pH 8.0, 200 mM NaCl, and 1 mM TCEP. Purified fractions were collected and dialyzed against a specific buffer designed for either biochemical or structural studies (See Fluorescence anisotropy binding assay or NMR experiments sections for specific buffer).
Fluorescence anisotropy binding assay
After the size-exclusion chromatography step to further purify tPTEN or VSP, the purified materials were dialyzed for 16 hours against a buffer containing 50 mM Tris pH 8.0, 50 mM NaCl, and 1 mM TCEP which is also the buffer used for the binding assay. A change in fluorescence anisotropy was measured using a Biotek Cytation 5 plate reader at 23°C (Ex: 495 nm, Em: 520 nm) to measure the binding affinity of 50 nM fluorescein labeled ligands (F-diC8PIP2 or F-4p-T4 peptide), 50 μg/mL ovalbumin, and varying concentrations of either tPTEN or the corresponding VSP. Soluble F-diC8PIP2 (fluorescein tagged dioctanoyl-phosphatidyl inositol 4,5-diphosphate) was purchased from Cayman Chemical. Anisotropy values were plotted versus the protein concentration and fitted to equation 1:
| Equation 1: |
where Y is the fluorescence anisotropy, X is the ligand concentration, and Kd is dissociation constant. Competition assays to displace the fluorescent ligand were also performed with various peptides. The same assay conditions were used except tPTEN or VSP were held at constant concentration (4 μM for F-4p-T4 peptide or 5 μM for F-diC8PIP2), with varying concentrations of competitor. A semi-log plot of the protein concentration and anisotropy was plotted and the data was fit to equation 2 to determine the IC50 value:
| Equation 2: |
with Y being the fluorescence anisotropy, X is the ligand concentration, and IC50 is the concentration of competitor required to displace 50% of the fluorescent ligand from protein. The IC50 value from equation 2 was used to calculate the affinity for the competitor with equation 364:
| Equation 3: |
where the I50 is the concentration of free competitor at 50% inhibited, L50 is concentration of free fluorescent ligand at 50% inhibition, Kd is the dissociation constant for the fluorescent ligand, Po is the concentration of protein (VSP or PTEN) at 0% inhibition, and Ki is the affinity of the competitor.
NMR experiments
VSP-C363S was expressed in minimal media (6 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 2 mM MgSO4, 0.1 mM CaCl2, and 40 μg/mL kanamycin) supplemented with 0.0002% (w/v) thiamine, 0.5% (w/v) glucose, and 1 g/L 15NH4Cl for 2D TROSY-HSQC experiments. The procedure for culturing and purification is the same as found in the “Expression and purification of VSP section” except that 5 mL of overnight culture in LB media was used to inoculate 1 L of fresh culture. For triple resonance experiments to express 2H, 13C, or 15N VSP-C363S the same composition of minimal media was used except all regents and culturing was executed in D2O supplemented with 1 g/L 15NH4Cl, 1 g/L U-13C6, 1,2,3,4,5,6,6-D7 D-glucose (Cambridge Isotope Laboratories, Cat. # CDLM-3813-1), and 1 g/L 13C, D, 15N, and celtone base powder (Cambridge Isotope Laboratories, Cat. # CGM-1030P-CDN-1). The culturing procedure for expression of deuterated VSP-C363S is as follows. A clump of single colonies from a fresh transformation was added to 25 mL LB media in D2O supplemented with 40 μg/mL kanamycin and incubated at 37°C for 12 h. The cell culture was then centrifuged to remove LB media and resuspend pellet in 50 mL of fresh minimal media supplemented for triple resonance experiments overnight at 37°C for 12 h. 50 mL of overnight cell culture was used to inoculate 1 L of fresh minimal media, supplemented with isotopes for triple resonance experiments, and incubated until OD600 of 0.6 at 37°C. The cell culture was then induced with 0.5 mM IPTG and incubated for another 20 h at 22°C. Purification of isotopically labeled VSP-C363S proceeds in the same way as previously discussed in the “Expression and purification of ciVSP” section. After purification by gel filtration, isotopically labeled VSP-C363S was dialyzed into 50 mM Tris pH 7.5, 50 mM NaCl, and 1 mM TCEP for NMR experiments.
