

Abstract
Autophagy is best known for its role in organelle and protein turnover, cell quality control, and metabolism. The autophagic machinery has, however, also adapted to enable protein trafficking and unconventional secretory pathways so that organelles (such as autophagosomes and multivesicular bodies) delivering cargo to lysosomes for degradation can change their mission from fusion with lysosomes to fusion with the plasma membrane, followed by secretion of the cargo from the cell. Some factors with key signalling functions do not enter the conventional secretory pathway but can be secreted in an autophagy-mediated manner.
Positive clinical results of some autophagy inhibitors are encouraging. Nevertheless, it is becoming clear that autophagy inhibition, even within the same cancer type, can affect cancer progression differently. Even next-generation inhibitors of autophagy can have significant non-specific effects, such as impacts on endosome-related secretory pathways and secretion of extracellular vesicles (EVs). Many studies suggest that cancer cells release higher amounts of EVs compared to non-malignant cells, which makes the effect of autophagy inhibitors on EVs secretion highly important and attractive for anticancer therapy. In this review article, we discuss how different inhibitors of autophagy may influence the secretion of EVs and summarize the non-specific effects of autophagy inhibitors with a focus on endosome-related secretory pathways. Modulation of autophagy significantly impacts not only the quantity of EVs but also their content, which can have a deep impact on the resulting pro-tumourigenic or anticancer effect of autophagy inhibitors used in the antineoplastic treatment of solid cancers.
Keywords: Autophagy, Autophagy inhibitors, Cancer, Endosomes, Multivesicular bodies, Extracellular vesicles, Exosomes, Amphisomes, Non-conventional secretory pathways
Introduction
Autophagy is a highly evolutionarily conserved mechanism best known for its role in organelle and protein turnover, cell quality control, and metabolism. Lysosome-mediated degradative autophagy provides a source of nutrients and energy by digestion of cytoplasmic elements and serves for the clearance of toxic protein aggregates and defective organelles [1]. This recycling pathway can also profoundly affect cellular specialization and differentiation [2], protein trafficking, and unconventional secretion [3, 4]. Three types of autophagy have been observed in mammalian cells: macroautophagy, microautophagy, and chaperone-mediated autophagy. During microautophagy the lysosomal membrane insulates the autophagic cargo directly, whereas, during macroautophagy, double-membrane structures called autophagosomes are formed to deliver autophagic cargo to endosomes or lysosomes. Macroautophagy also participates in the specific degradation of organelles during mitophagy, ribophagy, or pexophagy. Chaperone-mediated autophagy involves the selective degradation of KFERQ-like motif-bearing proteins supplied to the lysosomes via chaperone HSC70 and other cochaperones (e.g. CHIP, HOP, and heat shock protein 40). Internalization of cargo into lysosomes is managed via the receptor lysosome-associated membrane protein type 2A (LAMP2A) [5]. In this review article, we will focus on macroautophagy (hereafter referred to as autophagy).
Endocytosis is the process by which cells can sequester substances from the external environment by engulfing them in vesicles. Endocytosis includes the clathrin-dependent pathway as well as clathrin-independent pathways such as phagocytosis, pinocytosis, raft-mediated endocytosis, and ARF6-dependent internalization. As well as autophagy, endocytosis can culminate into lysosomal degradation, but here the cargo is internalized from the plasma membrane, not from the cytoplasm [6]. After internalization, the cargo is sorted by highly dynamic compartments, called early endosomes (EEs), marked by unique adaptor proteins, effector proteins, and small Rab GTPases such as RAB4, RAB5, early endosomal antigen-1 (EEA1), VPS34, and SNAREs. EEs are the major cellular sorting platform as they can mature into endosomes destined for various cellular fates. EE cargo can be recycled to the plasma membrane via recycling endosomes, transported to or from the Golgi apparatus via the retromer complex, or routed to lysosomes via multivesicular bodies (MVBs)/late endosomes [7]; see Fig. 1. Autophagy and endocytic pathways cooperate at some stages and share many components of the molecular machinery.
Fig. 1.

Autophagy and endocytic pathways can culminate in lysosomes. Endocytosis enables the transport of substances from the external environment and includes the clathrin-dependent pathway as well as clathrin-independent pathways such as phagocytosis. Phagocytosis is the endocytosis of large molecules or intact microorganisms. Protrusions of plasma membrane surround and internalize the extracellular cargo into single-membrane structures called phagosomes, which are then transported to the lysosome for degradation. Endocytosis (clathrin-mediated endocytosis is shown here) involves invagination of the plasma membrane and biogenesis of small intracellular vesicles that contain constituents of the plasma membrane and extracellular components. These small vesicles fuse and establish the compartment called the early endosome (EE). EE cargo can be recycled to the plasma membrane via recycling endosomes, transported to or from the Golgi apparatus via the retromer complex, or routed to lysosomes via multivesicular bodies (MVBs). During macroautophagy, double-membrane structures called autophagosomes are formed to deliver autophagic cargo to lysosomes or to fuse with MVBs. Autophagy and endocytic pathways cooperate at some stages and share many components of the molecular machinery
Recent studies also show that there are many interconnections between autophagy, exosome/amphisome biogenesis, and exocytosis of extracellular vesicles (EVs) [3, 4]. To release exosomes and/or amphisomes, several steps need to be performed such as the biogenesis of intraluminal vesicles (ILVs) in MVBs, transport of MVBs to autophagosomes or the plasma membrane and fusion of MVBs and/or amphisomes with the plasma membrane [8]. These steps are deeply affected by molecules of autophagy machinery [9, 10] including many Rab GTPases such as RAB7, RAB11, RAB35, RAB27A, RAB27B and the vesicle-associated membrane protein 7 (VAMP7) [11]. RAB7 and RAB11 also participate in autophagosome formation and RAB7 has a key role in autophagosome maturation (for further details see the chapter Autophagy and MVBs) [12]. Consequently, autophagy can have both stimulatory or inhibitory effects on the secretion of extracellular vesicles (EVs) and these effects will probably be deeply context-dependent. This can partially explain the double-edged sword character of autophagy in cancer progression. In this review article, we discuss how different inhibitors of autophagy may influence the secretion of EVs and summarize the non-specific effects of autophagy inhibitors with a focus on endosome-related secretory pathways. Modulation of autophagy significantly impacts not only the quantity of EVs but also their content which can have a deep impact on the resulting pro-tumourigenic or anticancer effect of autophagy inhibitors used in antineoplastic treatment of solid cancers.
Basic molecular mechanism of degradative macroautophagy
Macroautophagy (hereafter referred to as autophagy) is a process in which double-membrane vesicles (autophagosomes) are formed around a segment of the cytoplasm. Once autophagosomes are formed, they can either fuse with lysosomes and form autolysosomes, or they can bring together organelles of endosomal origin to form amphisomes with a single limiting membrane [13–15].
Autophagosome formation goes through five main stages — initiation, nucleation, elongation, fusion, and cargo degradation. The detailed molecular mechanism of autophagy has been extensively reviewed in Klionsky et al. [16], therefore, we present here only the basic molecular mechanisms important for the understanding of the effects of autophagy inhibitors on macroautophagy.
The initiation phase of autophagy is preceded by the inhibition of mTORC1. mTORC1 is inhibited by cellular and environmental stresses that are incompatible with continued growth, such as glucose or amino acid deprivation, DNA damage, or hypoxia. mTORC1 consists of three core components: mTOR (highly conserved serine/threonine-protein kinase belonging to the PI3K-related kinase family), RAPTOR (regulatory protein associated with mTOR responsible for mTORC1 localization and substrate recruitment), and mLST8. In addition to these three core components, mTORC1 also contains two inhibitory subunits DEPTOR (DEP domain-containing mTOR interacting protein) and PRAS40 (proline-rich Akt substrate of 40 kDa) [17]. A decrease in cellular energy activates the stress-responsive metabolic regulator AMPK (AMP-activated protein kinase), which inhibits mTORC1 indirectly through activation of Tuberous Sclerosis Complex (TSC), or directly through the phosphorylation of RAPTOR by protein kinase A (PKA) [18, 19]. TSC suppresses mTORC1 by converting Rheb GTPase from an active GTP-bound form to an inactive GDP-bound state. The TSC complex requires G3BPs (Ras GTPase-activating protein-binding proteins) as its lysosomal tether [20].
For sensing the levels of nutrients, the presence of mTORC1 on lysosomes is crucial [21]. In response to amino acids, mTORC1 present on lysosomes can be activated by Rag and Rheb guanosine triphosphatases (GTPases) and can trigger anabolic processes [22]. A key player in Rag-mTORC1 activation is the vacuolar H+ ATPase (V-ATPase) that couples ATP hydrolysis (peripheral V1 domain) to proton translocation through the lysosomal membrane (integral V0 domain) to acidify the lysosomes and enable their degradative functions. When the level of amino acids in the lumen of lysosomes is low, the V-ATPase turns off the activity of Rag GTPases. In contrast, when amino acids are abundant, the V-ATPase undergoes conformational changes leading to the activation of Rag heterodimers and the recruitment of mTORC1 to lysosomes [23] (see Fig. 2). Lysosomes are usually localized closer to the plasma membrane when amino acids and growth factors are abundant. On the contrary, when they are limited, the Rap1-GTPases imprison lysosomes in the perinuclear region and reduce lysosome abundance, therefore reducing the lysosomal surface available for mTORC1 activation, which suppresses mTORC1 signalling [24]. The inactivation of mTORC1 leads to rapid translocation of transcription factors TFEB and TFE3 to the nucleus. Active TFEB upregulates the expression of lysosomal genes and critical regulators of autophagy, including several proteins implicated in the formation of autophagosomes and autolysosomes. Therefore, TFEB contributes to the synchronization of autophagy and lysosomes [25]. TFEB can also mediate lysosomal exocytosis and secretion of their cargo including the proteolytic enzymes, such as cathepsins, which results in extracellular matrix remodelling and invasion of cancer cells [26].
Fig. 2.
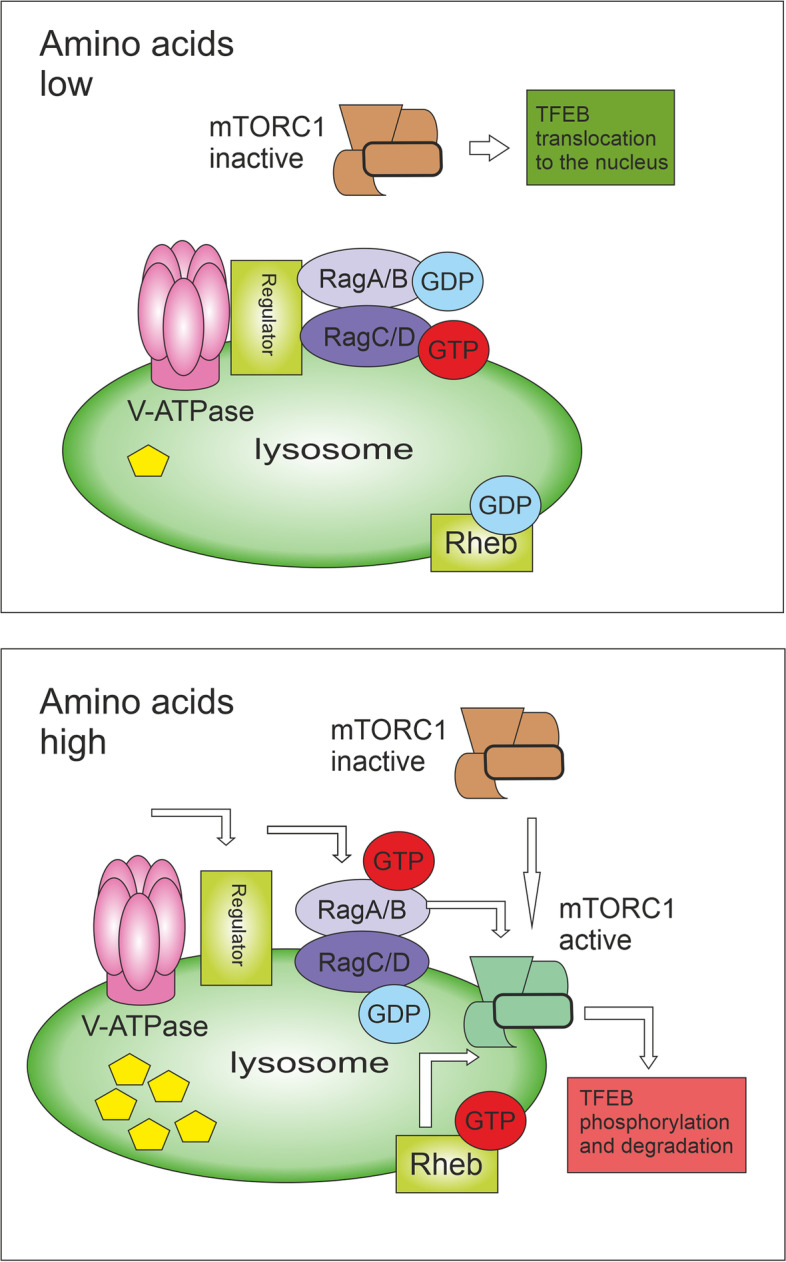
Amino acid-based mTORC1 activation. By amino acid starvation, the inactive V-ATPase-Ragulator complex is unable to activate Rag GTPases on the lysosomal surface, thus mTORC1 is not recruited to the lysosome. The inactivation of mTORC1 leads to rapid translocation of transcription factors TFEB and TFE3 to the nucleus. Active TFEB upregulates the expression of lysosomal genes and critical regulators of autophagy. By amino acid abundancy, the V-ATPase undergoes conformational changes leading to the activation of Regulator, which in turn promotes the Rag heterodimer activation. Active Rag heterodimer (RagA/B(GTP)-RagC/D(GDP)) then recruits mTORC1 to the lysosomal surface where Rheb is present. Rheb can directly bind and activate mTORC1. TFEB is recruited on the lysosomal membrane, phosphorylated by active mTORC1, and then degraded by the proteasome
The early stage of autophagy machinery is the activation of the ULK1 (unc51-like autophagy-activating kinase 1) complex. This complex consists of ULK1, FIP200, ATG13, and ATG101 (see Fig. 3). ULK1 complex forms puncta usually associated with the endoplasmic reticulum (ER). ER membrane provides local support for many (putatively different) membrane sources and initiates the formation of the isolation membrane (commonly referred to as the phagophore) from the omegasome (a phosphatidylinositide-3-phosphate (PI3P)-enriched subdomain of the ER membrane) [27, 28]. The omegasome serves as a cornerstone for the formation of the phagophore. To the omegasomes, ATG9 vesicles and coat protein complex II (COPII) vesicles are recruited to elongate the autophagosome. The origin of the rest of the lipid bilayers is currently unknown [29]. ATG9 migrates through the trans-Golgi network and the endosomal system under nutrient-rich conditions and transiently binds to the autophagosome in case of autophagy induction. The trafficking of ATG9 through the recycling endosomes may be a fundamental step for autophagosome genesis [30] as ATG9 meets and fuses with ATG16L1 in a VAMP3-dependent manner in recycling endosomes [31].
Fig. 3.

