

ABSTRACT
Enteric viruses infect the mammalian gastrointestinal tract and lead to significant morbidity and mortality worldwide. Data indicate that enteric viruses can utilize intestinal bacteria to promote viral replication and pathogenesis. However, the precise interactions between enteric viruses and bacteria are unknown. Here, we examined the interaction between bacteria and coxsackievirus B3, an enteric virus from the picornavirus family. We found that bacteria enhance the infectivity of coxsackievirus B3 (CVB3) in vitro. Notably, specific bacteria are required, as Gram-negative Salmonella enterica, but not Escherichia coli, enhanced CVB3 infectivity and stability. Investigating the cell wall components of both S. enterica and E. coli revealed that structures in the O-antigen or core of lipopolysaccharide, a major component of the Gram-negative bacterial cell wall, were required for S. enterica to enhance CVB3. To determine if these requirements were necessary for similar enteric viruses, we investigated if S. enterica and E. coli enhanced infectivity of poliovirus, another enteric virus in the picornavirus family. We found that while E. coli did not enhance the infectivity of CVB3, E. coli enhanced poliovirus infectivity. Overall, these data indicate that distinct bacteria enhance CVB3 infectivity and stability, and specific enteric viruses may have differing requirements for their interactions with specific bacterial species.
IMPORTANCE Previous data indicate that several enteric viruses utilize bacteria to promote intestinal infection and viral stability. Here, we show that specific bacteria and bacterial cell wall components are required to enhance infectivity and stability of coxsackievirus B3 in vitro. These requirements are likely enteric virus specific, as the bacteria for CVB3 differ from poliovirus, a closely related virus. Therefore, these data indicate that specific bacteria and their cell wall components dictate the interaction with various enteric viruses in distinct mechanisms.
KEYWORDS: bacteria-viral interactions, coxsackievirus, enteric viruses, intestinal bacteria, viral stability
INTRODUCTION
Enteric viruses are highly infectious pathogens that account for 1.3 million deaths in neonates and children on an annual basis worldwide (1–3). Enteric viruses initiate infection in the gastrointestinal tract and are spread by contamination in food and water and direct contact (4, 5). Many enteric viruses belong to the Picornaviridae family, whose members are characterized by a positive-sense, single-stranded RNA genome, encapsulated by an icosahedral capsid (6). Coxsackievirus is a common, clinically isolated picornavirus and acts as an etiological agent of hand, foot, and mouth disease, hemorrhagic conjunctivitis, and viral myocarditis (7–9). Because hygiene acts as an essential barrier and prevents viral fecal-oral route of transmission, the virus predominantly infects infants and young children, with coxsackievirus B3 (CVB3) infection accounting for an 11% fatality rate in neonates (5, 10, 11). Despite this, there are no vaccines or treatments for coxsackievirus infections.
The gastrointestinal tract is colonized by more than 1014 bacteria (12, 13), which aid in host digestion, regulate gut homeostasis, and protect from pathogenic bacteria (14–16). Imbalances in this microbial niche have been related to a variety of diseases such as diabetes (17), inflammatory bowel disease (IBD) (18), and obesity (19). Previous studies have shown that these intestinal microbes also aid in enhancing replication and pathogenesis of enteric viruses, including CVB3 (20–28), but the mechanism behind these interactions remains unclear.
Previous studies have shown that intestinal bacteria can aid in the stability of multiple enteric viruses (20, 22, 24, 29). Interestingly, the bacteria required to enhance viral stability may differ among enteric viruses (21, 30). Here, we show that species-specific bacteria improve CVB3 stability, and this stability is dependent on the structure of lipopolysaccharides present on the bacterial cell wall. Furthermore, we confirm that even closely related enteric viruses utilize different bacteria to enhance their infectivity. Overall, these data indicate that bacterial-mediated enhancement of enteric viral stability may be a broad, conserved feature of bacteria-virus interactions and that species-specific bacteria may regulate this mechanism in different viruses.
RESULTS
CVB3 infection is enhanced by intestinal bacteria ex vivo and in vitro.
Antibiotic treatment of interferon a/b receptor knockout (Ifnar−/−) mice before oral inoculation with CVB3 decreases fecal shedding and lethality compared to untreated mice (21). To assess if fecal bacteria could also enhance CVB3 infectivity in vitro, we treated male Ifnar−/− mice with an antibiotic cocktail for 10 days to deplete their intestinal bacteria (Fig. 1A). Following antibiotic treatment, fecal samples from antibiotic-treated mice, untreated mice, or phosphate-buffered saline (PBS) were incubated with 105 PFU of CVB3 at 37°C for 6 h, and CVB3 infectivity was measured by a standard plaque assay on HeLa cells. We found that CVB3 infectivity was significantly increased when incubated with feces from untreated mice compared to antibiotic-treated or PBS (Fig. 1B). Since previous data suggested that specific bacteria may be required to enhance CVB3 replication and lethality in vivo (21, 30), we next sought to determine if particular bacteria improve CVB3 infectivity in vitro. CVB3 was incubated with selected Gram-positive and Gram-negative bacteria at 37°C, followed by a viral plaque assay. CVB3 lost viral titer when incubated with Dulbecco's modified Eagle's medium (DMEM) but gained viral titer when incubated with Bacillus subtilis, Bacillus badius, Salmonella enterica, and Enterobacter cloacae (Fig. 1C). Lactococcus lactis, Enterococcus faecium, Escherichia coli, and Pseudomonas aeruginosa did not enhance the infectivity of CVB3. Bacteroides vulgatus and Klebsiella pneumoniae appeared to reduce the infectivity of CVB3; however, the data did not reach statistical significance compared to CVB3 incubated with DMEM. Overall, these data indicate that bacteria enhance CVB3 infectivity in a species-specific manner.
FIG 1.
