

ABSTRACT
Objectives:
Although a number of genetic forms of cholestasis have been identified, the genetic etiology of disease remains unidentified in a subset of cholestasis patients.
Methods:
Whole exome sequencing (WES) was performed in DNA from patients diagnosed with cholestasis, at different points on the continuum from progressive familial intrahepatic cholestasis to benign recurrent intrahepatic cholestasis, in whom no disease mutations in known cholestasis genes had been identified. Candidate genes were then assessed in a larger patient sample, by targeted next-generation sequencing (NGS). Disease features at presentation and follow-up were collected from available medical records.
Results:
By WES, we identified 3 patients with homozygous mutations in USP53. Screening of USP53 in a larger set of patients identified 4 additional patients with homozygous mutations in USP53. Six of the 7 patients had deletion mutations, and 1 had a missense mutation; 3 of the patients were siblings, all bearing a deletion that also disrupted neighboring MYOZ2. Age of onset ranged from early infancy to adolescence. Cholestasis tended to be biochemically mild and intermittent, and responsive to medication. Liver fibrosis was, however, present in all 4 patients who were biopsied, and splenomegaly was apparent in 5 of 7 at last ultrasound.
Conclusions:
Two groups recently identified patients with liver disease and mutation in USP53. We have now identified biallelic mutation in USP53 in 7 further patients with cholestasis, from 5 families. Most individuals had evidence of chronic liver disease, and long-term follow-up is recommended.
Keywords: genetics, hepatology, intrahepatic cholestasis, liver, MYOZ2
See “Progressive Familial Intrahepatic Cholestasis: Is It Time to Transition to Genetic Cholestasis?” by Squires and Monga on page 641.
What Is Known/What Is New
What Is Known
Several distinct mechanisms are known to underlie genetic cholestasis.
USP53 is related to a family of ubiquitin-specific proteases, but may not function as one.
Usp53 mutation underlies a mouse deafness model.
Mutations in USP53 have recently been reported in early onset cholestasis.
What Is New
Seven cholestasis patients with mutations in USP53.
USP53 deficiency may cause progressive liver disease.
Age of onset is highly variable.
Rifampicin may be useful as a treatment.
The manifestations of impaired bile formation are termed cholestasis; cholestasis can result from abnormalities in several different biological processes. The most prevalent defects are those affecting transporters involved in bile formation (1,2). Other cholestatic diseases are the result of abnormal development of the biliary tree (3,4). A third group of disorders are caused by ciliary dysfunction, reflecting the importance of cilia in regulation of bile flow (5–8). Patients with inborn errors of bile acid synthesis have been described (9). The normal organization and function of apical membranes is critical for bile formation and highly dependent on the correct trafficking of membrane constituents. Several cholestatic phenotypes are known to be the consequence of membrane abnormalities (10–14). Lastly, the structure of all epithelia is dependent on both cell/cell interactions and apical/basal polarization. Both functions are dependent on normal tight junction structures and tight junction-related forms of cholestasis have been described (15–17).
Most genetic causes of cholestasis (‘cholestasis genes’) have been identified through the investigation of children with early-onset disease. Many cholestasis genes so far identified, however, have now also been associated with later-onset forms of disease (18–21). In many cases, a degree of phenotype--genotype correlation has been found, with milder mutations leading to less severe phenotypes (22–25).
We performed whole exome sequencing (WES), and identified homozygous loss-of-function variants in Ubiquitin-specific protease 53 (USP53) in 3 cholestasis patients. Targeted screening of USP53 in 1815 patients with early-onset or later-onset cholestasis then identified 4 additional patients harboring biallelic USP53 variants likely to cause disease. Two groups have recently identified mutations in this gene, in patients with cholestasis (26,27).