Macroautophagy pathways. The autophagic process is divided into five stages including initiation, phagophore nucleation, phagophore formation, autophagosome-lysosome fusion, and cargo degradation in autolysosomes. Signals activating macroautophagy usually originate from starvation, hypoxia, oxidative stress, and stress of the endoplasmic reticulum (ER). These signals trigger the activity of Unc-51-like kinase 1 (ULK1) complex (consisting of ULK1, FIP200, ATG13, and ATG101), which then starts phosphorylation of components of the class III PI3K (PI3KC3) complex I (consisting of VPS34, VPS15, Beclin1, ATG14L, and NRBF2) enabling nucleation of the phagophore. VPS34 produces phosphatidylinositol-3-phosphate (PI3P) allowing the recruitment of autophagy-associated PI3P-binding proteins such as DFCP1 and WIPI mediating the initial stages of autophagosome formation by associating ATG2A stably to PI3P-containing areas. Expansion of the phagophore requires the ATG2A-WIPI complex mediating ER–phagophore association and establishing the transfer of lipid membranes from the ER and the vesicles to the phagophore. WIPI was also shown to bind ATG16L1, thus recruiting the ATG12–ATG5–ATG16L1 complex. Elongation of autophagosomes requires the ubiquitin-like conjugation system managing the orchestrated activity of ATG proteins and LC3 (microtubule-associated protein light chain 3) and/or GABARAP. The ATG12–ATG5–ATG16L1 complex enhances the final connection of phosphatidylethanolamine (PE) molecules resulting in the formation of membrane-bound LC3-II and/or GABARAP-PE. Cellular membranes, including the mitochondrial membrane, the plasma membrane, recycling endosomes, and the Golgi complex, contribute to the elongation of the phagophore by providing membrane material. Elongation of the phagophore gives rise to double-layered vesicles called autophagosomes. In addition to managing autophagy induction in complex I, complex VPS34-Beclin1 has also a role in the fusion of autophagosomes with lysosomes as complex II. UVRAG competes with ATG14L for binding to Beclin1. When bound to Beclin1, UVRAG stimulates RAB7 GTPase activity and autophagosome fusion with lysosomes. Autophagosome-lysosome fusion is managed by Syntaxin-17 (STX17) on autophagosomes, VAMP8 on lysosomes, and by accessory proteins such as ATG14 and homotypic fusion, and protein sorting (HOPS) tethering complex
Once activated, ULK1 phosphorylates the class III phosphoinositide 3-kinase (PI3K) complex I (consisting of VPS34, VPS15, Beclin1, ATG14L, and NRBF2 [16]; see Fig. 3). VPS34 generates PI3P enabling the recruitment of autophagy-related PI3P-binding proteins such as WIPI and DFCP1 [32]. In some circumstances, such as shear stress, PI3KC2α-dependent and VPS34-independent generation of PI3P can take place [33]. Expansion of the phagophore requires the ATG2A-WIPI4 complex mediating ER–phagophore association and establishing the transfer of lipid membranes from the ER and the vesicles to the phagophore [34, 35]. One of the key molecular events of autophagosome formation is the lipidation of ATG8-family proteins with phosphatidylethanolamine (PE). Mammals express 2 subfamilies of ATG8 proteins: the LC3 subfamily consisting of LC3A, LC3B, LC3B2, and LC3C (referred to here as LC3; microtubule-associated protein light chain 3) and the GABARAP subfamily consisting of GABARAP, GABARAPL1, and GABARAPL2 (referred to here as GABARAP). Lipidation of LC3 and GABARAP is a membrane-curvature dependent process [36] catalysed by E1-like activating enzyme ATG7, E2-like conjugating enzyme ATG3 and enhanced by the ATG12–ATG5–ATG16L1 system formed in the previous step [37, 38]. The cysteine protease ATG4B executes two LC3/GABARAP processing events: priming of newly synthesized pro-LC3/GABARAP to enable lipidation (newly translated LC3B, called pro-LC3, is cleaved by the cysteine protease ATG4B at the C-terminal section to give LC3-I) and deconjugation of lipidated LC3/GABARAP after cargo degradation in autolysosome [39] (see Fig. 4). ATG4B is considered to be the main isoform of ATG4 as it possessed the broadest spectrum against all substrates, followed by ATG4A, whereas ATG4C and ATG4D had minimal activity [40]. The final conjugation of LC3-I to PE molecules results in the formation of membrane-bound LC3-II. LC3-II is specifically targeted to the elongating phagophore and remains on autophagosomes until their fusion with lysosomes [37]. The proper closure of the autophagosomal membrane requires the ESCRT-III component CHMP2A and the activity of VPS34 [41]. While the LC3 subfamily mediates the elongation of the phagophore, GABARAP proteins probably function in the final sealing of the autophagosome [42]. GABARAP subfamily positively regulates ULK1 activity and phagophore formation in response to starvation. On the other hand, the LC3 subfamily regulates them negatively [43].
Fig. 4.

Autophagy with emphasis on the state of MAP1LC3B (LC3B). Newly translated LC3B, called pro-LC3, is cleaved by the cysteine protease ATG4B at the C-terminal end with subsequent exposure of glycine residues (G). A cleaved form of a protein (LC3-I; soluble LC3) is further processed by ATG7 and ATG3, which conjugates LC3-I to phosphatidylethanolamine (PE) molecules. The ATG12–ATG5–ATG16L1 complex enhances the final conjugation of LC3-I to PE molecules resulting in the formation of membrane-bound LC3-II specifically targeted to the elongating autophagosomal membrane. The two ends of the insulating membrane are subsequently joined together to form an autophagosome with a double membrane. STX17, which is located on the outer autophagosomal membrane (not on the isolation or lysosomal membrane), is required for the fusion of an autophagosome with a lysosome. In the resulting autolysosome, the material is cleaved by acid hydrolases. During internal degradation, STX17 proteins are released from the outer membrane of the autophagosome. LC3-II molecules conjugated to the inner autophagosomal membrane are degraded by acids hydrolases, while the LC3-II molecules on the outer membrane of the autophagosome are cleaved by ATG4B and recycled as LC3-I
Autophagosomes are transported along microtubules, which require the function of dynein. Consequently, inhibition of dynein-dependent transport or depolymerization of microtubules results in the inhibition of autophagy [44]. The degradative autophagy culminates in a fusion of closed autophagosomes with lysosomes where the cargo is eventually degraded. The autophagosome-lysosome fusion is managed by Syntaxin-17 (STX17) on autophagosomes (see Fig. 4), which binds the VAMP8 (vesicle-associated membrane protein 8) on the lysosomal membrane via the Qbc-SNARE SNAP29 (synaptosome-associated protein 29). Accessory proteins such as ATG14 and homotypic fusion, protein sorting (HOPS) tethering complex, ESCRT, RAB7, and the class C VPS proteins are also needed [45, 46]. Some studies indicate that the kinase ULK1 regulates STX17 engagement during autophagosome maturation. Unphosphorylated ULK1 recruits STX17 and increases its affinity towards SNAP29. Protein kinase C alpha (PKCα) mediated phosphorylation of ULK1 does not change its kinase activity but decreases autophagosome-lysosome fusion [47, 48]. Inactivation of mTORC1 might also be required to facilitate the fusion between autophagosomes and lysosomes [49]. In addition to managing autophagy induction in complex I, complex VPS34-Beclin1 has a role in the fusion of autophagosomes with lysosomes as complex II (see Fig. 3). UVRAG competes with ATG14L for binding to Beclin1 [50]. When bound to Beclin1, UVRAG stimulates RAB7 GTPase activity and autophagosome fusion with lysosomes or multivesicular bodies (late endosomes; MVBs) [51]. VPS34–Beclin1-UVRAG complex may also contribute to autophagy induction via Bif-1/Endophilin B1-mediated activation of VPS34 [52].
Autophagy in the progression and therapy of solid cancers
Autophagy is undoubtedly an important tumour suppressive mechanism maintaining cellular homeostasis by executing lysosomal degradation of toxic material in the cell, as well as mediating intercellular communication via proteins and hormones with signalling function that can be secreted in an autophagy-mediated manner [53]. During the early phases of cancerogenesis, autophagy has significant cytoprotective and tumour-suppressive potential. Dysfunction of this process is associated with an increased risk of cancer development. Haploinsufficiency of the BECN1 gene was observed in 40–75% of sporadic human breast and ovarian cancers [54, 55] and more than 25% of gastric and colorectal tumours are haploinsufficient in one of the ATG2B, ATG5, ATG9B, or ATG12 genes [56]. In multiple cancer types, ATG5 mutations and alternative mRNA splicing disrupt the ATG16L1-binding to ATG5 and impair the ATG12-ATG5 conjugation. Furthermore, ATG16L2 is overexpressed in several cancers and competes with ATG16L1 for binding to ATG5 resulting in proteasomal degradation of ATG16L1 and disruption of autophagy [57]. The tumour-suppressive effect of autophagy is also supported by the fact that autophagy is stimulated by some tumour suppressors, including PTEN, TSC, or DEPTOR [58–61] (Role of autophagy-related proteins in solid cancers is summarized in Table 1). Nevertheless, if tumourigenesis has been started up, autophagy can further support tumour progression. Many aggressive tumours need autophagy for important tumour-promoting processes (e.g. autophagy enables ERBB2 (Erb-B2 Receptor Tyrosine Kinase 2) trafficking and supports tumourigenesis in ERBB2-driven breast cancer [111]). Increased autophagic activity mediates an escape of premalignant cells from genotoxic stress or anoikis, suppresses immune surveillance and can result in intrinsic resistance against anticancer therapy [112–114]. Autophagy also increases the metabolic plasticity of tumour cells, allowing them to survive in adverse conditions and supports forming of cancer stem cells [115].
Table 1.
Autophagy-related proteins in cancer
| Autophagy-related protein | Type of aberration | Effect on | Type of solid cancer | Reference |
|---|---|---|---|---|
| AMPK | genetic and transcriptional aberrations | energy homeostasis; tissue-dependent pro- or anticancer impact | many cancer types | [62] |
| ATG101 | overexpression | immunotherapy response | many cancer types | [63] |
| ATG16L2 | overexpression | proteasomal degradation of ATG16L1 | many cancer types | [57] |
| ATG2B, ATG5, ATG9B, ATG12 | genetic aberrations, haploinsufficiency | cytoprotection | gastric and colorectal tumours | [56] |
| ATG9A | overexpression | proliferation | breast cancer | [64] |
| DEPTOR | reduced expression/overexpression | epithelial to mesenchymal transition (EMT) | low DEPTOR levels in cancer of pancreas, prostate, lungs, triple-negative and breast cancer; high DEPTOR levels in osteosarcoma and differentiated thyroid carcinoma | [60, 65–69] |
| FIP200 | aberrant activation | immune checkpoint therapy response | breast cancer; glioblastoma | [70, 71] |
| mLST8 | overexpression | cancer progression | hepatocellular carcinoma | [72] |
| mTORC1 | overactivation | survival of stem cells; reprogramming of metabolism; tumour invasion and metastasis | many cancer types | [73–75] |
| PRAS40 | overexpression | proliferation; enhanced NF-κB activity | hepatocellular carcinoma; lung adenocarcinoma; cutaneous melanoma | [76, 77] |
| RAP1 | changes in the RAP1 activation | formation of cell adhesions and junctions; migration and polarization; may suppress oncogenic Ras phenotype | Rap1 inhibits invasion and metastasis in the bladder, lung, and brain cancer; it has the opposite effect in melanoma and breast cancer or oesophageal squamous cell, head and neck squamous cell pancreatic, and non-small cell lung carcinomas | [78, 79] |
| RAPTOR | overexpression | proliferation and migration; the resistance to PI3K-mTOR inhibition | colorectal cancer; renal cancer; oropharyngeal squamous cell carcinoma | [80–82] |
| TFE3 | gene fusions | insulin-dependent metabolism; retinoblastoma-dependent cell cycle arrest | renal cell carcinoma | [83] |
| TFEB | gene fusions, transcriptional aberrations | biology of lysosomes; lysosomal exocytosis; proliferation; glutamine metabolism; regulator of tumour-associated macrophages; role in the TME; WNT and TGFβ signalling | Pancreatic, breast , and renal cancer; melanoma; colorectal cancer; gastric carcinoma; non-small cell lung cancer | [84–87] |
| TSC1/2 | genetic aberrations | anticancer impact | many cancer types | [88] |
| ULK1 | aberrant activation, genetic and transcriptional aberrations | antitumour immunity; NADPH production; innate immune response; cell cycle progression | LKB1-mutant lung cancer and many other types of cancer | [89–92] |
| V-ATPase | deregulation of some subunits, overexpression | biogenesis of endosomes and lysosomes; treatment resistance | breast cancer, lung and oesophagal tumours | [93–95] |
| VPS34 | aberrant activation | antitumour immunity; survival of cancer stem cells; activation of p62; antigen cross-presenting CD8α+ dendritic cells | melanoma; colorectal cancer; hepatocellular carcinoma | [96–99] |
| WIPI3 | genetic and transcriptional aberrations | cell cycle and spliceosome | hepatocellular carcinoma | [100] |
| VAMP3 | deletion of WDFY2 (negative regulator of VAMP3 recycling) | increase in extracellular matrix degradation | metastatic ovarian and prostate cancers | [101, 102] |
| GABARAP | transcriptional aberrations | tumour differentiation; EMT | colorectal cancer; breast cancer | [103, 104] |
| MAP1LC3A or B | transcriptional aberrations | cancer progression | breast cancer; renal cell carcinoma | [105, 106] |
| p62/SQSTM1 | accumulation | cancer progression | many cancer types | [107] |
| RAB7 | reduced expression | biogenesis of endosomes, autophagosomes, and lysosomes | highly invasive breast cancer | [93] |
| ESCRT | polymorphism in ESCRT-III (rs35094336, CHMP4C) | endosomal-sorting; cytokinetic abscission; genome instability | many cancer types | [108] |
| UVRAG | inhibition | cytokine production; oncogenic signalling; progression of age-related malignancies | many cancer types | [109] |
| Beclin1 | genetic and transcriptional aberrations | cytoprotection; proliferation under hypoxia and nutrient starvation | haploinsufficiency in breast and ovarian cancer; overexpression in colorectal and gastric carcinomas | [54, 55, 110] |
Because the induction of autophagy has been observed as a side effect of many cytotoxic anticancer therapies causing therapy resistance, a large number of strategies using autophagy inhibition have been proposed to increase the efficacy of these therapies [116]. Inhibition of autophagy in the tumour microenvironment can disrupt metabolic communication between tumour and stromal cells [117] and decrease cell motility of metastatic tumour cells as inhibition of autophagy reduces disassembly of focal adhesions at the leading edge of the cell [118]. Systemic acute deletion of ATG7 in adult mice with preexisting non-small cell lung cancer reversed lung adenocarcinomas initiated by the KRASG12D oncogenic mutation and p53 deficiency to a benign form of tumour (oncocytomas) and blocked cell proliferation and cancer cell survival [119]. Furthermore, loss of tuberous sclerosis complex 2 (TSC2) sensitizes cancer cells to nelfinavir−bortezomib therapy due to intensifying endoplasmic reticulum stress-induced cell death [61]. On the other hand, autophagy induction causes a decrease in the levels of transcription factors triggering EMT [120], reduces 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (Pfkfb3) expression and elicits metastatic dormancy in breast cancer stem cells [121]. Consequently, many studies are demonstrating the benefit of autophagy during cancer therapies, especially in inducing immunogenic cell death. Tumour cells dying of autophagic cell death leads to the recruitment of immune cells to the tumour site and the activation of a tumour-specific immune response. Accordingly, caloric restriction (which promotes autophagy by inactivating mTORC1) has resulted in increased control of the immune system over the tumour, but only in tumours capable of autophagy [122].
