

SUMMARY
Deregulation of oncogenic signals in cancer triggers replication stress. Immediate Early Genes (IEGs) are rapidly and transiently expressed following stressful signals, contributing to an integrated response. Here, we find that the orphan nuclear receptor NR4A1 localizes across the genebody and 3’-UTR of IEGs, where it inhibits transcriptional elongation by RNA Pol II, generating R-loops and accessible chromatin domains. Acute replication stress causes immediate dissociation of NR4A1 and a burst of transcriptionally poised IEG expression. Ectopic expression of NR4A1 enhances tumorigenesis by breast cancer cells, while its deletion leads to massive chromosomal instability and proliferative failure, driven by deregulated expression of its IEG target FOS. Approximately half of breast and other primary cancers exhibit accessible chromatin domains at IEG genebodies, consistent with this stress-regulatory pathway. Cancers that have retained this mechanism in adapting to oncogenic replication stress may be dependent on NR4A1 for their proliferation.
Graphical Abstract
The eTOC Blurb:
Studying tumorigenesis by patient-derived breast cancer cells, Guo et al. identify NR4A1 as a master regulator of replication stress-induced Immediate Early Gene expression, through a transcriptional processing checkpoint. Suppression of NR4A1 in cancer cells dependent on this pathway triggers mitotic catastrophe through unregulated expression of the IEG family member FOS.
INTRODUCTION
Cancer cells exist in a chronically stressed environment, including hypoxia, nutrient deprivation, replication and ER stress (Cubillos-Ruiz et al., 2017; Gaillard et al., 2015; Hanahan and Weinberg, 2011; Macheret and Halazonetis, 2015). Replication stress in cancer may be caused by DNA synthesis abnormalities that are lacking in normal cells (Bartkova et al., 2005; Gorgoulis et al., 2005). These include premature entry into S phase, oncogene-induced firing of non-canonical replication origins, nucleotide pool depletion, and stalled replication forks with collisions between transcriptional and replication machineries generating DNA: RNA hybrids (R-loops). All of these activate the S phase checkpoint, which in normal cells halts proliferation, allowing for DNA damage repair or induction of either apoptotic or senescence pathways (Kastan and Bartek, 2004; Mooi and Peeper, 2006; Serrano et al., 1997; Willis and Rhind, 2009). By contrast, in cancer cells with deficient checkpoints, aberrantly replicated DNA can lead to chromosomal missegregation and micronuclei, aneuploidy and gross chromosomal instability. As cancer cells adapt to replication stress to continue proliferating, genomic instability may contribute to tumor evolution. Pathways underlying such adaptation may present novel therapeutic opportunities (Dobbelstein and Sorensen, 2015; Ubhi and Brown, 2019).
Immediate Early Genes (IEGs), about 40 across the genome, exhibit rapid and transient transcript induction in response to acute stress or proliferation-inducing signals (Bahrami and Drablos, 2016; Morgan and Curran, 1995). Their promoters typically contain serum-response elements (SREs) and cAMP-response elements (CRE), which are bound by multi-protein complexes (SRF, CREB, ETS/TCF) upon phosphorylation by Protein Kinase A (PKA), ERK, p38 MAPK, RhoA-actin, PI3K and other stress-induced kinases. However, the immediate burst of stress-induced IEG RNAs suggests additional mechanisms that are more rapid than transcription factor-mediated gene activation. While many IEGs encode transcription factors (eg. FOS, FOSB, EGR1, NR4A1), their transcriptional targets and functions are highly context dependent. Of these, NR4A1 (also known as Nur77 or NGFIB) is an orphan nuclear receptor, which functions as ligand-independent transcription factor (Maxwell and Muscat, 2006). NR4A1 is expressed in multiple cancers (Lee et al., 2010; Wu et al., 2011; Zhou et al., 2014) and in cancer-associated stromal cells, where it mediates prostaglandin-driven paracrine signals (Zheng et al., 2019). NR4A1 has also been implicated as a master regulator of T cell exhaustion, a major limitation to effective cancer immunotherapy (Chen et al., 2019; Liu et al., 2019). While transcriptional regulation by NR4A1 of target genes has been presumed to contribute to its diverse functions, the precise mechanisms remain to be defined.
In tracing early steps of metastasis in mouse models using patient-derived circulating metastatic precursors (Circulating Tumor Cells; CTCs), we identified increased chromatin accessibility across the genebody and 3’-UTR of multiple IEGs. We show that NR4A1 binds to these genebodies and restrains baseline IEG expression; replication stress releases NR4A1, triggering a burst of IEG mRNAs. Overexpression or deletion of NR4A1 have opposing effects on genomic stability and tumorigenicity, pointing to a novel IEG regulatory pathway, with therapeutic implications.
RESULTS
Increased chromatin accessibility across the genebody and 3’-UTR of IEGs
Cultured CTCs preserve in vivo tumor genotypes and associated drug sensitivity profiles, providing unique models of metastastic precursors (Yu et al., 2014). In studying tumor initiation by two breast cancer patient-derived CTC lines (BRx142, BRx82) upon intracardiac injection of tagged cells into NSG mice and retrieval of single cancer cells from metastases, we observed major shifts in accessible chromatin patterns. While CTCs in vitro vs initial in vivo inoculation (day 0) display minimal differences in genome-wide ATAC-seq (Figures 1A, S1A and S1B), differences emerge as early as day 2 and are pronounced by day 30 (Figures 1B–D, S1C, S1D and Table S1, see Methods). Across the entire genome of both CTC lines, 37 gene loci show highly significant gains in chromatin accessibility (Figure 1D and Table S2), of which 31 (84%) reside within genebodies and 3’-UTRs, unlike the characteristic ATAC-seq signal within gene promoters and distal enhancers (Figure S1E). Gene ontology analysis nominates “Cellular response to stimulus” (P<7.7E-5) (Figure S1F), and 13/31 genes (42%) encode known IEGs, including NR4A1, FOS, FOSB (Figures 1C–E and 1D). Similar gain of accessible chromatin at IEG genebodies is evident during tumorigenesis by MDA-MB-231 (M231) breast cancer cells (Figures S1G–S1I). NR4A1 is thought to activate transcription by binding to NGFI-B-response elements at promoters (Pei et al., 2006; Winoto and Littman, 2002). However, under the conditions above, NR4A1 ChIP-seq reveals minimal promoter localization (<3%) (Figure 1F and Table S1). Single-cell RNA-seq (SMART-seq2) of CTCs recovered from metastases (259 single BRx142 cells and 123 single BRx82 cells) shows NR4A1 to be the IEG most highly induced in this in vivo model (Figure S1J). Using two different antibodies, we observe massive NR4A1 ChIP-seq signal across IEG genebodies and 3’-UTR, overlapping precisely with open chromatin domains identified by ATAC-seq (Figures 1E, 1F, S1K and Table S3). In contrast to NR4A1, the IEG transcription factor FOS exhibits the expected localization to promoter regions (42%), and it is not present at IEG genebodies or 3’-UTRs (Figures 1E and S1L). These findings reveal an early step in tumor formation where NR4A1 binding to the genebody and 3’-UTR of IEGs correlates with gain of open chromatin domains. To explore the functional significance of this phenomenon, we turned to in vitro models of IEG regulation.
Figure 1. Increased chromatin accessibility across IEG genebodies and 3’-UTRs during tumor initiation.

(A) Schematic showing intracardiac injection GFP-luciferase tagged CTCs, serial imaging of metastases, recovery of tagged single cells from mouse tissues and genomic characterization. (B) Heatmap of top 50 regions with concordant gain of chromatin accessibility during tumorigenesis in both CTC lines across all samples analyzed by ATAC-seq. Color key indicates chromatin accessibility (low, blue; high, red). (C) Volcano plots shows absent chromatin accessibility changes in BRx142s in vitro (pre-injection) vs those retrieved from mouse ovary at day 0; dramatic differences in pre-injection CTCs versus CTC from ovary metastasis at day260 (>day30). Dotted line indicates threshold (−log10(adjusted p-value)>40) used in analysis. (D) Scatter plot shows regions gaining or losing chromatin accessibility across BRx142 and BRx82s (>day30 versus pre-injection). Each dot represents a 2-kb tile in the genome. Green dots represent regions gaining chromatin accessibility during metastasis in both CTC lines, and red dots represent regions losing chromatin accessibility. IEGs gaining chromatin accessibility are identified. (E) IGV tracks show ATAC-seq (purple or cyan), NR4A1 ChIP-seq (yellow and green) and FOS ChIP-seq (blue) signal at three IEGs: NR4A1, FOS, FOSB (upper: BRx142; bottom: BRx82). Genebody and 3’-UTR regions are highlighted. For NR4A1 ChIP-seq, light and dark green tracks represent ChIP-seq signal generated using two different antibodies (see Methods). (F) Pie charts show distribution of NR4A1 ChIP-seq peaks among different genomic regions in CTC-derived metastases, with predominance at genebody regions.
In vitro modeling of stress-induced chromatin changes at IEG genebodies
To understand the role of NR4A1 and its unusual chromatin localization, we sought to recapitulate the tumor-associated phenotype using in vitro models of cellular stress. Among the various stimuli (DNA damage, heat shock, serum stimulation), serum starvation and replenishment, a classical IEG stimulation protocol recapitulates replication stress (Daigh et al., 2018), elicits NR4A1 induction and altered protein localization, analogous to that observed in vivo. Since CTCs are adapted to growth in serum-free medium, these experiments were conducted in M231 and MCF10A breast epithelial cells, both of which show tightly regulated serum-dependent proliferation. In these cells, expression of NR4A1 and other IEGs is minimal at baseline, serum-replete culture conditions (“B”). However, 24 hrs of serum starvation followed by serum replenishment results in rapid expression of IEGs, peaking at 30 min after replenishment for FOS and EGR1, and at 60 min for NR4A1. FOS induction begins at 24 hrs of serum starvation in MCF10A, whereas it is not initiated until 30 mins after serum replenishment in M231 (Figures S2A–S2D). “Serum stress”, encompassing both starvation and replenishment induces IEGs with a range of gene-specific patterns in different cell lines. NR4A1 protein staining shows strong induction in nuclei upon serum stress but chromatin-bound NR4A1 is not increased, suggesting the presence of different functional pools (Figure S2E).
To identify the timeline of NR4A1 chromatin localization in response to serum stress, we performed NR4A1 ChIP-seq on MCF10A and M231 cultured under B conditions, serum starvation, and at closely spaced sequential time points following serum replenishment. Using stringent statistical criteria for ChIP-seq peak calling (Table S1, see Methods), we identified 36 genes showing high-confidence NR4A1 binding under B conditions (Table S3). NR4A1 predominantly binds to gene bodies under these conditions, analogous to that observed in CTC-derived tumors (Figure S2F). In contrast, other IEG proteins, such as FOS and MYC, show characteristic chromatin localization to TSS (Figures S2F and S2G). Gene ontology analysis of NR4A1-bound genes reveals “Cellular response to stimulus” as the top pathway (P<4.0E-10) (Figure S2H). The six most highly significant genes bound by NR4A1 (ranked by p-values and FDR) are the IEGs EGR1 (P<1.1E-112), FOS (P<8.4E-59), FOSB (P<1.9E-43), JUND (P<4.0E-22), JUNB (P<8.6E-22) and JUN (P<3.9E-14). Expression of these IEGs, undetectable under B conditions, peaks 30 min after serum replenishment (Figure 2A). Conversely, NR4A1 binding to IEG genebodies, maximal under B conditions, begins to decline after 24 hrs of serum starvation with a nadir 1 hr after serum replenishment (Figures 2B, S2I and S2J). This tight sequential timing, together with the inverse relationship between NR4A1 chromatin binding and IEG expression, raises the possibility of an inhibitory role for NR4A1 on IEG expression.
Figure 2. In vitro modeling serum stress-induced chromatin changes at IEG genebodies.

(A) Boxplots show time course of IEG expression (FOS, FOSB, JUN, JUNB, JUND, EGR1) at serum-replete baseline (B), after 24 hour serum starvation (0h) and at serial intervals following serum replenishment (0.5–6h) in MCF10A. (B-C) Reduction in NR4A1 binding (B) and RNA Pol II p-S2 (C) at the 6 IEG genebodies from baseline to serum withdrawal and replenishment generated from ChIP-seq data. 6 randomly picked genebodies across the genome serve as control for RNA Pol II p-S2 binding. (D) Bar graphs of representative IEGs (FOS, FOSB and EGR1) show inverse correlation between ChIP-seq binding at genebodies by NR4A1 or RNA Pol II p-S2 (purple) and corresponding IEG mRNA expression (cyan). Error bar represents SD. (E) Timeline of PRO-seq signal at IEG genebodies (blue) and RNA Pol II p-S2 occupancy at the same region (red) for 6 IEGs (FOS, FOSB, EGR1, JUN, JUNB, JUND) from timepoints B, 0h and serial intervals after serum (1h and 3h) in MCF10A. Left y-axis: RNA Pol II p-S2 read coverage, right y-axis: PRO-seq genebody coverage of the corresponding IEG. (F) IGV tracks show nuclear run-on sequencing (PRO-seq) signal at IEG genebodies at timepoints B, 0h, and serum treatment (1h, 3h). GAPDH: non IEG control. (G) Transient decrease in calculated RNA Pol II Pausing Index (PI) at representative IEGs (FOS, FOSB and EGR1), during serum withdrawal and serum treatment. (H) Detection of chromatin-bound IEG pre-mRNAs (FOS, EGR1) in MCF10A under serum-replete conditions (B), with marked reduction following serum refeeding (1h). GAPDH: non-IEG control. Error bar, SD. (I) Co-immunoprecipitation followed by western blot shows protein association between NR4A1 and RNA Pol II p-S2 in MCF10A at baseline, but not uponserum withdrawal (0h). (J) Sequential ChIP-qPCR (initial RNA Pol II followed by NR4A1 immunoprecipitation), shows colocalization of RNA Pol II and NR4A1 at the same IEG (FOS, EGR1) genebodies at baseline (B) with reduction following serum starvation and replenishment (1h). OLIG2: non-IEG control. Error bar, SD. (K) Suppression of FOS mRNA induction by serum starvation/refeeding following treatment of MCF10A with AKT inhibitor (AKT-i), Ipatasertib. (L) ChIP-qPCR shows persistence of NR4A1 binding at FOS genebody in MCF10A treated with AKT-i, despite serum starvation/replenishment (1h), compared with stress-induced reduction in NR4A1 binding in controls. Error bar, SD. (M) Quantitative analysis of single cell staining (Nuc: nuclear, Cyto: cytoplasmic) of phospho-NR4A1 (serine 351) in MCF10A treated with AKT-i or DMSO at serial intervals following serum starvation/refeeding (B, 0h, 1h). Error bar, SEM. (N) Representative images show suppression of serum stress-inducible nuclear and cytoplasmic phospho-NR4A1 (ser351) by AKT-i treatment, compared with DMSO. Baseline (B) compared with serum starvation/refeeding (1h). DAPI indicates nuclei; scalebar, 10 μM. In panels A-C, H, J-M, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns, not significant, by two-tailed Student’s t-Test.
To correlate the sequential pattern of NR4A1 loading at IEG genebodies with that of RNA polymerase, we performed RNA Pol II ChIP-seq at the same serial intervals following serum starvation and refeeding (Table S1). RNA Pol II ChIP-seq reads are enriched at TSS, in both MCF10A and M231 under all culture conditions (Figure S2K). In contrast all the NR4A1-bound IEGs display broad RNA Pol II residency at genebodies and 3’-UTRs, in cells cultured under B conditions. RNA Pol II pile-ups at IEGs overlap precisely with those of NR4A1 (Figure S2I). This ChIP-seq pattern is observed using two different antibodies against the licensed S2-C-Terminal Domain (CTD)-phosphorylated form of the polymerase (RNA Pol II p-S2), as well as against total RNA Pol II (Figures S2L and S2M). The concordance in chromatin localization for RNA Pol II and NR4A1, as they pile up across IEG genebodies and 3’-UTRs, is clearly evident in CTC-derived tumors (>day 30), but not in preinoculation CTCs (Figures S2N and S2O), supporting the relevance of this in vitro model.