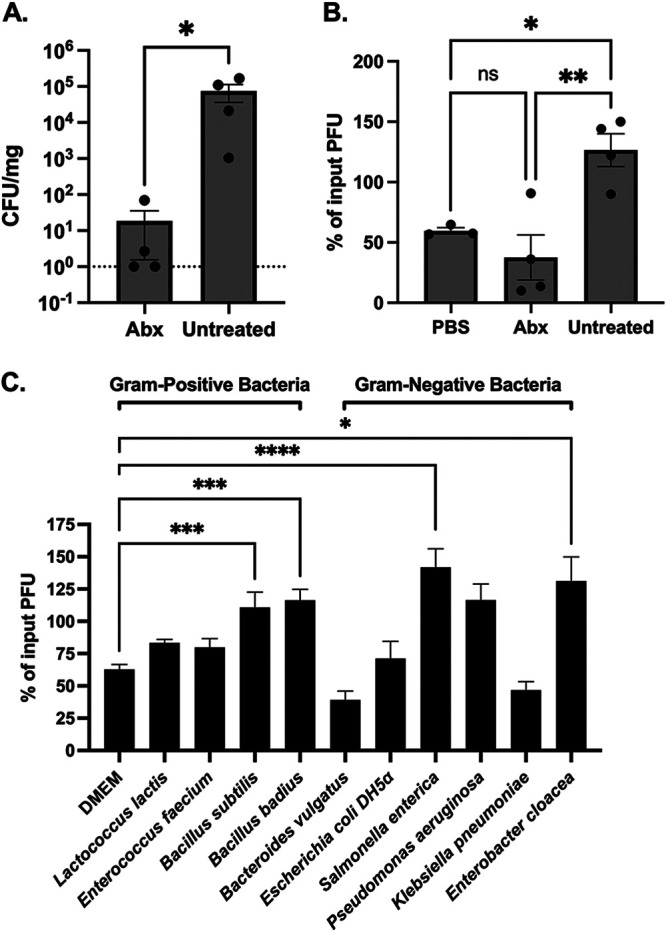
Fecal bacteria enhance the infectivity of CVB3 in vitro. Male C57BL/6 Pvr+/+ and Ifnar−/− mice were treated with an antibiotic cocktail for 10 days. (A) Bacterial load in the feces. Fecal pellets were collected from the antibiotic-treated and untreated group and plated to determine CFU per milligram of feces. Dashed line indicates the limit of detection; *, P < 0.05; Kolmogorov-Smirnov test. (B) CVB3 infectivity after exposure to PBS or feces from uninfected or antibiotic-treated mice (6 h at 37°C); n = 8 mice per group from 4 independent experiments. (C) CVB3 infectivity after exposure to selected Gram-positive and Gram-negative bacteria (6 h at 37°C). Data represent 2 to 6 independent experiments (n = 4 to 12). For all, bars represent mean ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA.
CVB3 can bind to both S. enterica and E. coli, but only S. enterica enhances CVB3 stability.
S. enterica and E. coli are both Gram-negative bacteria that are closely related (31). Interestingly, we found that S. enterica, but not E. coli, could enhance CVB3 infectivity in vitro. Previous data have shown that enteric viruses, including CVB3, could bind to bacteria (30); therefore, we investigated if S. enterica or E. coli could bind to the CVB3 virion. We incubated 105 PFU of CVB3 with 109 CFU of either S. enterica or E. coli at 37°C for 1 h to allow for binding. As a control, CVB3 was also incubated in PBS alone or with beads with a similar diameter to bacteria to rule out nonspecific binding as previously described (29). Following incubation, the samples were centrifuged at 5,000 × g for 10 min to pellet bacteria and washed, and the amount of CVB3-bound bacteria was quantified by a plaque assay on HeLa cells. We found a significantly larger amount of CVB3 in the S. enterica and E. coli pellet than PBS and beads (Fig. 2A). Since bacteria may enhance infectivity in HeLa cells, we confirmed these findings using a cell-free assay. We incubated 105 PFU of CVB3 with 109 CFU of either S. enterica or E. coli at 37°C for 1 h, and, following centrifugation, the amount of CVB3 bound bacteria was quantified by reverse transcription-quantitative PCR (qRT-PCR). We observed significantly more pelleted viral genome copies when CVB3 was incubated with S. enterica and E. coli pellets than CVB3 incubated with PBS or beads (Fig. 2B). These data indicate that CVB3 can bind to both S. enterica and E. coli.
FIG 2.
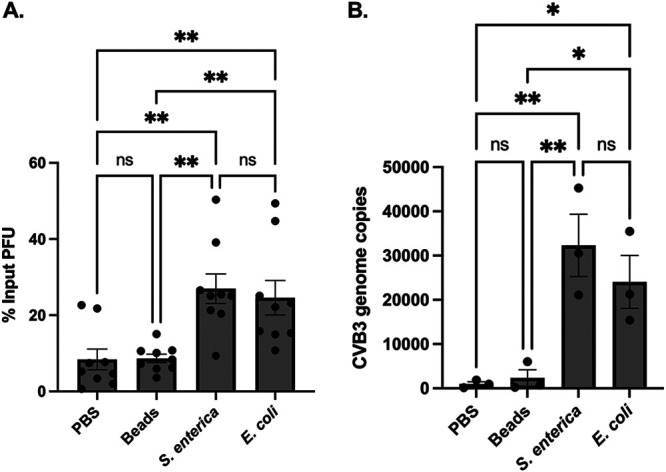
CVB3 bind to S. enterica and E. coli. We incubated 105 PFU of CVB3 with 109 CFU of either S. enterica or E. coli at 37°C for 1 h. After incubation, samples were spun down and washed to remove unbound virus. Bound virus was quantified by plaque assay on HeLa cells (A) and qRT-PCR (B). Data represent 3 to 4 independent experiments (n = 6 to 8). Bars represent ± SEM; *, P < 0.05; **, P < 0.01; one way ANOVA.