Autophagy may be one of the key modulators of the tumour microenvironment (TME). The autophagy-dependent secreted soluble factors or factors contained in EVs may enable metabolic manipulation of non-cancer cells in the tumour microenvironment, stimulate cellular proliferation, invasive phenotype, and promote immunosuppression [53]. This TME-modulating theory is supported by the fact that autophagy facilitates the selection of material for unconventional secretion of certain cytoplasmic proteins [123]. Intact autophagy machinery is required for the secretion of multiple factors favouring invasion, including interleukin-6 (IL-6), MMP2, and WNT5a [124]. Autophagy is also closely related to the biogenesis and secretion of exosomes and amphisomes.
Autophagy and MVBs
Autophagy and endocytic pathways are important in managing many aspects of homeostasis as both endosomes and autophagosomes are known to deliver cellular material to lysosomes for degradation. Autophagy and exocytosis seem to be largely interconnected as autophagy cargo can be released by amphisomes derived from multivesicular bodies (MVBs) and phosphoinositide-3-phosphate (PI3P) is essential for the genesis of both endosomes and autophagosomes, and their positioning as PI3P promotes the microtubule-dependent translocation of late endosomes and lysosomes to the cell periphery. This PI3P-dependent lysosome translocation to the cell periphery promotes mTORC1 activation [125]. The most of cellular PI3P is generated by class III PI3K VPS34 in complex II with a small contribution of class II PI3Ks [126]. Binding and activation of VPS34 on endosomes are initiated through the recruitment of RAB5 to endosomes by the guanine nucleotide exchange factor Rabex5 (RAB Guanine Nucleotide Exchange Factor 1) [127]. VPS34 then produces PI3P increasing the binding of RAB5 and other downstream effectors, including early endosome autoantigen 1 (EEA1), the hepatocyte growth factor-regulated tyrosine kinase substrate (HRS; ESCRT-0 subunit) regulating MVBs formation via ESCRT recruitment to endosomes [128]), and endosomal sorting nexin protein family (SNX) [129]. Overexpression of SNX3 may alter the morphology of endosomes and delay their transport to the lysosome [130]. SNX18 was identified as a positive regulator of autophagosome formation [131]. Consequently, VPS34 plays a crucial role in endosome biogenesis through EAA1 and other RAB5 effectors, vesicle invagination and cargo selection within MVBs, and the fusion of autophagosomes with lysosomes. Inhibition of VPS34 resulted in dysfunction in autophagy, vesicular trafficking, and endocytic recycling and sorting [132, 133]. Furthermore, proteins such as Beclin1 and ATG14L that regulate PI3P levels are positive modulators of autophagy [134]. Some data suggest that surface delivery of endosomal cargo requires hydrolysis of PI3P mediated by MTM1 as the endosomal accumulation of PI3P inhibits exocytosis. Defects caused by mutations in MTM1 can be partially reversed by pharmacological inhibition of VPS34 [135].
Endosomal maturation is accompanied by conversion from early endosomal RAB5 to late endosomal RAB7 and active cargo sorting into intraluminal vesicles (ILVs) by the ESCRT complex. The transition from early to late endosomes is complicated by a positive feedback loop between Rabex5 and RAB5. It was demonstrated that SAND1/MON1A is needed to interrupt this positive feedback loop by displacing Rabex5 from endosomal membranes. SAND1/MON1A also manages the recruitment of RAB7 (see Fig. 5) [127]. Then VPS34 recruits TBC1D2 protein to endosomes in a RAB7-dependent manner to further inactivate RAB5 and to facilitate early to late endosome maturation. VPS34 inhibition causes hyperactivation of RAB7, autolysosomal dysfunction, a phenotype with large late endosomes and an enhanced release of atypical exosomes harbouring poly-ubiquitinated proteins [136–138]. Interestingly, RAB7 can participate in both MVBs degradation and/or MVBs-related secretion as it regulates autolysosome maturation and simultaneously the secretion of syntenin and syndecan-containing exosomes [139]. RAB7-associated endosomal processes depend not only on RAB7 GTP-based state but also on modifications with ubiquitin [140]. Endosomal maturation during the late endosome/lysosome pathway is accompanied by conversion of PI3P to PI(3,5)P2 at the limiting membrane of late endosomes. This process depends on PIKfyve (phosphoinositide kinase, FYVE-type zinc finger containing). Consequently, the activity of PIKfyve is vital for the sorting of cargo into MVBs [141].
Fig. 5.

The transition from early to late endosomes. Binding and activation of VPS34 on endosomes are initiated through the recruitment of RAB5 to endosomes by the guanine nucleotide exchange factor Rabex5. VPS34 then produces PI3P increasing the binding of RAB5 and other downstream effectors. The transition from early to late endosomes is complicated by a positive feedback loop between Rabex5 and RAB5. MON1A is needed to interrupt this positive feedback loop by displacing Rabex5 from endosomal membranes. MON1A also manages the recruitment of RAB7. VPS34 recruits TBC1D2 protein to endosomes in a RAB7-dependent manner to further inactivate RAB5 and to facilitate early to late endosome maturation
In addition to the role in the degradation and recycling of cellular waste, autophagic and endo-lysosomal systems can play a key role in secretory pathways (see Fig. 6) as autophagy cargo can be released by amphisomes derived from multivesicular bodies (MVBs). MVBs are late endosomes containing many intraluminal vesicles (ILVs) formed by the invagination of the endosomal membrane. ILVs start to be generated in early endosomes and accumulate until the late endosomal stage. In vitro budding of ILVs into MVBs is regulated by syntenin-syndecan interactions requiring Alix which is known to interact with several ESCRT proteins including TSG101 and CHMP4 [142, 143]. MVBs can fuse with the plasma membrane to release intraluminal vesicles (ILVs) to the extracellular space as exosomes. During exosome biogenesis, Alix forms a complex with the scaffold protein syntenin, mediating the loading of cargo into exosomes and promoting exosome release. These Alix-dependent processes are controlled by ATG12–ATG3 and cells lacking ATG12–ATG3 showed reduced exosome biogenesis. Both ATG12–ATG3 and Alix promote basal, but not starvation-induced, autophagic flux [9]. Some results indicate that activated c-Src in the endosomal membrane promotes the secretion of exosomes. Alix was identified as a c-Src–interacting protein in exosomes, resulting in the upregulation of exosome secretion in c-Src–transformed cells [143]. The small GTPase ADP ribosylation factor 6 (Arf6) and its effector phospholipase D2 (PLD2) also regulate the syntenin pathway [144]. MVBs morphology and their docking to the plasma membrane is significantly disrupted by the loss of RAB27a and/or RAB27b activity [145]. Critical plasma membrane docking and secretion sites for MVBs are invasive actin structures called invadopodia. Invadopodia degrade the extracellular matrix through the local deposition of matrix metalloproteinases (MMPs) and manage cancer cell invasion [146]. Protein WDFY2 (WD Repeat and FYVE Domain Containing 2) is frequently lost in metastatic cancers (e.g. ovarian and prostate cancers). Through its interaction with VAMP3, WDFY2 restricts the budding of MMP14-containing VAMP3 vesicles from actin-stabilized endosomal tubules. Upon deletion of WDFY2, this negative control is disrupted and faster recycling of MMP14 to the plasma membrane leads to increased matrix degradation and cell invasion [102]. Furthermore, long non-coding RNA HOTAIR promotes the colocalization of VAMP3 with SNAP23 leading to MVBs fusion with the plasma membrane and exosome secretion in hepatocellular carcinoma [147]. The HOTAIR also activates autophagy by upregulating ATG3 and ATG7 [148]. Long intergenic noncoding RNA 00511 (LINC00511) promotes exosome secretion by regulation of the expression of RAB27B and the colocalization of VAMP7 and SNAP23, which are involved in MVBs trafficking and their fusion with the plasma membrane [149]. LINC00511 is highly expressed in diverse cancers and correlates with poor clinical outcomes [150]. LINC00511 knockdown suppressed proliferation, invasion and autophagy in trophoblast cells [151]. Exosome secretion was also promoted by cortactin through stabilizing cortical actin-rich MVBs docking sites [152].
Fig. 6.

Multivesicular bodies and autophagy. After maturation of early endosomes to multivesicular bodies (MVBs), MVBs can fuse with the plasma membrane to release intraluminal vesicles (ILVs) to the extracellular space as exosomes. With the help of specific proteins, MVBs are trafficked towards the plasma membrane and/or lysosome. Under certain conditions, MVBs can fuse with autophagosomes to generate hybrid organelles called amphisomes. Amphisomes contain typical autophagosomal markers such as lipidated LC3, and due to their origin from endosomes, they also contain endosomal markers such as CD63, RAB5, RAB7, and RAB11. The fusion of MVBs with the lysosome (direct or via autophagosome) results in autophagic degradation. The MVBs-related secretion and autophagy pathways are connected via many proteins, including RAB GTPases, ESCRTs, SNAREs, Beclin1, ATG proteins, and LC3
Nevertheless, in healthy cells, the majority of MVBs fuse with the lysosomes, resulting in degradation of their content. ISGylation of the MVB protein TSG101 by ISG15 (interferon-α/β-induced ubiquitin-like protein) induces its aggregation and degradation, being sufficient to impair exosome secretion. The secretion of exosomes is recovered when the fusion of MVBs with lysosomes or autophagosomes is inhibited, indicating that the inhibition of exosome secretion is mainly mediated by the induction of MVBs degradation by the lysosomes. ISGylation reduces the number of MVBs but does not inhibit the formation of ILVs. It also promotes selective autophagy and degradation of MVBs without inducing a global autophagy response [153]. The fusion of MVBs with lysosomes can be also triggered via the ubiquitination of TSG101 by MGRN1 [154].
Interestingly, ubiquitination of the cargo proteins can also profoundly influence the fate of MVBs. While MVBs containing ubiquitinated major histocompatibility complex (MHC-II) underwent lysosomal degradation in immature dendritic cells, non-ubiquitinated MHC-II placed into CD9-bearing MVBs was targeted for plasma membrane fusion and secretion in activated dendritic cells. Sorting of MHC II into exosomes correlated with its incorporation into CD9 containing detergent-resistant membranes [155]. Exosomes are probably preferentially secreted from cholesterol-rich detergent-resistant MVBs whereas the cholesterol-poor MVBs are subjected to degradation [156, 157]. The alternative fate of MVBs can be also governed by their connection to dynein or kinesin. Connection to dynein motor protein (minus end-directed transport) leads to perinuclear accumulation of MVBs and lysosomal degradation of cargo by recruitment of HOPS complex. By contrast, connection to kinesin (plus end-directed transport) causes accumulation of MVBs at cell periphery [158, 159].
Different autophagic effectors were shown to influence the biogenesis of extracellular vesicles and their secretion. Exosome production is strongly reduced in cells lacking ATG5 and ATG16L1 as these proteins protect MVBs from lysosomal degradation and direct them into the secretory pathway. ATG5 detaches V1V0-ATPase (vacuolar proton pump) from the MVBs via LC3 which specifically decreases acidification of MVBs lumen [160]. Emerging exosomes were strongly enriched in LC3-II (versus LC3-I) compared with the ratio of LC3-II to LC3-I in the corresponding cell lysates. This suggests that ATG5 and LC3 are sorted into these exosomes [160]. On the other hand, overexpression of LC3 or autophagy inducers such as starvation or rapamycin caused an enlargement of the vacuoles decorated with RAB11 and their colocalization with LC3. This situation led to the inhibition of exosome release. Even though RAB11 activity stimulates MVBs genesis and exosome release [161], the stimulatory effect of RAB11 in exosome secretion was nullified by overexpression of the autophagic protein LC3 [10].
Under certain conditions (such as shear stress [162]), autophagosomes fuse with MVBs to generate hybrid organelles called amphisomes [13]. Amphisomes contain typical autophagosomal markers such as lipidated LC3, and due to their origin from endosomes, they also contain endosomal markers such as CD63, RAB5, RAB7, and RAB11 [163]. Maturation of MVBs by ESCRT machinery is required for their fusion with autophagosomes [164] and the fusion of MVBs with the autophagosome compartment seems to be calcium- and VAMP3-dependent event [10, 163]. The resulting amphisomes are either degraded by fusion with lysosomes or released from the cell [162, 165]. ATG9 is required to form intraluminal vesicles in amphisomes and/or autolysosomes and is also needed for local acidification within amphisomes/autolysosomes [166]. The fusion between amphisomes and lysosomes requires VAMP7 but not VAMP3 [163]. VAMP7 is also needed for the homotypic fusion of ATG16L1 precursors, which is a key event in the early phases of autophagy enabling membrane acquisition and autophagosome biogenesis [167]. VAMP7-labelled vesicles can be loaded with ATP and starvation triggers the delivery of the ATP-containing amphisomes toward the plasma membrane, their release, and the import of ATP to the extracellular space [168].
Amphisomes, but not exosomes, were shown to be vehicles for the active release of DNA from the cell [169]. The formation of an amphisome may negatively regulate the coordination between exosome secretion and autophagy. For example, rapamycin or starvation promoted MVBs-autophagosome fusion and reduced exosome secretion in the K562 cell line [10]. On the other hand, the exosome release of toxic/damaged material may provide a cellular mechanism bypassing the autophagic defects caused by ageing or different pathological states [170].
Amphisomes can participate in immune response as they can function as anti-viral machinery by sequestering and exporting viral proteins from the cell [171]. IFNγ is a cytokine critical for innate and adaptive immunity against viral infections. Upon IFNγ-induced autophagy in lung epithelial cells, amphisomes containing annexin 2 (ANXA2) were released. This process was dependent on ATG5, RAB11, and RAB27A [165].
Interleukins IL-1β and IL-18 are potent pro-inflammatory cytokines crucial for responses to infection and injury. Secretion levels of these cytokines increased when low-autophagy cells were treated with the autophagy-inducing tat-Beclin1 peptide and decreased when ATG7 was silenced in high-autophagy cells [172]. An unobstructed autophagy pathway and functional MVBs are necessary for inflammasome-dependent IL-1β and IL-18 secretion [173, 174]. Nevertheless, it is not completely clear if IL-1β is secreted by amphisomes or by modified autophagosomes. Similarly to the degradative autophagosomes, modified secretory autophagosomes have a double membrane decorated with LC3-II. In degradative autophagosomes, STX17 manages the fusion with the lysosome. However, in secretory autophagosomes, SEC22B in combination with plasma membrane syntaxin 3 and syntaxin 4 as well as SNAP23 and SNAP29 facilitate fusion with the plasma membrane and cargo secretion [175, 176]. Autophagy-dependent secretion of IL-1β, IL-6, CSF3/G-CSF, CXCL1, TREM1, CCL2, CCL3/MIP-1α, and CXCL2 in response to UVB radiation was also described. Secretion of these cytokines was blocked by conditional ATG7 depletion [177].