Acute stress releases stalled transcriptional elongation at IEG genebodies
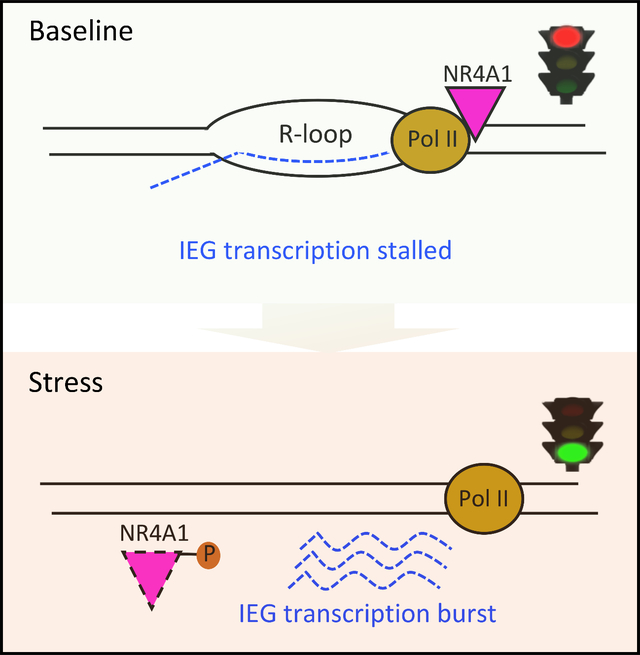
Applying the in vitro model of acute cellular replication stress, we observe that pile-up of RNA Pol II along the IEG genebodies resolves within 30–60 min after serum stress, concordant with the erasure of NR4A1 occupancy from the same sites (Figures 2C, 2D and S2H). The rapid completion of RNA Pol II transcription across these typically short IEG genebodies after NR4A1 dissociation likely precludes detection of polymerase translocation across the genebodies using RNA Pol II ChIP-seq. Both NR4A1 and RNA Pol II pile-ups at IEG genebodies are inversely correlated with IEG RNA expression: IEG mRNAs are undetectable at baseline (B) when both NR4A1 and RNA Pol II localize to IEG genebodies, and they peak promptly after serum stress, as the NR4A1 and RNA Pol II genebody pile-ups are resolved (Figures 2D and S2I). This inverse correlation between IEG expression and RNA Pol II occupancy at IEG genebodies suggests that the massive RNA Pol II pile-ups reflect paused transcription. This is consistent with a model in which cellular stress triggers NR4A1 dissociation from IEG genebodies, with RNA Pol II stalling resolved, and rapid release of poised RNAs.
The immediate induction of IEG mRNAs following acute serum stress is matched by its very rapid resolution (i.e., a single burst of expression). Interestingly, RNA Pol II does not reload at either the TSS or genebodies of IEGs within 6 hours of serum stimulation (Figures 2C, 2D and S2H–S2M). This raises the possibility that rapid completion of stalled transcriptional elongation may lead to both precipitous onset and rapid termination of IEG expression. To further define the NR4A1-associated phenomenon, we applied Precision nuclear Run-On followed by sequencing (PRO-seq), which measures nascent transcription and RNA Pol II pause sites at single-base resolution (Mahat et al., 2016), at serial intervals following serum stress (Table S1). Under B and serum stress conditions in MCF10A, PRO-seq shows an inverse relationship between RNA Pol II chromatin occupancy and RNA polymerase transcription at IEG genebodies, consistent with the RNA polymerase stalling model (Figure 2E). Coincident with serum stress and with the reduction in RNA Pol II and NR4A1 pile-ups, PRO-seq shows a transient increase in nascent transcription across the entire genebody of IEGs (including FOS, FOSB, EGR1), with a predominance of reads increasing at the 3’-end, rather than the TSS (Figures 2E and 2F).
We next used PRO-seq reads to calculate the RNA polymerase pausing index (PI). Low PI indicates active transcription and high PI is characteristic of stalled RNA Pol II (Day et al., 2016; Rahl et al., 2010). Comparing PRO-seq read densities between promoter and genebody regions for multiple IEGs, at different time points following serum stress, shows a reduction in PI as cells transition from B to serum-stress conditions (Figure 2G). Finally, RNA Pol II complex stalling along the genebody is likely to associate with unprocessed RNA transcripts. We therefore isolated chromatin-associated RNA from MCF10A by cellular fractionation: unspliced FOS and EGR1 pre-RNAs are evident in the chromatin fraction under B conditions and reduced upon serum stress. The accumulation of chromatin-bound unspliced IEG mRNAs in unstressed cells is consistent with stress-induced burst of IEGs being derived from rapid completion of transcriptional elongation and processing, rather than new transcriptional initiation (Figure 2H).
Recapitulating acute stress-induced IEG expression thus uncovers a gene-specific colocalization of NR4A1 and RNA Pol II that is inhibitory to transcriptional elongation across genebody and 3’-UTR of IEGs. The selective stress-induced NR4A1-associated effect on transcriptional elongation differs from the well-defined MYC-dependent “pause and release” mechanism, which is localized to promoter proximal sites and TSS of multiple genes (Rahl et al., 2010).
NR4A1 interacts with the RNA Pol II complex under unstressed conditions
While NR4A1 binds to DNA with sequence specificity, this consensus sequence does not mediate its localization along IEG genebodies as determined by ChIP-seq. We therefore postulated that NR4A1 recruitment to genebodies may be mediated in part by protein-protein interactions, potentially with RNA Pol II itself. To test this, we immunoprecipitated NR4A1 from MCF10A, followed by western blot against total RNA Pol II. Under B conditions, RNA Pol II co-immunoprecipitates with NR4A1, but this protein association is not detectable upon serum stress, as NR4A1 dissociates from IEG genebodies (Figure 2I). Thus, NR4A1 dynamically interacts with the RNA polymerase elongation complex, as a function of cellular stress. The basal association between NR4A1 and the RNA Pol II complex is evident by western blot using antibodies against the polymerase itself, the licensing modification on the polymerase (phospho-S2 CTD), and the genebody elongation factor ELL2 (Figures 2I and S3A) (Chen et al., 2018). A similar NR4A1/RNA Pol II protein association is evident in vivo in CTC-derived tumors (Figure S3B).
To confirm that NR4A1-RNA Pol II interactions occur within the same IEG genebody region denoted by their respective pile-ups, rather than on parallel molecules, we performed sequential immunoprecipitation in MCF10A under B conditions. Initial RNA Pol II ChIP pull-down, followed by release of immunoprecipitated proteins, sequential NR4A1 pull-down, and then qPCR for IEG genebody sequences, confirms that NR4A1 and RNA Pol II bind concurrently at the same IEG template. This protein-protein-DNA interaction is evident at baseline, but it is reduced upon serum stress (Figure 2J). To test whether NR4A1 binding at IEG genebodies is dependent on RNA Pol II, we treated MCF10A with actinomycin D (ActD), which triggers the dissociation of RNA Pol II from chromatin (Bensaude, 2011). ActD resolves RNA Pol II pile-ups at IEG genebodies and reduces IEG expression under B conditions (Figures S3C and S3D), but it has limited effect on NR4A1 binding to IEG genebodies (Figure S3E), indicating that the polymerase is not required to maintain NR4A1 binding.
Phosphorylation-dependent release of NR4A1 from IEG genebodies
Serum stimulation triggers complex kinase cascades, including ERK, p38 MAPK, PI3K and JNK, which phosphorylate and activate transcription factor/cofactor complexes including CREB, SRF, ELK1, ETS/TCF and MLK1, triggering IEG transcription (Hotamisligil and Davis, 2016; Ronkina et al., 2011). We tested whether these kinases could play a role in NR4A1 relocalization at IEG genebodies. NR4A1 is phosphorylated by AKT on serine 351, within the DNA binding domain, which mediates its relocalization from nucleus to cytoplasm (Pekarsky et al., 2001). Among a panel of kinase inhibitors (see Methods), the AKT inhibitor Ipatasertib suppresses IEG expression upon serum stress in MCF10A (Figures 2K, S3F and S3G). This reduced stress-induced IEG expression is accompanied by striking persistence of NR4A1 pile-ups at IEG genebodies despite acute serum stress (Figures 2L and S3H). High throughput single-cell imaging with two independent phospho-NR4A1 antibodies (ser351) confirms loss of both nuclear and cytoplasmic NR4A1 (Figures 2M, 2N and S3I). Thus, stress-induced phosphorylations activating IEG transcription also modulate their more rapid expression due to release of NR4A1 from IEG genebodies.
NR4A1 deletion disrupts IEG transcriptional regulation
To test whether NR4A1 has a direct role in RNA polymerase stalling at IEG genebodies, we used CRISPR/Cas9 with different pairs of guide RNAs to generate multiple NR4A1 knockout (KO) M231 clones (see Methods). Genomic PCR shows deletion of the NR4A1 gene, and immunoblot confirms lack of NR4A1 protein (Figures 3A and S3J). Consistent with NR4A1 suppressing IEG expression, FOS, FOSB and EGR1 are significantly elevated in NR4A1-null cells under B conditions (Figures 3B and S3K). ChIP-seq of NR4A1-null M231 shows virtual absence of RNA Pol II pile-ups at genebody regions of IEGs. In contrast, RNA Pol II binding at randomly selected genebody regions across the genome is unaffected by NR4A1 deletion (Figures 3C and 3D). These findings were verified using PRO-seq of NR4A1-null versus parental M231 under B conditions (Table S1): a 4.5-fold transcription increase across the entire genebody region of IEGs is observed in NR4A1-null cells (P=0.018) (Figures 3E and S3L). Increased IEG transcription is correlated with reduced RNA Pol II pile-ups at IEG genebodies, consistent with the release of transcriptional elongation arrest and rapid processing by RNA polymerase (Figures 3C–3E). The transcriptional Pausing Index, calculated from PRO-seq data, indicates that the transcriptional elongation arrest observed in controls is reduced upon NR4A1-KO (Figure 3F). Unlike M231, MCF10A do not tolerate CRISPR-mediated deletion of NR4A1. We therefore achieved shRNA-mediated NR4A1 knockdown (KD) in these cells using two different hairpin constructs (77.7% and 91.4% knockdown, respectively) (Figure S3M). RNA Pol II ChIP-seq of MCF10A with NR4A1-KD shows reduced RNA Pol II pile-ups at IEG gene bodies under B conditions, with increased expression of the IEGs FOS and EGR1 (Figures S3N and S3O).
Figure 3. NR4A1 restrains IEG transcriptional elongation under unstressed conditions.

(A) Western blot shows absence of serum stress-induced NR4A1 in M231 with CRISPR-mediated NR4A1 deletion. Vinculin, loading control. (B) Increased IEG expression (FOS, FOSB, EGR1) in NR4A1-null M231, versus parental controls, at baseline (B) and upon serum replenishment of starved cells (0.5h). Error bar, SD. (C) IGV tracks show increased nuclear run-on signal (PRO-seq) across genebody of IEGs (FOS, FOSB, EGR1) in NR4A1-null M231 at baseline compared with parental M231. GAPDH, control locus. (D) Quantitation of reduced RNA Pol II ChIP-seq signal at genebodies of 6 IEGs (FOS, FOSB, JUN, JUNB, JUND and EGR1) in NR4A1-null cells, compared with parental cells. No difference in RNA Pol II genebody coverage is observed following NR4A1 deletion at 6 randomly selected genomic loci. (E) Increased PRO-seq coverage at 6 IEG genebodies in NR4A1-null versus parental cells. No increase is evident at 6 randomly selected genebodies. (F) Reduction in calculated RNA Pol II Pausing Index (PI) at six IEGs in NR4A1-null M231 versus parental controls. P=0.03, by Wilcoxon test. (G) Design of NR4A1 deletion constructs without transactivation domain (TAD), DNA binding domain (DBD), or ligand binding domain (LBD), compared with the full length (FL) construct. (H) Suppression of deregulated IEG mRNA expression (FOS, EGR1) in NR4A1-null M231, following inducible (Dox) re-expression of NR4A1-FL, but not NR4A1-ΔDBD, despite comparable expression. Error bar, SD. (I) ChIP-qPCR quantitation showing enrichment of NR4A1 binding at IEG genebodies (FOS, FOSB, EGR1) in NR4A1-null M231, following inducible (Dox) re-expression of NR4A1-FL, but not NR4A1-ΔDBD. In panels B, D, E, H and I, *P<0.05, **P<0.01, ***P<0.001, ns, not significant, by two-tailed Student’s t-Test.
Like other orphan nuclear receptors, NR4A1 encodes DNA-binding (DBD), transactivation (TAD) and putative ligand-binding (LBD) domains (Kumar and Thompson, 1999). To map the noncanonical function of NR4A1 as a modulator of transcriptional elongation, we introduced into NR4A1-null M231, tetracycline-inducible full length NR4A1 (FL) or constructs with in-frame deletions of DBD, TAD, or LBD (see Methods, Figure 3G). Expression of the truncated constructs was confirmed by immunoblot (Figure S4A). Re-expression of FL-NR4A1 significantly reduces the deregulated baseline expression of FOS (29% reduction, P=0.005), EGR1 (55% reduction, P=0.003) and endogeneous NR4A1 (82% reduction, P=0.011) in NR4A1-null M231 at baseline (Figure 3H). NR4A1 mutants lacking either TAD or LBD domains have comparable reduction in IEG expression (Figure S4B). In contrast, NR4A1 lacking DBD fails to suppress IEG expression in NR4A1-null cells (Figure 3H). ChIP-qPCR shows significant enrichment of NR4A1 binding at IEG genebodies after ectopic expression of FL-NR4A1 in NR4A1-null M231, whereas no IEG genebody enrichment is observed upon expression of NR4A1 with DBD deletion (Figure 3I). These studies support a direct functional role for NR4A1 as a negative regulator of transcriptional elongation by RNA polymerase at IEGs, dependent upon NR4A1 DBD.
NR4A1-dependent R-loops at IEG genebodies
Stalled RNA polymerase elongation is associated with hybridization of the nascent transcript with the unwound matching antisense DNA strand, forming DNA: RNA hybrid structures called R-loops (Aguilera and Garcia-Muse, 2012). R-loops may result from RNA Pol II pausing (Allison and Wang, 2019; Skourti-Stathaki and Proudfoot, 2014) and may contribute to the chromatin accessibility at IEG genebodies. We tested whether NR4A1-mediated RNA Pol II pausing is associated with R-loop formation using DNA-RNA immunoprecipitation (DRIP) with the S9.6 monoclonal antibody, which recognizes DNA-RNA hybrids at subnanomolar affinity (Ginno et al., 2012; Sanz and Chedin, 2019). Using DRIP followed by either sequencing (DRIP-seq) or qPCR (DRIP-qPCR), we observe strong R-loop signals across IEG genebodies, including FOS and EGR1, in both MCF10A and M231, at baseline, with erasure of the R-loop signal following serum stress (Figures 4A, 4B, S4C and Table S1). We confirmed R-loop specificity of the DRIP signal in vitro using RNase H digestion of immunoprecipitates (see Methods). R-loops at IEG genebodies are reduced in NR4A1-null M231 or NR4A1-KD MCF10A, compared to parental cells (Figures 4C and 4D). Cellular fractionation demonstrates that retention of unspliced chromatin-bound IEG precursor transcripts in parental cells, is reduced upon NR4A1-KO in M231 or NR4A1-KD MCF10A (Figures 4E and 4F). To confirm that bound NR4A1 and R-loop structures are colocalized on the same molecules, we performed sequential NR4A1 ChIP and S9.6 DRIP assays (see Methods). Immunoprecipitation of NR4A1 followed by release of immunoprecipitates, S9.6 antibody pulldown, and qPCR demonstrates that NR4A1 protein and R-loops are co-localized at IEG genebodies in MCF10As. This co-localization is reduced upon serum stress (Figure 4G). NR4A1-R-loop co-localization likely results from RNA Pol II elongation arrest by NR4A1, rather than a direct consequence of NR4A1 binding; treatment of NR4A1-null M231s with the transcriptional inhibitor 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) leads to comparable RNA Pol II pausing and R-loop signal along IEG genebodies in the absence of NR4A1 (Figures S4D and S4E).
Figure 4. NR4A1-dependent R-loops and transcriptional elongation arrest at IEG genebodies.

(A) IGV tracks show S9.6 DRIP-seq signals from DNA:RNA hybrids (R-loops) across the genebody of IEGs (FOS, FOSB, EGR1) in M231 under serum replete conditions (B), and their time-dependent resolution upon 24 hour serum starvation (0h) and serum replenishment (0.5, 1, 3, 6h). For each IEG, the neighboring gene shows no R-loops, confirming gene-specific signal. (B) Quantitation of R-loops signal at IEG genebodies (FOS, FOSB, EGR1, JUN, JUNB and JUND) upon serum stress, compared with the same analysis for 6 randomly selected genebodies. (C) IGV tracks show loss of the S9.6 DRIP-seq signal across three IEG loci in three different CRISPR NR4A1-KO M231 clones, versus control, all cultured under serum-replete conditions (red). Specificity of R-loop signal is demonstrated by its disappearance upon in vitro RNaseH1 treatment (+RH). S9.6 DRIP-seq is also shown for MCF10A (green) following NR4A1-KD, compared with parental cells, again with RNase H1 treatment of parental cell lysates to confirm specificity (+RH). The neighboring gene within each IEG locus shows no R-loop signal. (D) Quantitative analysis of R-loop signal reduction at 6 IEG genebodies in 3 NR4A1-KO M231 clones and in MCF10A after NR4A1-KD. (E-F) Reduction in chromatin-associated FOS and EGR1 pre-mRNAs in NR4A1-KO M231, compared with parental controls (panel E) and in MCF10A with NR4A1-KD (panel F). Error bar, SD. (G) Sequential ChIP immunoprecipitations of NR4A1, followed by S9.6, demonstrates colocalization of NR4A1 and R-loops across the same locus in serum-replete MCF10A (B), but not upon serum replenishment (1h). ChIP-qPCR quantitation across genebodies of two IEGs (FOS, EGR1). GAPDH: non-IEG control. Error bar, SD. (H-I) Quantitation of R-loops (S9.6 DRIP-qPCR) and mRNA expression (qPCR) at IEGs FOS (panel H) and EGR1 (panel I), following Dox-inducible in vivo expression of wild-type (WT) RNase H1, versus an inactive RNase H1 mutant. Error bar, SD. (J) Reduction of chromatin accessibility (ATAC-seq) at 6 IEG genebodies upon ectopic inducible expression in MCF10A of RNase H1 (OE), versus uninduced control (K) Reduced chromatin accessibility at IEG genebodies (FOS, FOSB, EGR1) upon NR4A1-KD in MCF10A under basal conditions. Error bar, SD. (L) S9.6 DRIP-qPCR assay shows enrichment of R-loop signal at IEG genebody regions (FOS, FOSB, EGR1) in BRx82 metastases, compared with a control genebody locus (GAPDH). Error bar, SD. (M) Schematic model for NR4A1-mediated regulation of IEG expression: (i) Under unstressed conditions, NR4A1 restrains RNA Pol II transcriptional elongation across the IEG genebody, retaining pre-mRNA transcripts, generating R-loops and halting gene expression; (ii) Acute replication stress-induced phosphorylation of NR4A1 leads to its dissociation from the IEG genebody, releasing transcriptional elongation by RNA Pol II, resolving R-loops and triggering an acute burst of IEG expression from the rapid completion of poised pre-mRNA transcripts. In panels B, D-L, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns, not significant, by two-tailed Student’s t-Test.