PARTICIPANTS AND METHODS
Ethical Considerations
The study was approved by the King's College Hospital (KCH) Paediatric Liver Biobank, Childhood Liver Disease Research Network (ChiLDReN) site institutional review boards including UCSF, and the Ethics Committee of the Children's Memorial Health Institute (CMHI). The study was conducted in accordance with the guidelines of the 1975 Declaration of Helsinki. Informed consent was obtained from adult participants and parents of each child.
Participants
WES was undertaken using DNA from 72 patients with genetically unexplained cholestasis, on the continuum from progressive familial intrahepatic cholestasis (PFIC) to benign recurrent intrahepatic cholestasis (BRIC). Forty-one of these patients were from the Longitudinal Study of Genetic Causes of Intrahepatic Cholestasis (LOGIC—NCT00571272) protocol. This study is organized under ChiLDReN, an NIDDK/National Institutes of Health-funded network of 16 pediatric academic medical centers across North America. LOGIC is a longitudinal observational study of the natural history of intrahepatic cholestasis. Details of enrollment criteria have been published, but included cholestasis, and allowed study entry between birth and 25 years of age (28). DNA from 14 children from KCH, and 17 from CMHI, also underwent WES; all were under 18 years of age, and had intrahepatic cholestasis, with no other diagnosis at enrollment. Of the 72 patients, 7 were previously reported to have disease-causing mutations in Tight Junction Protein 2 (TJP2) (17). For the targeted sequencing of USP53, 1249 children and 566 adults being investigated for cholestasis at KCH were examined. Clinical data of patients bearing USP53 mutation was extracted from medical records or the ChiLDReN database.
Genetic Analysis
Genomic DNA was isolated from peripheral blood, or EBV-transfected cell lines, of patients and available parents. WES was performed, along with annotation and analysis as described previously (17). Subsequent analysis of variants identified by WES and targeted sequencing incorporated the use of Sorting Intolerant from Tolerant (SIFT), PolyPhen-2, Combined Annotation Dependent Depletion (CADD), and the Genome Aggregation Database (gnomAD) resources (29–32).
Once the initial patients with USP53 mutation had been identified by WES, USP53 was added to the NGS targeted gene panel used in the diagnostic/research laboratory at KCH. USP53 was not analyzed for clinical diagnostic purposes, as it remained unproven as a cause of cholestasis; however, the USP53 sequence data were extracted and analyzed as part of this project. Large deletions, identified by NGS, were confirmed by quantitative PCR (qPCR) of genomic DNA.
Histologic and Immunohistochemical Studies of Liver
Archival material obtained for the purposes of clinical diagnosis and treatment, fixed in formalin and processed into paraffin wax by routine methods, from individuals with mutation in USP53, was sectioned at 4 μm. Routine immunostaining was performed (BondMax, Leica Biosystems, Buffalo Grove, IL) with the antibodies used against USP53 (Sigma, Gillingham, Dorset, UK), TJP2 (LS Bioscience, Seattle, WA), gamma-glutamyl transpeptidase (GGT) (Abnova, Taipei City, Taiwan), and bile salt export pump (BSEP) (Sigma) proteins, using hematoxylin counterstaining.
Ultrastructure Study
Tissue specimens were primarily fixed in paraformaldehyde and glutaraldehyde, post-fixed in osmium tetroxide and embedded in resin. Ultrathin sections were stained with uranyl acetate and lead citrate.
RESULTS
Whole-exome Sequencing
Potentially disease-causing variants in USP53 were identified by WES in 3 patients. A homozygous >105 kb deletion was identified in 2 siblings (patients 1 and 2). The deletion breakpoints have not been sequenced but this deletion included the first coding exon of USP53, and all 5 coding exons of MYOZ2. Quantitative PCR (qPCR) of genomic DNA at multiple sites across the region confirmed homozygosity for this deletion in patients 1 and 2, and demonstrated heterozygosity in both parents. A third sibling (patient 3), born subsequent to the above discoveries, was found to be homozygous for the same deletion by qPCR and NGS panel analysis of the target regions (Table 1).
TABLE 1.