Another factor interconnecting cell stress and immune response with autophagy and amphisome genesis is IκB kinase (IKKβ). IKKβ is the predominant catalytic subunit of the IKK complex and is required for the activation of the canonical NF-κB signalling pathways. IKKβ activation also induces the fusion of MVBs with autophagosomes to form amphisomes and promotes their secretion in tumour cells. This secretion was absent or strongly reduced when autophagosome formation was impaired by 3-methyladenine or ATG7 inactivation [178]. In breast cancer cells independent of autophagy, ATG7 inhibition by shRNAs increased IL-6 secretion. On the other hand, in autophagy-dependent cells, ATG7 inhibition decreased IL-6 secretion, cell survival and mammosphere formation [179]. Besides RNA interference-mediated ATG7 depletion, pyrazolopyrimidine sulfamate compounds were found to be potent selective inhibitors of ATG7 [180].
In cancer cells, selective and non-selective autophagy and EVs secretion are often amplified because of harsh conditions in TME, such as hypoxia or ER stress [181–183]. Some regulators such as GAIP interacting protein C terminus (GIPC) control both EVs and autophagy pathways [184]. GIPC knockdown led to significant inhibition of pancreatic carcinoma growth in an orthotopic mouse model [185]. The crosstalk between endosome-related secretory pathways and autophagy orchestrates the intratumoural communication as autophagy significantly impacts not only the quantity of EVs but also their content. On the other hand, EVs can significantly influence the dynamic of autophagy in TME [186, 187]. Exosomes derived from breast cancer cells stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote cancer progression [187]. It was also shown that hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction [188] and MCF-7 breast cancer cells with undetectable MMP2 protein acquired expression of MMP2 and corresponding gelatinase activity after stimulation with exosomes derived from mesenchymal stromal stem cells [189].
Autophagy modulators and their effect on EVs secretion
Autophagy is a multi-step process. Each step can potentially be inhibited. Progress within the field has led to the development of agents targeting almost all phases of this process (see Fig. 7). Targeting the specific stage of autophagy may profoundly influence resulting secretory pathways as the early-stage autophagy inhibition does not seem to be equivalent to the late-stage inhibition. Furthermore, one compound (such as tacrine-melatonin heterodimer C10) can induce the early stages of autophagy and inhibit it at the late stages. Transitory treatment by C10 induced secretion of proinflammatory cytokine IL-6, proving interconnection between autophagy and secretory pathway [190]. Some amount of IL-6 produced was found to be secreted by exosomes [191, 192]. On the other hand, IL-6 inhibits starvation-induced autophagy and activates stress-induced autophagy via the STAT3 signalling pathway [193, 194].
Fig. 7.

Modulators of autophagy and their effect on EVs release. In nutrient-rich conditions, mTORC1 constitutively blocks the ULK complex and autophagy. mTORC1 signals can be inhibited directly by C10, rapamycin, exercise, or starvation and indirectly by bafilomycin A1 (BAFA1) through lysosomal inhibition. Physical exercise was shown to induce the release of small extracellular vesicles (EVs) into the circulation [195]. The ULK complex activates the VPS34 complex. VPS34 is a class III phosphatidylinositol 3-phosphate-kinase (PI3KC3). A group of PI3K inhibitors, including 3-methyladenine (3-MA), wortmannin, and synthetic inhibitor LY294002, inhibits both class I as well as class III PI3Ks. VPS34 inhibitors include Spautin-1, autophinib, SAR405, and VPS34-IN1. Spautin-1 initiates the degradation of Beclin1 due to the inhibition of two of its deubiquitinases. SAR405 and VPS34-IN1 are highly potent inhibitors of VPS34 selective for the VPS34 and not affecting the closely related class I and class II PI3Ks. Autophinib is an ATP-competitive inhibitor of VPS34 decreasing the accumulation of the lipidated protein LC3 on the autophagosomal membrane. The late stages of the autophagic machinery include fusion and degradation. During fusion, the mature autophagosome fuses with lysosomes creating an autolysosome. PIKfyve (phosphoinositide kinase, FYVE-type zinc finger containing) inhibitors and EACC block autophagosome-lysosome fusion. BAFA1 inhibits the acidification of the autolysosome by blocking the V-ATPase while chloroquine (CQ) and 3-hydroxychloroquine (HCQ) impair the maturation of autolysosomes. All drugs are depicted within the rectangles. Effects of modulators activating autophagy are green, inhibitory effects are red
ULK1 inhibitors
ULK1 is a serine/threonine-protein kinase. In most cell lines, loss of ULK1 is sufficient to disrupt autophagy [196]. However, ULK2 acts with a degree of redundancy with ULK1 [197]. MRT68921 potently inhibits both ULK1 and ULK2. ULK1 inhibition results in the accumulation of immature early autophagosomal structures, indicating a role for ULK1 in the initiation and maturation of autophagosomes [198]. ULK1 activity also manages the trafficking of ATG9 [199] which is required for intraluminal vesicle formation within amphisomes and autolysosomes [166]. Consequently, inhibition of ATG9 causes a reduced capacity to degrade endosomal cargo, which may result in enhanced EVs secretion. Amino acid starvation or rapamycin causes a redistribution of ATG9 from the trans-Golgi network to peripheral, endosomal membranes. The redistribution of ATG9 requires PI3K activity and is reversed after the restoration of amino acids [199]. On the other hand, AMPK-ULK1-mediated but mTOR-independent signalling plays an important role in the induction of autophagy-mediated PARK7 secretion. 6-hydroxydopamine-induced oxidative stress triggered PARK7 secretion which was suppressed by co-treatment with MRT68921 [200]. Nevertheless, PARK7 seems not to be secreted by classical exosomes [169]. MRT68921 can also disrupt the signals between lysosomes and autophagic machinery. The lysosomal calcium channel TRPML1 connects lysosomal calcium to autophagosome biogenesis through the triggering of the CaMKKβ/VPS34 pathway. TRPML1-mediated generation of PI3P requires functional VPS34 and ULK1 [201]. RNAi-mediated knockdown of ULK1 and/or ULK2 resulted in impaired endocytosis of nerve growth factor (NGF) [202]. MRT68921 was also shown to be a potent inhibitor of NUAK1 (NUAK family SNF1-like kinase 1) which is a critical component of the antioxidant defence necessary for the survival of tumour cells during cytotoxic therapy and EMT. As cytotoxic therapy induces elevated ROS levels and triggers the ULK1 pathway to activate protective autophagy and mitophagy, dual targeting of NUAK1 and ULK1 by MRT68921 can be beneficial in tumour management [203]. ULK1 inhibition also overcomes compromised antigen presentation in LKB1 (liver kinase B1)-mutant lung cancer [89].
Pan-PI3K inhibitors
Phosphoinositide 3-kinases (PI3Ks) are divided into three classes: class I, class II, and class III. Class I PI3Ks predominantly produce PI(3,4,5)P3 (and indirectly, PI(3,4)P2), class III PI3Ks synthesise PI3P, and to a lesser extent, class II PI3Ks synthesise PI3P and PI(3,4)P2). PI(3,4)P2 was described as a key mediator of the late stages of clathrin-mediated endocytosis [126]. PI(3,4,5)P3 at the plasma membrane recruits effectors such as the protein kinase B/Akt. Active Akt controls many downstream pathways such as the mTORC1 pathway. Activated mTORC1 destabilizes the ULK1 complex and inhibits autophagy [204]. The role of PI3P has been discussed above.
A group of PI3K inhibitors includes 3-methyladenine (3-MA), wortmannin, and synthetic inhibitor LY294002. These compounds inhibit class I PI3Ks as well as class III PI3Ks. Wortmannin and 3-MA have been routinely used as autophagy inhibitors due to their suppressive effect on class III PI3Ks. Nevertheless, 3-MA with a prolonged period of treatment (up to 9 hours) was also found to promote autophagy even under nutrient-rich conditions. 3-MA also does not inhibit Beclin1-independent autophagy [205], however, it can still suppress starvation-induced autophagy [206]. In contrast, wortmannin suppresses autophagy regardless of the nutrient status [206]. The effect of 3-MA (but not wortmannin) is further complicated by differential temporal effects on class I and class III PI3Ks. 3-MA persistently blocks class I PI3Ks, whereas its impact on class III PI3Ks is only transient [206]. Furthermore, activation of autophagy was shown in gastric cancer cell line SGC7901 due to LY294002 treatment [207]. Another problem is that LY294002, and to a lesser extent wortmannin (but not 3-MA [208]), can inhibit proteosynthesis [209] and 3-MA can induce a decrease in cell viability not driven by the inhibition of the Akt/mTOR axis. The cytotoxicity induced by 3-MA correlated with massive DNA damage monitored by γ-H2AX and was observed using the 10 mM concentration, the usual concentration used to inhibit autophagy [210].
Off-target activities of PI3K inhibitors influence proteasome degradation, PI3K/Akt signalling pathways, endocytosis, lysosomal acidification, mitochondrial permeability transition, and glycogen metabolism [211, 212]. Moreover, it is known that PI3Ks also participate in the biogenesis of MVBs and their activity is required for the correct maturation of the ILVs [10, 129]. As would be expected, factors having some effect on the formation of MVBs also affect the secretion of exosomes. Consequently, conventional exosome production can be decreased by PI3K inhibitors [213–215]. Accordingly, wortmannin reduced the secretion of prostasomes from PC-3 cells [216]. On the other hand, inhibition of autophagy by wortmannin or CRISPR/Cas9-mediated knockout of ATG5 greatly increased the release of exosome-associated prions [217]. In fibroblasts, wortmannin caused swollen endosome phenotype coupled with the failure of membrane recycling but not the inhibition of MVBs biogenesis [218]. Nevertheless, wortmannin can change the inner content of ILVs [129]. The efficacy of wortmannin may be diminished by its covalent binding to free amino acids [219].
Inhibition of autophagy with 3-MA or wortmannin can have profound effects on cytokine secretion. In macrophages, toll-like receptor ligands initiate sequestration of pro-IL-1β into autophagosomes and activation of autophagy with rapamycin triggered the degradation of this sequestrated pro-IL-1β and blocked secretion of the mature cytokine. When treated with 3-MA or wortmannin, LPS-stimulated bone marrow-derived macrophages (iBMM) and dendritic cells (BMDC) secreted increased levels of IL-1β. At the 10 mM concentration, 3-MA induced IL-1β but inhibited IL-6 secretion from BMDC and iBMM. In contrast, 3-MA and wortmannin markedly reduced IL-1β secretion induced by LPS + ATP in human neutrophils [220]. 3-MA, in combination with LPS, increased IL-1α secretion by BMDC and IL-18 secretion by iBMM. 3-MA also activates the inflammasome through inhibition of type III PI3Ks [221]. Wortmannin was found to enhance IL-12 production in dendritic cells [222]. In contrast, LY294002 prevented IL-12 secretion in dendritic cells [209].
VPS34 inhibitors
VPS34 is involved not only in autophagy but also in the endosomal trafficking of receptors such as the epidermal growth factor receptor (EGFR) [129]. In addition to managing autophagy induction in complex I, VPS34 also has a role in the fusion of autophagosomes with lysosomes. Disruption of neuronal VPS34 function impairs autophagy, lysosomal degradation, as well as lipid metabolism, causing endolysosomal membrane damage. PI3P deficiency caused by a malfunction of VPS34 also promotes the secretion of unique exosomes enriched for undigested lysosomal substrates [223]. Considering the key role of VPS34 in autophagy, many compounds aim to target this kinase. The following section will present the characterization of VPS34 inhibitors Spautin-1, autophinib, SAR405, VPS34-IN1, and Cpd18.
Spautin-1 does not directly affect the catalytic activity of VPS34 but promotes the degradation of VPS34 complexes by inhibiting ubiquitin-specific peptidases USP10 and USP13. Under glucose-free conditions, Spautin-1 supports the ubiquitination of Beclin1 and triggers its degradation leading to destabilization and degradation of VPS34 complexes and inhibition of autophagy. VPS34 complexes also provide a molecular mechanism for class III PI3K to control the levels of p53. Therefore, levels of p53 are reduced in the tissues of BECN1+/− mice [224]. Exosome production was found to be regulated by the p53 response as up-regulation of TSAP6 transcription by activated p53 can increase exosome release [225]. The destabilization of the PI3K complex that occurs upon suppressing Beclin1 (either via siRNA-mediated knockdown or Spautin-1 treatment) reduced both exosome release and autophagy flux in chronic myeloid leukaemia cells [226]. Alternatively, Beclin1 may regulate autophagosome formation [227] and fusion of endosomes and autophagosomes leading to amphisome formation [228, 229]. In addition, it was also found that Beclin1 is needed for autophagosome fusion with lysosomes [230].
Autophinib is an ATP-competitive inhibitor of VPS34 decreasing the accumulation of the lipidated protein LC3 on the autophagosomal membrane. It inhibits autophagy induced by rapamycin or by amino acid starvation. The in vitro IC50 value for VPS34 is 19 nM [231]. Since VPS34 has also a role in the fusion of autophagosomes with lysosomes, VPS34 inhibition causes autolysosomal dysfunction, a phenotype with large late endosomes and an enhanced release of atypical exosomes harbouring poly-ubiquitinated proteins [136–138].
SAR405 and VPS34-IN1 are highly potent and selective inhibitors of VPS34 not affecting the closely related class I and class II PI3Ks. SAR405 and VPS34-IN1 cause defects in autophagosome formation and endosomal trafficking [232]. SAR405 can inhibit autophagy induced by starvation and/or mTOR inhibition [233]. SAR405 prevents the catalytic activity of ATG14L and UVRAG-containing VPS34 complexes, induces late endosome swelling, and affects late endosome-lysosome compartments [233]. Using SAR405 decreased the tumour growth and improved mouse survival in multiple tumour models by inducing tumour infiltration of NK, CD8+, and CD4+ T effector cells [96] and repressed viability of liver cancer stem cells [97]. Nevertheless, VPS34 function is critical in dendritic cells where it controls antigen cross-presenting. Consequently, VPS34 inhibition may lead to impaired T-cell–mediated immunity that may limit the use of SAR405 in anticancer therapy [99].
VPS34-IN1 selectively decreased PI3P levels, increased Beclin1 levels, but did not downregulate its other interacting partners from complex I, namely ATG14L, and VPS15 (in contrast to knockout of VPS34), ruling out indirect effects of destabilization of these proteins [223]. PI3P deficiency was shown to promote the secretion of unique exosomes enriched in undigested lysosomal substrates, including amyloid precursor protein C-terminal fragments, specific sphingolipids, and the phospholipid bis(monoacylglycero)phosphate. Secretion of these exosomes needs neutral sphingomyelinase 2 and sphingolipid synthesis. It was noted that proteins typically associated with exosomes such as Alix and CD63, or with ESCRT-dependent ILV sorting (TSG101 and Hrs) were minimally affected, suggesting that VPS34-IN1 likely affects composition rather than quantity of extracellular vesicles [223]. Neurons treated with VPS34-IN1 showed delayed degradation of EGFR following EGF stimulation. The remaining degradation was blocked by V-ATPase inhibitor bafilomycin A1, suggesting that VPS34 inhibition only partially impairs lysosomal function [223].
Cpd18 and 3-MA are structurally related compounds that differ only in a methyl piperidine group at the C6 of the adenine, but unlike 3-MA, Cpd18 does not inhibit class I PI3Ks. Cpd18 inhibits omegasome formation [234]. Nevertheless, the concentrations of Cpd18 that presented a greater attenuation of the autophagic flux are associated with cytotoxicity [210] and decreased ubiquitin/proteasome-dependent proteolysis in living cells [234].