R-loops have primarily been studied in processes such as DNA damage exemplified by collisions of transcriptional and replication machineries (Aguilera and Garcia-Muse, 2012). In contrast, NR4A1-triggered R-loops at IEGs occur under physiological unstressed culture conditions. To test the consequences of suppressing R-loops, we ectopically expressed in M231 tretracycline-inducible RNase H1, which degrades RNA in DNA: RNA hybrids and resolves R-loops in vivo (Figures S4F–S4H, see Methods) (Akman et al., 2016; Ginno et al., 2012; Parajuli et al., 2017). S9.6 DRIP-qPCR at baseline shows abrogation of R-loop signals at the FOS (31.5% reduction, P=0.0002) and EGR1 (48.9% reduction, P=0.01) genebodies in RNase H1 expressing cells (Figures 4H and 4I). Ectopic RNase H1 expression significantly increases FOS (3.1-fold, P=0.0001), and EGR1 (2.2-fold, P=0.007) expression (Figures 4H and 4I), indicating that R-loops may contribute to transcriptional suppression. As control, a tetracycline-inducible nonfunctional mutant RNase H1 has no effect on IEG R-loops and RNA expression (Figures 4H, 4I and S4H). The effect of RNase H1 is also observed by direct staining of both MCF10A and M231 (Figure S4I).
We tested if R-loop accumulation along IEG genebodies explains the open chromatin genebody configuration identified by ATAC-seq. MCF10A at baseline shows ATAC-seq signal across IEG genebodies, which is reduced by RNase H1 expression in vivo (Figures 4J and S4J). Similarly, NR4A1-KD in MCF10A reduces ATAC-seq signal at IEG genebodies (Figure 4K). Thus, NR4A1 binding is required for R-loop formation at IEG genebodies, contributing to accumulation of open chromatin domains at these loci. Of note, in CTC-derived tumors with high chromatin accessibility and strong NR4A1 and RNA Pol II occupancy at IEG genebodies, S9.6 DRIP-qPCR reveals R-loop enrichment at the same IEG genebodies (Figures 4L and S4K). In both BRx82 and BRx142-derived tumors, the H3K27me3 repressive histone mark is also enriched at these IEG genebodies (Figure S4L).
Taken together, we propose a model whereby NR4A1 interacts with RNA Pol II to arrest transcriptional elongation specifically at IEG genebodies, generating extensive R-loops and chromatin accessibility. Arrested IEG transcripts along the genebody are “poised”, such that stress-induced NR4A1 dissociation releases an immediate coordinated burst of mRNAs (Figure 4M). This unique IEG transcriptional elongation checkpoint may play a role in cancer cell adaptation to chronic oncogenic replication stress
NR4A1 suppresses replication stress and genomic instability
NR4A1-null M231 display increased apoptosis and impaired proliferation (Figures S5A–S5C). This is also observed in NR4A1-KD MCF10A (Figure S5D). Dual staining of NR4A1-null M231 with α-tubulin and DAPI reveals multiple chromosomal defects: 39.5% (mean, range: 34.1%−44.9%) of cells have multiple nuclei or gross chromosomal fragmentation, compared to parental M231 (2.1% mean, range: 1.1%−3.0%) (Figures 5A and 5B). Similar chromosomal instability, including cells with multiple nuclei and micronuclei, is observed in NR4A1-KD MCF10A (Figures 5C and 5D). DNA content analysis reveals extensive aneuploidy and genome duplications in NR4A1-deficient cells, compared with parental cells (Figure 5E). Single-cell karyotypes show increased chromosome number per cell, from a median of 59 in parental M231 to 87, 132 and 182 in three independent NR4A1-null clones (P=0.0115, P<0.0001 and P<0.0001, respectively) (Figure 5F). NR4A1-null M231 also show prolonged G2/M phase consistent with mitotic arrest, both under baseline conditions and upon serum replenishment (Figure S5E). A prolonged G2/M is also observed in NR4A1-KD MCF10A (Figures S5F and S5G). In addition to chromosomal defects evident in the NR4A1-null cells, acute NR4A1-KD using three different shRNAs in M231 also triggers genomic duplications and G2/M arrest (Figures S5H and S5I). Similar chromosomal defects are observed upon NR4A1-KD in CTC lines (Figures S5H and S5I).
Figure 5. NR4A1 deletion triggers FOS-mediated replication stress and genomic instability.

(A) Representative images of dual α-tubulin and nuclear (DAPI) staining, shows increase of multinucleated cells and multinuclei in NR4A1-null M231 (arrows). Right inset, high magnification. Scalebar, 50 μM. (B) Quantitation of cells with gross mitotic defects in two M231 NR4A1-null clones, compared with parental control. Error bar, SD. (C) α-tubulin and DAPI staining shows multiple micronuclei (arrows) in NR4A1-KD MCF10A versus scrambled control (shScr). Scalebar, 50 μM. (D) Quantitation of NR4A1-KD MCF10A with mitotic defects. Error bar, SD. (E) Increased DNA ploidy (flow cytometry) in three independent NR4A1-null M231 clones, compared with controls. X-axis: relative DNA content (PI stain). (F) Single-cell karyotyping shows chromosome gain in three NR4A1-null M231 clones, compared with controls. (G) Quantitation of cells positive for phospho-Chk1 and phospho-γH2AX foci in two NR4A1-null M231 clones, versus parental cells. Error bar, SEM. (H) Resolution of aneuploidy (flow cytometry) in NR4A1-null M231, following suppression of FOS by siRNA (technical replicates: #1–2) versus siRNA control. No such rescue following knockdown of IEGs MYC, BHLHE40 or FOSB. X-axis: relative DNA content. (I) Reduction in phospho-Chk1 and phospho-γH2AX foci in M231 upon simultaneous siRNA knockdown of NR4A1 and FOS. Error bar, SEM. In panels G and I, image quantification was carried out by ImageJ. In panels B, D, F, G and I, *P<0.05, **P<0.01, ****P<0.0001, by two-tailed Student’s t-Test.
RNA-seq in NR4A1-null M231 compared with parental reveals 501 downregulated genes (fold change>2, P<0.05) (Figure S6A, Tables S1 and S4), with the top pathways related to DNA replication (P<3.0E-5) and DNA repair (P<5.7E-4) (Figure S6B). NR4A1-null cells show increased phosphorylated Chk1, γ-H2AX, RPA32 and 53BP1, evidence for replication stress and activated DNA-damage response (Figures 5G and S6C–S6E). Increased Chk1 in NR4A1-null cells is associated with increased sensitivity to the Chk1 inhibitor MK-8776, compared with control (Figure S6F). Thus, NR4A1 is critical to maintain genomic stability and its deletion causes chromosomal defects that compromise proliferation and survival.
Replication stress in cancer cells results from deregulated oncogenic proliferative signals, in the absence of either negative feedback or checkpoint controls (Kotsantis et al., 2018; Macheret and Halazonetis, 2015). To test if IEG deregulation contributes to NR4A1-dependent genomic instability, we tested the consequences of individual IEG knockdown in NR4A1-null M231. Remarkably, FOS-KD by either siRNA or shRNA in NR4A1-depleted cells reverses their mitotic defects and genome duplication, and attenuates markers of replication stress and DNA damage (Figures 5H, 5I and S6G–S6I). No such effect is observed upon knockdown of other IEGs, including FOSB, BHLHE40 and MYC (Figure 5H). The reversal of genome duplication in NR4A1-null cells is not due to cell cycle arrest, since NR4A1-null/FOS-KD cells recover from the proliferation defect of NR4A1-null cells (Figure S6J). Instead this rescue likely reflects the death of mitotically compromised cells (rather than resolution of their chromosomal defects) and with the suppression of further genomic instability mediated by ongoing deregulated FOS expression. By analogy, ectopic FOS expression in parental M231 triggers replication stress and mitotic defects (Figures S6K–S6O).
These observations indicate that NR4A1 ensures genomic stability in proliferating cells, protecting against replication stress, “mitotic catastrophe” and proliferative failure. The rescue of the NR4A1-null phenotype by FOS-KD directly links the role of NR4A1 in genomic stability to its suppression of deregulated and sustained FOS expression.
Targeting NR4A1 in breast cancer patient-derived circulating tumor cells
Publicly available RNA-seq databases show that NR4A1 is upregulated >18-fold in breast cancer, compared with normal breast epithelial cells (Chung et al., 2017; Nguyen et al., 2018). Previous studies have correlated NR4A1 expression with a poor prognosis (Lee et al., 2010; Lee et al., 2012; Wu et al., 2011). We find that NR4A1 expression is highly correlated with adverse outcomes in TP53-mutant metastatic breast cancer [(https://kmplot.com/analysis/); Figures S7A and S7B)]. Interrogating our single cell RNA-seq of 109 individual breast CTCs freshly isolated from 41 women with metastatic breast cancer (Jordan et al., 2016), we find a positive correlation between NR4A1 expression and proliferative signatures (r=0.63, P<3.1E-13). Among these 41 patients, those having one or more CTCs with high NR4A1 expression (top decile) are more likely to have developed brain metastases (P=0.031, Fisher exact test) or meningeal metastases (P=0.0029, Fisher exact test), features of highly invasive metastatic cancer.
Despite the negative prognosis of elevated NR4A1 expression in cancer, functional studies have produced conflicting pro- and anti-proliferative phenotypes, depending on experimental contexts (Safe and Karki, 2021). We therefore tested the impact of NR4A1 on the ability of patient-derived breast CTCs (BRx142, BRx82) to generate tumors in NSG mice. NR4A1-KD in both CTC lines reduces in vitro proliferation and completely abrogates in vivo tumorigenesis upon orthotopic mammary gland inoculation (Figures 6A, S7C and S7D). NR4A1-null M231 also show reduced tumorigenesis in vivo (Figures 6B and S7E). Overexpression of NR4A1 in the two CTC lines (40- and 30-fold, respectively) enhances both orthotopic tumorigenesis and cardiogenic metastasis (Figures 6C and 6D), while only modestly increasing in vitro proliferation (Figures S7F and S7G). NR4A1-overexpressing tumors display reduced apoptosis (Figures S7H and S7I). Thus, NR4A1 expression appears to be rate-limiting for in vivo tumorigenesis.
Figure 6. NR4A1 contributes to breast cancer tumorigenesis.

(A) Reduced orthotopic mammary tumorigenesis in mice by BRx142 with NR4A1-KD (cyan), compared with scrambled control (black), plotted as average bioluminescence (luciferase) signal over time. Representative mouse with paired tumors (right gland, shNR4A1; left, shScr) at day 75. (B) Reduced mammary tumors from NR4A1-KO M231, compared with parental control. (bioluminescence signal). Error bar, SD. (C) Increased orthotopic mammary tumorigenesis by BRx142 overexpressing NR4A1 (OE) versus control. (D) Increased metastases by NR4A1-OE BRx142 following intracardiac inoculation, compared with control. (E) Suppression of BRx142-derived orthotopic tumors, following treatment with NR4A1 inhibitor DIM-C-pPhCO2Me (NR4A1-i; 20 or 40 mg/kg/day). Average bioluminescence signal over time. (F) Immunohistochemistry staining (IHC) shows elevated FOS and phospho-γH2AX, but decreased Ki-67 in tumors following NR4A1-i (Day 29, 40 mg/kg/day). Scalebar, 100 μM. (G) Quantitation of tumor cells per field positive for FOS, phospho-γH2AX or Ki-67, after treatment with either vehicle or NR4A1-i (Day 29, 40 mg/kg/day). Error bar, SD. In panels A-E and G, **P<0.01, ***P<0.001, by two-tailed Student’s t-Test.
We treated BRx-142-generated orthotopic tumors with the bis-indole-derived NR4A1 antagonist DIM-C-pPhCO2Me (NR4A1-i), which suppresses migration and induces cell death in breast cancer cells (Hedrick et al., 2015). Compared with vehicle, NR4A1-i caused tumor shrinkage (Figure 6E). Residual tumors in NR4A1-i-treated mice have elevated FOS expression, increased levels of the DNA damage marker phospho γH2AX, and decreased expression of Ki-67 (Figures 6F and 6G). Testing for potential interactions with drugs used to treat metastatic breast cancer, we observed cooperative sensitivity in BRx142 and BRx82 treated with NR4A1-i and the DNA damage-inducing doxorubicin, but not with the CDK4/6 inhibitor palbociclib (Figure S7J). The mitotic disruption that accompanies NR4A1 inhibition in cancer cells may thus sensitize them to DNA damage-inducing agents, providing added therapeutic opportunities in advanced breast cancers.
Prevalence of IEG genebody accessibility across human cancers
To extend our observations on the NR4A1-IEG pathway beyond cultured breast cancer cells, we determined whether this phenomenon is observed in primary human cancers by reanalyzing a recent ATAC-seq analysis of 404 TCGA human primary cancers across 23 cancer types (Corces et al., 2018). We calculated the ATAC-seq read density ratio of genebody and 3’-UTR to promoter regions for IEGs, including NR4A1, FOS and EGR1; tumors can be readily classified as NR4A1 genebody accessibility-high (genebody/promoter ATAC-seq ratio of NR4A1 >=1) or NR4A1 genebody accessibility-low (genebody/promoter ATAC-seq ratio of NR4A1 <1) (Figures 7A–7C). Across all tumor types, approximately 62.4% of all primary cancers analyzed are NR4A1 genebody accessibility-high. However, this fraction varies by cancer type, ranging from 100% high in prostate cancers to 59.5% in breast cancers and 11.8% in liver cancers (Figures 7A–7C). Across all TCGA cancers studied, ATAC-seq signal shows strong concordance across the genebodies of NR4A1, FOS, EGR1 and other IEGs (Figures 7B and S7K). Among primary breast cancers, NR4A1 genebody accessibility is high in 65.9% of luminal A/B cancers, which are characteristically HR-positive and well differentiated, versus 22.7% of basal cancers, which include the more aggressive TNBC, and 9.1% of HER2-amplified tumors. Similarly, FOS genebody accessibility is high in 72.1% of luminal A/B, 26.6% of basal subtype and 18.2% of HER2-enriched breast cancers (Figures 7D and S7L). In contrast to the correlation between total cellular NR4A1 expression and poor clinical outcome, IEG genebody accessibility status is highly correlated with a favorable clinical outcome in breast cancer (logrank P=0.013 for NR4A1 genebody accessibility; logrank P=0.025 for FOS genebody accessibility), as well as with lower tumor stages (Figures 7E–7G and S7M). Across all cancer types, increased IEG genebody chromatin accessibility is correlated with reductions in apoptosis signaling, stress response and DNA damage response signaling and increases in developmental growth signaling pathways, including WNT, NOTCH and TGF-β signaling (Figures 7H and S7N).
Figure 7. Prevalence of NR4A1 and IEG genebody chromatin accessibility in primary human cancers.

(A) Heatmap shows ATAC-seq signal at three IEG loci across TCGA tumor types, highlighting the prevalence of accessible chromatin at genebody and 3’-UTR regions. Color key from either blue to yellow (genebody) or white to black (other loci) indicates normalized read density from low to high. Original ATAC-seq data derived from (Corces et al., 2018). TCGA tumor study abbreviations are referenced from https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations. (B) Representative IGV tracks show normalized ATAC-seq signal at IEG genebody (highlighted) in prostate (PRAD), breast (BRCA) and liver (LIHC) cancers (Corces et al., 2018). (C) Percentage of patients shown in (A) with primary tumors showing high (red bars) or low (blue bars) ATAC-seq signal at the NR4A1 genebody across TCGA cancer types. Only cancer types with more than 10 ATAC-seq samples are shown. Patients are classified into high and low groups according to the NR4A1 genebody/promoter ATAC-seq signal ratio (see Methods). A higher prevalence of favorable breast cancer histology (PAM50 molecular subtypes) (D) and lower pathologic tumor stages (AJCC) (E) is evident in patients with high NR4A1 genebody ATAC-seq signal. *P<0.05, **P<0.01 by Fisher exact test. (F-G) Kaplan-Meier plot showing improved overall survival in breast cancer patients with high accessibility (genebody ATAC-seq signal) at NR4A1 (panel F) and FOS (panel G). Statistical significance assessed by logrank test. (H) Box plots showing GSVA analysis of different pathways in all analyzed TCGA primary tumors (Corces et al., 2018) with high (n=237) or low (n=140) NR4A1 genebody accessibility. **P<0.01; ***P<0.001, by two-sided Welch’s t-test. (I) Schematic representation of the proposed non-canonical role of NR4A1 in cancer progression: effective NR4A1-mediated regulation of IEG expression favors adaptation of cancer cells to chronic replication stress and continued proliferation, whereas genetic or drug-induced suppression of NR4A1 triggers mitotic catastrophe and proliferative failure.