Previous data indicate that viral stability is a mechanism for bacterial-mediated enhancement of viral infectivity (20, 29, 30). Since CVB3 bound to both S. enterica and E. coli but only S. enterica enhanced infectivity (Fig. 1C), we next quantified the effect of these bacteria on CVB3 stability. To remain infectious, CVB3, like other viruses, must maintain its capsid integrity (6, 32, 33). Previous data indicate that bacteria can improve enteric viruses' stability, including poliovirus, reovirus, and CVB3 (20, 24, 30); therefore, we sought to determine CVB3 stability by assessing first-order viral decay in the presence of S. enterica and E. coli (34). We incubated 105 PFU of CVB3 with PBS or 108 CFU of S. enterica at 37°C for 0 to 72 h. At various time points, samples were collected, and CVB3 was quantified by a standard plaque assay. We found that CVB3 incubated with S. enterica significantly reduced the decay rate of CVB3 compared to the virus incubated in PBS (Fig. 3A and C). Further, we found that similar incubation with E. coli did not decrease the viral decay rate for CVB3 compared to PBS, indicating that E. coli does not significantly increase viral stability (Fig. 3B and C). To examine if this effect was dose dependent, we incubated 105 PFU of CVB3 at 37°C with PBS or increasing concentrations (102 to 108 CFU) of bacteria and quantified viral titers at various time points between 0 and 72 h. We observed a significant dose-dependent effect of S. enterica on CVB3 stability; however, this stability effect was not observed when CVB3 was incubated with E. coli (Fig. 3D and E). These data suggest that S. enterica, but not E. coli, enhances CVB3 stability in a dose-dependent manner.
FIG 3.

S. enterica, but not E. coli, enhances CVB3 stability. CVB3 decay at 37°C when incubated with DMEM (slope, −0.06651; R2, 0.6940) of 108 of S. enterica (slope, 0.001960; R2, 0.9337) (A) or 108 of E. coli (slope, −0.06700; R2, 0.8773) (B). The first-order decay rate for CVB3 was determined by a best-fit line generated by fitted linear regression as previously described (47). (C) Rates of CVB3 decay in either PBS, 108 CFU of S. enterica, or 108 CFU of E. coli. ***, P < 0.001; ****, P < 0.0001; one-way ANOVA. (D) Rates of CVB3 decay in either PBS or 108, 106, 104, or 102 CFU of S. enterica. (E) Rates of CVB3 decay in either PBS or 108, 106, 104, or 102 CFU of E. coli. For all, the data represent 4 to 6 independent experiments (n = 8 to 12); bars represent mean ± SEM; **, P < 0.01; one-way ANOVA.
S. enterica LPS, but not E. coli LPS, enhances CVB3 stability.
Components of the bacterial cell wall, such as lipopolysaccharide (LPS) and peptidoglycan (PGN), have been shown to bind and stabilize enteric viruses (20, 24). To determine whether bacterial cell wall components could stabilize CVB3, we tested whether LPS from S. enterica or E. coli could enhance CVB3 stability in vitro. We incubated 105 PFU of CVB3 with 1 mg/ml of LPS from each bacterium or PBS at 42°C for 1 h and quantified the viral infection on HeLa cells. We used 1 mg/ml of each bacterial cell component to imitate physiological concentrations observed in the gut (35, 36). We also increased the temperature of our stability assays to 42°C to induce a faster inactivation, which has been previously shown as a method to determine the stability of other heat-stable picornaviruses (20). When CVB3 was incubated with either S. enterica LPS or E. coli K-12 LPS, we found the exposure of CVB3 to S. enterica LPS, but not E. coli LPS, enhanced stability at 42°C (Fig. 4A). We next examined if PGN from E. coli could improve CVB3 stability. We found that, similar to E. coli LPS, PGN from E. coli and mannan, a control polysaccharide from Saccharomyces cerevisiae, could not enhance CVB3 stability. However, like S. enterica LPS, PGN from Gram-positive B. subtilis could improve CVB3 stability, suggesting a bacterial species-specific effect of cell wall components. Finally, we examined if the stability effect of S. enterica LPS was dose dependent. We incubated 105 PFU of CVB3 with dilutions of S. enterica LPS at 42°C for 1 h, followed by a plaque assay. We observed a dose-dependent effect of S. enterica LPS induced enhancement of CVB3 stability (Fig. 4B). Overall, these data show that bacterial cell wall components LPS and PGN can enhance CVB3 stability in a bacterial species-dependent manner.
FIG 4.