All NMR data were acquired using TopSpin (Bruker), processed with NmrPipe65 and analyzed with CCPNmr66. Structural biology applications were compiled by the SBGrid consortium67. 3D NMR experiments were conducted on Bruker spectrometers operating at 800 or 900 MHz, equipped with TCI cryoprobes and z-shielded gradients, and Avance III and NEO consoles, respectively. Samples of ca. 180 μM 2H, 13C, 15N-labeled VSP (aa 241-576) and 4 molar equivalents of unlabeled tetra-phosphorylated PTEN C-tail (aa 379-395) in 50 mM Tris pH 7.5, 50 mM NaCl, 1 mM TCEP and 5% v/v 2H2O were used. Temperature was set to 20°C. In those conditions, sample stability was approximately 2 days, about the time necessary for each one of the experiments. Initially, standard triple resonance backbone experiments using TROSY selection37 were used: HNCOCA, HNCO, HNCACO, HNCACB. For all the backbone triple resonance experiments 10% of the Nyquist grid, in the indirect dimension, was sampled using Poisson-Gap sampling. The resulting non-uniformly sampled spectra were reconstructed using the hmsIST algorithm68. Spectra were acquired with recycle delays of 1 s, ca. 64 transients were accumulated with 20 and 128 complex points in the 15N and 13C dimension, respectively. Measurement time was ca. 2 days and 7 hrs. We complemented those experiments with a series of HNCA acquisitions using different BADCOP decoupling schemes. BADCOP38 uses a set of optimized decoupling schemes to modulate NMR peak shapes. In order to remove ambiguities in the sequential assignments due to degeneracies in Cα resonances, we recorded HNCA with BADCOP pulses that decouples Cα depending on the chemical shift of the attached Cβ (and thus amino acid type). We measured 4 such spectra with decoupling patterns using 5% NUS, 96 transients and 20x320 (15Nx13C) complex points. Measurement times were 1 day 20 hrs and Cα resolution ca. 18 Hz. Finally, we complemented the 3D NMR data set with multiple 2D spectra of Arg, Asn, His, Lys and Gln unlabeled VSP39 as well as R13G, R46G, H122G, R158A, K276G, R280G and K314A point mutants. 90 of 325 non-proline backbone resonances could be assigned. Chemical shift assignments as well as chemical shift perturbations due to the presence of 4 eq 4P-tail are deposited in the Biological Magnetic Resonance Bank (BMRB) under accession code 50381.
2D spectra were acquired using TROSY-HSQC69 on a Bruker 800 MHz Avance III spectrometer equipped with a TXO cryoprobe and z-shielded gradients. Samples of either 50 μM 15N-labeled VSP (aa 241-576) in 50 mM Tris pH 7.5, 50 mM NaCl, 1 mM TCEP and 5% v/v 2H2O were used. Samples with different amounts of tri- or tetra-phosphorylated PTEN C-tail were measured. Recycle delays of 1 s, 64 transients and 128 complex points in the indirect dimension were set for a total experimental time of 5 hrs. Chemical shifts were calibrated using internal water calibration in NmrPipe. Combined chemical shift perturbations and associated standard deviations were calculated according to the protocol from Schumann et al.70.
FloppyTail modeling of PTEN
Modeling was started with a previously published PTEN crystal structure (PDB:1D5R). First, the structure was energy-minimized in Rosetta with the FastRelax protocol71 with the “talaris2014” scoring function. Following energy minimization, the disordered C-terminus that was not observed in the crystal structure was re-introduced in the extended (φ=−135°, ψ=135°) conformation. From this starting point, two sets of simulations were run. The first set followed the standard FloppyTail protocol that is fully detailed elsewhere and has been shown to make accurate predictions of disordered region interactions40,41. The second set used 3-residue fragment insertions to better sample backbone dihedral space (Box 1) and introduced constraints derived from a cross-linking experiment (Box 2) in the low-resolution stage. In both cases, after 10,000 models were generated PyRosetta was used to re-introduce all side-chains atoms and to phosphorylate residues S380, T382, T383, S385. Each one of these models was used to seed a high-resolution FloppyTail simulation producing a single final model (Box 3).