Late autophagy inhibitors
Another way to inhibit autophagy is to target the later stages of the autophagy machinery, such as the fusion of autophagosomes with lysosomes and the degradation of autolysosome content by the lysosomal enzymes. Autophagosome–lysosome fusion involves the action of SNAREs. Autophagosome-lysosome fusion is orchestrated by the autophagosomal SNAREs STX17 and SNAP29, lysosomal R-SNARE VAMP8, HOPS tethering complex, small GTPase RAB7, and accessory proteins such as ATG14. Interestingly, the translocation of STX17 occurs only on complete autophagosomes and not on partially formed autophagosomes. EACC blocks autophagosome-lysosome fusion by preventing STX17 and SNAP29 loading onto autophagosomes without impeding the completion of autophagosomes. It also causes an accumulation of LC3-II and reduces the interaction of STX17 with the HOPS subunit VPS33A and the lysosomal VAMP8. On the other hand, autophagy induction, the number of autophagosome biogenesis sites, expansion of the phagophore, lysosomal pH, localization of lysosomal SNAREs or RABs, and cargo recognition remain unaltered in the presence of EACC [235] although STX17 was also shown to be involved in autophagy initiation [46]. Interestingly, STX17 depletion increased the production of exosomes in A549 cells [236] and effectively blocks the formation of axonal amphisomes after 3 hours starvation in dorsal root ganglion neurons [237].
Chloroquine (CQ) and its derivatives (such as 3-hydroxychloroquine HCQ or Lys05) inhibit the maturation of autolysosomes and block late steps of autophagy. In contradiction with previous studies [238], some more recent studies indicate that CQ does not substantially decrease lysosomal acidity, and the lysosomes retain their capacity to degrade delivered material [239]. Although CQ may induce a temporal elevation of lysosomal pH, this elevation may be only transient and can be followed by reacidification of the lysosomes [240]. The kinetics of this transient phase may differ between cell types [239]. The greater part of the confusion about CQ effects on lysosomal pH might be attributed to how it was measured because LysoTracker Red dye, often used to estimate lysosomal pH, is not a pH sensor and the intensity of its fluorescence signal does not correlate with the lysosomal pH [239]. Nevertheless, CQ behaves as a weak base and accumulates in the lysosomes causing lysosomal stress. Lysosomal stress may cause the release of EVs to eliminate cellular waste [223]. Accordingly, the production of exosomes was increased due to CQ treatment [236]. CQ treatment led to marked lysosomal swelling and recruitment of Galectin-3 to sites of membrane damage [241]. In response to lysosomal damage, IL-1β can be recognized by secretory autophagy cargo receptor TRIM16 [176]. Strikingly, glucose starvation or hexokinase inhibition by 2-deoxyglucose prevented CQ from inducing lysosomal damage and subsequent cell death [241]. Accordingly, IL-1β release correlates with the degree of lysosome damage [242] and glucose is required for LPS-induced IL-1β production by monocytes [243]. Furthermore, autophagy inhibition with CQ also induced the secretion of pro-inflammatory cytokines MIF (Macrophage migration inhibitory factor) and IL-6 in triple-negative breast cancer cells [244].
Although CQ and HCQ are indisputably impairing the autophagic flux, their use entails multiple side effects including the disorganization of the Golgi and endo-lysosomal networks, dysregulation of STX17 and SNAP29 targeting, and even a temporary induction of autophagic sequestration activity (probably by inhibiting mTORC1 in a Rag-dependent manner [245]) and a drop in ATP content [227, 239]. Furthermore, during starvation or CQ-induced lysosomal stress, TFEB and TFE3 rapidly translocate to the nucleus to initiate lysosomal biogenesis [240]. CQ was also shown to increase amphisome and IFN-α production in human plasmacytoid dendritic cells stimulated by the Herpes simplex virus. On the other hand, when Beclin1 was knocked down, virus-induced IFN-α production was significantly suppressed [246].
Bafilomycin A1 (BAFA1) is a V-ATPase inhibitor that blocks the autophagic flux by inhibiting autophagosome-lysosome fusion (possibly by inhibiting the ATP2A/SERCA pump [247]) and autolysosomal and/or lysosomal acidification. It also reduces the delivery of internalized molecules from MVBs to lysosomes [248]. On the other hand, trafficking through early and late endosomes continues upon BAFA1 treatment [249]. BAFA1-treated cells display phenotypes associated with an inhibition of the degradation capacity of lysosomes such as the presence of intact cytoplasm in the lysosomal lumen and a loss of acidity [239]. BAFA1 inhibits lysosomal degradation and thereby negatively affects the amino acid efflux from the lysosomes, possibly impairing mTOR signalling which is dependent on this organelle (mTOR localizes to lysosomes and its activation depends on amino acids inside the lysosomal lumen) [245]. mTOR inhibition strongly decreased exosomal prion release [217]. In contrast, starvation stimulated exosome release through a RAB27a-dependent mechanism but did not significantly alter exosomal cargo content [250]. BAFA1 also triggers BAX- and/or BAK-dependent cytotoxicity and caspase activation [210] and has indirect effects on Golgi trafficking [251]. According to some indications, BAFA1 can also inhibit signals from the lysosomal P5-type ATPase ATP13A2 (also known as PARK9) [252]. ATP13A2 has been found to regulate both autophagic degradation and exosomal release [253]. Elevated levels of ATP13A2 enhance the externalization of α-synuclein through amphisomal structures, which is proposed to be accomplished through ATP13A2-mediated modulation of intraluminal zinc ion levels in MVBs [254, 255]. On the other hand, the exosomal release was enhanced due to BAFA1 treatment [153, 236]. Inhibition of autophagy with BAFA1 markedly reduced IL-1β secretion induced by LPS+ATP in human neutrophils [220].
The principal enzymatic activity of PIKfyve (phosphoinositide kinase, FYVE-type zinc finger containing) is to phosphorylate PI3P to PI(3,5)P2. PIKfyve inhibitors, such as YM201636, vacuolin-1, and apilimod mesylate, disrupt lysosome turnover, the heterotypic fusion of lysosomes with autophagosomes and/or MVBs, and the formation of autolysosomes resulting in autophagy inhibition [256]. Acute inhibition of PIKfyve also blocks protein sorting and their turnover in late endosomes. Both PI(3,5)P2-deficient cells and cells that lack TRPML1 exhibited enlarged endolysosomes and trafficking defects in the late endocytic pathway [257]. Furthermore, PI(3,5)P2 is required for lysosomal acidification by the V-ATPase [258]. Consequently, PIKfyve inhibition enhances exosome release and triggers secretory autophagy in PC-3 cells (probably to relieve stress caused by disruption of recycling pathways). These exosomes bear the typical exosomal markers (TSG101, Alix) and a subset of ATGs [259].
Conclusion
Autophagy and MVBs-related secretory pathways are interconnected at many levels. These pathways, collectively with the ubiquitin-proteasome system, orchestrate the dynamics of intracellular waste removal, where each pathway may complement the deficiencies of the other. In other words, exosome secretion can reduce stress when degradative and recycling pathways are disrupted. On the other hand, unwanted MVBs with damaged material may be directed to lysosomes. Furthermore, some parts of functional autophagy machinery are important for the genesis of endosomes and amphisomes. Consequently, autophagy inhibition can both promote and/or decrease EVs release. The resulting effect is largely context-dependent and could be significantly affected by different kinds of autophagy modulators. Moreover, modulation of autophagy significantly impacts not only EVs quantity but probably also their content. Late and early autophagy inhibitors can have a profoundly different effect on secretion. For example, Spautin-1 and CQ are both autophagy inhibitors but have nearly opposite effects on EVs release. Many studies suggest that cancer cells release higher amounts of EVs compared to non-malignant cells, which makes the effect of autophagy inhibitors on EVs secretion highly important and attractive for anticancer therapy. In future studies, it should be carefully assessed how exactly autophagy could be targeted (late versus early autophagy inhibitors) to maximize patient benefit and improve cancer therapy. For safe and successful clinical use of autophagy inhibitors, we need to carefully explore the molecular mechanisms underlying the effects of autophagy on tumour progression and possibly discover all pathways affected by particular autophagy inhibitors (see Table 2).
Table 2.
Inhibitors of autophagy
| Drug | Systematic name | Known targets | Status | Limitations | References |
|---|---|---|---|---|---|
| MRT68921 | N-[3-[[5-Cyclopropyl-2-[(1,2,3,4-tetrahydro-2-methyl-6-isoquinolinyl)amino]-4-pyrimidinyl]amino]propyl]-cyclobutanecarboxamide dihydrochloride | ULK1, ULK2, NUAK1 | Preclinical | Mitotic dysregulation; crosstalk with endocytic pathways | [166, 202, 203, 260] |
| 3-MA | 3-Methyladenine | PI3Ks | preclinical | non-selectivity; activation of autophagy in a longer period of treatment; cytotoxicity; crosstalk with endocytic pathways | [206, 210–212] |
| Wortmannin | (1alpha,11alpha)-11-(Acetyloxy)-1-(methoxymethyl)-2-oxaandrosta-5,8-dieno(6,5,4-bc)furan-3,7,17-trione | PI3Ks, DNA-PK | preclinical | failed clinical translation due to drug-delivery challenges; proteosynthesis inhibition; crosstalk with endocytic pathways | [209, 211, 261] |
| LY294002 | 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one | PI3Ks | preclinical | activation of autophagy; proteosynthesis inhibition; crosstalk with endocytic pathways | [207, 209, 211, 212] |
| Spautin-1 | C43, 6-Fluoro-N-[(4-fluorophenyl)methyl]-4-quinazolinamine | USP10, USP13 | preclinical | ROS-mediated DNA damag; Beclin1 degradation; crosstalk with endocytic pathways | [224, 228, 229, 262] |
| Autophinib | 6-Chloro-N-(5-methyl-1H-pyrazol-3-yl)-2-(4-nitrophenoxy)-pyrimidinamine | VPS34 | preclinical | pleiotropic impact of VPS34 inhibition; impaired T-cell–mediated immunity | [99, 231] |
| SAR405 | (8S)-9-[(5-chloro-3-pyridinyl)methyl]-6,7,8,9-tetrahydro-2-[(3R)-3-methyl-4-morpholinyl]-8-(trifluoromethyl)-4H-pyrimido[1,2-a]pyrimidin-4-one | VPS34 | preclinical | pleiotropic impact of VPS34 inhibition; defects in endosomal trafficking; impaired T-cell–mediated immunity | [99, 232, 233] |
| VPS34-IN1 | 1-((2-((2-chloropyridin-4-yl)amino)-4'-(cyclopropylmethyl)-[4,5'-bipyrimidin]-2'-yl)amino)-2-methylpropan-2-ol | VPS34 | preclinical | pleiotropic impact of VPS34 inhibition; defects in endosomal trafficking; impaired T-cell–mediated immunity | [99, 232] |
| Cpd18 | 3-methyl-6-(3-methylpiperidin-1-yl)-3H-purine | omegasomes | preclinical | toxicity; decreased ubiquitin/proteasome-dependent proteolysis | [210, 234] |
| Chloroquine | 4-N-(7-chloroquinolin-4-yl)-1-N,1-N-diethylpentane-1,4-diamine | autolysosomes; lysosomes; endosomes | 21 clinical trials phase 1 or 2; only one phase 3 study (NCT00224978) | lysosomal stress; crosstalk with endocytic pathways; uptake may differ based on pH | [223, 236, 241, 244, 246, 263] |
| HCQ | 2-[4-[(7-chloroquinolin-4-yl)amino]pentyl-ethylamino]ethanol | autolysosomes; lysosomes; endosomes | 94 clinical trials; only one phase 2/3 study (NCT03008148) | uptake may differ based on pH | [227, 239, 264] |
| Bafilomycin A1 | (3Z,5E,7R,8S,9S,11E,13E,15S,16R)-16-[(2S,3R,4S)-4-[(2R,4R,5S,6R)-2,4-dihydroxy-5-methyl-6-propan-2-yloxan-2-yl]-3-hydroxypentan-2-yl]-8-hydroxy-3,15-dimethoxy-5,7,9,11-tetramethyl-1-oxacyclohexadeca-3,5,11,13-tetraen-2-one | V-ATPase | preclinical | cytotoxicity and caspase activation; effects on Golgi trafficking; crosstalk with endocytic pathways | [210, 251, 265] |
| YM201636 | 6-amino-N-[3-(6-morpholin-4-yl-8-oxa-3,5,10-triazatricyclo[7.4.0.02,7]trideca-1(9),2(7),3,5,10,12-hexaen-4-yl)phenyl]pyridine-3-carboxamide | PIKfyve | preclinical | block of protein sorting; crosstalk with endocytic pathways and exocytosis | [257, 259] |
| Vacuolin-1 | 2-N-[(E)-(3-iodophenyl)methylideneamino]-6-morpholin-4-yl-4-N,4-N-diphenyl-1,3,5-triazine-2,4-diamine | PIKfyve | preclinical | block of protein sorting; crosstalk with endocytic pathways and exocytosis | [257, 259] |
| Apilimod mesylate | methanesulfonic acid;N-[(E)-(3-methylphenyl)methylideneamino]-6-morpholin-4-yl-2-(2-pyridin-2-ylethoxy)pyrimidin-4-amine | PIKfyve | preclinical | block of protein sorting; crosstalk with endocytic pathways and exocytosis | [257, 259] |
| EACC | ethyl (2-(5-nitrothiophene-2-carboxamido) thiophene-3-carbonyl) carbamate. | STX17 | preclinical | crosstalk with endocytic pathways and exocytosis | [236, 237] |
Acknowledgements
Not applicable
Abbreviations
- 3-MA
3-methyladenine
- AKT
Rac-alpha serine/threonine-protein kinase
- Alix
Apoptosis-linked gene 2-interacting protein X
- AMBRA1
Autophagy and Beclin 1 regulator 1
- AMPK
AMP-activated protein kinase
- ANXA2
Annexin 2
- Arf6
ADP ribosylation factor 6
- ATG
Autophagy-related genes
- ATP
Adenosine triphosphate
- ATP13A2
ATPase cation transporting 13A2
- Beclin1
Coiled-coil, moesin-like BCL2 interacting protein
- Bif-1
Bax-interacting factor 1
- BMDC
Bone marrow-derived dendritic cells
- CaMKKβ
Calcium/calmodulin-dependent protein kinase kinase β
- CCL
C-C motif chemokine ligand
- COPII
Coat protein complex II
- CQ
Chloroquine
- CSF3/G-CSF
Granulocyte colony-stimulating factor
- c-Src
Proto-Oncogene Tyrosine-Protein Kinase Src
- CXCL
C-X-C motif chemokine ligand,
- DEPTOR
DEP domain-containing mTOR interacting protein
- DFCP1
Double FYVE-containing protein 1
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial to mesenchymal transition
- ER
Endoplasmic reticulum
- ESCRT
Endosomal sorting complexes required for transport
- EVs
Extracellular vesicles
- FIP200
FAK family-interacting protein of 200 kDa
- G3BP
Ras GTPase-activating protein SH3-domain-binding protein
- GDP
Guanosine diphosphate
- GTP
Guanosine triphosphate
- GTPase
Guanosine triphosphatase
- HCQ
Hydroxychloroquine
- HOPS
Homotypic fusion, and protein sorting complex
- CHMP
Charged multivesicular body protein
- iBMM
Bone marrow-derived macrophages
- IFN-α
Interferon alpha
- IKKβ
IκB kinase
- IL
Interleukin
- ILVs
Intraluminal vesicles
- ISG15
Interferon (IFN)-α/β-induced ubiquitin-like protein
- LAMP2
Lysosome-associated membrane protein 2
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- LKB1
Liver kinase B1
- LPS
Bacterial lipopolysaccharides
- MGRN1
Mahogunin ring finger 1
- MHC
Major histocompatibility complex (MHC-II)
- MIF
Macrophage migration inhibitory factor
- mLST8
Mammalian lethal with Sec13 protein 8, alias GβL
- MMP
Matrix metalloproteinase
- mTOR
Mammalian target of rapamycin
- mTORC
Mammalian target of rapamycin complex
- MVBs
Multivesicular bodies
- NF-κB
Nuclear factor-kappa B
- NUAK1
NUAK family SNF1-like kinase 1
- p115
Vesicular transport factor p115
- PARK7
Parkinsonism-associated deglycase 7
- PI(3,4,5)P3
Phosphatidylinositol (3,4,5)-trisphosphate
- PI3K
Phosphoinositide 3-kinase
- PI3P
Phosphatidylinositide-3-phosphate
- PIKfyve
Phosphoinositide kinase, FYVE-type zinc finger containing
- PKA
Protein kinase A
- PLD2
Phospholipase D2
- PRAS40
Proline-rich Akt substrate of 40 kDa
- Rab
Ras-associated GTP-binding protein
- Rag
Ras-related GTP-binding protein
- RAPTOR
Regulatory-associated protein of mTOR also known as KIAA1303
- Rheb
Ras homolog enriched in brain
- SEC22b
SEC22 vesicle-trafficking protein homolog B
- shRNA
Small hairpin RNA
- siRNA
Small interfering RNA
- SNAP29
Synaptosome-associated protein 29
- SNARE
Soluble N-ethylmaleimide-sensitive fusion attachment protein receptors
- STAT3
Signal transducer and activator of transcription 3
- STX17
Syntaxin-17
- TBC-2
TBC domain-containing protein 2
- TFE3
Transcription factor binding to IGHM Enhancer 3
- TFEB
Transcription factor EB
- TME
Tumour microenvironment
- TREM1
Triggering receptor expressed on myeloid cells 1
- TRPML1
Transient receptor potential channel mucolipin 1
- TSC
Tuberous sclerosis complex
- TSG101
Tumuor susceptibility 101
- ULK
Unc51-like autophagy-activating kinase
- UVB
Ultraviolet light B
- UVRAG
UV resistance-associated gene
- VAMP
Vesicle-associated membrane protein
- V-ATPase
V1V0-vacuolar-type ATPase
- VPS
Vacuolar protein sorting
- WIPI
WD repeat domain phosphoinositide-interacting protein
- γ-H2AX
Phosphorylation of the Ser-139 of the histone variant H2AX
Authors’ contributions
MR wrote the manuscript, JB conceived the structure and revised the manuscript, MM revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Ministry of Health of the Czech Republic (NU20J-08-00018), by Grant Agency of the Czech Republic (GACR - 21-06873S), by funds from Specific University Research Grant, as provided by the Ministry of Education, Youth and Sports of the Czech Republic in the year 2021 (MUNI/A/1698/2020 and MUNI/A/1246/2020), by the “Center for Tumor Ecology – Research of the Cancer Microenvironment Supporting Cancer Growth and Spread” (reg. no. CZ.02.1.01/0.0/0.0/16_019/0000785) supported by the Operational Program Research, Development and Education and by the Ministry of Education, Youth and Sports of the Czech Republic, project Advanced Functional Nanorobots reg. No. CZ.02.1.01/0.0/0.0/15_003/0000444.