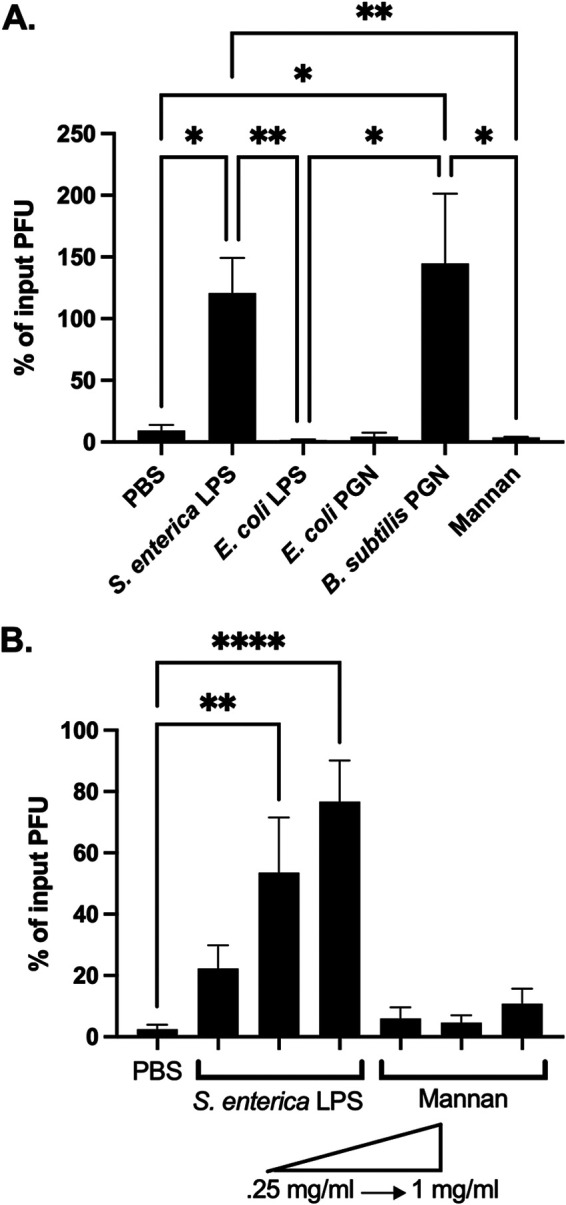
Specific bacterial cell wall components stabilize CVB3. (A) We incubated 105 PFU of CVB3 in 1-mg/ml concentration of either LPS or PGN from indicated bacterial species at 42°C for 1 h. PBS and mannan were used as controls. (B) We incubated 105 PFU of CVB3 in increasing concentrations from 0.25 mg/ml to 1 mg/ml of LPS or mannan at 42°C for 1 h. For all, data represent 4 to 5 experiments (n = 8 to 10), and bars represent mean ± SEM. *, P < 0.05; **, P < 0.01; ****, P < 0.0001; one-way ANOVA.
LPS from S. enterica that lacks the outer core and O-antigen does not enhance CVB3 stability.
LPS consists of three components: a conserved hydrophobic lipid A moiety, a semiconserved hydrophilic core polysaccharide chain, and a polymorphic hydrophilic O-antigen side chain as the outermost exposed region of LPS (Fig. 5A) (37–40). The O-antigen chain exhibits a strain-specific structural diversity based on the number of repeating oligosaccharide units (41). Smooth Gram-negative bacterial strains contain all three components of the LPS structure, while rough bacterial isolates have truncated LPS that lack O-antigen and core components (37, 41) (Fig. 5A). Our initial experiments with E. coli utilized the rough, lab-adapted DH5α and K-12 strains that lack the core and O-antigen components (Fig. 3 and 4). In contrast, our S. enterica strain was a smooth, full-length LPS molecule. Therefore, we reasoned that the lack of an O-antigen in E. coli might influence CVB3 stability. To test this, we examined the stability of CVB3 in the presence of LPS from two smooth strains of E. coli, both having an intact O-antigen: E. coli 0127 and E. coli 0111. We incubated 105 PFU of CVB3 with 1 mg/ml of LPS from E. coli 0127, E. coli 0111, or PBS at 42°C for 1 h and quantified the viral infection on HeLa cells. Similar to rough strains of E. coli, we found that LPS from smooth strains of E. coli did not enhance CVB3 stability (Fig. 5B). Further, CVB3 incubated with whole bacteria from smooth strains of E. coli also did not improve infectivity as seen in S. enterica (Fig. 5C).
FIG 5.

Smooth LPS from S. enterica enhance CVB3 stability. (A) Basic structure of bacterial lipopolysaccharide. The O-antigen can have repeating oligosaccharide units (designated n). (B) We incubated 105 PFU of CVB3 in 1 mg/ml concentration of LPS from a smooth strain of S. enterica, E. coli (0127), E. coli (0111), a rough strain of E. coli (DH5α), or mannan. (C) We incubated 105 PFU of CVB3 with 107 CFU of rough E. coli (DH5α, K-12) or smooth E. coli (O157) at 37°C for 6 h. (D) We incubated 105 PFU of CVB3 with 1 mg/ml of LPS from a smooth or a rough strain of S. Minnesota. Data represent 4 independent experiments (n = 8). Bars represent mean ± SEM; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA.
Since the O-antigen from E. coli did not impact CVB3 stability, we next tested whether the O-antigen or core polysaccharide from S. enterica could affect CVB3. We incubated 105 PFU of CVB3 with 1 mg/ml of LPS from smooth or rough strains of S. enterica serovar Minnesota at 42°C for 1 h, and then CVB3 was quantified by a plaque assay. We found that LPS with a fully intact O-antigen from smooth S. Minnesota enhanced CVB3 stability, while LPS from the rough strain did not (Fig. 5D). Overall, these results indicate that polysaccharides in the O-antigen or core of S. enterica LPS are required to promote CVB3 stability.
S. enterica and E. coli enhance the infectivity of poliovirus, a closely related enteric virus.
Poliovirus and CVB3 are both members of the Picornaviridae family and share approximately 62.3% sequence identity (21). Previous studies have shown that both poliovirus and CVB3 (20, 22, 29) utilize bacteria to promote infection in vivo; however, the specific bacteria required may differ between the two viruses (21). Further, previous studies indicate that E. coli enhances the infectivity of poliovirus (20, 22). Since our data suggest that CVB3 infectivity is enhanced by S. enterica and not E. coli (Fig. 1C), we tested whether S. enterica has an effect on poliovirus. We incubated 105 PFU of poliovirus with S. enterica or E. coli at 37°C for 6 h, followed by a plaque assay on HeLa cells. We found that poliovirus gained viral titer when incubated with S. enterica and E. coli; thus, infectivity was significantly enhanced by both bacteria (Fig. 6). These data further indicate that highly similar enteric viruses may utilize different bacteria and/or mechanisms to enhance their infectivity.
FIG 6.