(Box 1). Low-resolution flags (run as FloppyTail.linuxgccrelease @ lr.flags. To reproduce the simulation without constrains and fragments remove the –frag3 and – cst_* flags):
–s Lowres_Input/1D5R_extend.pdb
–FloppyTail::flexible_start_resnum 305
–FloppyTail::flexible_chain A
–FloppyTail::shear_on 0.333
–FloppyTail::short_tail::short_tail_fraction 1
–FloppyTail::short_tail::short_tail_off 0
–packing::repack_only
–FloppyTail::perturb_temp 0.8
–FloppyTail::perturb_cycles 10000
–FloppyTail::refine_temp 0.8
–FloppyTail::refine_cycles 10
–FloppyTail::refine_repack_cycles 5
–in::file::frag3 aat000_03_05.200_v1_3.txt
–run::min_type dfpmin_armijo_nonmonotone
–cst_file Lowres_Input/photocrosslinking.cst.txt
–cst_weight 1
–cst_fa_file Lowres_Input/photocrosslinking.cst.txt
–cst_fa_weight 1
–nstruct 10000
–multiple_processes_writing_to_one_directory
–out:path:pdb ./lowres_models
(Box 2). Constraints definitions (Rosetta numbered):
AmbiguousConstraint
AtomPair CA 31A CA 348A FLAT_HARMONIC 5 5 5
AtomPair CA 32A CA 348A FLAT_HARMONIC 5 5 5
AtomPair CA 33A CA 348A FLAT_HARMONIC 5 5 5
AtomPair CA 34A CA 348A FLAT_HARMONIC 5 5 5
AtomPair CA 35A CA 348A FLAT_HARMONIC 5 5 5
AtomPair CA 36A CA 348A FLAT_HARMONIC 5 5 5
AtomPair CA 37A CA 348A FLAT_HARMONIC 5 5 5
AtomPair CA 31A CA 348A TOPOUT 1 5 6
AtomPair CA 32A CA 348A TOPOUT 1 5 6
AtomPair CA 33A CA 348A TOPOUT 1 5 6
AtomPair CA 34A CA 348A TOPOUT 1 5 6
AtomPair CA 35A CA 348A TOPOUT 1 5 6
AtomPair CA 36A CA 348A TOPOUT 1 5 6
AtomPair CA 37A CA 348A TOPOUT 1 5 6
End
(Box 3). High-resolution flags (run as FloppyTail.linuxgccrelease @ hr.flags):
–l lowres_models_cst_frag3.list
–FloppyTail::flexible_start_resnum 305
–FloppyTail::flexible_chain A
–FloppyTail::shear_on 0.333
–FloppyTail::short_tail::short_tail_fraction 1
–FloppyTail::short_tail::short_tail_off 0
–packing::repack_only
–FloppyTail::perturb_temp 0.8
–FloppyTail::perturb_cycles 0
–FloppyTail::refine_temp 0.8
–FloppyTail::refine_cycles 6000
–FloppyTail::refine_repack_cycles 60
–run::min_type dfpmin_armijo_nonmonotone
–nstruct 1
–out:path:pdb ./highres_models_cst_frag3
–out:file:scorefile highres_cst_frag3_score.tsv
–multiple_processes_writing_to_one_directory
The final models were analyzed with a custom PyRosetta script and pairwise energies were calculated between individual pairs of phosphorylated residues and crystallized protein core residues. Two-body interactions included electrostatics, van der Waals, hydrogen bonding, and solvation. Residues were defined to be interacting with the phosphorylated region of the tail if their pairwise energy was non-zero. The interacting fraction was calculated as the number of interactions with a particular core residue divided by the total number of models. The enrichment of each core–tail interaction was calculated by comparing the fraction of interactions in the top 100 models (lowest energy, computed without constraints) versus the rest of models. All Rosetta calculations were performed with version 3.6, and PyRosetta calculations were performed with the Python 2.7 bindings of the same Rosetta-version source code.