Finally, reanalyzing a published dataset of ATAC-seq from normal mouse mammary gland development (Dravis et al., 2018), we find IEG genebody accessibility in normal mouse mammary progenitor cells (Figure S7O). Thus, the regulation of IEG expression by NR4A1, as scored by the presence of open ATAC-seq across IEGs genebodies, likely reflects a physiological mechanism contributing to normal and tightly coordinated induction of IEG expression in response to cellular stress. A large fraction of human cancers, across all histological types, appears to have preserved this NR4A1-IEG pathway, raising the possibility that this reflects a cellular dependency that may be exploited for therapeutic benefit.
DISCUSSION
We uncovered a NR4A1-mediated transcription elongation checkpoint, specific to IEGs, whose rapid expression involves immediate release of poised transcripts. This non-canonical NR4A1 function involves binding to IEG genebodies and 3’-UTRs, restraining RNA Pol II transcriptional elongation at baseline. Upon acute replication stress, NR4A1 dissociates from genebodies, releasing a burst of IEG transcripts. NR4A1 overexpression enhances breast tumorigenesis and its suppression triggers profound mitotic dysfunction and proliferative failure, suggesting that it may play a critical role in cancer cell adaptation to chronic replication stress (Figure 7I). ‘Non-oncogene addiction’ of cancer cells depends on genes that are not oncogenic drivers, but whose function is critical to cells experiencing deregulated pathways (Luo et al., 2009; Solimini et al., 2007) including replication stress triggered oncogenic signals and aneuploidy; attenuation of replication stress by NR4A1 may therefore present therapeutic opportunities.
Acute stress and NR4A1-mediated regulation of IEGs
NR4A1 regulates a wide range of processes including metabolism, angiogenesis inflammation and immune cell differentiation (Maxwell and Muscat, 2006). While NR4A1-mediated transcriptional activation of promoters is well-described, its unexpected non-canonical function points to a novel role as a master regulator of stress-induced genes. Our PRO-seq data link transcriptional stalling with accumulation of both RNA Pol II and NR4A1 at IEG gene bodies, and the stress-induced release of poised mRNAs, accounting for an immediate sharp peak of expression. IEG expression is also known to be regulated by phoshophorylation of transcriptional regulators, poised bivalent domains within promoters, downregulation of siRNA inhibitors, and transcript stabilization (Bahrami and Drablos, 2016; Dean et al., 2004; Degese et al., 2015). Among these, the NR4A1 effect may drive the most rapid stress-induced burst of expression.
The genome-wide specificity of NR4A1 tethering to IEG genebodies, requiring its DNA binding domain, but independent of its canonical DNA recognition site, is unexplained and raises the intriguing possibility of RNA-based zinc finger interactions. Whether the unspliced FOS and EGR1 transcripts detected within their respective NR4A1-associated chromatin complexes play such a role remains to be shown. The release of NR4A1 from IEG genebodies mediated by AKT signaling provides a potential mechanism by which stress-induced phosphorylation initiates the burst of IEG expression.
NR4A1-mediated arrest of transcriptional elongation is distinct from that of MYC in releasing promoter-proximal pausing of RNA Pol II (Adelman and Lis, 2012; Eberhardy and Farnham, 2002; Rahl et al., 2010). Whereas MYC targets protein complexes localizating 30–50 bp downstream of the TSS to prevent transcription elongation, NR4A1 is localized across a large stretch of genebody and 3’-UTR. While MYC-regulated promoter-proximal pausing may serve both to prevent ubiquitous leaky transcription of many genes and as a rheostat to broadly increase transcription under proliferative conditions, the more targeted effect of NR4A1 on transcriptional stalling across IEG genebodies may serve to enable synchronous bursts of gene expression in response to stress.
The triggering of R-loops by transcriptional inhibitors along genebodies of highly expressed genes (Zatreanu et al., 2019) is consistent with the physiological R-loops seen across gene bodies of IEGs whose transcription is blocked by NR4A1. Indeed, the presence of abundant R-loops at IEG loci contributes to their characteristic open chromatin domains, which are resolved either by NR4A1 deletion or by ectopic expression of RNase H1.
Together, we identify a novel transcriptional elongation checkpoint mechanism, providing a distinct level of regulation for immediate stress response signaling.
NR4A1 and adaptation to chronic replication stress in cancer
The physiological rapid induction of IEGs in response to stress ensures coordinated proliferative and damage responses (Galluzzi et al., 2018). Chronic replication stress in cancer cells may be exacerbated by deregulation of IEGs, including FOS, which itself can trigger proliferation (Shaulian and Karin, 2001). A striking observation here is that, among the IEGs dysregulated in NR4A1-null cells, FOS-KD is unique in rescuing the mitotic abnormalities triggered by NR4A1 deletion. Indeed, NR4A1 inhibition in CTC-derived tumors increases FOS expression and markers of genomic instability and DNA damage, consistent with the importance of NR4A1-mediated FOS regulation.
While not studied here, NR4A1 serves as a key regulator of T cell exhaustion, the unresponsive phenotype following excessive stimulation in antigen-reactive T cells, and in synthetic CAR-T cells (Chen et al., 2019; Liu et al., 2019). IEG induction accompanies T cell activation, and NR4A1 blocks FOS-JUN (AP1)-mediated transcription. However, even in T cells (Liu et al., 2019), NR4A1 binding is restricted to IEG genebodies rather than to promoters or TSS, consistent with the transcriptional elongation control described here. ATAC-seq datasets of human CD8 positive T cells (Calderon et al., 2019) show striking chromatin accessibility at IEG genebodiess under unstimulated condition, with it disappearance upon cytokine stimulation (Figure S7P).
The relevance to cancer progression of IEG regulation by NR4A1 is suggested by three lines of evidence: first, the acquisition of NR4A1-associated open chromatin domains at IEGs during early tumor formation and increased tumorigenesis of breast CTCs with NR4A1 overexpression; second, the mitotic defects and proliferative failure upon NR4A1 suppression, a phenomenon linked to dysregulation of FOS expression; and third, a large fraction of primary human cancers having retained open chromatin domains at IEG genebodies, a hallmark of NR4A1-dependent regulatory pathway, indicating that this phenomenon may contribute to tumorigenesis across multiple different types of cancer.
TCGA and other public clinical databases show distinct associations between NR4A1 gene expression and NR4A1-IEG chromatin accessibility, the former associated with adverse prognosis and the latter with favorable outcome. There are significant anti-correlations between expression of NR4A1 and patient survival in carcinomas of the bladder, esophagus, lung, ovary, stomach and thyroid (Lee et al., 2010; Lee et al., 2012; Nagy et al., 2018; Wu et al., 2011). In TP53-mutated breast cancer, where genomic instability is more prevalent, this is particularly significant. In contrast, NR4A1-mediated IEG transcriptional elongation, as measured by open chromatin accessibility at IEG genebodies and 3’-UTRs, denotes less invasive cancers that are more well-differentiated, with a favorable clinical prognosis. Tumors with high NR4A1 and FOS genebody chromatin accessibility encompass over half of all primary human cancers. This includes virtually all prostate cancers and the more favorable subset of breast cancer histologies, and it is rare in aggressive tumor types such as hepatobiliary cancers. Thus, the preservation of the NR4A1-IEG axis may denote retention of a physiological replication stress checkpoint, which may be lost in aggressive cancers.
Targeting of cancer-associated stress is emerging as a therapeutic consideration (Dobbelstein and Sorensen, 2015; Ubhi and Brown, 2019). Irrespective of their initial underlying oncogenic drivers, advanced cancers accumulate multiple genetic and epigenetic abnormalities that may both accelerate their invasive properties and present homeostatic challenges. These include deregulated proliferative signals, aneuploidy and unstable gene amplifications, overproduced and poorly folded proteins, high levels of reactive oxygen species, and altered mitochondrial metabolism, all of which are hallmarks of cancer progression (Hanahan and Weinberg, 2011). Physiological checkpoints that are normally activated to halt proliferation under such conditions are commonly mutated or altered as cancer cells adapt to these persistent stresses (Dobbelstein and Sorensen, 2015). At the same time, non-essential regulatory pathways may become rate-limiting in cancer cells, as they acquire dependence on alternative mechanisms to ensure survival (i.e., non-oncogene addiction), thereby presenting a therapeutic opportunity (Gaillard et al., 2015; Pfau and Amon, 2012). Further studies are required to test whether cancers with preserved NR4A1-IEG regulatory axis are susceptible to therapeutic targeting of NR4A1, either alone or in combination with DNA-damaging agents that enhance replication stress.
Limitations of the study
Our observations raise unanswered questions, including the mechanism by which NR4A1 is specifically recruited to multiple IEG genebodies across the entire genome. The physiological significance NR4A1-mediated IEG regulation is complex, and total NR4A1 expression is clearly distinct from NR4A1 genebody localization, with respect to prognostic significance in cancer. The distinction between primary cancers with high degrees of IEG open chromatin domains, and those lacking such markers is unexplained. While the former appear to have a more differentiated phenotype and a favorable prognosis, the drivers of this heterogeneity among tumors, their differences among different types of cancer, and whether they can be exploited therapeutically remain to be studied.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel A. Haber (dhaber@mgh.harvard.edu).
Materials availability
All unique reagents generated in the study are available on request to Lead Contact.
Data and code availability
All raw and processed sequencing data in this study, including ATAC-seq, RNA-seq, ChIP-seq, DRIP-seq and PRO-seq are available at NCBI Gene Expression Omnibus (GEO) database under accession number: GSE161853. All the original western blot images and microscopy images have been deposited at Mendeley and will be shared by the lead contact upon request.
All original code has been deposited at GitHub (https://github.com/ggolczer/nr4a1_ieg_cancer) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
BRx82 and BRx142 CTC cell lines were established from primary CTCs, viably isolated from blood specimen of patients with metastatic hormone-receptor positive breast cancer, following microfluidic enrichment using the CTC-iChip (Jordan et al., 2016; Ozkumur et al., 2013). CTC cultures were maintained as anchorage independent clusters under hypoxia (4% O2), in tumor sphere medium as previously described (Yu et al., 2014). MDA-MB-231 (M231), SKBR3, MCF7, T47D and MCF10A cells were all purchased from ATCC. M231 and MCF7 cells were grown under adherent condition with 10% FBS (Gibco) in DMEM high glucose medium (Gibco). SKBR3 cells were grown in McCoy’s 5a medium (ATCC) with 10% FBS (Gibco); while T47D cells were cultured under RPMI-1640 medium (ATCC), supplemented with 10% FBS (Gibco). MCF10A cells were grown in DMEM/F12 (Gibco) with 5% horse serum, 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml Cholera Toxin and 10 μg/ml insulin. All the cell lines used in this study were routinely checked for mycoplasma using Mycoalert kit (Lonza) and their cell of origin was confirmed using genetic fingerprinting analysis.
METHOD DETAILS
Serum deprivation and replenishment study
Both M231 and MCF10A cells were routinely maintained in serum replete media, as described above. For serum deprivation studies, serum-containing medium was removed and cells were washed three times in PBS and cultured in serum-free medium for 24 hours. After 24 hours of serum starvation, serum-containing medium was added back to the cells and cells were harvested for RNA/protein extraction or ChIP-seq or PRO-seq at 30 minutes, 1 hour, 3 hours and 6 hours.
Plasmid construction
RNase H1, either wild type or containing WKKD mutant fragments, in addition to the V5 tag, were PCR amplified from plasmids ppyCAG_RNaseH1_WT (addgene #111906) and ppyCAG_RNaseH1_WKKD (addgene #111905), respectively, and subsequently cloned into the lentivial tetracycline-inducible pLVX-tetOne-Puro plasmid (Clontech, Cat# 631849) using EcoRI and BamHI sites. Different human NR4A1 truncation fragments and the full length NR4A1 were amplified from plasmid pLenti-C-Myc-DDK-P2A-Puro-NR4A1 (Origene, Cat# RC202202L1) and then also cloned into pLVX-tetOne-Puro plasmid (Clontech, Cat# 631849) with EcoRI and BamHI. All the sequences of constructs were validated by Sanger sequencing. Induction of gene expression using doxycline-regulated constructs was confirmed at 48h following drug exposure (1 μg/ml; Clontech).
Lentiviral transduction and shRNA knockdown
Early passage 293T cells were co-transfected with either pLKO.1 shRNA knockdown constructs or FOS or NR4A1 overexpression constructs (Origene), combined with pMD2.G and psPAX2 plasmids (Addgene). After 48–72 hours, viral supernatants were collected, concentrated using LentiX concentrator (Clontech), and suspended in PBS. CTC cell lines, M231 or MCF10A cells were transduced with the virus in the presence of polybrene (Santa Cruz, 8 μg/ml as final concentration) overnight. After transduction, fresh culture medium containing puromycin (InvivoGen, 2 μg/ml as final concentration) was added to the cells for 4–7 days. Cells were harvested for both RNA and protein extractions to evaluate the efficiency of knockdown or overexpression. All the shRNA constructs, including control shRNA constructs were obtained from the TRC collection (Broad Institute).
siRNA knockdown
All the siRNA used in this study, along with their non-targeting control siRNAs, were purchased from Dhamacom in smartpool format, with each pool containing four optimized oligonucleotides with distinct sequences against the target gene. M231 cells were seeded in 6-well plates one day before transfection with 30 nM siRNA smart pools in presence of Lipofectamine RNAimax (Invitrogen), medium was replaced 12–24 hours later. RT-qPCR was performed 72 hours after transfection to confirm the knockdown efficiency.
Mouse xenograft assays
All the animal experiments were performed according to the animal protocol reviewed and approved by MGH Subcommittee on Research Animal Care (IACUC). All the animals were maintained under a 12h light and 12 h dark cycle in the animal facility at MGH. 6–8 week-old female immunodeficient NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (Jackson Laboratory) were used for mammary fat pad injections and intracardiac injections. Mice were anaesthetized with isoflurane prior to tumor cell injections. For mammary fat pad injections, 20,000 BRx142, BRx82 or M231 cells were suspended in 1:1 mix with PBS/growth factor reduced Matrigel (BD), and injected into the fourth mammary fat pad. For intracardiac injections, the left side of the mouse chest was opened to expose the rib cage and 500,000 BRx142, BRx82 or M231 cells suspended in 100 μl PBS were gently injected into the left ventricle of the heart. The skin was closed and sutured and a 90-day estrogen release pellet (Innovative Research of America) was placed subcutaneously behind the neck of each mouse. Mice were routinely monitored under the IVIS Lumina II imaging system (PerkinElmer) after intraperitoneal injections of D-luciferin substrate (Sigma), and tumor derived bioluminescent signal was measured. For mouse xenograft experiments involving treatment with NR4A1 antagonist, mice carrying orthotopic mammary fat pad tumors were randomized into control and treatment groups (6 mice per group), and treated with either vehicle (corn oil, Sigma) or NR4A1 antagonist DIM-C-pPhCO2Me (NR4A1-i; Sigma; 20 mg/kg/day as low dose group; 40 mg/kg/day as high dose) by oral gavage every other day for 50 days (Hedrick et al., 2015). Tumor derived bioluminescent signal was measured weekly (IVIS).
Fluorescence-activated cell sorting (FACS)
Tumors tissue was dissected immediately after sacrificing the mice. Tissues were washed with PBS to remove excessive blood and stromal tissue and were minced with a scalpel. Tissue digestion was performed with collagenase/hyaluronidase cocktail (Stemcell) in DMEM medium at 37°C on a rocker for 2–4 hours. Samples were vortexed every 30 minutes to enhance tissue dissociation/digestion. After digestion, the tissue solution was passed through a 70 μm filter to obtain a single cell suspension, which was then stained with PE-anti-human CD298 antibody (Biolegend) together with DAPI for 30 minutes at 4°C in the dark. Unbound antibody and DAPI were removed by washing with DMEM medium. In addition to the anti-human antibody CD298, CTCs were also tagged with endogenous GFP, and human CTC-derived cells recovered from mouse tissues were identified by double staining for both PE and GFP, as well as being low for DAPI staining (indicating cell viability). Cells were sorted using a FACS Fusion Cell Sorter (BD) into an 1.5 ml Eppendorf tube for bulk ChIP-seq or RNA extraction.