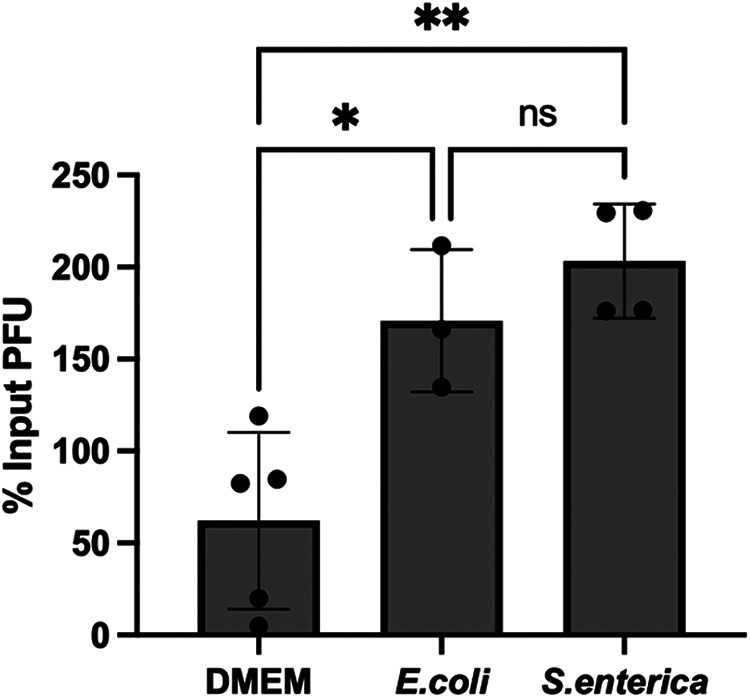
S. enterica and E. coli enhance poliovirus's infectivity, a closely related enteric virus. We incubated 105 PFU of poliovirus with DMEM or 107 of E. coli or S. enterica at 37°C for 6 h, followed by a plaque assay on HeLa cells. Data represent 3 independent experiments (n = 6); bars represent mean ± SEM, *, P < 0.05; **, P < 0.01; one-way ANOVA.
DISCUSSION
Coxsackieviruses are spread through the fecal-oral route, where they are exposed to a community of intestinal bacteria. Previous studies have shown the pivotal role intestinal bacteria play in the replication and pathogenesis of several enteric viruses (22–24, 27, 28). Further, data indicate that bacteria can enhance the stability of different enteric viruses, which suggests that this may be a broad, conserved mechanism of viral-bacterial interactions (21, 30). However, the precise mechanisms underlying the different bacterial requirements for interacting with CVB3 remain unclear. Here, we show that interactions with specific bacteria and lipopolysaccharide structures enhance CVB3 infectivity and stability.
Our data indicate that Gram-positive and Gram-negative bacteria can enhance CVB3 infectivity in vitro. We found that CVB3 gained viral titer when incubated Bacillus subtilis, Bacillus badius, Salmonella enterica, and Enterobacter cloacae (Fig. 1C). While the mechanism is unclear, previous studies have shown that bacterial LPS can enhance poliovirus attachment to host cells and promote coinfection, leading to enhanced recombination which could rescue defective viral particles (20, 22, 29). CVB3 may utilize similar mechanisms. Additionally, Bacteroides vulgatus and Klebsiella pneumoniae appeared to reduce the infectivity of CVB3. While these data did not reach statistical significance, it is interesting to speculate that specific bacteria may also destabilize CVB3. However, further work is required to determine the mechanism of bacterial species-specific effects on CVB3 infectivity.
We found that closely related Gram-negative bacteria, S. enterica and E. coli, had different abilities to impact CVB3. In contrast to infectivity, our data indicate that both S. enterica and E. coli bind to CVB3 (Fig. 2), even though only S. enterica could enhance CVB3 stability (Fig. 3). Further, we determined that CVB3 stability was increased when incubated with LPS from S. enterica, but not E. coli (Fig. 4A and B). We hypothesize that the mechanism(s) that facilitate bacterial binding and influence viral stability may differ. In agreement with this hypothesis, data from Erickson et al. (29) show that poliovirus binding to specific bacteria does not always correlate with an enhancement of infectivity. Of the 32 strains of bacteria that were bound to poliovirus, less than half enhanced poliovirus infection compared to control (29). It is unclear if variances in bacterial cell wall structure account for this observation with poliovirus; however, poliovirus requires at least six subunits of N-acetylglucosamine to stabilize the virion (20, 42). The size of glycan necessary to bind poliovirus differs from the size needed to stabilize, where longer glycans promote virion stability. This length requirement is hypothesized to allow the glycan to bridge two adjacent 5-fold axes to help retain capsid stability (42). Since we found that a smooth E. coli strain, which contains N-acetylglucosamine and is of sufficient length, was unable to stabilize CVB3 (Fig. 5C), these data suggest differences between CVB3 and poliovirus in their interactions with bacteria. In agreement, we found that E. coli enhanced the infectivity of poliovirus (Fig. 6), but not CVB3. Since previous data revealed that CVB3 and poliovirus have different bacterial requirements to improve replication in vivo (21) and E. coli has been shown to enhance reovirus stability (24), these data indicate the bacterial requirements to bind and stabilize enteric viruses may be distinct, and different enteric viruses exploit specific bacterial species to enhance their infection.