Molecular docking
Molecular docking was performed using the web server of the software Haddock2.4 in Guru mode42,43 (https://bianca.science.uu.nl/haddock2.4). The crystal structure of ciVSP (PDB:3V0G)35 and the lowest energy tetra-phosphorylated PTEN C-tail (aa 353-403) model generated with Rosetta FloppyTail72 (see “FloppyTail modelling of PTEN” section) were used as starting material. Active residues were defined on VSP as residues with significant chemical shift perturbations: 250, 260, 268, 269, 270, 284, 362, 363, 430, 431, 432, 447, 456, 457, 458, 487, 506, 507, 514, 515, 520, 536, 537, 545, 547, 553 and 567. In the absence of NMR data of the C-tail, the 4 phosphorylation sites (380, 382, 383, 385) as well as Phe392 which was previously shown to cross-link to PTEN21 were defined as active. Passive residues were defined automatically. The C-terminal residue of VSP (Asp575) and the N-terminal residue of the C-tail (Glu353 in PTEN) were constrained together by a set of hard distance restraints corresponding to the geometry of a peptide bond (e.g. 3.6 Å between Cα atoms). The cross-linking restraint was defined as a distance ranging from 5 to 15 Å between Phe392 (PTEN) and Leu284 (VSP) Cγ atoms. An additional network of soft restraints was defined based on our tri-phosphorylated NMR data. Thr514, Ser515 and Arg520 were constrained to the vicinity of pSer380 and pThr382 while Arg545, Glu547 and Lys553 were constrained near pThr383 and pSer385 (3 to 10 Å between N atoms). The C-tail was left entirely flexible (backbone and side chains) throughout the docking procedure. Some parts of VSP known to interact with the C-tail and to be more flexible were also left flexible. That includes the N-terminus (aa 246-263), the site of cross-linking previously reported (aa 278-287), the active site loop (aa 362-367), the loop at aa 448-459, the CBRIII loop (aa 514-526) and the Cα2 loop (aa 550-563). In elements of secondary structure only the side chains were left flexible and for unstructured residues the backbone as well. 2,000 starting poses were subjected to solvated docking and flexible refinement, then the top 190 structures were refined using short molecular dynamics simulations in explicit water shells.
Two clusters emerged as possible models. Cluster 1 contained 91% of the refined structures and had a HADDOCK score of −453. Cluster 2 contained 9% of the structures and a score of −406 (the lower the better). More detailed scoring parameters and models can be found in Fig. S7.
Supplementary Material
Acknowledgements
We would like to thank NIH K99GM130961 (D.R.D), NIH F32GM120855 (D.R.D), NIH R01CA74305 (P.A.C), T32 GM080189 (S.H.), TM32-GM008403 (J.R.J.) and R01-GM078221 (J. R. J. and J. J. G.) for generous financial support. H. A. acknowledges funding from the Claudia Adams Barr Program for Innovative Cancer Research. Maintenance of the NMR equipment was supported by NIH grant no. EB002026. The authors would like to acknowledge the use of the Eukaryotic Tissue Culture Facility at JHU and its manager Yana Li for her expertise in protein expression using insect cells and Michelle Miller for her expertise with XDS.
Footnotes
Code availability
Custom FloppyTail code/flags used in this study can be found in methods section.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing Interests
The authors report no competing interests with this work.
Data availability
The PDBs of n-crPTEN-13sp-T1, 4p-crPTEN-13sp-T2, 4p-crPTEN-20sp-T3, and , 4p-crPTEN-20sp-T3 have been deposited with the following accession codes 7JUL, 7JUK, 7JVX, and 7JTX. The chemical shift assignments and perturbations of VSP have been deposited in the BMRB under accession code 50381. Additional raw data are deposited in the Dryad database https://doi.org/10.5061/dryad.dfn2z34zh.