RNA isolation, reverse transcription and quantitative PCR (qRT-PCR)
Bulk RNA was extracted from either cultured cells or sorted cells using RNeasy Micro kit (QIAGEN) with on-column DNA digestion. cDNA was synthesized from 50–200 ng total RNA using SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (ThermoFisher). Quantitative PCR was performed on ABI 7500 real time PCT system using comparative Ct method with SYBR green reagents (ThermoFisher). All of the Ct values for target gene expression were normalized to GAPDH.
Cell proliferation assays
Cell proliferation assays were carried out using CellTiterGlo 2.0 Viability assay (Promega) following the manufacturer’s instructions. Cells were seeded in 96-well plates at a density of 1,000 cells per well in quadruplicates, and data from four to five time points (every other day) was collected to generate the growth curve.
Annexin V Apoptosis Assay
Annexin V apoptosis assay was carried out following the instructions of Annexin V-PE apoptosis detection kit (Abcam, Cat#ab14155). Briefly, 100,000 cells (M231 control cells or NR4A1-null cells) collected from the fresh culture (cell confluency no more than 80%) were first washed with PBS and resuspended in 1x binding buffer, 5 μl Annexin V-PE probe together with DAPI (1:5,000 diluted) were added into the cell suspension and cells were incubated for 5–10 minutes before flow cytometry analysis. Dead cells or apoptotic cells were stained positive for both Annexin V-PE and DAPI.
Western blotting
Cells or tumor tissues were lysed in Laemmli buffer (Sigma) and passed through a 28G needle to enhance lysis. Protein concentration was determined using DC protein assay (Bio-rad). Proteins (15–20 μg) were separated on precast NuPAGE 4–12% Bis-Tris protein gels (ThermoFisher), and transferred onto nitrocellulose membranes (Bio-Rad). After blocking with 5% BSA buffer for 1 hour at room temperature, membranes were incubated with primary antibodies overnight at the recommended concentrations. HRP conjugated secondary antibodies (1:10,000; Bio-rad; Cat#5196–2504) were applied, and ultra-sensitive autoradiography film (Amersham) was used to detect the chemiluminescence signal. GAPDH or Vinculin was used as loading control. ImageJ software (https://imagej.nih.gov/ij/) was used to quantify the protein bands. Primary antibodies used for western blots in this study are: NR4A1 ab#1 (to detect total NR4A1 protein, and also NR4A1 protein with transavtivation domain or DNA binding domain deletion; 1:1,000; Cell Signaling technology; Cat#3960S); NR4A1 ab#2 (to detect NR4A1 protein with ligand binding domain deletion; 1:500; Santa Cruz Biotechnology; Cat#sc-365113); FOS ab#1 (1:1,000; Cell Signaling technology; Cat#2250S); phospho-Chk1 (1:1,000; Cell Signaling technology; Cat#2348S); total Chk1 (1:1,000; Santa Cruz Biotechnology; Cat#sc-8408); phospho-γH2AX ab#1 (1:1,000; Abcam; Cat#ab2893); phospho RNA polymerase II S2 (1:1,000; Abcam; Cat#ab5095); total RNA polymerase II (1:1,000; Active motif; Cat#61667); ELL2 (1:1,000; Santa Cruz Biotechnology; Cat#sc-515276X); RNase H1 (1:3,000; Abcam; Cat#ab56560); SP1 (1:1,000; Cell Signaling technology; Cat#5931S); H3K27me3 (1:1,000; Active motif; Cat#39155); GAPDH (1:2,000; Cell Signaling technology; Cat#2118S) and Vinculin (1:2,000; Cell Signaling technology; Cat#13901S). For nucleoplasmic and chromatin-bound protein analyses, subcellular fractionation of MCF10A and M231 cells under baseline culture condition and under serum replenishment conditions was performed following the instructions of Subcellular Protein Fractionation Kit (ThermoFisher, Cat#78840). Nucleoplasm and chromatin proteins were then subjected to western blot analysis.
Co-immunoprecipitation (Co-IP)
Co-IP was performed using a universal magnetic Co-IP kit (Active motif) per manufacturer’s protocol. In brief, 5–10 million cells were collected and lysed in lysis buffer. Whole cell lysate was incubated with 5 μg NR4A1 ab#3 antibody (R&D systems; Cat#PP-H1648–00) overnight at 4°C with rotation. Protein A/G magnetic beads were added to the reaction for at least three hours at 4°C. Beads were washed extensively on a magnetic rack, and protein was eluted by heating for 15 minutes at 95°C in reducing sample buffer (Bio-rad) for western blot analysis. Mouse IgG (Millipore; Cat#12–371) was used as negative control for immunoprecipitation. All the co-IP reactions were carried out in the presence of ethidium bromide (1:1000 dilution; ThermoFisher) to avoid possible indirect association with DNA.
Immunohistochemistry (IHC) staining
Tumor tissues from mouse xenografts were harvested and formalin-fixed before embedding into tissue blocks with paraffin. 10 μM thick sections were cut and stained for FOS ab#2 (1:125; Sigma; Cat#HPA018531); phospho-γH2AX ab#2 (1:480; Cell Signaling technology; Cat#9718S) and Ki-67 (ready-to-use; clone 30–9; Ventana Medical System) using epitope retrieval either through heat induced epitope retrieval (HIER) method or submerging in EDTA buffer (pH 9.0). High-quality images were acquired using the Aperio Scanscope slide scanner (Leica Biosystems). Positive foci were quantified by counting on at least 4 independent tissue areas using ImageJ software (https://imagej.nih.gov/ij/).
Immunofluorescence (IF) staining
M231 or MCF10A cells were seeded in 12-well or 24-well glass-bottomed plates (MatTek Corporation) overnight, fixed with 4% paraformaldehyde (Electron Microscopy Sciences) and permeabilized with 0.5% Triton X-100 in PBS. Cells were blocked in 5% BSA solution before incubating with primary antibody at 4°C overnight. Alexa Fluor dye conjugated secondary antibodies (1:500; Invitrogen) were applied at room temperature for 1–2 hours in the dark, and cells were counterstained with DAPI (1:10,000; Invitrogen) for 10 minutes before imaging under the confocal microscope (Zeiss LSM170) in the MGH Cancer Cancer and Molecular Pathology Confocal Core at 20x (0.80 NA) magnification. Nuclear signal intensity analysis and quantification of images were carried out with ImageJ software (https://imagej.nih.gov/ij/) and cell segmentation and staining intensity in the nuclear and cytoplasmic compartments was quantified using Halo (Indica Labs) digital image analysis software using the HighPlex FL v3.2.1 module. Primary antibodies used for staining are: NR4A1 ab#1 (1:100; Cell Signaling technology; Cat#3960S); FOS ab#1 (1:1,000; Cell Signaling technology; Cat#2250S); phospho-γH2AX ab#1 (1:500; Abcam; Cat#ab2893); phospho-53BP1 (1:100; Cell Signaling technology; Cat#2675S); 53BP1 (1:500; Cell Signaling technology; Cat#88439S); phospho-RPA32 (1:500; Bethy Laboratories; Cat#A300–245A); phospho-Chk1 (1:50; Cell Signaling technology; Cat#2348S); S9.6 (1:150; Kerafast; Cat#ENH001) phospho-NR4A1 ab#1 (Ser351; 1:400; Abcam; Cat#ab111962); phospho-NR4A1 ab#2 (Ser351; 1:100; ThermoFisher; Cat#PA5–37594) and α-tubulin (1:250; ThermoFisher; Cat#PA5–16891).
Apoptosis assay
Cells were grown under serum replete medium and cells were washed and collected when they reached 90% confluency. 100,000 cells were resuspended in 500 μl 1x Binding buffer, together with 5 μl Annexin V-PE reagent (Abcam) and 1 μl DAPI (1:50,000 dulited, ThermoFisher). After 10 minutes incubation at room temperature in the dark, samples were analyzed by flow cytometry.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining assay
TUNEL assay for in situ apoptosis detection was carried out following the protocol of Click-iT Plus TUNEL assay kit (ThermoFisher). Briefly, formalin fixed, paraffin embedded (FFPE) tumor tissues were first sectioned onto the slide and then deparaffinized in xylene followed by decreased concentrations of ethanol solutions. Terminal deoxynucleotidyl transferase (TdT) reaction was performed to each slide for 10 minutes at 37°C; Click-iT reaction was further performed for 30 minutes at 37°C in dark. DNA was stained by Hoechst 33342 dye (ThermoFisher), slides were washed and then imaged under the confocal microscope (Zeiss LSM170). Analysis and quantification were carried out using ImageJ software (https://imagej.nih.gov/ij/).
EdU incorporation and cell cycle analysis
EdU incorporation and cell cycle analysis were carried out per instructions of the Click-iT plus EdU flow cytometry assay kit (ThermoFisher). Briefly, cells were seeded one day prior, then treated with 10 μM EdU for 2 hours at 37°C before being fixed at room temperature for 15 minutes. After washing, cells were labeled with fluorescent dye picoyl azide by Click-iT reaction. DNA was stained with PI dye (ThermoFisher) and analyzed using LSRFortessa (BD). Cell cycle and DNA content analysis was performed using FlowJo software (https://www.flowjo.com/).
In vitro drug sensitivity assay
Cultured CTCs or M231 cells were washed with PBS and suspended in fresh culture medium. Cells were then seeded into 96-well plates at a concentration of 1,000 cells per well. Serial dilutions of Chk1 inhibitor (MK-8776, Selleckchem) were added to cells and cell viability was assayed 7 days after drug treatment using CellTiterGlo 2.0 reagent (Promega) in quadruplicate. Drug combination assays were carried out by adding serial dilutions of two drugs: NR4A1 inhibitor (Sigma) and doxorubicin (Sigma), or NR4A1 inhibitor (Sigma) and Palbociclib (Selleckchem), either alone or in combination into cell cultures, and assaying cell viability using CellTiterGlo 2.0 reagent (Promega) in quadruplicate. Data were analyzed with GraphPad Prism software (https://www.graphpad.com/).
Chromosomal karyotyping
Karyotyping was carried out as reported previously (Comaills et al., 2016). Briefly, NR4A1-null and control M231 cells were seeded one day prior to being treated with colcemid (Roche) for 2 hours. Colcemid was used at a final concentration of 1 μg/ml. The cells were then trypsinized and resuspended gently in 0.075 M hypotonic KCl buffer (Sigma) and incubated at 37°C for 15 minutes. After centrifugation, cells were fixed in methanol: acetic acid (3:1), and cell suspensions were dropped from a 12-inch distance onto a slide (prewashed with 100% ethanol) to spread the chromosomes. Slides were dried, mounted with mounting medium containing DAPI (Vectorlabs), and scanned under oil immersion at 60x magnification (Nikon). The number of chromosomes of each spread was counted using ImageJ software (https://imagej.nih.gov/ij/).
Actinomycin D treatment
Both MCF10A cells or SKBR3 cells were maintained in serum-containing media, and actinomycin D (ActD; Gibco) was added to the media at the concentration of 10 μg/ml, cells were collected at timepoints of 0h, 1h, 2h 4h and 6h. RT-qPCR was performed to assess IEG gene expression; total RNA Pol II ChIP-qPCR and NR4A1 ChIP-qPCR were carried out to assess RNA Pol II genebody binding and NR4A1 genebody binding.
DRB treatment
One day before treatment with 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB, Sigma), NR4A1-null M231 cells were replenished with fresh medium, and DRB was directly added at the final concentration of 100 μM for 1–3 hours. Cells were washed with 1x PBS, 5 times to ensure the complete removal of DRB, and cultured in fresh medium for another 1–2 hours before collection. Cells were subjected to total RNA Pol II ChIP, followed by qPCR assay using primers against the genebody regions of IEGs.
Kinase inhibitor treatment
A series of well characterized kinase inhibitors, including U0126 (Promega), SB203580 (Selleckchem), Ipatasertib (Selleckchem) and SP600125 (Selleckchem) were used to target MEK1/2, p38/MAPK, AKT and JNK signaling pathways, respectively. Inhibitors were supplemented into MCF10A cell culture medium individually after serum deprivation and also after serum replenishment for 30 minutes, 1 hour, 3 hours and 6 hours, all at the concentration of 10 μM. Cells were further collected for both RT-qPCR assay and NR4A1 ChIP-qPCR assays to assess IEG gene expression changes and NR4A1 genebody localization alterations upon serum stimulation.
CRISPR-Cas9 mediated gene knockout
Guide RNAs (gRNAs) were designed using CRISPR design online tool (http://crispr.mit.edu/), and then cloned into CRISPR/Cas9 plasmids: pX458 (Addgene#48138), pX459 (Addgene#62988) or lentiCRISPRv2-mCherry (Addgene#99154). Sequences of the gRNA were further confirmed using Sanger sequencing. For each gene, at least two gRNAs were designed to target the 5’ exonic region and introduce a frame shift mutation (sgRNA1+sgRNA3 as pair#1; sgRNA2+sgRNA3 as pair#2). Cas9 and gRNA plasmids were delivered into cell lines either through Lipofectamine 3000 (Invitrogen, for pX458/pX459 plasmids) or lentiviral transduction (for lentiCRISPRv2-mCherry). For pX458 and lentiCRISPRv2-mCherry plasmids, cells were sorted to enrich the population, while for pX459, cells were further selected using puromycin (InvivoGen, 2 μg/ml as final concentration). Genomic DNA was isolated followed by PCR and Sanger sequencing to verify the genome editing efficiency.
Chromatin-associated RNA isolation and quantification
Cellular fractionation and isolation of chromatin associated RNA was carried out following the protocol published previously (Conrad and Orom, 2017). Briefly, 1–3 millions MCF10A or M231 cells were first collected and lysed in 0.15% NP40 lysis buffer on ice for 5 minutes to disrupt the plasma membrane while leaving the nuclear membrane intact. The cell lysate was gently added on top of the ice-cold 24% sucrose buffer (“sucrose cushion”) followed by the centrigugation at 3,500g for 10 minutes. Supernatant (cytoplamic fraction) was collected and the resulting pellet (cell nuclei fraction) was briefly rinsed with ice-cold PBS buffer, and suspended in glycerol buffer. Urea buffer (1 M Urea and 1% NP-40) was immediately added to the suspension followed by vortexing for 5 sec and incubation on ice for 2 minutes. After 13,000g centrifugation, supernatant (nucleoplasmic fraction) was collected for later RNA extraction, and the pellect (containing the chromatin-RNA complex) was rinsed with ice-cold PBS buffer again and resuspended in 1 ml Trizol (ThermoFisher) for RNA extraction. A 21 gauge needle and syringe were used to fully solubilize the pellet in Trizol. Purified RNA was further digested with two rounds of on-column DNAse I digestion (QIAGEN), and qPCR was performed to confirm complete subcellular fractionation and absence of genomic DNA contamination. The enrichment of chromatin-associated IEG transcript across different conditions was normalized against either intronic GAPDH or XIST expression.
Assay for Transposase Accessible Chromatin with sequencing (ATAC-seq)
ATAC-seq was carried out according to the Omni-ATAC-seq protocol published previously (Corces et al., 2017). Briefly, cultured or sorted cells were washed with PBS once and subjected to nuclei preparation in ATAC resuspension buffer containing 0.1% NP-40 (Sigma), 0.1% Tween-20 (Sigma), and 0.01% digitonin (Promega) on ice. Transposition reaction was carried out in 50 μl transposition mix containing 25 μl 2x TD buffer (Illumina), 2.5 μl Tn5 transposase (Illumina), 16.5 μl PBS (Gibco), 0.5 μl 1% digitonin (Promega), 0.5 μl 10% Tween-20 (Sigma) and 5 μl H2O at 37°C for 30 minutes with 1000 RPM mixing in a thermomixer. Transposed DNA fragments were amplified with 6–15 cycles on Thermocycler using 2x NEBNext MasterMix (New England Biolabs) together with sample-specific barcoded primers. After purification with AMPure XP beads (Beckman Coulter), samples were pooled and sequenced on HiSeq X sequencer with 150 bp paired-end reads.
Chromatin immunoprecipitation (ChIP)
Cell lines cultured in vitro, or tumor xenograft-derived FACS-sorted single cell suspensions were washed with ice-cold PBS, and crosslinked with 1% formaldehyde (Sigma) for 10 minutes (for histone modifications and RNA pol II ChIP) or 15 minutes (for transcription factor ChIP) at room temperature and quenched with glycine. ChIP was performed using EZ-Magna ChIP HiSens Chromatin Immunoprecipitation Kit (Millipore) following the recommended protocol. Briefly, nuclei were isolated using cold nuclei isolation buffer and sheared using Covaris S2 instrument with an optimized program (Duty Cycle: 5%; Intensity: 4; Cycles/Burst: 200; treatment time: 360s) to yield an average fragment size of approximately 300 bp. Chromatin was incubated with Protein A/G Dynabeads (ThermoFisher) together with 5 μg ChIP-grade antibody overnight at 4°C, with rotation. The beads were then collected and washed extensively with high- and low-salt buffers. DNA was de-crosslinked and purified through ethanol precipitation. Before immunoprecipitation with Dynabeads and antibody, 10% of chromatin was saved as input control. qPCR was performed to confirm the enrichment before library preparation and sequencing. Antibodies used for ChIP assays were: NR4A1 (ChIP antibody#1; Abcam; Cat#ab13851); NR4A1 (ChIP antibody#2; Novus Biologicals; Cat#NB100–56745); FOS (Santa Cruz Biotechnology; Cat#sc-271243X); MYC (Santa Cruz Biotechnology; Cat#sc-40X); MAX (Santa Cruz Biotechnology; Cat#sc-8011X); total RNA polII (ChIP antibody#1; Diagenode; Cat#C15200004); phospho RNA polymerase II S2 (ChIP antibody#1; Abcam; Cat#ab5095); total RNA polII (ChIP antibody#2; Active motif; Cat#61667); phospho RNA polymerase II S2 (ChIP antibody#2; Active motif; Cat# 61984); H3K27me3 (Diagenode, Cat#C15410195); normal rabbit IgG (Cell Signaling technology; Cat#2729) and normal mouse IgG (Millipore; Cat#12–371).