E. coli and S. enterica share several O serogroups that have identical structures (31, 43, 44); however, there are numerous distinct O-antigens present in both species of S. enterica and E. coli due to lateral gene transfer and side chain modifications (44). Our data also show that the O-antigen or core polysaccharide from S. enterica LPS plays a critical role in stabilizing the CVB3 capsid (Fig. 5B and D). In contrast to S. enterica, our data suggest that smooth E. coli LPS does not stabilize CVB3 (Fig. 4A and 5B). Data from Aguilera et al. (30) found that LPS from E. coli 0127:B8 stabilizes CVB3 Nancy, which contrasts with our data. However, differences in the incubation temperature may account for the discrepancy, as it has been previously shown that incubation temperature can impact LPS stability on an LPS-binding mutant of poliovirus (20). We also found that another smooth LPS from E. coli 0111 was unable to stabilize CVB3, consistent with LPS from E. coli 0127. Further, Aguilera et al. found that smooth E. coli 1470 was unable to stabilize CVB3 Nancy, which provides additional evidence that specific polysaccharides in LPS are required to promote CVB3 stability. These data also indicate that enteric viral interactions with specific LPS structures may be similar to how bacteriophages bind to LPS as a viral receptor. Bacteriophages bind to unique sugars in LPS, which can dictate bacterial tropism (45). We hypothesize that enteric viruses may act similarly; however, it remains to be determined how this impacts infection in the intestine. Further work is needed to determine whether distinct O-antigen and O serogroups of S. enterica have differing impacts on CVB3 and other enteric viruses.
In conclusion, our data suggest that binding to specific bacteria via their LPS structures stabilizes CVB3 virions. We hypothesize that this interaction may help retain infectivity and promote transmission in the environment. Future work will be needed to deduce the specific bacterial LPS sugar(s) required to interact with CVB3. Further, since S. enterica is not a common commensal bacterium, the conserved features of the bacterial cell wall will need to be evaluated to identify other bacterial members of the intestinal microbiota that may directly interact with CVB3. Overall, understanding the specific mechanisms for how various enteric viruses utilize bacteria will help develop novel therapeutics and limit future transmission events.
MATERIALS AND METHODS
Cells and virus.
HeLa cells, grown in Dulbecco's modified Eagle's medium (DMEM), were supplemented with 10% calf serum and 1% penicillin-streptomycin at 37°C with 5% CO2. CVB3 Nancy infectious clone (IC) was obtained from Marco Vignuzzi (Pasteur Institute, Paris, France) and propagated in HeLa cells as described previously (46). CVB3 was quantified by plaque assays in HeLa cells. Poliovirus (serotype 1, Mahoney) infections and plaque assay were performed using HeLa cells as previously described (20).
Bacterial strains and cell wall components.
Bacillus strains, Pseudomonas aeruginosa, Enterobacter cloacae, Salmonella enterica Lactococcus lactis, Enterococcus faecium, Klebsiella pneumoniae, and Escherichia coli were kindly provided by Ryan Relich (Indiana University School of Medicine). LPS molecules from E. coli O127 (L3129), E. coli 0111(L2630), E. coli K-12 (tlrl-eklps), Salmonella enterica (L6011), Salmonella enterica serotype Minnesota (L6261, L9764), and mannan from Saccharomyces cerevisiae (M7504) were obtained from Sigma-Aldrich, and PGN molecules from E. coli O111 (tlrl-pgneb) and Bacillus subtilis (L3265) obtained from Invivogen and Sigma-Aldrich, respectively.
Mouse experiments.
Male mice (C57BL/6 PVR+/+ Ifnar−/−) were administered a combination of 4 antibiotics by oral gavage (ampicillin, neomycin, metronidazole, and vancomycin; 10 mg of each antibiotic per day) for 5 days, followed by the administration in drinking water available ad libitum (for ampicillin, neomycin, and metronidazole, 1 g/liter; for vancomycin, 500 mg/liter) for additional 5 days. As a control, a group of mice was left untreated. To confirm bacterial depletion, feces were collected on day 10 of antibiotic treatment. Fecal pellets were weighed and resuspended in 5 volumes of 1× PBS. Fecal bacteria were then quantified by determining CFU on LB agar plates in aerobic conditions and on blood agar plates in anaerobic conditions.
Viral infectivity assay.
We incubated 105 PFU of CVB3 Nancy with fecal slurry or 1× PBS at 37°C for 6 h. Following incubation, 1/10 volume of chloroform was added to the samples and centrifuged at 13,000 relative centrifugal force (rcf) for 5 min, and the supernatant was collected. The viral supernatants were then quantified by a plaque assay on HeLa cells. For incubation with bacteria, specific bacteria were grown in either LB or brain heart infusion (BHI) media in a 5-ml culture incubated at 37°C for 16 to 20 h. CFU per milliliter was quantified by absorbance measurements at optical density at 600 nm (OD600). Bacterial cultures were pelleted at 3,250 rpm for 30 min, resuspended in 1× PBS, and centrifuged again. The pellet was homogenized in 500 μl of DMEM, and 107 CFU of each bacterium, normalized to 200 μl with 1× PBS, was added to respective tubes. We added 105 PFU of CVB3 to each bacterial tube, and they were incubated at 37°C for 6 h. We used 105 PFU of CVB3 in PBS pre- and postincubation as controls. Following incubation, the mixture was chloroform extracted and quantified by a plaque assay on HeLa cells.
Viral pulldown assays.
We incubated 105 PFU of CVB3 Nancy with 1 × 109 CFU of bacteria (as determined by OD600 values) or ∼3.5 × 106 inert beads (Dynabeads M-280 streptavidin; Invitrogen) for 1 h at 37°C to facilitate viral binding. Following incubation, samples were centrifuged at 5,000 × g for 10 min and washed with PBS, and virus was quantified by a plaque assay on HeLa cells or by qRT-PCR to measure viral genome copies.
Viral stability and decay rate.