REFERENCES
- 1.Maehama T & Dixon JE The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem 273, 13375–13378 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Worby CA & Dixon JE Pten. Annu. Rev. Biochem 83, 641–669 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Li J et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Yuan TL & Cantley LC PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalhoub N & Baker SJ PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol 4, 127–150 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sansal I & Sellers WR The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol 22, 2954–2963 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Toker A & Cantley LC Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387, 673–376 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Manning BD & Toker A AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu P, Cheng H, Roberts TM & Zhao JJ Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov 8, 627–644 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janku F, Yap TA & Meric-Bernstam F Targeting the PI3K pathway in cancer: are we making headway? Nat. Rev. Clin. Oncol 15, 273–291 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Campbell RB, Liu F & Ross AH Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem 278, 33617–33620 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Walker SM, Leslie NR, Perera NM, Batty IH & Downes CP The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem. J 379, 301–307 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iijima M, Huang YE, Luo HR, Vazquez F & Devreotes PN Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J. Biol. Chem 279, 16606–16613 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Lee JO et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Vazquez F, Ramaswamy S, Nakamura N & Sellers WR Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell Biol 20, 5010–5018 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres J & Pulido R The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem 276, 993–998 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Henager SH et al. Enzyme-catalyzed expressed protein ligation. Nat. Methods 13, 925–927 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolduc D et al. Phosphorylation-mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife 2, e00691 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Khouri AM, Ma Y, Togo SH, Williams S & Mustelin T Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J. Biol. Chem 280, 35195–35202 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Dempsey DR & Cole PA Protein Chemical Approaches to Understanding PTEN Lipid Phosphatase Regulation. Methods Enzymol. 607, 405–422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z et al. Molecular Features of Phosphatase and Tensin Homolog (PTEN) Regulation by C-terminal Phosphorylation. J .Biol. Chem 291, 14160–14169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masson GR, Perisic O, Burke JE & Williams RL The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity. Biochem. J 473, 135–144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das S, Dixon JE & Cho W Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. U. S. A 100, 7491–7496 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z et al. Enzymatic Analysis of PTEN Ubiquitylation by WWP2 and NEDD4-1 E3 Ligases. Biochemistry 55, 3658–3666 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen HN et al. Mechanism of human PTEN localization revealed by heterologous expression in Dictyostelium. Oncogene 33, 5688–5696 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odriozola L, Singh G, Hoang T & Chan AM Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J. Biol. Chem 282, 23306–23315 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Rahdar M et al. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. U. S. A 106, 480–485 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dempsey DR, Jiang H, Kalin JH, Chen Z & Cole PA Site-Specific Protein Labeling with N-Hydroxysuccinimide-Esters and the Analysis of Ubiquitin Ligase Mechanisms. J .Am. Chem. Soc 140, 9374–9378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muir TW, Sondhi D & Cole PA Expressed protein ligation: a general method for protein engineering. Proc. Natl. Acad. Sci. U. S. A 95, 6705–6710 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henager SH, Henriquez S, Dempsey DR & Cole PA Analysis of Site-Specific Phosphorylation of PTEN by Using Enzyme-Catalyzed Expressed Protein Ligation. Chembiochem 21, 64–68 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murata Y, Iwasaki H, Sasaki M, Inaba K & Okamura Y Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Okamura Y & Dixon JE Voltage-sensing phosphatase: its molecular relationship with PTEN. Physiology (Bethesda) 26, 6–13 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Iwasaki H et al. A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. U. S. A 105, 7970–7975 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuda M et al. Crystal structure of the cytoplasmic phosphatase and tensin homolog (PTEN)-like region of Ciona intestinalis voltage-sensing phosphatase provides insight into substrate specificity and redox regulation of the phosphoinositide phosphatase activity. J. Biol. Chem 286, 23368–23377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L et al. A glutamate switch controls voltage-sensitive phosphatase function. Nat. Struct. Mol. Biol 19, 633–641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei Y, Stec B, Redfield AG, Weerapana E & Roberts MF Phospholipid-binding sites of phosphatase and tensin homolog (PTEN): exploring the mechanism of phosphatidylinositol 4,5-bisphosphate activation. J. Biol. Chem 