Genetic and clinical findings in 7 patients with USP53 deficiency
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||||||
| USP53 variant | Deletion of first coding exon | Deletion of first coding exon | Deletion of first coding exon | c.145-11_167del | c.145-11_167del | c.725C>T | c.510delA | |||||||
| Predicted effect | Deletion | Deletion | Deletion | Deletion | Deletion | p.(Pro242Leu) | p.(Ser171ArgfsTer62) | |||||||
| Zygosity | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | |||||||
| Sex | Female | Male | Female | Female | Male | Male | Male | |||||||
| Ethnicity | Turkish | Turkish | Turkish | Middle Eastern | Middle Eastern | North African | South Asian | |||||||
| Onset of cholestasis | 3 months, recurred at 11 months and 6 years | 2 months | 5 months, approximately 18 and 21 months | 7 years, plus gallstones | Neonatal then at 2, 3, and 13 years | 15, 16, and 18 years | Approximately 4 years | |||||||
| Earliest available biochemistry documenting cholestasis | ||||||||||||||
| Age | 3 months | 2 months | 7 months | N/A | 13 years | 15 years | 21 years | |||||||
| TB/DB, mg/dL | 6.6/6.1 | 5.2/3.8 | 18.4/14.6 | NK | 3.9/1.9 | 3.4/- | 6.3/2.8 | |||||||
| ALT/AST, U/L | −/163 | 169/222 | 61/103 | NK | 22/38 | 78/74 | 84/46 | |||||||
| GGT, U/L | 41 | 62 | 46 | “Normal” | 35 | 25 | 34 | |||||||
| TBA, μmol/L | 219 | ND | ND | NK | 37 | ND | 15 | |||||||
| Audiometry | Normal | Normal | Normal | NK | NK | ND | NK | |||||||
| Liver and spleen USS | Liver normal Spleen 12.5 cm @11.5 years (enlarged) | Liver normal Spleen 10.2 cm @8.5 years ULN | Liver normal Spleen 9.8 cm @2 years (enlarged) | Both normal @ 10 years | Liver normal Spleen 10.5 cm @14 years (enlarged for weight) | Liver normal Spleen 15.5 cm @16 years (enlarged) | Liver normal, GB wall thickened, contains sludge Spleen normal @ 20 years | |||||||
| Last follow-up | ||||||||||||||
| Age | 11 years | 8 years | 2 years | 10 years | 13 years | 18 years | 21 years | |||||||
| Medication | None | None | Rifampicin, UDCA. Relapses off Rifampicin | UDCA | Rifampicin | None | Rifampicin, UDCA and cholestyramine | |||||||
| TB, mg/dL | 0.3 | 0.1 | 0.1 | 0.6 | 1.2 | 6.4 | As above | |||||||
| ALT, U/L | 40 | 48 | 40 | 47 | 17 | 50 | ||||||||
| TBA, μmol/L | 7 | 6 | ND | ND | 35 | ND | ||||||||
ALT = alanine aminotransferase; AST = aspartate aminotransferase; DB = direct bilirubin; GB = gall bladder; GGT = gamma-glutamyl transpeptidase; ND = not done; NK = not known; TB = total bilirubin; TBA = total bile acids; UDCA = ursodeoxycholic acid; ULN = upper limit of normal; USS = ultrasound scan. All USP53 variant positions are annotated on transcript: NM_019050.2.
By WES, patient 7 was found to have a homozygous single nucleotide deletion in USP53, (c.510delA), predicted to result in a frameshift and subsequent protein truncation, p.(Ser171ArgfsTer62).