S. enterica and E. coli DH5α were grown in BHI and LB media, respectively, for 16 h at 37°C, and CFU were quantified by plating on BHI and LB agar, as well as by measuring OD600 absorbance values. Bacteria were centrifuged at 3,250 rpm for 30 min to remove media and then washed in 1× PBS and centrifuged again. The pellet was resuspended in 500 μl of 1× PBS. Based on CFU and OD600 values, 1 × 108, 1 × 106, 1 × 104, and 1 × 102 CFU of bacteria were added to tubes for each of the following time points and normalized to 200 μl using 1× PBS. We added to 105 PFU of CVB3 to each tube, and they were incubated at 37°C for 0, 2, 6, 16, 24, 48, and 72 h. Following incubation, 1/10 volume of chloroform was added to the samples and centrifuged at 13,000 rcf for 5 min, and supernatant was collected. The supernatant was used to quantify viral titers by a plaque assay on HeLa cells. For stability experiments, 105 PFU of CVB3 was incubated with 200 μl of either 1 mg/ml, 0.50 mg/ml, or 0.25 mg/ml of LPS, PGN, 1× PBS, or mannan at 42°C for 1 h and then used to quantify viral titers by a plaque assay on HeLa cells.
Statistical analysis.
Comparisons between control and study groups were analyzed using either Mann-Whitney U test or one-way analysis of variance (ANOVA). Kolmogorov-Smirnov test was used to compare CFU/mg between feces from antibiotic-treated and untreated mice. For our stability experiments, a simple linear regression was used to determine the best-fit line, and the decay rate of CVB3 was determined using the slope of each individual experiment.
ACKNOWLEDGMENTS
We thank Ryan Relich at Indiana University School of Medicine for providing bacterial isolates for these studies.
This work is funded by a K01 DK110216, R03 DK124749, and a Showalter Trust Award from the Indiana Clinical and Translational Sciences Institute to C.M.R.
Contributor Information
Christopher M. Robinson, Email: cmrobin@iu.edu.
Susana López, Instituto de Biotecnologia/UNAM.
REFERENCES
- 1.Desselberger U. 2014. Global issues related to enteric viral infections. Virusdisease 25:147–149. doi: 10.1007/s13337-014-0223-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Page NA, Nadan S, Mans J. 2019. Viral gastroenteritis, p 135–149. In Eslick GD (ed), Gastrointestinal diseases and their associated infections. Elsevier, Philadelphia, PA. doi: 10.1016/B978-0-323-54843-4.00011-8. [DOI] [Google Scholar]
- 3.Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, Rudan I, Campbell H, Cibulskis R, Li M, Mathers C, Black RE, Child Health Epidemiology Reference Group of WHO and UNICEF. 2012. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet 379:2151–2161. doi: 10.1016/S0140-6736(12)60560-1. [DOI] [PubMed] [Google Scholar]
- 4.Sin J, Mangale V, Thienphrapa W, Gottlieb RA, Feuer R. 2015. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 484:288–304. doi: 10.1016/j.virol.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abedi GR, Watson JT, Pham H, Nix WA, Oberste MS, Gerber SI. 2015. Enterovirus and human parechovirus surveillance - United States, 2009–2013. MMWR Morb Mortal Wkly Rep 64:940–943. doi: 10.15585/mmwr.mm6434a3. [DOI] [PubMed] [Google Scholar]
- 6.Hogle JM. 2002. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol 56:677–702. doi: 10.1146/annurev.micro.56.012302.160757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhoades RE, Tabor-Godwin JM, Tsueng G, Feuer R. 2011. Enterovirus infections of the central nervous system. Virology 411:288–305. doi: 10.1016/j.virol.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garmaroudi FS, Marchant D, Hendry R, Luo H, Yang D, Ye X, Shi J, McManus BM. 2015. Coxsackievirus B3 replication and pathogenesis. Future Microbiol 10:629–653. doi: 10.2217/fmb.15.5. [DOI] [PubMed] [Google Scholar]
- 9.Montes M, Artieda J, Pineiro LD, Gastesi M, Diez-Nieves I, Cilla G. 2013. Hand, foot, and mouth disease outbreak and coxsackievirus A6, northern Spain, 2011. Emerg Infect Dis 19:676–678. doi: 10.3201/eid1904.121589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khetsuriani N, Lamonte A, Oberste MS, Pallansch M. 2006. Neonatal enterovirus infections reported to the National Enterovirus Surveillance System in the United States, 1983–2003. Pediatr Infect Dis J 25:889–893. doi: 10.1097/01.inf.0000237798.07462.32. [DOI] [PubMed] [Google Scholar]
- 11.Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA, Centers for Disease Control and Prevention. 2006. Enterovirus surveillance–United States, 1970–2005. MMWR Surveill Summ 55:1–20. [PubMed] [Google Scholar]
- 12.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. 2011. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci USA 108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kline KA, Falker S, Dahlberg S, Normark S, Henriques-Normark B. 2009. Bacterial adhesins in host-microbe interactions. Cell Host Microbe 5:580–592. doi: 10.1016/j.chom.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar Reddy D. 2015. Role of the normal gut microbiota. World J Gastroenterol 21:8787–8803. doi: 10.3748/wjg.v21.i29.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, Tuohy K. 2018. Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr 57:1–24. doi: 10.1007/s00394-017-1445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, Rosenbaum M, Gordon JI. 2013. The long-term stability of the human gut microbiota. Science 341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Upadhyaya S, Banerjee G. 2015. Type 2 diabetes and gut microbiome: at the intersection of known and unknown. Gut Microbes 6:85–92. doi: 10.1080/19490976.2015.1024918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knox NC, Forbes JD, Van Domselaar G, Bernstein CN. 2019. The gut microbiome as a target for IBD treatment: are we there yet? Curr Treat Options Gastroenterol 17:115–126. doi: 10.1007/s11938-019-00221-w. [DOI] [PubMed] [Google Scholar]
- 19.Thursby E, Juge N. 2017. Introduction to the human gut microbiota. Biochem J 474:1823–1836. doi: 10.1042/BCJ20160510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson CM, Jesudhasan PR, Pfeiffer JK. 2014. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 15:36–46. doi: 10.1016/j.chom.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson CM, Acevedo MAW, McCune BT, Pfeiffer JK. 2019. Related enteric viruses have different requirements for host microbiota in mice. J Virol 93:e01339-19. doi: 10.1128/JVI.01339-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. 2011. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uchiyama R, Chassaing B, Zhang B, Gewirtz AT. 2014. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J Infect Dis 210:171–182. doi: 10.1093/infdis/jiu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger AK, Yi H, Kearns DB, Mainou BA. 2017. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLoS Pathog 13:e1006768. doi: 10.1371/journal.ppat.1006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amarasiri M, Sano D. 2019. Specific interactions between human norovirus and environmental matrices: effects on the virus ecology. Viruses 11:224. doi: 10.3390/v11030224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, Golovkina TV. 2011. Successful transmission of a retrovirus depends on the commensal microbiota. Science 334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinje J, Tibbetts SA, Wallet SM, Karst SM. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karst SM. 2016. The influence of commensal bacteria on infection with enteric viruses. Nat Rev Microbiol 14:197–204. doi: 10.1038/nrmicro.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erickson AK, Jesudhasan PR, Mayer MJ, Narbad A, Winter SE, Pfeiffer JK. 2018. Bacteria facilitate enteric virus co-infection of mammalian cells and promote genetic recombination. Cell Host Microbe 23:77–88.e5. doi: 10.1016/j.chom.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguilera ER, Nguyen Y, Sasaki J, Pfeiffer JK. 2019. Bacterial stabilization of a panel of picornaviruses. mSphere 4:e00183-19. doi: 10.1128/mSphere.00183-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu B, Perepelov AV, Liu B, Shevelev SD, Guo D, Senchenkova SN, Shashkov AS, Feng L, Knirel YA, Wang L. 2010. Structural and genetic evidence for the close relationship between Escherichia coli O71 and Salmonella enterica O28 O-antigens. FEMS Immunol Med Microbiol 59:161–169. doi: 10.1111/j.1574-695X.2010.00676.x. [DOI] [PubMed] [Google Scholar]
- 32.Baggen J, Thibaut HJ, Strating JRPM, van Kuppeveld FJM. 2018. The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol 16:368–381. doi: 10.1038/s41579-018-0005-4. [DOI] [PubMed] [Google Scholar]
- 33.Baker TS, Olson NH, Fuller SD. 1999. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol Mol Biol Rev 63:862–922, table of contents. doi: 10.1128/MMBR.63.4.862-922.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carson SD, Chapman NM, Hafenstein S, Tracy S. 2011. Variations of coxsackievirus B3 capsid primary structure, ligands, and stability are selected for in a coxsackievirus and adenovirus receptor-limited environment. J Virol 85:3306–3314. doi: 10.1128/JVI.01827-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bates JM, Akerlund J, Mittge E, Guillemin K. 2007. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2:371–382. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freudenberg MA, Meier-Dieter U, Staehelin T, Galanos C. 1991. Analysis of LPS released from Salmonella abortus equi in human serum. Microb Pathog 10:93–104. doi: 10.1016/0882-4010(91)90070-q. [DOI] [PubMed] [Google Scholar]
- 37.Voss BJ, Trent MS. 2018. LPS transport: flipping out over MsbA. Curr Biol 28:R30–R33. doi: 10.1016/j.cub.2017.10.067. [DOI] [PubMed] [Google Scholar]
- 38.Alexander C, Rietschel ET. 2001. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res 7:167–202. doi: 10.1179/096805101101532675. [DOI] [PubMed] [Google Scholar]
- 39.Weinstein JR, Swarts S, Bishop C, Hanisch UK, Moller T. 2008. Lipopolysaccharide is a frequent and significant contaminant in microglia-activating factors. Glia 56:16–26. doi: 10.1002/glia.20585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nilsson C, Skoglund A, Moran AP, Annuk H, Engstrand L, Normark S. 2008. Lipopolysaccharide diversity evolving in Helicobacter pylori communities through genetic modifications in fucosyltransferases. PLoS One 3:e3811. doi: 10.1371/journal.pone.0003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nnalue NA. 1999. All Accessible Epitopes in the Salmonella lipopolysaccharide core are associated with branch residues. Infect Immun 67:998–1003. doi: 10.1128/IAI.67.2.998-1003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu H, Lehrman MA, Pfeiffer JK. 2020. Use of a glycan library reveals a new model for enteric virus oligosaccharide binding and virion stabilization. J Virol 94:e01894-19. doi: 10.1128/JVI.01894-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winfield MD, Groisman EA. 2004. Phenotypic differences between Salmonella and Escherichia coli resulting from the disparate regulation of homologous genes. Proc Natl Acad Sci USA 101:17162–17167. doi: 10.1073/pnas.0406038101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L, Reeves PR. 2000. The Escherichia coli O111 and Salmonella enterica O35 gene clusters: gene clusters encoding the same colitose-containing O antigen are highly conserved. J Bacteriol 182:5256–5261. doi: 10.1128/JB.182.18.5256-5261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Y, Roznowski AP, Tokuda JM, Klose T, Mauney A, Pollack L, Fane BA, Rossmann MG. 2017. Structural changes of tailless bacteriophage ΦX174 during penetration of bacterial cell walls. Proc Natl Acad Sci USA 114:13708–13713. doi: 10.1073/pnas.1716614114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson CM, Wang Y, Pfeiffer JK. 2017. Sex-dependent intestinal replication of an enteric virus. J Virol 91:e02101-16. doi: 10.1128/JVI.02101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carson SD, Cole AJ. 2020. Albumin enhances the rate at which coxsackievirus B3 strain 28 converts to A-particles. J Virol 94:e01962-19. doi: 10.1128/JVI.01962-19. [DOI] [PMC free article] [PubMed] [Google Scholar]