Sequential ChIP of RNA Pol II and NR4A1 was carried out following the protocol published (Yan et al., 2016). Briefly, 50 million MCF10A or SKBR3 cells were fixed and a first round of ChIP pulldown was performed using 20 μg total RNA Pol II antibody (Diagenode; Cat#C15200004) at 4°C overnight. Beads were collected, washed, and eluted in ChIP elution buffer (part of EZ-Magna ChIP HiSens Chromatin Immunoprecipitation Kit) supplemented 10 mM DTT (Promega). The eluate was further diluted 20-fold with SCW buffer (also part of EZ-Magna ChIP HiSens Chromatin Immunoprecipitation Kit) and a second round of ChIP was carried out using 5 μg NR4A1 antibody (Novus Biologicals; Cat#NB100–56745) at 4°C overnight. The final DNA product was de-crosslinked and purified through ethanol precipitation. qPCR was performed to assess the enrichment of both RNA Pol II and NR4A1 at genebodies.
DNA-RNA hybrid immunoprecipitation (DRIP)
DRIP was performed as described elsewhere (Sanz and Chedin, 2019). Briefly, DNA from cultured cells was isolated using phenol/chloroform extraction with phase lock tubes, followed by ethanol precipitation. DNA was digested with a restriction enzyme cocktail (BsrgI, EcoRI, HindIII, SspI and XbaI, all from New England Biolabs) overnight at 37°C. For RNase H in vitro digestion controls, 8 μg digested DNA was incubated with RNase H (New England Biolabs) for 6–8 hours at 37°C before purifying with phenol/chloroform. 1 % of digested DNA was saved as input control. 4 μg DNA (with or without RNase H treatment) was incubated with 10 μg S9.6 antibody (Kerafast; Cat#ENH001) in 1x DRIP binding buffer (10 mM NaPO4 pH 7, 140 mM NaCl, 0.05% Triton X-100) overnight at 4°C, followed by incubation with protein A/G magnetic beads (Invitrogen) for an additional 2 hours. Bound beads were collected using a magnetic rack, washed 3 times with 1x DRIP binding buffer, and eluted with DRIP elution buffer (50 mM Tris pH 8, 10 mM EDTA, 0.5% SDS, Proteinase K) for 45 minutes at 55°C. DNA was purified using phenol/chloroform extraction coupled with ethanol precipitation. qPCR was performed to confirm the R-loop enrichment at the FOS and EGR1 genebodies. The same qPCR primers used for ChIP were used for the DRIP assay. Only the samples with confirmed enrichment by qPCR assay were selected to sonication using Covaris S2 instrument (average DNA fragment size into around 300bp), and then subjected to library construction and sequencing.
Sequential pulldown of NR4A1 and S9.6 was performed using the sequential ChIP-qPCR protocol with minor modifications. 50 million MCF10A cells were first fixed and chromatin was isolated and sonicated before NR4A1 antibody (20 μg; Novus Biologicals; Cat#NB100–56745) pulldown. The ChIP precipitated DNA was purified through protease K digestion, followed by ethanol precipitation, and subjected to S9.6 antibody (5 μg; Kerafast; Cat#ENH001) pulldown without any restriction enzyme digestion. The final DNA was extracted using phenol/chloroform extraction coupled with ethanol precipitation. qPCR was carried out to assess the enrichment of both NR4A1 and R-loops.
Library construction and sequencing
Immunoprecipitated DNA from ChIP and DRIP experiment, together with input DNA, were subjected to end repair, dA-tailing and ligation of illumina sequencing adapters according to the instructions from KAPA Hyper Prep Kits (Kapa Biosystems). After ligation, the final PCR amplification was performed with KAPA Hifi Hotstart Readymix (2x, Kapa Biosystems), Illumina universal primer and Illumina index primer. RNA samples extracted from RNeasy Micro kit (QIAGEN) were subjected to RNA-seq library construction following the instructions of NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Cat#E7530L). All the barcoded libraries were pooled and sequenced on Illumina HiSeq X platform with 150 bp paired-end reads.
Precision nuclear Run-On and sequencing (PRO-seq)
Precision nuclear run on was performed according the published protocol (Mahat et al., 2016). Briefly, MCF10A or M231 cells were first washed with cold PBS to remove dead cells and harvested by scraping and centrifugation at 4°C. Cell pellets were resuspended in ice-cold permeabilization buffer (10 mM Tris-HCl, pH 7.4 (Sigma), 300 mM sucrose (Sigma), 10 mM KCl (Sigma), 5 mM MgCl2 (Ambion), 1 mM EGTA (Sigma), 0.05% Tween-20 (Sigma), 0.1% NP40 substitute (Roche), 0.5 mM DTT (Promega), one tablet of protease inhibitors cocktail (Roche) per 50 ml and 4 units/ml RNase inhibitor (Ambion)) on ice for 5 minutes. After centrifugation, cells were washed twice with ice-cold permeabilization buffer, resuspended in storage buffer (10 mM Tris-HCl, pH 8.0 (Sigma), 25% (vol/vol) glycerol (Sigma), 5 mM MgCl2 (Ambion), 0.1 mM EDTA (Sigma) and 5 mM DTT (Promega)) and immediately frozen in liquid nitrogen.
Nuclear run on reactions were carried out on permeabilized cells by adding the same volume of pre-warmed 2x nuclear run on mix (10 mM Tris-HCl pH 8.0 (Sigma), 300 mM KCl (Sigma), 1% Sarkosyl (Sigma), 5 mM MgCl2 (Ambion), 1 mM DTT (Promega), 200 mM biotin-11-A/C/G/UTP (Perkin-Elmer), 0.8 units/mL RNase inhibitor (Ambion)) and incubating at 37°C for 3 minutes. Total RNA was extracted using Trizol LS reagent (Invitrogen) and further fragmented by base hydrolysis in NaOH solution (Sigma) on ice for 10 minutes. Fragmented RNA was bound with prewashed streptavidin-coated magnetic beads (M280, Invitrogen) in binding buffer (10 mM Tris-HCl, pH 7.4 (Sigma), 300 mM NaCl (Sigma) and 0.1% (vol/vol) Triton X-100 (Sigma) in nuclease-free H2O). After washing with ice-cold high-salt wash buffer and low-salt wash buffer, bound RNA was extracted from beads with Trizol reagent (Invitrogen). RNA was then subjected to 3’-RNA adapter ligation (IDT, 5’-GAUCGUCGGACUGUAGAACUCUGAAC-/inverted dT/−3’) in the presence of T4 RNA ligase I (New England Biolabs). Ligated RNA was further enriched by another round of streptavidin-coated magnetic beads incubation, followed by washing and extraction using Trizol reagent (Invitrogen). RNA was then treated with RNA 5’-Pyrophosphohydrolase (RppH, New England Biolabs) at 37°C for 1 hour, followed by treatment with Polynucleotide Kinase (PNK, New England Biolabs) at 37°C for 1 hour. After another round of streptavidin beads incubation, and washing and extraction with Trizol reagent (Invitrogen), 5’RNA adapter (IDT, 5’-CCUUGGCACCCGAGAAUUCCA-3’) was ligated to the RNA molecules using T4 RNA ligase I (New England Biolabs), and the ligated RNA was enriched using incubation with streptavidin beads, washed and again extracted using Trizol reagent (Invitrogen). Reverse transcription was performed with RP1 primer (IDT, 5’-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3’) in the presence of Superscript III RT enzyme (Invitrogen). PCR amplification was carried out with Phusion DNA polymerase (Invitrogen) together with RP1 primer and RPI-n primer (IDT, 5’-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’, where the six N’s indicate barcodes for Illumina TRUseq mutiplexing) for 16 cycles. qPCR was performed against IEG and non IEG genebody regions after PCR amplification (same primers for NR4A1 ChIP) and nuclear run on signal enrichment was calculated based on qPCR results. Only the qPCR confirmed samples were subjected to size selection by running an 8% native PAGE gel (ThermoFisher), and 140–350 bp DNA fragments were excised and recovered. Final libraries were sequenced in Illumina HiSeq X platform with 150 bp paired-end reads.
ATAC-seq and ChIP-seq data analysis
Reads from ATAC-seq and ChIP-seq samples were first trimmed for adapter pollutions and low quality using Trimgalore (v0.4.3) (https://github.com/FelixKrueger/TrimGalore) and then mapped to the human genome (hg19 assembly) using BWA (v0.7.15) (Li and Durbin, 2009). Reads with valid alignment were converted to BigWig files for visualization using deeptools (Ramirez et al., 2014). Quality controls for ATAC-seq samples including removal of mitochondrial reads were performed using the R package ATACseqQC (Ou et al., 2018). PCR duplicates were removed using sambamba (Tarasov et al., 2015) and potential mouse reads were further removed using a custom Java script with the criteria of (1) more than 2 mismatches; (2) human genome alignment score ratio less than 0.67 and mapping quality less than 30. BigWig files were generated using deeptools as described previously (Ramirez et al., 2014), differential peaks were called using MACS2 (v2.0.10) with a q value cutoff 0.05 (Zhang et al., 2008). Binning of the genome into 2 kb segments and subsequent coverage calculation, as well as coverage of each peak were computed using bedtools (v2.27.1) (Quinlan and Hall, 2010). Annotation of each peak was carried out with R package ChIPseeker (Yu et al., 2015).
PRO-seq data analysis
Reads from PRO-seq analyses were trimmed and aligned using the same pipeline described for the ATAC-seq data analysis. Quality controls including removal of mitochondrial reads and duplicated reads were performed using the R package ATACseqQC (Ou et al., 2018). Reads with valid alignment were converted to BigWig files for visualization using deeptools (Ramirez et al., 2014). Peak calling was performed using MACS2 (v2.0.10) (Zhang et al., 2008). Human genome was further segmented into 2 kb bins using bedtools (v2.27.1) (Quinlan and Hall, 2010), and the reads per million for each bin was computed using customized R scripts for downstream analysis. Regions with either gain or loss of PRO-seq signal were annotated using R package ChIPseeker (Yu et al., 2015). Pausing index (PI) based on the PRO-seq data was calculated using the same method published elsewhere (Day et al., 2016).
RNA-seq data analysis
Reads from bulk RNA-seq were first cleaned using Trimgalore (v0.4.3) (https://github.com/FelixKrueger/TrimGalore), and the trimmed reads were further aligned to hg19 assembly using Tophat2 (v2.1.1) with parameter of no-novel-juncs (Kim et al., 2013). Gene expression was determined using Htseq (v2.1.1) with parameter count (Anders et al., 2015), and differentially expressed genes (DEG) analysis was carried out with R package DEseq2 using count matrices (Love et al., 2014).
Gene ontology (GO) and gene set variation analysis (GSEA)
Gene ontology analysis was performed using online tool DAVID (Huang da et al., 2009), and only the GO terms with adjusted p-value less than 0.05 were selected to plot. Gene set variation analysis (GSVA) was performed using R package “gsva” with the settings of maximum difference true, parallel size of 1 and minimum size of 10 (Hanzelmann et al., 2013). Logfold changes, as well as p-values and adjusted p-values (FDR corrected) of the GSVA results were computed using the function lmFit and eBayes from the R package “limma” (Ritchie et al., 2015).
Annotations of genomic elements
Promoter regions were defined as −3 kb to transcription start site (TSS); downstream regions were within 3 kb after transcription end site (TES); genebody was the region between TSS and TES; while intergenic regions were defined as regions 3 kb downstream of TES and 3 kb upstream of TSS. All the annotation of TSS and TES of each gene was downloaded from UCSC (http://genome.ucsc.edu/cgi-bin/hgTables).
Reanalysis of TCGA primary tumor ATAC-seq data
ATAC-seq data (BigWig files) of 404 primary tumors were first downloaded from website (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG) (Corces et al., 2018), read density was recalculated based on 200 bp window. IEG genebody and promoter ATAC-seq signal was computed and genebody/promoter ratio was calculated. Genebody/promoter ratio was used for pairwise correlation analysis across different IEGs, including NR4A1, FOS and EGR1. All the primary tumor samples were ranked based on the NR4A1 (or FOS) genebody/promoter ATAC-seq ratio from high to low within each cancer type, and further classified into NR4A1 (or FOS) genebody accessibility-high (genebody/promoter ATAC-seq ratio >=1) and NR4A1 (or FOS) genebody accessibility-low (genebody/promoter ATAC-seq ratio <1) groups. The corresponding clinical and molecular features for each BRCA tumor sample, including tumor purity, PAM50 subtyping, AJCC pathological staging, patient overall survival, mutation status and transcriptional profiling were downloaded from TCGA (Cancer Genome Atlas, 2012) and further used to compare the difference between NR4A1 (or FOS) genebody accessibility-and NR4A1 (or FOS) genebody accessibility-low patients. TCGA tumor study abbreviations are referenced from https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations.
Supplementary Material
Table S1: Number of sequencing reads in ATAC-seq, ChIP-seq, PRO-seq, DRIP-seq and RNA-seq samples used in this study. Related to Figures 1, 2, 3, 4, S1, S2 and S4.
Table S2: List of genes showing significant gain or loss of chromatin accessibility in BRx142 and BRx82 CTC-derived tumor cells recovered after intracardiac injection. When scoring across the entire genome in both CTC lines, 37 gene loci show highly significant gains in chromatin accessibility at >day30 (combined log2 fold change in BRx142 and BRx82 CTCs >2.5). In contrast to the 37 genes with increased ATAC-seq genebody signal, 100 gene loci lose chromatin accessibility during early tumor formation (combined log2 fold change < −2.5), and none of them are IEGs. Genes with loss of chromatin accessibility are involved in diverse pathways, including vesicle docking (P<0.004) and protein transport (P<0.002). Related to Figures 1 and S1.
Table S3: List of NR4A1-bound genes in BRx142 and BRx82 CTC-derived metastases and in MCF10A cells identified by NR4A1 ChIP-seq analysis. Related to Figures 2 and S2.