290, 1592–1606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salzmann M, Pervushin K, Wider G, Senn H & Wuthrich K TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. U. S. A 95, 13585–13590 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coote PW et al. Optimal control theory enables homonuclear decoupling without Bloch-Siegert shifts in NMR spectroscopy. Nat. Commun 9, 3014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubey A, Kadumuri RV, Jaipuria G, Vadrevu R & Atreya HS Rapid NMR Assignments of Proteins by Using Optimized Combinatorial Selective Unlabeling. Chembiochem 17, 334–40 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Kleiger G, Saha A, Lewis S, Kuhlman B & Deshaies RJ Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell 139, 957–968 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santiago-Frangos A, Jeliazkov JR, Gray JJ & Woodson SA Acidic C-terminal domains autoregulate the RNA chaperone Hfq. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Vries SJ, van Dijk M & Bonvin AM The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc 5, 883–897 (2010). [DOI] [PubMed] [Google Scholar]
- 43.van Zundert GCP et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol 428, 720–725 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Manning G, Whyte DB, Martinez R, Hunter T & Sudarsanam S The protein kinase complement of the human genome. Science 298, 1912–1934 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Rabenstein DL Heparin and heparan sulfate: structure and function. Nat. Prod. Rep 19, 312–331 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Johnston SB & Raines RT Catalysis by the tumor-suppressor enzymes PTEN and PTEN-L. PLoS One 10, e0116898 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen HN et al. Engineering ePTEN, an enhanced PTEN with increased tumor suppressor activities. Proc. Natl. Acad. Sci. U. S. A 111, E2684–E2693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gil A et al. A functional dissection of PTEN N-terminus: implications in PTEN subcellular targeting and tumor suppressor activity. PLoS One 10, e0119287 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maehama T, Taylor GS & Dixon JE PTEN and myotubularin: novel phosphoinositide phosphatases. Annu. Rev. Biochem. 70, 247–279 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Mighell TL, Evans-Dutson S & O’Roak BJ A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am. J. Hum. Genet 102, 943–955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Padua RAP et al. Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat. Commun 9, 4507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whittier SK & Loria JP (1)H, (1)(5)N, and (1)(3)C backbone resonance assignments for the Yersinia protein tyrosine phosphatase YopH. Biomol. NMR Assign 8, 387–389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choy MS et al. Conformational Rigidity and Protein Dynamics at Distinct Timescales Regulate PTP1B Activity and Allostery. Mol. Cell 65, 644–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McConnachie G, Pass I, Walker SM & Downes CP Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN: evidence for activation by anionic phospholipids. Biochem. J 371, 947–955 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller MS et al. Getting the Most Out of Your Crystals: Data Collection at the New High-Flux, Microfocus MX Beamlines at NSLS-II. Molecules 24(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winter G & McAuley KE Automated data collection for macromolecular crystallography. Methods 55, 81–93 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Kabsch W Xds. Acta Crystallogr. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallog.r 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mishto M et al. An in silico—in vitro Pipeline Identifying an HLA-A*02:01+ KRAS G12V+ Spliced Epitope Candidate for a Broad Tumor-Immune Response in Cancer Patients. Frontiers in Immunology 10(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallog.r D Biol. Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murshudov GN, Vagin AA & Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krissinel E & Henrick K Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Nikolovska-Coleska Z et al. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem 332, 261–273 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Vranken WF et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Morin A et al. Collaboration gets the most out of software. Elife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hyberts SG, Milbradt AG, Wagner AB, Arthanari H & Wagner G Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 52, 315–327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pervushin K, Riek R, Wider G & Wuthrich K Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U. S. A 94, 12366–12371 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schumann FH et al. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 39, 275–289 (2007). [DOI] [PubMed] [Google Scholar]
- 71.Khatib F et al. Algorithm discovery by protein folding game players. Proc. Natl. Acad. Sci. U. S. A 108, 18949–18953 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kleiger G, Saha A, Lewis S, Kuhlman B & Deshaies RJ Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell 139, 957–968 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PDBs of n-crPTEN-13sp-T1, 4p-crPTEN-13sp-T2, 4p-crPTEN-20sp-T3, and , 4p-crPTEN-20sp-T3 have been deposited with the following accession codes 7JUL, 7JUK, 7JVX, and 7JTX. The chemical shift assignments and perturbations of VSP have been deposited in the BMRB under accession code 50381. Additional raw data are deposited in the Dryad database https://doi.org/10.5061/dryad.dfn2z34zh.