Availability of data and materials
Not applicable
Declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Martina Raudenska and Jan Balvan contributed equally to this work.
References
- 1.Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. doi: 10.1007/978-3-642-00302-8_3. [DOI] [PubMed] [Google Scholar]
- 2.Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. 2019;19(3):170–183. doi: 10.1038/s41577-018-0095-2. [DOI] [PubMed] [Google Scholar]
- 3.Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012;22(8):397–406. doi: 10.1016/j.tcb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salimi L, et al. Synergies in exosomes and autophagy pathways for cellular homeostasis and metastasis of tumor cells. Cell Biosci. 2020;10(1):64. doi: 10.1186/s13578-020-00426-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365–381. doi: 10.1038/s41580-018-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jovic M, et al. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol. 2010;25(1):99–112. doi: 10.14670/hh-25.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barysch SV, et al. Sorting in early endosomes reveals connections to docking- and fusion-associated factors. Proc Natl Acad Sci. 2009;106(24):9697–9702. doi: 10.1073/pnas.0901444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. doi: 10.1007/s00018-017-2595-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murrow L, Malhotra R, Debnath J. ATG12–ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat Cell Biol. 2015;17(3):300–310. doi: 10.1038/ncb3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fader CM, et al. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 cells. Traffic. 2008;9(2):230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- 11.Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol. 2016;36(3):301–312. doi: 10.1007/s10571-016-0366-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014;21(3):348–358. doi: 10.1038/cdd.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fengsrud M, et al. Ultrastructural characterization of the delimiting membranes of isolated autophagosomes and amphisomes by freeze-fracture electron microscopy. Eur J Cell Biol. 2000;79(12):871–882. doi: 10.1078/0171-9335-00125. [DOI] [PubMed] [Google Scholar]
- 14.Lucocq J, Walker D. Evidence for fusion between multilamellar endosomes and autophagosomes in HeLa cells. Eur J Cell Biol. 1997;72(4):307–313. [PubMed] [Google Scholar]
- 15.Klionsky DJ, Eskelinen E-L, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I'm confused. Autophagy. 2014;10(4):549–551. doi: 10.4161/auto.28448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1) Autophagy. 2021;17(1):1–382. doi: 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jewell JL, et al. GPCR signaling inhibits mTORC1 via PKA phosphorylation of Raptor. Elife. 2019;8. [DOI] [PMC free article] [PubMed]
- 19.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prentzell MT, et al. G3BPs tether the TSC complex to lysosomes and suppress mTORC1 signaling. Cell. 2021. [DOI] [PMC free article] [PubMed]
- 21.González A, Hall MN. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017;36(4):397–408. doi: 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sancak Y, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mutvei AP, et al. Rap1-GTPases control mTORC1 activity by coordinating lysosome organization with amino acid availability. Nat Commun. 2020;11(1):1416. doi: 10.1038/s41467-020-15156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kundu ST, et al. TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat Commun. 2018;9(1):2731. doi: 10.1038/s41467-018-05013-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karanasios E, et al. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun. 2016;7:12420. doi: 10.1038/ncomms12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karanasios E, et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci. 2013;126(22):5224–5238. doi: 10.1242/jcs.132415. [DOI] [PubMed] [Google Scholar]
- 29.Shima T, Kirisako H, Nakatogawa H. COPII vesicles contribute to autophagosomal membranes. J Cell Biol. 2019;218(5):1503–1510. doi: 10.1083/jcb.201809032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai K, et al. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J Cell Sci. 2016;129(20):3781–3791. doi: 10.1242/jcs.196196. [DOI] [PubMed] [Google Scholar]
- 31.Puri C, et al. ATG16L1 meets ATG9 in recycling endosomes: additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy. 2014;10(1):182–184. doi: 10.4161/auto.27174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 33.Boukhalfa A, et al. PI3KC2α-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat Commun. 2020;11(1):294. doi: 10.1038/s41467-019-14086-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chowdhury S, et al. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc Natl Acad Sci. 2018;115(42):E9792. doi: 10.1073/pnas.1811874115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. Elife. 2019;8. [DOI] [PMC free article] [PubMed]
- 36.Dancourt J, Melia TJ. Lipidation of the autophagy proteins LC3 and GABARAP is a membrane-curvature dependent process. Autophagy. 2014;10(8):1470–1471. doi: 10.4161/auto.29468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36(12):2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kharaziha P, Panaretakis T. Dynamics of Atg5-Atg12-Atg16L1 Aggregation and Deaggregation. Methods Enzymol. 2017;587:247–255. doi: 10.1016/bs.mie.2016.09.059. [DOI] [PubMed] [Google Scholar]
- 39.Agrotis A, et al. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy. 2019;15(6):976–997. doi: 10.1080/15548627.2019.1569925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M, et al. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286(9):7327–7338. doi: 10.1074/jbc.M110.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takahashi Y, et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun. 2018;9(1):2855. doi: 10.1038/s41467-018-05254-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weidberg H, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29(11):1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grunwald DS, et al. GABARAPs and LC3s have opposite roles in regulating ULK1 for autophagy induction. Autophagy. 2020;16(4):600–614. doi: 10.1080/15548627.2019.1632620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamoto M, Suzuki SO, Himeno M. The effects of dynein inhibition on the autophagic pathway in glioma cells. Neuropathology. 2010;30(1):1–6. doi: 10.1111/j.1440-1789.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- 45.Tong J, Yan X, Yu L. The late stage of autophagy: cellular events and molecular regulation. Protein Cell. 2010;1(10):907–915. doi: 10.1007/s13238-010-0121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar S, et al. Phosphorylation of Syntaxin 17 by TBK1 controls autophagy initiation. Dev Cell. 2019;49(1):130–144. doi: 10.1016/j.devcel.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat Commun. 2018;9(1):3492. doi: 10.1038/s41467-018-05449-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viret C, Faure M. Regulation of syntaxin 17 during autophagosome maturation. Trends Cell Biol. 2019;29(1):1–3. doi: 10.1016/j.tcb.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Choi YJ, et al. Inhibitory effect of mTOR activator MHY1485 on autophagy: suppression of lysosomal fusion. PLoS One. 2012;7(8):e43418. doi: 10.1371/journal.pone.0043418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Itakura E, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–5372. doi: 10.1091/mbc.e08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9(10):1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keulers TG, Schaaf MBE, Rouschop KMA. Autophagy-dependent secretion: contribution to tumor progression. Front Oncol. 2016;6(251). [DOI] [PMC free article] [PubMed]
- 54.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 55.Yue Z, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang MR, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217(5):702–706. doi: 10.1002/path.2509. [DOI] [PubMed] [Google Scholar]
- 57.Wible DJ, et al. ATG5 cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell Discov. 2019;5:42. doi: 10.1038/s41421-019-0110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ueno T, et al. Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy. 2008;4(5):692–700. doi: 10.4161/auto.6085. [DOI] [PubMed] [Google Scholar]
- 59.Hou W, et al. Mutation analysis of key genes in RAS/RAF and PI3K/PTEN pathways in Chinese patients with hepatocellular carcinoma. Oncol Lett. 2014;8(3):1249–1254. doi: 10.3892/ol.2014.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen X, et al. DEPTOR is an in vivo tumor suppressor that inhibits prostate tumorigenesis via the inactivation of mTORC1/2 signals. Oncogene. 2020;39(7):1557–1571. doi: 10.1038/s41388-019-1085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson CE, et al. Loss of tuberous sclerosis complex 2 sensitizes tumors to nelfinavir-bortezomib therapy to intensify endoplasmic reticulum stress-induced cell death. Oncogene. 2018;37(45):5913–5925. doi: 10.1038/s41388-018-0381-2. [DOI] [PubMed] [Google Scholar]
- 62.Chang WH, Lai AG. An integrative pan-cancer investigation reveals common genetic and transcriptional alterations of AMPK pathway genes as important predictors of clinical outcomes across major cancer types. BMC Cancer. 2020;20(1):773. doi: 10.1186/s12885-020-07286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zijian Z, et al. Research square. 2021. [Google Scholar]
- 64.Claude-Taupin A, et al. ATG9A is overexpressed in triple negative breast cancer and its in vitro extinction leads to the inhibition of pro-cancer phenotypes. Cells. 2018;7(12):248. doi: 10.3390/cells7120248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caron A, et al. DEPTOR at the nexus of cancer, metabolism, and immunity. Physiol Rev. 2018;98(3):1765–1803. doi: 10.1152/physrev.00064.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu B, et al. Downregulation of DEPTOR inhibits the proliferation, migration, and survival of osteosarcoma through PI3K/Akt/mTOR pathway. Onco Targets Ther. 2017;10:4379–4391. doi: 10.2147/OTT.S143518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen R, Yang Q, Lee J-D. BMK1 kinase suppresses epithelial–mesenchymal transition through the Akt/GSK3β signaling pathway. Cancer Res. 2012;72(6):1579–1587. doi: 10.1158/0008-5472.CAN-11-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parvani JG, et al. Deptor enhances triple-negative breast cancer metastasis and chemoresistance through coupling to survivin expression. Neoplasia. 2015;17(3):317–328. doi: 10.1016/j.neo.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pei L, et al. Overexpression of DEP domain containing mTOR-interacting protein correlates with poor prognosis in differentiated thyroid carcinoma. Mol Med Rep. 2011;4(5):817–823. doi: 10.3892/mmr.2011.503. [DOI] [PubMed] [Google Scholar]
- 70.Okamoto T, et al. FIP200 suppresses immune checkpoint therapy responses in breast cancers by limiting AZI2/TBK1/IRF signaling independent of its canonical autophagy function. Cancer Res. 2020;80(17):3580–3592. doi: 10.1158/0008-5472.CAN-20-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang D, et al. Downregulation of FIP200 induces apoptosis of glioblastoma cells and microvascular endothelial cells by enhancing Pyk2 activity. PLoS One. 2011;6(5):e19629. doi: 10.1371/journal.pone.0019629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu X-N, et al. Enhanced mLST8 expression correlates with tumor progression in hepatocellular carcinoma. Ann Surg Oncol. 2020;27(5):1546–1557. doi: 10.1245/s10434-020-08263-6. [DOI] [PubMed] [Google Scholar]
- 73.Chen S, et al. Inhibition of PI3K/Akt/mTOR signaling in PI3KR2-overexpressing colon cancer stem cells reduces tumor growth due to apoptosis. Oncotarget. 2017;8(31):50476–50488. doi: 10.18632/oncotarget.9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18(12):744–757. doi: 10.1038/s41568-018-0074-8. [DOI] [PubMed] [Google Scholar]
- 75.Zou Z, et al. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10(1):31. doi: 10.1186/s13578-020-00396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qi Z, et al. PRAS40 hyperexpression promotes hepatocarcinogenesis. EBioMedicine. 2020;51. [DOI] [PMC free article] [PubMed]
- 77.Zhu G, et al. PRAS40 promotes NF-κB transcriptional activity through association with p65. Oncogenesis. 2017;6(9):e381. doi: 10.1038/oncsis.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nussinov R, et al. The mystery of Rap1 suppression of oncogenic ras. Trends Cancer. 2020;6(5):369–379. doi: 10.1016/j.trecan.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y-L, et al. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol Med. 2017;14(1):90–99. doi: 10.20892/j.issn.2095-3941.2016.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Earwaker P, et al. RAPTOR up-regulation contributes to resistance of renal cancer cells to PI3K-mTOR inhibition. PLoS One. 2018;13(2):e0191890. doi: 10.1371/journal.pone.0191890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang T, et al. RAPTOR promotes colorectal cancer proliferation by inducing mTORC1 and upregulating ribosome assembly factor URB1. Cancer Med. 2020;9(4):1529–1543. doi: 10.1002/cam4.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kondo S, et al. Raptor and rictor expression in patients with human papillomavirus-related oropharyngeal squamous cell carcinoma. BMC Cancer. 2021;21(1):87. doi: 10.1186/s12885-021-07794-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kauffman EC, et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol. 2014;11(8):465–475. doi: 10.1038/nrurol.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim JH, et al. TFEB supports pancreatic cancer growth through the transcriptional regulation of glutaminase. Cancers. 2021;13(3):483. doi: 10.3390/cancers13030483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Y, et al. TFEB is a master regulator of tumor-associated macrophages in breast cancer. J ImmunoTher Cancer. 2020;8(1):e000543. doi: 10.1136/jitc-2020-000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Medina DL, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21(3):421–430. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Astanina E, Bussolino F, Doronzo G. Multifaceted activities of transcription factor EB in cancer onset and progression. Mol Oncol. 2021;15(2):327–346. doi: 10.1002/1878-0261.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Astrinidis A, et al. Tuberin, the tuberous sclerosis complex 2 tumor suppressor gene product, regulates Rho activation, cell adhesion and migration. Oncogene. 2002;21(55):8470–8476. doi: 10.1038/sj.onc.1205962. [DOI] [PubMed] [Google Scholar]
- 89.Deng J, et al. ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1-mutant lung cancer. Nat Can. 2021;2(5):503–514. doi: 10.1038/s43018-021-00208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kumar M, Papaleo E. A pan-cancer assessment of alterations of the kinase domain of ULK1, an upstream regulator of autophagy. Sci Rep. 2020;10(1):14874. doi: 10.1038/s41598-020-71527-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.TAN L, TAN Y, LIU D. Functions of ULK1 in autophagy and non-autophagy pathways and its implications in human physiology and disease. Biocell. 2020;44(4):535–543. doi: 10.32604/biocell.2020.09171. [DOI] [Google Scholar]
- 92.Li Z, et al. ULK1-ATG13 and their mitotic phospho-regulation by CDK1 connect autophagy to cell cycle. PLoS Biol. 2020;18(6):e3000288. doi: 10.1371/journal.pbio.3000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Luca M, Romano R, Bucci C. Role of the V1G1 subunit of V-ATPase in breast cancer cell migration. Sci Rep. 2021;11(1):4615. doi: 10.1038/s41598-021-84222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang L, et al. ABCG2/V-ATPase was associated with the drug resistance and tumor metastasis of esophageal squamous cancer cells. Diagn Pathol. 2012;7:180. doi: 10.1186/1746-1596-7-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu Q, et al. The expression of V-ATPase is associated with drug resistance and pathology of non-small-cell lung cancer. Diagn Pathol. 2013;8:145. doi: 10.1186/1746-1596-8-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Noman MZ, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti–PD-1/PD-L1 immunotherapy. Sci Adv. 2020;6(18):eaax7881. doi: 10.1126/sciadv.aax7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu F, et al. PIK3C3 regulates the expansion of liver CSCs and PIK3C3 inhibition counteracts liver cancer stem cell activity induced by PI3K inhibitor. Cell Death Dis. 2020;11(6):427. doi: 10.1038/s41419-020-2631-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang X, et al. VPS34 stimulation of p62 phosphorylation for cancer progression. Oncogene. 2017;36. [DOI] [PMC free article] [PubMed]