Table S4: List of genes showing significant up- or down-regulation by RNA-seq in NR4A1-null M231 cells. Related to Figure S6.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-anti-human CD298 | Biolegend | Cat# 341704, RRID:AB_2274458 |
| NR4A1 ab#1 | Cell Signaling technology | Cat#3960S, RRID:AB_2153738 |
| NR4A1 ab#2 | Santa Cruz Biotechnology | Cat#sc-365113, RRID:AB_10709310 |
| FOS ab#1 | Cell Signaling technology | Cat#2250S, RRID:AB_2247211 |
| phospho-Chk1 | Cell Signaling technology | Cat#2348S, RRID:AB_331212 |
| total Chk1 | Santa Cruz Biotechnology | Cat#sc-8408, RRID:AB_627257 |
| phospho-γH2AX ab#1 | Abcam | Cat#ab2893, RRID:AB_303388 |
| phospho RNA polymerase II S2 | Abcam | Cat#ab5095, RRID:AB_304749 |
| total RNA polymerase II | Active motif | Cat#61667, RRID:AB_2687513 |
| ELL2 | Santa Cruz Biotechnology | Cat#sc-515276X |
| RNase H1 | Abcam | Cat#ab56560, RRID:AB_945244 |
| SP1 | Cell Signaling technology | Cat#5931S, RRID:AB_10621245 |
| H3K27me3 | Active motif | Cat#39155, RRID:AB_2561020 |
| GAPDH | Cell Signaling technology | Cat#2118S, RRID:AB_561053 |
| Vinculin | Cell Signaling technology | Cat#13901S, RRID:AB_2728768 |
| NR4A1 ab#3 | R&D systems | Cat#PP-H1648–00, RRID:AB_1964307 |
| Mouse IgG | Millipore | Cat#12–371, RRID:AB_145840 |
| FOS ab#2 | Sigma | Cat#HPA018531, RRID:AB_1846576 |
| phospho-γH2AX ab#2 | Cell Signaling technology | Cat#9718S, RRID:AB_2118009 |
| phospho-53BP1 | Cell Signaling technology | Cat#2675S, RRID:AB_490917 |
| 53BP1 | Cell Signaling technology | Cat#88439S |
| phospho-RPA32 | Bethy Laboratories | Cat#A300–245A, RRID:AB_210547 |
| phospho-Chk1 | Cell Signaling technology | Cat#2348S, RRID:AB_331212 |
| S9.6 | Kerafast | Cat#ENH001, RRID:AB_2687463 |
| phospho-NR4A1 (Ser351) #ab1 | Abcam | Cat#ab111962, RRID:AB_10867229 |
| phospho-NR4A1 (Ser351) #ab2 | ThermoFisher | Cat#PA5–37594, RRID:AB_2554202 |
| Alpha-tubulin | ThermoFisher | Cat#PA5–16891, RRID:AB_10984790 |
| NR4A1 ChIP ab#1 | Abcam | Cat#ab13851, RRID:AB_300679 |
| NR4A1 ChIP ab#2 | Novus Biologicals | Cat#NB100–56745, RRID:AB_2153757 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| MK-8776 | Selleckchem | Cat#S2735, CAS: 891494-63-6 |
| DIM-C-pPhCO2Me | Sigma | Cat# SML1976, CAS: 151358-48-4 |
| Doxorubicin | Sigma | Cat#D1515, CAS: 25316-40-9 |
| Palbociclib | Selleckchem | Cat#S1116, CAS: 827022-32-2 |
| Actinomycin D | Gibco | Cat# 11805017, CAS: 9048-46-8 |
| 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) | Sigma | Cat#D1916, CAS: 53-85-0 |
| U0126 | Promega | Cat# V112A, CAS: 109511-58-2 |
| SB203580 | Selleckchem | Cat#S1076, CAS: 152121-47-6 |
| Ipatasertib | Selleckchem | Cat#S2808, CAS: 1001264-89-6 |
| SP600125 | Selleckchem | Cat#S1640, CAS: 129-56-6 |
| Critical commercial assays | ||
| CellTiterGlo 2.0 Viability assay | Promega | Cat#G9241 |
| Annexin V-PE apoptosis detection kit | Abcam | Cat#ab14155 |
| Subcellular Protein Fractionation Kit | ThermoFisher | Cat#78840 |
| Universal magnetic Co-IP kit | Active motif | Cat#54002 |
| Click-iT Plus TUNEL assay kit | ThermoFisher | Cat#C10619 |
| Click-iT plus EdU flow cytometry assay kit | ThermoFisher | Cat#C10636 |
| EZ-Magna ChIP HiSens Chromatin Immunoprecipitation Kit | Millipore | Cat#17–10461 |
| KAPA Hyper Prep Kits | Kapa Biosystems | Cat#KK8505 |
| NEBNext Ultra RNA Library Prep Kit for Illumina | NEB | Cat#E7530L |
| Nextera DNA Library Prep Kit | Illumina | Cat# FC-121–1030 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE161853 |
| Experimental models: Cell lines | ||
| BRx82 | Patient derived CTC culture | Jordan et al., 2016 |
| BRx142 | Patient derived CTC culture | Jordan et al., 2016 |
| MDA-MB-231 | ATCC | Cat#HTB-26 |
| SKBR3 | ATCC | Cat#HTB-30 |
| MCF7 | ATCC | Cat#HTB-22 |
| T47D | ATCC | Cat#HTB-133 |
| MCF10A | ATCC | Cat#CRL-10317 |
| 293T | ATCC | Cat#CRL-3216 |
| Experimental models: Organisms/strains | ||
| Female NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice | Jackson Laboratory | https://www.jax.org/strain/005557 |
| Oligonucleotides | ||
| PRO-seq 3’-RNA adapter: 5’-GAUCGUCGGACUGUAGAACUCUGAAC-/inverted dT/-3’ | This paper | N/A |
| PRO-seq 5’-RNA adapter: 5’-CCUUGGCACCCGAGAAUUCCA-3’ | This paper | N/A |
| PRO-seq RP1 primer: 5’-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3’ | This paper | N/A |
| PRO-seq RPI-n primer: 5’-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’ | This paper | N/A |
| Plasmid cloning EcoRI-RNaseH1-F: 5’-CAAAGAATTCCTCGAGACCATGC-3’ | This paper | N/A |
| Plasmid cloning BamHI-RNaseH1-R: 5’-CGCGGATCCTCGATATCAAGCTTATCGAGCGG-3’ | This paper | N/A |
| Plasmid cloning EcoRI-NR4A1-deltaTAD-F: 5’-CCCTCGTAAAGAATTCATGGAAGGCCGCTGTGCTGTG-3’ | This paper | N/A |
| Plasmid cloning BamHI-NR4A1-deltaTAD-R: 5’- GAGGTGGTCTGGATCCTCAGAAGGGCAGCGTGTCCATGAAGATCT-3’ | This paper | N/A |
| Plasmid cloning EcoRI-NR4A1-deltaLBD-F: 5’-CCCTCGTAAAGAATTCATGCCCTGTATCCAAGCCCAATATGGG-3’ | This paper | N/A |
| Plasmid cloning BamHI-NR4A1-deltaLBD-R: 5’- GAGGTGGTCTGGATCCTCATGTTCGGACAACTTCCTTCACCATGCC-3’ | This paper | N/A |
| Plasmid cloning EcoRI-NR4A1-onlyTAD-F: 5’- CCCTCGTAAAGAATTCATGCCCTGTATCCAAGCCCAATATGGG-3’ | This paper | N/A |
| Plasmid cloning AgeI-NR4A1-onlyTAD-R: 5’- GAGGTGGTCTACCGGTACTTCCACCTGGGGCCCC-3’ | This paper | N/A |
| Plasmid cloning AgeI-NR4A1-onlyLBD-F: 5’- GGTCTACCGGTCCTGCCAATCTCCTCACTTCCCTGG-3’ | This paper | N/A |
| Plasmid cloning BamHI-NR4A1-onlyLBD-R: 5’- GAGGTGGTCTGGATCCTCAGAAGGGCAGCGTGTCCATGAAGATCT-3’ | This paper | N/A |
| Target sequence of NR4A1 shRNA #1: 5’- TCCTTCCACATGTACATAAAC-3’ | This paper | N/A |
| Target sequence of NR4A1 shRNA #2: 5’- GTCACGTCTGTTGGGCAAACT-3’ | This paper | N/A |
| Target sequence of NR4A1 shRNA #3: 5’- CAACGCTTCATGCCAGCATTA-3’ | This paper | N/A |
| Target sequence of FOS shRNA #1: 5’- TCTCCAGTGCCAACTTCATTC-3’ | This paper | N/A |
| Target sequence of FOS shRNA #2: 5’- GCGGAGACAGACCAACTAGAA-3’ | This paper | N/A |
| siRNA sequences of NR4A1 smart pool: 5’-UCGAGGACUUCCAGGUGUA-3’, 5’-GGACAGAGCAGCUGCCCAA-3’, 5’-GAAGGCCGCUGUGCUGUGU-3’, 5’-CGGCUACACAGGAGAGUUU-3’ | Dharmacon | Cat#L-003426-00-0005 |
| siRNA sequences of MYC smart pool: 5’-ACGGAACUCUUGUGCGUAA-3’, 5’-GAACACACAACGUCUUGGA-3’, 5’-AACGUUAGCUUCACCAACA-3’, 5’-CGAUGUUGUUUCUGUGGAA-3’ | Dharmacon | Cat#L-003282-02-0005 |
| siRNA sequences of FOS smart pool: 5’-GGGAUAGCCUCUCUUACUA-3’, 5’-ACAGUUAUCUCCAGAAGAA-3’, 5’-GAACCUGUCAAGAGCAUCA-3’, 5’-GCAAUGAGCCUUCCUCUGA-3’ | Dharmacon | Cat#L-003265-02-0005 |
| siRNA sequences of FOSB smart pool: 5’-CGUGAGUGUGUGAGCGCUU-3’, 5’-GUUGUUAACCCUUCGUACA-3’, 5’-CCGGGAACGAAAUAAACUA-3’, 5’-AGUGAGGGGCUGUGACAUU-3’ | Dharmacon | Cat#L-010086-01-0005 |
| siRNA sequences of BHLHE40 smart pool: 5’-AAAGAGACGUGACCGGAUU-3’, 5’-GAGAAAGGAUCGGCGCAAU-3’, 5’-CCGAACAUCUCAAACUUAC-3’, 5’-CCCGGGAGGUGCUUCAGUA-3’ | Dharmacon | Cat#L-010318-00-0005 |
| siRNA sequences of non-targeting smart pool: 5’-UGGUUUACAUGUCGACUAA-3’, 5’-UGGUUUACAUGUUGUGUGA-3’, 5’-UGGUUUACAUGUUUUCUGA-3’, 5’-UGGUUUACAUGUUUUCCUA-3’ | Dharmacon | Cat#D-001810-10-05 |
| Human NR4A1 total qPCR-F: 5’- GTGCAGAAAAACGCCAAGTACA-3’ | This paper | N/A |
| Human NR4A1 total qPCR-R: 5’- TCTGTTCGGACAACTTCCTTCA-3’ | This paper | N/A |
| Human NR4A1 endo. qPCR-F: 5’- CGTGCTGTAAATAAGCCCAGTG-3’ | This paper | N/A |
| Human NR4A1 endo. qPCR-R: 5’- ACATGTGGAAGGAGGGTAGGAA-3’ | This paper | N/A |
| Human RNaseH1 endo. qPCR-F: 5’- TCTGTCAAGTCGGCCGTTTTAT-3’ | This paper | N/A |
| Human RNaseH1 endo. qPCR-R: 5’- GCTTGCAAATATGGACCAGGAC-3’ | This paper | N/A |
| Human RNaseH1 exo. qPCR-F: 5’- GGAGCTAAACAATCGGAAGACG-3’ | This paper | N/A |
| Human RNaseH1 exo. qPCR-R: 5’- CGTAGAATCGAGACCGAGGAGA-3’ | This paper | N/A |
| Human FOS qPCR-F: 5’- TCCGGAGATGTAGCAAAACGCA-3’ | This paper | N/A |
| Human FOS qPCR-R: 5’- GGCCTGGCTCAACATGCTACT-3’ | This paper | N/A |
| Human FOSB qPCR-F: 5’- AACTGCTTCTAGAAACTCTGGCTCA-3’ | This paper | N/A |
| Human FOSB qPCR-R: 5’- GAGAAAAGACAGAGGGAGAGAGACC-3’ | This paper | N/A |
| Human EGR1 qPCR-F: 5’- TGGGAGCAAAATAAGGAAGAGG-3’ | This paper | N/A |
| Human EGR1 qPCR-R: 5’- ACATTCTGGAGAACCGAAGCTC-3’ | This paper | N/A |
| Human JUN qPCR-F: 5’- GGAGGACCGGAGACAAGTGG-3’ | This paper | N/A |
| Human JUN qPCR-R: 5’- CGCCGTGGAGAAGCCTAAGA-3’ | This paper | N/A |
| Human JUNB qPCR-F: 5’- GGAAAAGAAACACGCACTTAGTCTC-3’ | This paper | N/A |
| Human JUNB qPCR-R: 5’- AACACACACAAACACAAACACGTC-3’ | This paper | N/A |
| Human JUND qPCR-F: 5’- CATTCCTGTTTGTAATCCTTGGTTC-3’ | This paper | N/A |
| Human JUND qPCR-R: 5’- GGCGTAACGAGACTTTACTGAAAAC-3’ | This paper | N/A |
| Human GAPDH qPCR-F: 5’- CCTCAACGACCACTTTGTCAAG-3’ | This paper | N/A |
| Human GAPDH qPCR-R: 5’- TGTTGCTGTAGCCAAATTCGTT-3’ | This paper | N/A |
| NR4A1 sgRNA #1: 5’-GCATTATGGTGTCCGCACAT-3’ | This paper | N/A |
| NR4A1 sgRNA #2: 5’- ATATGGGACACCAGCACCGA-3’ | This paper | N/A |
| NR4A1 sgRNA #3: 5’- CCAGACTTACGAAGGCCTGC-3’ | This paper | N/A |
| Scramble sgRNA: 5’- GCACTACCAGAGCTAACTCA-3’ | This paper | N/A |
| NR4A1 gDNA PCR (sgRNA pair#1) F: 5’- CCCAGCCAGACTTACGAAGG-3’ | This paper | N/A |
| NR4A1 gDNA PCR (sgRNA pair#1) R: 5’- CCTCTCTCCATTTCCAACAAAA-3’ | This paper | N/A |
| NR4A1 gDNA PCR (sgRNA pair#2) F: 5’- GGCCCAAATACTTTGAGACAAG-3’ | This paper | N/A |
| NR4A1 gDNA PCR (sgRNA pair#2) R: 5’- ACTGAAGGAAAAGAAGGCTGGA-3’ | This paper | N/A |
| Chromatin associated RNA quantification GAPDH intronic F: 5’- CCCACCTTTCTCATCCAAGACT-3’ | This paper | N/A |
| Chromatin associated RNA quantification GAPDH intronic R: 5’- TCCCCACCTTGAAAGGAAATTA-3’ | This paper | N/A |
| Chromatin associated RNA quantification GAPDH span intron F: 5’- CAACTTTGGTATCGTGGAAGGA-3’ | This paper | N/A |
| Chromatin associated RNA quantification GAPDH span intron R: 5’- TAGAGGCAGGGATGATGTTCTG-3’ | This paper | N/A |
| Chromatin associated RNA quantification GAPDH exon-intron F: 5’- GAGGGAGGTAGAGGGGTGATGT-3’ | This paper | N/A |
| Chromatin associated RNA quantification GAPDH exon-intron R: 5’- TAGAGGCAGGGATGATGTTCTG-3’ | This paper | N/A |
| Chromatin associated RNA quantification XIST intronic F: 5’- GATCAGTTGCCATGGTTCACAT-3’ | This paper | N/A |
| Chromatin associated RNA quantification XIST intronic R: 5’- CCCTAGTGTTCAGCAGTGGATG-3’ | This paper | N/A |
| Chromatin associated RNA quantification FOS intronic F: 5’- ATGCTTTCAGACTGGGCTCTTC-3’ | This paper | N/A |
| Chromatin associated RNA quantification FOS intronic R: 5’- TGCAGTTGCAGACTAGGTGTGA-3’ | This paper | N/A |
| Chromatin associated RNA quantification FOS span intron F: 5’- CTTACTACCACTCACCCGCAGA-3’ | This paper | N/A |
| Chromatin associated RNA quantification FOS span intron R: 5’- GGAATGAAGTTGGCACTGGAG-3’ | This paper | N/A |
| Chromatin associated RNA quantification FOS exon-intron F: 5’- AGCTGCAGGAGCCAGTTCTACT-3’ | This paper | N/A |
| Chromatin associated RNA quantification FOS exon-intron R: 5’- CTTTCCCTTCGGATTCTCCTTT-3’ | This paper | N/A |
| Chromatin associated RNA quantification EGR1 intronic F: 5’- AGAGGACCGAGCTTTTGTTTTG-3’ | This paper | N/A |
| Chromatin associated RNA quantification EGR1 intronic R: 5’- AAGGCAAGGCAAGTCTTACTGG-3’ | This paper | N/A |
| Chromatin associated RNA quantification EGR1 span intron F: 5’- AGCAGCAGCAGCACCTTCA-3’ | This paper | N/A |
| Chromatin associated RNA quantification EGR1 span intron R: 5’- CAGCACCTTCTCGTTGTTCAGA-3’ | This paper | N/A |
| Chromatin associated RNA quantification EGR1 exon-intron F: 5’- TACCTCGGGAGTCAATGGTAGC-3’ | This paper | N/A |
| Chromatin associated RNA quantification EGR1 exon-intron R: 5’- GCACCTTCTCGTTGTTCAGAGA-3’ | This paper | N/A |
| Chromatin associated RNA quantification Intergenic region 1 F: 5’- ACTGAAGGGTCAACCTGTTGGT-3’ | This paper | N/A |
| Chromatin associated RNA quantification Intergenic region 1 R: 5’- GGGGTGTTCTGATGAGCCTAAC-3’ | This paper | N/A |
| Chromatin associated RNA quantification Intergenic region 2 F: 5’- CAAGCCCATTCTCCTTAGCACT-3’ | This paper | N/A |
| Chromatin associated RNA quantification Intergenic region 2 R: 5’- CCACCATACACAGCCCTCAATA-3’ | This paper | N/A |
| ChIP FOS genebody region 1 qPCR-F: 5’- GGGTCCTTCTATGCAGCAGACT-3’ | This paper | N/A |
| ChIP FOS genebody region 1 qPCR-R: 5’- GTAAGCAGTGCAGCTGGGAGTA-3’ | This paper | N/A |
| ChIP FOS genebody region 2 qPCR-F: 5’- GGCAGGATCGTTTCTCTTCACT-3’ | This paper | N/A |
| ChIP FOS genebody region 2 qPCR-R: 5’- CCGTGCAGAAGTCCTAGAACAA-3’ | This paper | N/A |
| ChIP EGR1 genebody region 1 qPCR-F: 5’- GTCAGTGGCCTAGTGAGCATGA-3’ | This paper | N/A |
| ChIP EGR1 genebody region 1 qPCR-R: 5’- GTAAATGGGACTGCTGTCGTTG-3’ | This paper | N/A |
| ChIP EGR1 genebody region 2 qPCR-F: 5’- CGACTACCTGTTTCCACAGCAG-3’ | This paper | N/A |
| ChIP EGR1 genebody region 2 qPCR-R: 5’- TAGACAGAGGGGTTAGCGAAGG-3’ | This paper | N/A |
| ChIP FOSB genebody region 1 qPCR-F: 5’- TCTGAGATGGAAAAGGCTCACA-3’ | This paper | N/A |
| ChIP FOSB genebody region 1 qPCR-R: 5’- ATTGCAAACATCTCCCGACTCT-3’ | This paper | N/A |
| ChIP FOSB genebody region 2 qPCR-F: 5’- GGGGGTGTGTATGTGTGTGAGT-3’ | This paper | N/A |
| ChIP FOSB genebody region 2 qPCR-R: 5’- GAGGGAGAAGGAGAGGCCTTAG-3’ | This paper | N/A |
| ChIP GAPDH control genebody qPCR-F: 5’- TCTCTGCTGTAGGCTCATTTGC-3’ | This paper | N/A |
| ChIP GAPDH control genebody qPCR-R: 5’- CCTGATGATCTTGAGGCTGTTG-3’ | This paper | N/A |
| ChIP OLIG2 control genebody qPCR-F: 5’-GGGTCAATCCACACCCTCTTAG-3’ | This paper | N/A |
| ChIP OLIG2 control genebody qPCR-R: 5’- TCTCCCAAATCAACGAGAGACA-3’ | This paper | N/A |
| ChIP FOS promoter qPCR-F: 5’- GCCCGTGACGTTTACACTCATT-3’ | This paper | N/A |
| ChIP FOS promoter qPCR-R: 5’- CAGATGCGGTTGGAGTACGAG-3’ | This paper | N/A |
| ChIP EGR1 promoter qPCR-F: 5’- GTCCTGCCATATTAGGGCTTCC-3’ | This paper | N/A |
| ChIP EGR1 promoter qPCR-R: 5’- TCCGCCTCTATTTGAAGGGTCT-3’ | This paper | N/A |
| ChIP FOSB promoter qPCR-F: 5’-CGGGGAGAAAGTTTTTGGAAAG-3’ | This paper | N/A |
| ChIP FOSB promoter qPCR-R: 5’- TCGGTCCTCCAAAAGAAAATGA-3’ | This paper | N/A |
| ChIP GAPDH control promoter qPCR-F: 5’- CACGTAGCTCAGGCCTCAAGAC-3’ | This paper | N/A |
| ChIP GAPDH control promoter qPCR-R: 5’- GTCGAACAGGAGGAGCAGAGAG-3’ | This paper | N/A |
| ChIP Intergenic negative region 1 qPCR-F: 5’- AGGTTCCTCCAAATCAGAGCAG-3’ | This paper | N/A |
| ChIP Intergenic negative region 1 qPCR-R: 5’- GCTCCTCTCCAGCTAACGTCTC-3’ | This paper | N/A |
| ChIP Intergenic negative region 2 qPCR-F: 5’- CTACAGAAGCCAGCAAGCAAAA-3’ | This paper | N/A |
| ChIP Intergenic negative region 2 qPCR-R: 5’- GCCAGCATGGTTCTTATGTTGA-3’ | This paper | N/A |
| Recombinant DNA | ||
| ppyCAG_RNaseH1_WT | Addgene | Cat#111906 |
| ppyCAG_RNaseH1_WKKD | Addgene | Cat#111905 |
| pLVX-tetOne-Puro | Clontech | Cat# 631849 |
| pLenti-C-Myc-DDK-P2A-Puro-NR4A1 | Origene | Cat# RC202202L1 |
| pLVX-tetOne-NR4A1-FL | This paper | N/A |
| pLVX-tetOne-NR4A1-deltaTAD | This paper | N/A |
| pLVX-tetOne-NR4A1-deltaLBD | This paper | N/A |