Targeted Gene Panel Sequencing
In a large group of patients with cholestasis in whom targeted gene panel sequencing had been performed, USP53 sequence data were retrospectively analyzed. To note, USP53 was evaluated in all patients from whom data were available, including patients who received diagnoses of other genetic forms of cholestasis, through analysis of the gene panel results. Of 566 adults tested, none showed likely disease-causing variants. In 1249 children with data available, 3 further individuals were found to have potentially disease-causing variants in USP53. None of these 3 patients had likely disease-causing mutations in the known cholestasis genes included on the gene panel. Two unrelated individuals (patients 4 and 5) were both found to be homozygous for a deletion spanning an intron/exon boundary; c.145-11_167del. Patient 6 was homozygous for c.725C>T, predicted to result in a missense change, p.(Pro242Leu). This variant is not present in the gnomAD database. SIFT predicts it to be deleterious, PolyPhen-2 predicts it to be damaging, and the scaled CADD score of 31 indicates that this mutation is very likely to deleteriously impact protein function. Pro242 is conserved, and only 14 amino acids distal to Cys228, the amino acid altered in the mambo mouse deafness model (33). Both of these amino acid residues are present in the fingers subdomain of USPs, considered the primary site of ubiquitin interaction (34).
Phenotype of USP53-Deficient Patients
Patients 1, 2, 3, and 5 came to medical attention with cholestasis in early infancy. Patient 2 also had heart failure, attributed to extremely low vitamin D levels. Patients 4 and 6 had their first episodes of cholestasis at age 7 and 15 years. In patients 1–6 the initial cholestasis resolved, although the time to so doing varied between 2 and 6 months. Patients 1, 3, 5, and 6 have had several episodes of cholestasis. In patients 1, 2, 3 and 4, cholestasis had resolved biochemically at last follow-up, but patients 5 and 6 were last seen during a cholestatic episode. Patient 7 presented with jaundice, pruritus, diarrhea, and abnormal liver function tests at approximately 4 years of age, and was jaundiced with raised transaminases when seen at 21 years of age; information on disease course between ages 4 and 21 is not available (Table 1).
Biochemically, the earliest documented cholestasis was associated with hyperbilirubinemia between 3.4 and 18.4 mg/dL with minimal or moderate elevation of transaminases. GGT levels were normal. Serum bile acid levels were not evaluated systematically; a level of 219 μmol/L was recorded in patient 1 in infancy, 37 μmol/L in patient 5 at 13 years of age during an episode, and 15 μ mol/L in patient 7 at 21 years of age. Patient 4 was noted to have gallstones when she presented at 7 years. At last follow-up, 4 patients had enlarged spleen, by ultrasound measurement.
The only extrahepatic manifestation noted in these patients was the infant (patient 2) with heart failure. This child was 1 of 3 siblings whose homozygous disease-causing mutation also included complete loss of the gene MYOZ2. All 3 siblings had subsequent echocardiograms, which were normal. All available serum calcium levels at presentation and during follow-up were normal. Three of the patients had hearing tests with normal results. None of the patients have reported hearing problems.
A variety of treatments were employed. At last follow-up, 4 of 7 patients were still receiving medication. One was on rifampicin alone, 1 was on ursodeoxycholic acid (UDCA) alone, 1 was on UDCA and rifampicin, and 1 was on rifampicin, UDCA and cholestyramine. The patient on UDCA and rifampicin had relapse of pruritus twice when medication was withdrawn, with prompt amelioration of pruritus on reintroduction of rifampicin.
Histopathologic, Immunohistologic, and Ultrastructural Findings
Liver biopsy material was available from 4 patients, who underwent liver biopsy ages 2 months and 19 days (patient 2), 4 months and 23 days (patient 1), 13 years 2 months (patient 5), and 16 years 6 months (patient 6). All biopsies demonstrated fibrosis, ranging from portoseptal fibrosis in patients 1, 2, and 6 (Figs. 1 and 2) to portoportal bridging fibrosis (patient 5, Fig. 2). Portal tracts in all patients demonstrated a mild ductular reaction. Cholangiopathic features were not identified. One patient (patient 2, Fig. 1) demonstrated neonatal giant cell hepatitis. In the remaining patients, the liver lobule showed minimal disorganization, with the exception of biliary rosette formation in patient 1. All demonstrated mild lobular activity. Canalicular and hepatocellular cholestasis was variably seen. Canalicular proteins GGT and BSEP were expressed in a pan-lobular distribution in all patients. TJP2 was expressed in all patients. USP53 immunostaining was attempted but a meaningful expression pattern was not achieved.