- 99.Parekh VV, et al. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8α<sup>+</sup> dendritic cells. Proc Natl Acad Sci. 2017;114(31):E6371–E6380. doi: 10.1073/pnas.1706504114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liang T-T, et al. Systemic expression analysis reveals prognostic significance of WIPI3 in hepatocellular carcinoma. Front Genet. 2020;11(847). [DOI] [PMC free article] [PubMed]
- 101.Kean MJ, et al. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J Cell Sci. 2009;122(Pt 22):4089–4098. doi: 10.1242/jcs.052761. [DOI] [PubMed] [Google Scholar]
- 102.Sneeggen M, Schink KO, Stenmark H. Tumor suppression by control of matrix metalloproteinase recycling. Mol Cell Oncol. 2019;6(6):e1646606. doi: 10.1080/23723556.2019.1646606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miao Y, et al. GABARAP is overexpressed in colorectal carcinoma and correlates with shortened patient survival. Hepatogastroenterology. 2010;57(98):257–261. [PubMed] [Google Scholar]
- 104.Liu Y, et al. GABARAP suppresses EMT and breast cancer progression via the AKT/mTOR signaling pathway. Aging. 2021;13(4):5858–5874. doi: 10.18632/aging.202510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Othman EQ, et al. Immunohistochemical expression of MAP1LC3A and MAP1LC3B protein in breast carcinoma tissues. J Clin Lab Anal. 2009;23(4):249–258. doi: 10.1002/jcla.20309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kang HM, et al. Ubiquitination of MAP1LC3B by pVHL is associated with autophagy and cell death in renal cell carcinoma. Cell Death Dis. 2019;10(4):279. doi: 10.1038/s41419-019-1520-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19(1):12. doi: 10.1186/s12943-020-1138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sadler JBA, et al. A cancer-associated polymorphism in ESCRT-III disrupts the abscission checkpoint and promotes genome instability. Proc Natl Acad Sci U S A. 2018;115(38):E8900–E8908. doi: 10.1073/pnas.1805504115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Song Y, Quach C, Liang C. UVRAG in autophagy, inflammation, and cancer. Autophagy. 2020;16(2):387–388. doi: 10.1080/15548627.2019.1709768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ahn CH, et al. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS. 2007;115(12):1344–1349. doi: 10.1111/j.1600-0463.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- 111.Hao M, Yeo SK, Guan J-L. Autophagy inhibition perturbs ERBB2 trafficking and abolishes tumorigenesis in ERBB2-driven breast cancer. Autophagy. 2021;17(4):1059–1060. doi: 10.1080/15548627.2021.1907168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cararo-Lopes E, et al. Autophagy buffers Ras-induced genotoxic stress enabling malignant transformation in keratinocytes primed by human papillomavirus. Cell Death Dis. 2021;12(2):194. doi: 10.1038/s41419-021-03476-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang G-M, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18(1):17. doi: 10.1186/s12943-019-0944-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fung C, et al. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19(3):797–806. doi: 10.1091/mbc.e07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nazio F, et al. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019;26(4):690–702. doi: 10.1038/s41418-019-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pérez-Hernández M, et al. Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers. 2019;11(10):1599. doi: 10.3390/cancers11101599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maes H, et al. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med. 2013;19(7):428–446. doi: 10.1016/j.molmed.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 118.Sharifi MN, et al. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep. 2016;15(8):1660–1672. doi: 10.1016/j.celrep.2016.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karsli-Uzunbas G, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Catalano M, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9(8):1612–1625. doi: 10.1016/j.molonc.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.La Belle Flynn A, et al. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun. 2019;10(1):3668. doi: 10.1038/s41467-019-11640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pietrocola F, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30(1):147–160. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kimura T, et al. Cellular and molecular mechanism for secretory autophagy. Autophagy. 2017;13(6):1084–1085. doi: 10.1080/15548627.2017.1307486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lock R, et al. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4(4):466–479. doi: 10.1158/2159-8290.CD-13-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hong Z, et al. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J Cell Biol. 2017;216(12):4217–4233. doi: 10.1083/jcb.201611073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marat AL, Haucke V. Phosphatidylinositol 3-phosphates—at the interface between cell signalling and membrane traffic. EMBO J. 2016;35(6):561–579. doi: 10.15252/embj.201593564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Poteryaev D, et al. Identification of the Switch in early-to-late endosome transition. Cell. 2010;141(3):497–508. doi: 10.1016/j.cell.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 128.Bache KG, et al. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol. 2003;162(3):435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Futter CE, et al. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J Cell Biol. 2001;155(7):1251–1264. doi: 10.1083/jcb.200108152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xu Y, et al. SNX3 regulates endosomal function through its PX-domain-mediated interaction with PtdIns(3)P. Nat Cell Biol. 2001;3(7):658–666. doi: 10.1038/35083051. [DOI] [PubMed] [Google Scholar]
- 131.Knævelsrud H, et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J Cell Biol. 2013;202(2):331–349. doi: 10.1083/jcb.201205129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bechtel W, et al. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol. 2013;24(5):727–743. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Siddhanta U, et al. Distinct roles for the p110alpha and hVPS34 phosphatidylinositol 3'-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol. 1998;143(6):1647–1659. doi: 10.1083/jcb.143.6.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nascimbeni AC, Codogno P, Morel E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017;284(9):1267–1278. doi: 10.1111/febs.13987. [DOI] [PubMed] [Google Scholar]
- 135.Ketel K, et al. A phosphoinositide conversion mechanism for exit from endosomes. Nature. 2016;529(7586):408–412. doi: 10.1038/nature16516. [DOI] [PubMed] [Google Scholar]
- 136.Miranda AM, Di Paolo G. Endolysosomal dysfunction and exosome secretion: implications for neurodegenerative disorders. Cell Stress. 2018;2(5):115–118. doi: 10.15698/cst2018.05.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Law F, et al. The VPS34 PI3K negatively regulates RAB-5 during endosome maturation. J Cell Sci. 2017;130(12):2007–2017. doi: 10.1242/jcs.194746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jaber N, et al. Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase-activating protein Armus. J Cell Sci. 2016;129(23):4424–4435. doi: 10.1242/jcs.192260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Baietti MF, et al. Syndecan–syntenin–ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14(7):677–685. doi: 10.1038/ncb2502. [DOI] [PubMed] [Google Scholar]
- 140.Sapmaz A, et al. USP32 regulates late endosomal transport and recycling through deubiquitylation of Rab7. Nat Commun. 2019;10(1):1454. doi: 10.1038/s41467-019-09437-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 1998;95(6):847–858. doi: 10.1016/S0092-8674(00)81707-9. [DOI] [PubMed] [Google Scholar]
- 142.Falguières T, et al. In vitro budding of intralumenal vesicles into late endosomes is regulated by Alix and Tsg101. Mol Biol Cell. 2008;19(11):4942–4955. doi: 10.1091/mbc.e08-03-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hikita T, et al. Src in endosomal membranes promotes exosome secretion and tumor progression. Sci Rep. 2019;9(1):3265. doi: 10.1038/s41598-019-39882-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ghossoub R, et al. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat Commun. 2014;5:3477. doi: 10.1038/ncomms4477. [DOI] [PubMed] [Google Scholar]
- 145.Ostrowski M, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(1):19–30. doi: 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- 146.Hoshino D, et al. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 2013;5(5):1159–1168. doi: 10.1016/j.celrep.2013.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang L, et al. Long non-coding RNA HOTAIR promotes exosome secretion by regulating RAB35 and SNAP23 in hepatocellular carcinoma. Mol Cancer. 2019;18(1):78. doi: 10.1186/s12943-019-0990-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yang L, et al. The long noncoding RNA HOTAIR activates autophagy by upregulating ATG3 and ATG7 in hepatocellular carcinoma. Mol BioSyst. 2016;12(8):2605–2612. doi: 10.1039/C6MB00114A. [DOI] [PubMed] [Google Scholar]
- 149.Peng X, et al. LINC00511 drives invasive behavior in hepatocellular carcinoma by regulating exosome secretion and invadopodia formation. J Exp Clin Cancer Res. 2021;40(1):183. doi: 10.1186/s13046-021-01990-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Agbana YL, et al. LINC00511 as a prognostic biomarker for human cancers: a systematic review and meta-analysis. BMC Cancer. 2020;20(1):682. doi: 10.1186/s12885-020-07188-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dong X, et al. Long noncoding RNA LINC00511 regulates the proliferation, apoptosis, invasion and autophagy of trophoblast cells to mediate pre-eclampsia progression through modulating the miR-31-5p/homeobox protein A7 axis. J Obstet Gynaecol Res. 2020;46(8):1298–1309. doi: 10.1111/jog.14344. [DOI] [PubMed] [Google Scholar]
- 152.Sinha S, et al. Cortactin promotes exosome secretion by controlling branched actin dynamics. J Cell Biol. 2016;214(2):197–213. doi: 10.1083/jcb.201601025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Villarroya-Beltri C, et al. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat Commun. 2016;7:13588. doi: 10.1038/ncomms13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Majumder P, Chakrabarti O. Mahogunin regulates fusion between amphisomes/MVBs and lysosomes via ubiquitination of TSG101. Cell Death Dis. 2015;6(11):e1970. doi: 10.1038/cddis.2015.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Buschow SI, et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic. 2009;10(10):1528–1542. doi: 10.1111/j.1600-0854.2009.00963.x. [DOI] [PubMed] [Google Scholar]
- 156.Möbius W, et al. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J Histochem Cytochem. 2002;50(1):43–55. doi: 10.1177/002215540205000105. [DOI] [PubMed] [Google Scholar]
- 157.Möbius W, et al. Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic. 2003;4:222–231. doi: 10.1034/j.1600-0854.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 158.Jordens I, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol. 2001;11(21):1680–1685. doi: 10.1016/S0960-9822(01)00531-0. [DOI] [PubMed] [Google Scholar]
- 159.Jongsma ML, et al. SKIP-HOPS recruits TBC1D15 for a Rab7-to-Arl8b identity switch to control late endosome transport. EMBO J. 2020;39(6):e102301. doi: 10.15252/embj.2019102301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Guo H, et al. Atg5 disassociates the V1V0-ATPase to promote exosome production and tumor metastasis independent of canonical macroautophagy. Dev Cell. 2017;43(6):716–730.e7. doi: 10.1016/j.devcel.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 161.Savina A, et al. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic. 2005;6(2):131–143. doi: 10.1111/j.1600-0854.2004.00257.x. [DOI] [PubMed] [Google Scholar]
- 162.Wang K, et al. Mechanical stress-dependent autophagy component release via extracellular nanovesicles in tumor cells. ACS Nano. 2019;13(4):4589–4602. doi: 10.1021/acsnano.9b00587. [DOI] [PubMed] [Google Scholar]
- 163.Fader CM, et al. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793(12):1901–1916. doi: 10.1016/j.bbamcr.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 164.Lefebvre C, Legouis R, Culetto E. ESCRT and autophagies: Endosomal functions and beyond. Semin Cell Dev Biol. 2018;74:21–28. doi: 10.1016/j.semcdb.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 165.Chen Y-D, et al. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci Rep. 2017;7(1):5676. doi: 10.1038/s41598-017-06076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bader CA, et al. Atg9 is required for intraluminal vesicles in amphisomes and autolysosomes. Biol Open. 2015;4(11):1345–1355. doi: 10.1242/bio.013979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Moreau K, et al. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011;146(2):303–317. doi: 10.1016/j.cell.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fader CM, Aguilera MO, Colombo MI. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy. 2012;8(12):1741–1756. doi: 10.4161/auto.21858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jeppesen DK, et al. Reassessment of exosome composition. Cell. 2019;177(2):428–445.e18. doi: 10.1016/j.cell.2019.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Baixauli F, López-Otín C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014;5:403. doi: 10.3389/fimmu.2014.00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Omi J, et al. The inducible amphisome isolates viral hemagglutinin and defends against influenza A virus infection. Nat Commun. 2020;11(1):162. doi: 10.1038/s41467-019-13974-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kraya AA, et al. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy. 2015;11(1):60–74. doi: 10.4161/15548627.2014.984273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dupont N, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30(23):4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhang M, et al. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife. 2015;4. [DOI] [PMC free article] [PubMed]
- 175.New J, Thomas SM. Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy. 2019;15(10):1682–1693. doi: 10.1080/15548627.2019.1596479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kimura T, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36(1):42–60. doi: 10.15252/embj.201695081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Qiang L, et al. Autophagy gene ATG7 regulates ultraviolet radiation-induced inflammation and skin tumorigenesis. Autophagy. 2017;13(12):2086–2103. doi: 10.1080/15548627.2017.1380757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Peng X, et al. IKKβ activation promotes amphisome formation and extracellular vesicle secretion in tumor cells. Biochim Biophys Acta Mol Cell Res. 1868;2021(1):118857. doi: 10.1016/j.bbamcr.2020.118857. [DOI] [PubMed] [Google Scholar]
- 179.Maycotte P, et al. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13(4):651–658. doi: 10.1158/1541-7786.MCR-14-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Huang S-C, et al. Discovery and optimization of pyrazolopyrimidine sulfamates as ATG7 inhibitors. Bioorg Med Chem. 2020;28(19):115681. doi: 10.1016/j.bmc.2020.115681. [DOI] [PubMed] [Google Scholar]