| pLVX-tetOne-NR4A1-deltaDBD | This paper | N/A |
| pLVX-tetOne- RNaseH1_WT | This paper | N/A |
| pLVX-tetOne- RNaseH1_WKKD | This paper | N/A |
| pMD2.G | Addgene | Cat#12259 |
| psPAX2 | Addgene | Cat#12260 |
| pLKO.1-scramble | Addgene | Cat#136035 |
| pX458 | Addgene | Cat#48138 |
| pX459 | Addgene | Cat#62988 |
| lentiCRISPRv2-mCherry | Addgene | Cat#99154 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Halo | Indica Labs | https://indicalab.com/halo/ |
| FlowJo | BD | https://www.flowjo.com/ |
| Prism | GraphPad | https://www.graphpad.com/ |
| Trimgalore (v0.4.3) | https://github.com/FelixKrueger/TrimGalore | |
| BWA (v0.7.15) | Li and Durbin, 2009 | https://github.com/lh3/bwa |
| deeptools | Ramirez et al., 2014 | https://deeptools.readthedocs.io/en/develop/ |
| ATACseqQC | Ou et al., 2018 | https://github.com/jianhong/ATACseqQC/blob/master/R/ATACseqQC.R |
| sambamba | Tarasov et al., 2015 | https://lomereiter.github.io/sambamba/ |
| MACS2 (v2.0.10) | Zhang et al., 2008 | https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html |
| Bedtools (v2.27.1) | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| ChIPseeker | Yu et al., 2015 | https://guangchuangyu.github.io/software/ChIPseeker/ |
| Tophat2 (v2.1.1) | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Htseq (v2.1.1) | Anders et al., 2015 | https://htseq.readthedocs.io/_/downloads/en/release_0.10.0/pdf/ |
| DEseq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| DAVID | Huang da et al., 2009 | https://david.ncifcrf.gov/summary.jsp |
| GSVA | Hanzelmann et al., 2013 | http://bioconductor.org/packages/release/bioc/html/GSVA.html |
| Other | ||
| ATAC-seq of TCGA primary tumor samples | Corces et al., 2018 | https://gdc.cancer.gov/about-data/publications/ATACseq-AWG |
Highlights:
NR4A1 specifically localizes to IEG genebodies suppressing transcriptional elongation
Acute replication stress triggers NR4A1 release, triggering burst of IEG expression
NR4A1 deletion deregulates expression of the IEG FOS, triggering mitotic catastrophe
The NR4A1-IEG pathway denotes differentiated cancers with a favorable prognosis
ACKNOWLEDGMENTS
This work was supported by grants from NIH (2RO1CA129933 to D.A.H, 2U01EB012493 to M.T., D.A.H., S.M., 5U01EB012493 to M.T., 5P41EB002503 to M.T., R01CA197779 to L.Z. and R01GM128448 to R.M.), HHMI (to D.A.H.), ESSCO (to S.M.), BCRF (to D.A.H.) and NFCR (to D.A.H.).
Footnotes
DECLARATION OF INTERESTS
M.T., D.A.H. and S.M. are cofounders and have equity in the CTC-Chip companyTell-Bio, which is not related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adelman K, and Lis JT (2012). Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, and Garcia-Muse T (2012). R loops: from transcription byproducts to threats to genome stability. Mol Cell 46, 115–124. [DOI] [PubMed] [Google Scholar]
- Akman G, Desai R, Bailey LJ, Yasukawa T, Dalla Rosa I, Durigon R, Holmes JB, Moss CF, Mennuni M, Houlden H, et al. (2016). Pathological ribonuclease H1 causes R-loop depletion and aberrant DNA segregation in mitochondria. Proc Natl Acad Sci U S A 113, E4276–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DF, and Wang GG (2019). R-loops: formation, function, and relevance to cell stress. Cell Stress 3, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrami S, and Drablos F (2016). Gene regulation in the immediate-early response process. Adv Biol Regul 62, 37–49. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Bensaude O (2011). Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon D, Nguyen MLT, Mezger A, Kathiria A, Muller F, Nguyen V, Lescano N, Wu B, Trombetta J, Ribado JV, et al. (2019). Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat Genet 51, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Smith ER, and Shilatifard A (2018). Born to run: control of transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 19, 464–478. [DOI] [PubMed] [Google Scholar]
- Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, and Rao A (2019). NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH, et al. (2017). Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 8, 15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comaills V, Kabeche L, Morris R, Buisson R, Yu M, Madden MW, LiCausi JA, Boukhali M, Tajima K, Pan S, et al. (2016). Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep 17, 2632–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, and Orom UA (2017). Cellular Fractionation and Isolation of Chromatin-Associated RNA. Methods Mol Biol 1468, 1–9. [DOI] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz JR, Bettigole SE, and Glimcher LH (2017). Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 168, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigh LH, Liu C, Chung M, Cimprich KA, and Meyer T (2018). Stochastic Endogenous Replication Stress Causes ATR-Triggered Fluctuations in CDK2 Activity that Dynamically Adjust Global DNA Synthesis Rates. Cell Syst 7, 17–27 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day DS, Zhang B, Stevens SM, Ferrari F, Larschan EN, Park PJ, and Pu WT (2016). Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome Biol 17, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JL, Sully G, Clark AR, and Saklatvala J (2004). The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal 16, 1113–1121. [DOI] [PubMed] [Google Scholar]
- Degese MS, Tanos T, Naipauer J, Gingerich T, Chiappe D, Echeverria P, LaMarre J, Gutkind JS, and Coso OA (2015). An interplay between the p38 MAPK pathway and AUBPs regulates c-fos mRNA stability during mitogenic stimulation. Biochem J 467, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelstein M, and Sorensen CS (2015). Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 14, 405–423. [DOI] [PubMed] [Google Scholar]
- Dravis C, Chung CY, Lytle NK, Herrera-Valdez J, Luna G, Trejo CL, Reya T, and Wahl GM (2018). Epigenetic and Transcriptomic Profiling of Mammary Gland Development and Tumor Models Disclose Regulators of Cell State Plasticity. Cancer Cell 34, 466–482 e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardy SR, and Farnham PJ (2002). Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem 277, 40156–40162. [DOI] [PubMed] [Google Scholar]
- Gaillard H, Garcia-Muse T, and Aguilera A (2015). Replication stress and cancer. Nat Rev Cancer 15, 276–289. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Yamazaki T, and Kroemer G (2018). Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol 19, 731–745. [DOI] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, and Chedin F (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr., Kastrinakis NG, Levy B, et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick E, Lee SO, Doddapaneni R, Singh M, and Safe S (2015). Nuclear receptor 4A1 as a drug target for breast cancer chemotherapy. Endocr Relat Cancer 22, 831–840. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, and Davis RJ (2016). Cell Signaling and Stress Responses. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, Yu M, Sundaresan TK, Licausi JA, Desai R, et al. (2016). HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 537, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, and Bartek J (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Petermann E, and Boulton SJ (2018). Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov 8, 537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, and Thompson EB (1999). The structure of the nuclear hormone receptors. Steroids 64, 310–319. [DOI] [PubMed] [Google Scholar]
- Lee SO, Abdelrahim M, Yoon K, Chintharlapalli S, Papineni S, Kim K, Wang H, and Safe S (2010). Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res 70, 6824–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SO, Andey T, Jin UH, Kim K, Singh M, and Safe S (2012). The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 31, 3265–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, Hao J, Alekseev A, Khong H, Chen T, et al. (2019). Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, and Elledge SJ (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell MA, and Muscat GE (2006). The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal 4, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooi WJ, and Peeper DS (2006). Oncogene-induced cell senescence--halting on the road to cancer. N Engl J Med 355, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Morgan JI, and Curran T (1995). Immediate-early genes: ten years on. Trends Neurosci 18, 66–67. [PubMed] [Google Scholar]
- Nagy A, Lanczky A, Menyhart O, and Gyorffy B (2018). Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep 8, 9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QH, Pervolarakis N, Blake K, Ma D, Davis RT, James N, Phung AT, Willey E, Kumar R, Jabart E, et al. (2018). Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat Commun 9, 2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J, Liu H, Yu J, Kelliher MA, Castilla LH, Lawson ND, and Zhu LJ (2018). ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genomics 19, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, Yu M, Chen PI, Morgan B, Trautwein J, et al. (2013). Inertial focusing for tumor antigen-dependent and - independent sorting of rare circulating tumor cells. Sci Transl Med 5, 179ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli S, Teasley DC, Murali B, Jackson J, Vindigni A, and Stewart SA (2017). Human ribonuclease H1 resolves R-loops and thereby enables progression of the DNA replication fork. J Biol Chem 292, 15216–15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Waki H, Vaitheesvaran B, Wilpitz DC, Kurland IJ, and Tontonoz P (2006). NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat Med 12, 1048–1055. [DOI] [PubMed] [Google Scholar]
- Pekarsky Y, Hallas C, Palamarchuk A, Koval A, Bullrich F, Hirata Y, Bichi R, Letofsky J, and Croce CM (2001). Akt phosphorylates and regulates the orphan nuclear receptor Nur77. Proc Natl Acad Sci U S A 98, 3690–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau SJ, and Amon A (2012). Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep 13, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronkina N, Menon MB, Schwermann J, Arthur JS, Legault H, Telliez JB, Kayyali US, Nebreda AR, Kotlyarov A, and Gaestel M (2011). Stress induced gene expression: a direct role for MAPKAP kinases in transcriptional activation of immediate early genes. Nucleic Acids Res 39, 2503–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S, and Karki K (2021). The Paradoxical Roles of Orphan Nuclear Receptor 4A (NR4A) in Cancer. Mol Cancer Res 19, 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, and Chedin F (2019). High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat Protoc 14, 1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Shaulian E, and Karin M (2001). AP-1 in cell proliferation and survival. Oncogene 20, 2390–2400. [DOI] [PubMed] [Google Scholar]
- Skourti-Stathaki K, and Proudfoot NJ (2014). A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28, 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini NL, Luo J, and Elledge SJ (2007). Non-oncogene addiction and the stress phenotype of cancer cells. Cell 130, 986–988. [DOI] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi T, and Brown GW (2019). Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res 79, 1730–1739. [DOI] [PubMed] [Google Scholar]
- Willis N, and Rhind N (2009). Regulation of DNA replication by the S-phase DNA damage checkpoint. Cell Div 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winoto A, and Littman DR (2002). Nuclear hormone receptors in T lymphocytes. Cell 109 Suppl, S57–66. [DOI] [PubMed] [Google Scholar]
- Wu H, Lin Y, Li W, Sun Z, Gao W, Zhang H, Xie L, Jiang F, Qin B, Yan T, et al. (2011). Regulation of Nur77 expression by beta-catenin and its mitogenic effect in colon cancer cells. FASEB J 25, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Guo H, Hu B, Li R, Yong J, Zhao Y, Zhi X, Fan X, Guo F, Wang X, et al. (2016). Epigenomic Landscape of Human Fetal Brain, Heart, and Liver. J Biol Chem 291, 4386–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, et al. (2014). Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatreanu D, Han Z, Mitter R, Tumini E, Williams H, Gregersen L, Dirac-Svejstrup AB, Roma S, Stewart A, Aguilera A, et al. (2019). Elongation Factor TFIIS Prevents Transcription Stress and R-Loop Accumulation to Maintain Genome Stability. Mol Cell 76, 57–69 e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Comaills V, Burr R, Boulay G, Miyamoto DT, Wittner BS, Emmons E, Sil S, Koulopoulos MW, Broderick KT, et al. (2019). COX-2 mediates tumor-stromal prolactin signaling to initiate tumorigenesis. Proc Natl Acad Sci U S A 116, 5223–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Drabsch Y, Dekker TJ, de Vinuesa AG, Li Y, Hawinkels LJ, Sheppard KA, Goumans MJ, Luwor RB, de Vries CJ, et al. (2014). Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-beta signalling. Nat Commun 5, 3388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Number of sequencing reads in ATAC-seq, ChIP-seq, PRO-seq, DRIP-seq and RNA-seq samples used in this study. Related to Figures 1, 2, 3, 4, S1, S2 and S4.
Table S2: List of genes showing significant gain or loss of chromatin accessibility in BRx142 and BRx82 CTC-derived tumor cells recovered after intracardiac injection. When scoring across the entire genome in both CTC lines, 37 gene loci show highly significant gains in chromatin accessibility at >day30 (combined log2 fold change in BRx142 and BRx82 CTCs >2.5). In contrast to the 37 genes with increased ATAC-seq genebody signal, 100 gene loci lose chromatin accessibility during early tumor formation (combined log2 fold change < −2.5), and none of them are IEGs. Genes with loss of chromatin accessibility are involved in diverse pathways, including vesicle docking (P<0.004) and protein transport (P<0.002). Related to Figures 1 and S1.
Table S3: List of NR4A1-bound genes in BRx142 and BRx82 CTC-derived metastases and in MCF10A cells identified by NR4A1 ChIP-seq analysis. Related to Figures 2 and S2.
Table S4: List of genes showing significant up- or down-regulation by RNA-seq in NR4A1-null M231 cells. Related to Figure S6.
Data Availability Statement
All raw and processed sequencing data in this study, including ATAC-seq, RNA-seq, ChIP-seq, DRIP-seq and PRO-seq are available at NCBI Gene Expression Omnibus (GEO) database under accession number: GSE161853. All the original western blot images and microscopy images have been deposited at Mendeley and will be shared by the lead contact upon request.
All original code has been deposited at GitHub (https://github.com/ggolczer/nr4a1_ieg_cancer) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.