FIGURE 1.
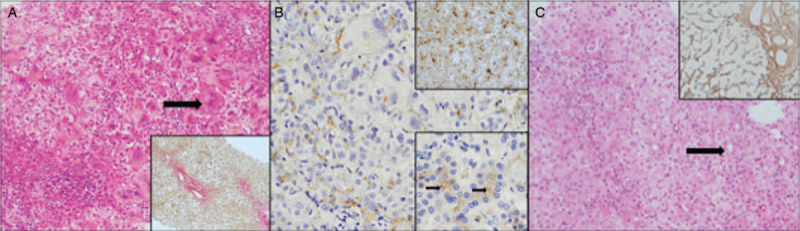
Patient 2 underwent liver biopsy ages 2 months and 19 days. Portoseptal fibrosis was present (A, inset, picro sirius red ×100 magnification). The liver demonstrated hepatocyte giant cell change (A, main image, H&E ×200 magnification, arrow) canalicular and hepatocellular cholestasis and mild lobular activity. Immunostaining demonstrated panlobular GGT expression (B, main image, ×200 magnification), panlobular BSEP expression (B, upper inset ×200 magnification) and canalicular TJP2 (B, lower inset ×200 magnification, arrows demonstrating accentuation at the tight junction). Patient 1 underwent biopsy ages 4 months and 23 days of age. The liver demonstrated portoseptal and perisinusoidal fibrosis (C, inset, reticulin ×100 magnification). Biliary rosette formation was present, but the lobule was otherwise unremarkable (C, main image, H&E ×100 magnification, arrow).
FIGURE 2.
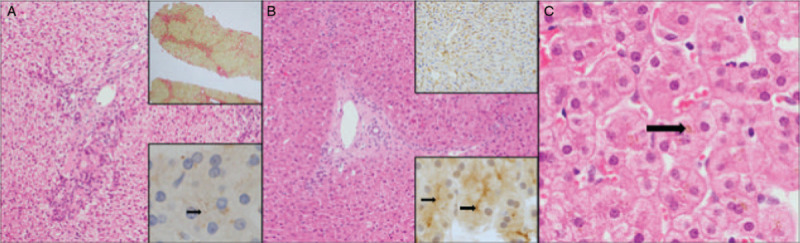
Patient 5 was biopsied ages 13 years and 2 months. The liver demonstrated portoportal bridging fibrosis (A upper inset, picro Sirius red ×40 magnification). There was no significant disorganization of the lobular architecture (A main image, H&E ×100 magnification). TJP2 expression was focal and of low intensity (A inset, ×400 magnification). Patient 6 was biopsied ages 16 years and 6 months of age. The liver demonstrated portoseptal fibrosis (B, main image, H&E ×100 magnification). There was panlobular expression of canalicular markers (B, upper inset shows GGT expression ×100 magnification) and TJP2 was expressed at the canalicular membrane (B, lower inset, TJP2 immunostain ×400 magnification). Very focal hepatocellular and canalicular cholestasis was seen (C, H&E ×200 magnification with arrow pointing to hepatocellular cholestasis).
Material was available for electron microscopy from patients 1, 2, and 5. A number of features were found (Fig. 3). In all 3 cases, some canaliculi were distended with bile whilst others contained healthy looking microvilli. In patient 1, the retained bile was coarsely granular bile (panel A), but that seen in patients 2 and 5 was finely filamentous. Patient 1 showed marked peri-canalicular filamentous actin (panel A). Abnormality of some desmosomes was noted in patient 2. They were either strikingly abnormal in length, or apparently detached from the plasma membrane (panel B). No abnormalities of tight junction morphology were identified in any patient. It is not clear if the changes noted are primary consequences of USP53 deficiency, or are secondary to the disease.
FIGURE 3.
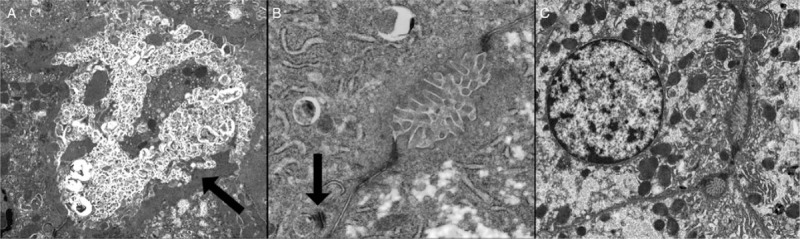
Transmission electron micrograph of biopsy taken from patient 1. Showing coarsely granular bile and pericanalicular filamentous actin, indicated by arrow (A, original magnification ×2600). Biopsy from patient 2 showing relatively normal canaliculus and desmosome apparently detached from plasma membrane, indicated by arrow (B, original magnification ×9200). Biopsy from patient 5 showing unremarkable canaliculi and tight junctions (C, original magnification ×2600).
DISCUSSION
On the basis of sequence homology, USP53 is predicted to encode a member of a family of proteins involved in de-ubiquitination, and therefore, important for regulation of protein degradation (35). USP53, however, lacks a histidine residue essential for de-ubiquitination activity, and is thought to have completely lost this function (35,36). In mice, Usp53 has been shown to colocalize and interact with Tjp2 (ZO-2) and, at least in the ear, contribute to tight junction structures (33). TJPs are cytoplasmic proteins that appear to link the intrinsic membrane tight junction proteins to the cytoskeleton (37,38). Variants in TJP2 have been shown to underlie a range of liver phenotypes, from familial hypercholanemia to severe progressive cholestatic liver disease (15,17,39). In addition, loss of claudin 1, an intrinsic tight junction protein, has been shown to lead to a cholangiopathy (16).
The patients identified in this study presented with normal-GGT cholestasis, at a variety of ages from early infancy to adolescence. Four patients were demonstrated to have had recurrent episodes of cholestasis for which there have been no obvious precipitating factors, except patient 3, who relapsed twice after stopping rifampicin administration. All patients were homozygous for their disease-causing variants. In 6 of 7 patients, the variant was predicted to cause a complete loss of function. One patient was, however, homozygous for a missense change; this individual presented at the oldest age, not manifesting any symptoms until 15 years of age.
Patients 1, 2, and 3 were homozygous for a deletion including exon 1 of USP53 and the whole of MYOZ2. Heterozygous missense changes in MYOZ2 have been associated with hypertrophic cardiomyopathy (HCM) (40). Mice heterozygous for the same changes also developed HCM (41). Mice with homozygous knockout of Myoz2 did not show HCM, but manifested abnormalities of calcineurin signaling (42). One of the 3 siblings with the large deletion presented with heart failure. At the time this was attributed to hypovitaminosis D. All 3 siblings have subsequently had cardiac investigations with normal results. It is not clear whether the MYOZ2 deletion and/or low vitamin D levels (even in presence of normal serum calcium levels) were the precipitant(s) of the heart failure seen in patient 2.
A Middle Eastern family has recently been published in which 3 children with cholestasis, presenting at 1 year or less, were homozygous for a protein-truncating variant in USP53 p.(Phe317LeufsTer6) (26). Subsequently, 7 Chinese patients with onset of cholestasis <7 months of age, and biallelic mutations in USP53, were reported (27). In the former article, 2 of the children had bilateral hearing loss, all 3 had notable hypocalcemia, and 1 child received a liver transplant for intractable pruritus at age 6 years. In the latter article, 6 of 7 patients had normal bilirubin at last follow-up; 1 had severe early-onset deafness. Two Middle Eastern patients appeared responsive to rifampicin treatment, and 4 of 7 Chinese patients were on UDCA and/or cholestyramine at last follow-up. The Mambo mouse deafness model, harboring a homozygous missense change in Usp53, manifests cochlear hair cell loss (33). No other phenotype was described in these mice. Neither hearing loss nor hypocalcemia has been noted in any of our patients.
Our data, and the 2 previous reports, convincingly indicate that loss of USP53 function is associated with normal-GGT cholestatic liver disease. In general, the clinical features appear relatively mild, and can be relapsing. The fibrosis seen in the liver biopsies, and the enlarged spleens in 4 patients, however, suggest that long-term follow-up is essential.
Footnotes
Drs Rebecca Ellmers and Pierre Foskett contributed equally to this work.
This work was supported by National Institutes of Health (NIH) R01 DK094828 (to L.N.B. and R.J.T.), NIH U01 DK062500 to P.R., NIH U01 DK062453 and UL1 TR002535 to R.J.S. and NIH U01 DK062456 to J.C.M. and R.M.M. Whole-exome sequencing was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by NHGRI and NHLBI grants UM1 HG006493 and U24 HG008956. Further whole-exome sequencing was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the National Institutes of Health, NHS, the NIHR, UK Department of Health or Illumina Inc.
R.J.T. consults for Albireo, Mirum, GenerationBio, Alnylam, Qing Bile, Horizon, Sana, and Retrophin and has share options in Qing Bile and GenerationBio. Other authors report no conflicts of interest. M.S. is now an employee of Illumina Inc.
REFERENCES
- 1.Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998; 20:233–238. [DOI] [PubMed] [Google Scholar]
- 2.de Vree JM, Jacquemin E, Sturm E, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A 1998; 95:282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oda T, Elkahloun AG, Meltzer PS, et al. Identification and cloning of the human homolog (JAG1) of the rat Jagged1 gene from the Alagille syndrome critical region at 20p12. Genomics 1997; 43:376–379. [DOI] [PubMed] [Google Scholar]
- 4.Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997; 16:243–251. [DOI] [PubMed] [Google Scholar]
- 5.Grammatikopoulos T, Sambrotta M, Strautnieks S, et al. Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J Hepatol 2016; 65:1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strongin A, Heller T, Doherty D, et al. NISC Comparative Sequencing Program. Characteristics of liver disease in 100 individuals with Joubert syndrome prospectively evaluated at a single center. J Pediatr Gastroenterol Nutr 2018; 66:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oud MM, Lamers IJ, Arts HH. Ciliopathies: genetics in pediatric medicine. J Pediatr Genet 2017; 6:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong MY, McCaughan GW, Strasser SI. An update on the pathophysiology and management of polycystic liver disease. Expert Rev Gastroenterol Hepatol 2017; 11:569–581. [DOI] [PubMed] [Google Scholar]
- 9.Heubi JE, Setchell KDR, Bove KE. Inborn errors of bile acid metabolism. Clin Liver Dis 2018; 22:671–687. [DOI] [PubMed] [Google Scholar]
- 10.Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 1998; 18:219–224. [DOI] [PubMed] [Google Scholar]
- 11.Gonzales E, Taylor SA, Davit-Spraul A, et al. MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Hepatology 2017; 65:164–173. [DOI] [PubMed] [Google Scholar]
- 12.Qiu YL, Gong JY, Feng JY, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low gamma-glutamyltransferase cholestasis. Hepatology 2017; 65:1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet 2004; 36:400–404. [DOI] [PubMed] [Google Scholar]
- 14.Cullinane AR, Straatman-Iwanowska A, Zaucker A, et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat Genet 2010; 42:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlton VE, Harris BZ, Puffenberger EG, et al. Complex inheritance of familial hypercholanemia with associated mutations in Tjp2 and BAAT. Nat Genet 2003; 34:91–96. [DOI] [PubMed] [Google Scholar]
- 16.Hadj-Rabia S, Baala L, Vabres P, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology 2004; 127:1386–1390. [DOI] [PubMed] [Google Scholar]
- 17.Sambrotta M, Strautnieks S, Papouli E, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet 2014; 46:326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tygstrup N, Steig BA, Juijn JA, et al. Recurrent familial intrahepatic cholestasis in the Faeroe Islands. Phenotypic heterogeneity but genetic homogeneity. Hepatology 1999; 29:506–508. [DOI] [PubMed] [Google Scholar]
- 19.van Mil SW, van der Woerd WL, van der Brugge G, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology 2004; 127:379–384. [DOI] [PubMed] [Google Scholar]
- 20.Rosmorduc O, Hermelin B, Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 2001; 120:1459–1467. [DOI] [PubMed] [Google Scholar]
- 21.Pasmant E, Goussard P, Baranes L, et al. First description of ABCB4 gene deletions in familial low phospholipid-associated cholelithiasis and oral contraceptives-induced cholestasis. Eur J Hum Genet 2012; 20:277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004; 40:27–38. [DOI] [PubMed] [Google Scholar]
- 23.Pawlikowska L, Strautnieks S, Jankowska I, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol 2010; 53:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schatz SB, Jungst C, Keitel-Anselmo V, et al. Phenotypic spectrum and diagnostic pitfalls of ABCB4 deficiency depending on age of onset. Hepatol Commun 2018; 2:504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sticova E, Jirsa M. ABCB4 disease: many faces of one gene deficiency. Ann Hepatol 2020; 19:126–133. [DOI] [PubMed] [Google Scholar]
- 26.Maddirevula S, Alhebbi H, Alqahtani A, et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet Med 2019; 21:1164–1172. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Yang Y, Gong JY, et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: clinical, histological, and ultrastructural characterization. Liver Int 2020; 40:1142–1150. [DOI] [PubMed] [Google Scholar]
- 28.Loomes KM, Spino C, Goodrich NP, et al. Childhood Liver Disease Research Network. Bone density in children with chronic liver disease correlates with growth and cholestasis. Hepatology 2019; 69:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sim NL, Kumar P, Hu J, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012; 40 (Web Server issue):W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kazmierczak M, Harris SL, Kazmierczak P, et al. Progressive hearing loss in mice carrying a mutation in Usp53. J Neurosci 2015; 35:15582–15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye Y, Scheel H, Hofmann K, et al. Dissection of USP catalytic domains reveals five common insertion points. Mol Biosyst 2009; 5:1797–1808. [DOI] [PubMed] [Google Scholar]
- 35.Quesada V, Diaz-Perales A, Gutierrez-Fernandez A, et al. Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem Biophys Res Commun 2004; 314:54–62. [DOI] [PubMed] [Google Scholar]
- 36.Hu M, Li P, Li M, et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 2002; 111:1041–1054. [DOI] [PubMed] [Google Scholar]
- 37.González-Mariscal L, Gallego-Gutiérrez H, González-González L, et al. ZO-2 Is a master regulator of gene expression, cell proliferation, cytoarchitecture, and cell size. Int J Mol Sci 2019; 20: 4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roehlen N, Roca Suarez AA, El Saghire H, et al. Tight junction proteins and the biology of hepatobiliary disease. Int J Mol Sci 2020; 21: 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Liu LL, Gong JY, et al. TJP2 hepatobiliary disorders: novel variants and clinical diversity. Hum Mutat 2020; 41:502–511. [DOI] [PubMed] [Google Scholar]
- 40.Osio A, Tan L, Chen SN, et al. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res 2007; 100:766–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruggiero A, Chen SN, Lombardi R, et al. Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovasc Res 2013; 97:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frey N, Barrientos T, Shelton JM, et al. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med 2004; 10:1336–1343. [DOI] [PubMed] [Google Scholar]