- 181.King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer. 2012;12:421. doi: 10.1186/1471-2407-12-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and selective autophagy in cancer development and therapy. Front Cell Dev Biol. 2018;6(104). [DOI] [PMC free article] [PubMed]
- 183.Kanemoto S, et al. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem Biophys Res Commun. 2016;480(2):166–172. doi: 10.1016/j.bbrc.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 184.Bhattacharya S, et al. GAIP interacting protein C-terminus regulates autophagy and exosome biogenesis of pancreatic cancer through metabolic pathways. PLoS One. 2014;9(12):e114409. doi: 10.1371/journal.pone.0114409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Muders MH, et al. Targeting GIPC/synectin in pancreatic cancer inhibits tumor growth. Clin Cancer Res. 2009;15(12):4095–4103. doi: 10.1158/1078-0432.CCR-08-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dutta S, et al. Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS One. 2014;9(5):e97580. doi: 10.1371/journal.pone.0097580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu Q, et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J Exp Clin Cancer Res. 2019;38(1):223. doi: 10.1186/s13046-019-1210-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 188.Xu J, et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 2021;12(4):373. doi: 10.1038/s41419-021-03664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yang Y, et al. Acquisition of new tumor cell properties by MSC-derived exosomes. Int J Oncol. 2015;47(1):244–252. doi: 10.3892/ijo.2015.3001. [DOI] [PubMed] [Google Scholar]
- 190.Kucharewicz K, et al. Simultaneous induction and blockade of autophagy by a single agent. Cell Death Dis. 2018;9(3):353. doi: 10.1038/s41419-018-0383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chen Y, et al. Increased interleukin-6 levels in the astrocyte-derived exosomes of sporadic amyotrophic lateral sclerosis patients. Front Neurosci. 2019;13(574). [DOI] [PMC free article] [PubMed]
- 192.Im K, et al. The comparison of exosome and exosomal cytokines between young and old individuals with or without gastric cancer. Int J Gerontol. 2018;12(3):233–238. doi: 10.1016/j.ijge.2018.03.013. [DOI] [Google Scholar]
- 193.Qin B, et al. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci Rep. 2015;5:15701. doi: 10.1038/srep15701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Linnemann A, et al. Interleukin 6 protects pancreatic β cells from apoptosis by stimulation of autophagy. FASEB J. 2017;31. [DOI] [PMC free article] [PubMed]
- 195.Frühbeis C, et al. Physical exercise induces rapid release of small extracellular vesicles into the circulation. J Extracell Vesicles. 2015;4(1):28239. doi: 10.3402/jev.v4.28239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Follo C, et al. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol Carcinog. 2018;57(3):319–332. doi: 10.1002/mc.22757. [DOI] [PubMed] [Google Scholar]
- 197.Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61(6):585–596. doi: 10.1042/EBC20170021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Petherick KJ, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290(18):11376–11383. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Young ARJ, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119(18):3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 200.Urano Y, et al. 6-Hydroxydopamine induces secretion of PARK7/DJ-1 via autophagy-based unconventional secretory pathway. Autophagy. 2018;14(11):1943–1958. doi: 10.1080/15548627.2018.1493043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Scotto Rosato A, et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat Commun. 2019;10(1):5630. doi: 10.1038/s41467-019-13572-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhou X, et al. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc Natl Acad Sci. 2007;104(14):5842–5847. doi: 10.1073/pnas.0701402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chen Y, et al. Dual targeting of NUAK1 and ULK1 using the multitargeted inhibitor MRT68921 exerts potent antitumor activities. Cell Death Dis. 2020;11(8):712. doi: 10.1038/s41419-020-02885-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Palamiuc L, Ravi A, Emerling BM. Phosphoinositides in autophagy: current roles and future insights. FEBS J. 2020;287(2):222–238. doi: 10.1111/febs.15127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhou C, et al. The heme oxygenase-1 inhibitor ZnPPIX induces non-canonical, Beclin 1-independent, autophagy through p38 MAPK pathway. Acta Biochim Biophys Sin. 2012;44:815–822. doi: 10.1093/abbs/gms064. [DOI] [PubMed] [Google Scholar]
- 206.Wu YT, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Xing C, et al. Class I phosphatidylinositol 3-kinase inhibitor LY294002 activates autophagy and induces apoptosis through p53 pathway in gastric cancer cell line SGC7901. Acta Biochim Biophys Sin Shanghai. 2008;40(3):194–201. doi: 10.1111/j.1745-7270.2008.00393.x. [DOI] [PubMed] [Google Scholar]
- 208.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79(6):1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lelouard H, et al. Regulation of translation is required for dendritic cell function and survival during activation. J Cell Biol. 2007;179(7):1427–1439. doi: 10.1083/jcb.200707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chicote J, et al. Cell death triggered by the autophagy inhibitory drug 3-methyladenine in growing conditions proceeds with DNA damage. Front Pharmacol. 2020;11:580343. doi: 10.3389/fphar.2020.580343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Caro LH, et al. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem. 1988;175(2):325–329. doi: 10.1111/j.1432-1033.1988.tb14200.x. [DOI] [PubMed] [Google Scholar]
- 212.Korolchuk VI, et al. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33(4):517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhang H-G, Grizzle WE. Exosomes: a novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am J Pathol. 2014;184(1):28–41. doi: 10.1016/j.ajpath.2013.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang J, et al. Exosomes/tricalcium phosphate combination scaffolds can enhance bone regeneration by activating the PI3K/Akt signaling pathway. Stem Cell Res Ther. 2016;7:136. doi: 10.1186/s13287-016-0391-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Clayton A, et al. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J Immunol Methods. 2001;247(1-2):163–174. doi: 10.1016/S0022-1759(00)00321-5. [DOI] [PubMed] [Google Scholar]
- 216.Llorente A, de Marco MAC, Alonso MA. Caveolin-1 and MAL are located on prostasomes secreted by the prostate cancer PC-3 cell line. J Cell Sci. 2004;117(22):5343–5351. doi: 10.1242/jcs.01420. [DOI] [PubMed] [Google Scholar]
- 217.Abdulrahman BA, Abdelaziz DH, Schatzl HM. Autophagy regulates exosomal release of prions in neuronal cells. J Biol Chem. 2018;293(23):8956–8968. doi: 10.1074/jbc.RA117.000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bright NA, et al. The relationship between lumenal and limiting membranes in swollen late endocytic compartments formed after wortmannin treatment or sucrose accumulation. Traffic. 2001;2(9):631–642. doi: 10.1034/j.1600-0854.2001.20906.x. [DOI] [PubMed] [Google Scholar]
- 219.Isosaki M. Inhibition of wortmannin activities by amino compounds. Biochem Biophys Res Commun. 2004;324(4):1406–1412. doi: 10.1016/j.bbrc.2004.09.200. [DOI] [PubMed] [Google Scholar]
- 220.Iula L, et al. Autophagy mediates interleukin-1β secretion in human neutrophils. Front Immunol. 2018;9:269. doi: 10.3389/fimmu.2018.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Harris J, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Fukao T, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3(9):875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 223.Miranda AM, et al. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat Commun. 2018;9(1):291. doi: 10.1038/s41467-017-02533-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Liu J, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Yu X, Harris SL, Levine AJ. The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 2006;66(9):4795–4801. doi: 10.1158/0008-5472.CAN-05-4579. [DOI] [PubMed] [Google Scholar]
- 226.Liu J, et al. Distinct dasatinib-induced mechanisms of apoptotic response and exosome release in imatinib-resistant human chronic myeloid leukemia cells. Int J Mol Sci. 2016;17(4):531. doi: 10.3390/ijms17040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang C, et al. Autophagic lipid metabolism sustains mTORC1 activity in TSC-deficient neural stem cells. Nat Metab. 2019;1(11):1127–1140. doi: 10.1038/s42255-019-0137-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.McKnight NC, et al. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014;10(10):e1004626. doi: 10.1371/journal.pgen.1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.McKnight NC, Yue Z. Beclin 1, an essential component and master regulator of PI3K-III in health and disease. Curr Pathobiol Rep. 2013;1(4):231–238. doi: 10.1007/s40139-013-0028-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gladue DP, et al. Foot-and-mouth disease virus nonstructural protein 2C interacts with Beclin1, modulating virus replication. J Virol. 2012;86(22):12080–12090. doi: 10.1128/JVI.01610-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Robke L, et al. Phenotypic identification of a novel autophagy inhibitor chemotype targeting lipid kinase VPS34. Angew Chem Int Ed. 2017;56(28):8153–8157. doi: 10.1002/anie.201703738. [DOI] [PubMed] [Google Scholar]
- 232.Marsh T, Debnath J. Ironing out VPS34 inhibition. Nat Cell Biol. 2014;17:1–3. doi: 10.1038/ncb3089. [DOI] [PubMed] [Google Scholar]
- 233.Ronan B, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10(12):1013–1019. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- 234.Wu Y, et al. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy. 2013;9(4):595–603. doi: 10.4161/auto.23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Vats S, Manjithaya R. A reversible autophagy inhibitor blocks autophagosome–lysosome fusion by preventing Stx17 loading onto autophagosomes. Mol Biol Cell. 2019;30(17):2283–2295. doi: 10.1091/mbc.E18-08-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Keller MD, et al. Decoy exosomes provide protection against bacterial toxins. Nature. 2020;579(7798):260–264. doi: 10.1038/s41586-020-2066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Cheng X-T, et al. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209(3):377–386. doi: 10.1083/jcb.201412046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Al-Bari A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. 2015;70. [DOI] [PMC free article] [PubMed]
- 239.Mauthe M, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Lu S, et al. Lysosomal adaptation: how cells respond to lysosomotropic compounds. PLoS One. 2017;12(3):e0173771. doi: 10.1371/journal.pone.0173771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gallagher LE, et al. Lysosomotropism depends on glucose: a chloroquine resistance mechanism. Cell Death Dis. 2017;8(8):e3014. doi: 10.1038/cddis.2017.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Davis MJ, Swanson JA. Technical advance: Caspase-1 activation and IL-1β release correlate with the degree of lysosome damage, as illustrated by a novel imaging method to quantify phagolysosome damage. J Leukoc Biol. 2010;88(4):813–822. doi: 10.1189/jlb.0310159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Orlinska U, Newton RC. Role of glucose in interleukin-1 beta production by lipopolysaccharide-activated human monocytes. J Cell Physiol. 1993;157(1):201–208. doi: 10.1002/jcp.1041570126. [DOI] [PubMed] [Google Scholar]
- 244.Cotzomi-Ortega I, et al. Autophagy inhibition induces the secretion of macrophage migration inhibitory factor (MIF) with autocrine and paracrine effects on the promotion of malignancy in breast cancer. Biology. 2020;9(1):20. doi: 10.3390/biology9010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Li M, et al. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J Biol Chem. 2013;288(50):35769–35780. doi: 10.1074/jbc.M113.511212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Deng J, et al. The role of autophagy and amphisomes in virus recognition, TLR 9 recruitment and virus-stimulated IFN-α production by human plasmacytoid dendritic cells (pDC) (45.19) J Immunol. 2012;188(1 Supplement):45.19. [Google Scholar]
- 247.Mauvezin C, et al. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. doi: 10.1038/ncomms8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.van Deurs B, Holm PK, Sandvig K. Inhibition of the vacuolar H(+)-ATPase with bafilomycin reduces delivery of internalized molecules from mature multivesicular endosomes to lysosomes in HEp-2 cells. Eur J Cell Biol. 1996;69(4):343–350. [PubMed] [Google Scholar]
- 249.van Weert AW, et al. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J Cell Biol. 1995;130(4):821–834. doi: 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zou W, et al. Exosome release is regulated by mTORC1. Adv Sci. 2019;6(3):1801313. doi: 10.1002/advs.201801313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Palokangas H, et al. Retrograde transport from the pre-Golgi intermediate compartment and the Golgi complex is affected by the vacuolar H+-ATPase inhibitor bafilomycin A1. Mol Biol Cell. 1998;9(12):3561–3578. doi: 10.1091/mbc.9.12.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Chen Q, et al. Knockdown of Parkinson’s disease-related gene ATP13A2 reduces tumorigenesis via blocking autophagic flux in colon cancer. Cell Biosci. 2020;10(1):144. doi: 10.1186/s13578-020-00506-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Bento CF, et al. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun. 2016;7(1):11803. doi: 10.1038/ncomms11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kong SM, et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum Mol Genet. 2014;23(11):2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- 255.Minakaki G, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14(1):98–119. doi: 10.1080/15548627.2017.1395992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Sharma G, et al. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy. 2019;15(10):1694–1718. doi: 10.1080/15548627.2019.1586257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Dong XP, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1(4):38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Li SC, et al. The signaling lipid PI(3,5)P2 stabilizes V1-V(o) sector interactions and activates the V-ATPase. Mol Biol Cell. 2014;25(8):1251–1262. doi: 10.1091/mbc.e13-10-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hessvik NP, et al. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell Mol Life Sci. 2016;73(24):4717–4737. doi: 10.1007/s00018-016-2309-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ji X, Zhang X, Li Z. ULK1 inhibitor induces spindle microtubule disorganization and inhibits phosphorylation of Ser10 of histone H3. FEBS Open Biol. 2020;10(11):2452–2463. doi: 10.1002/2211-5463.13000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Karve S, et al. Revival of the abandoned therapeutic wortmannin by nanoparticle drug delivery. Proc Natl Acad Sci U S A. 2012;109(21):8230–8235. doi: 10.1073/pnas.1120508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Guo J, et al. Potent USP10/13 antagonist spautin-1 suppresses melanoma growth via ROS-mediated DNA damage and exhibits synergy with cisplatin. J Cell Mol Med. 2020;24(7):4324–4340. doi: 10.1111/jcmm.15093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pellegrini P, et al. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy. 2014;10(4):562–571. doi: 10.4161/auto.27901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Collins KP, Jackson KM, Gustafson DL. Hydroxychloroquine: A Physiologically-Based Pharmacokinetic Model in the Context of Cancer-Related Autophagy Modulation. J Pharmacol Exp Ther. 2018;365(3):447–459. doi: 10.1124/jpet.117.245639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Bayer N, et al. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J Virol. 1998;72(12):9645–9655. doi: 10.1128/JVI.72.12.9645-9655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable