

ABSTRACT
The viral fusion protein glycoprotein B (gB) is conserved in all herpesviruses and is essential for virus entry. During entry, gB fuses viral and host cell membranes by refolding from a prefusion to a postfusion form. We previously introduced three structure-based mutations (gB-I671A/H681A/F683A) into the domain V arm of the gB ectodomain that resulted in reduced cell-cell fusion. A virus carrying these three mutations (called gB3A) displayed a small-plaque phenotype and remarkably delayed entry into cells. To identify mutations that could counteract this phenotype, we serially passaged the gB3A virus and selected for revertant viruses with increased plaque sizes. Genomic sequencing revealed that the revertant viruses had second-site mutations in gB, including E187A, M742T, and S383F/G645R/V705I/V880G. Using expression constructs encoding these mutations, only gB-V880G was shown to enhance cell-cell fusion. In contrast, all of the revertant viruses showed enhanced entry kinetics, underscoring the fact that cell-cell fusion and virus-cell fusion are different. The results indicate that mutations in three different regions of gB (domain I, the membrane proximal region, and the cytoplasmic tail domain) can counteract the slow-entry phenotype of gB3A virus. Mapping these compensatory mutations to prefusion and postfusion structural models suggests sites of intramolecular functional interactions with the gB domain V arm that may contribute to the gB fusion function.
IMPORTANCE The nine human herpesviruses are ubiquitous and cause a range of diseases in humans. Glycoprotein B (gB) is an essential viral fusion protein that is conserved in all herpesviruses. During host cell entry, gB mediates virus-cell membrane fusion by undergoing a conformational change. Structural models for the prefusion and postfusion forms of gB exist, but the details of how the protein converts from one to the other are unclear. We previously introduced structure-based mutations into gB that inhibited virus entry and fusion. By passaging this entry-deficient virus over time, we selected second-site mutations that partially restore virus entry. The locations of these mutations suggest regulatory sites that contribute to fusion and gB refolding during entry. gB is a target of neutralizing antibodies, and defining how gB refolds during entry could provide a basis for the development of fusion inhibitors for future research or clinical use.
KEYWORDS: herpes simplex virus 1, glycoprotein gB, fusion, entry, revertant, glycoproteins
INTRODUCTION
Herpesviruses establish lifelong latent infections and are ubiquitous in the human population. Herpes simplex virus 1 (HSV-1) causes mucocutaneous lesions that typically localize to the face, mouth, cornea, or genitalia. Serious complications from HSV infection include perinatal infection that can cause lethal disseminated disease, HSV-induced keratitis, and, in rare occurrences, encephalitis or meningitis.
Herpesviruses infect cells through membrane fusion, and the viral fusion protein glycoprotein B (gB) is conserved in all herpesviruses. Entry requires the coordinated interaction of several essential glycoproteins that comprise the fusion machinery: gD, gH, gL, and gB (1, 2). HSV-1 entry is initiated by gD binding to a cellular receptor. Receptor binding transmits a signal to the heterodimer gHgL. Activated gHgL subsequently triggers gB to insert into the host cell membrane and refold to merge the viral and cellular membranes.
Structures of the postfusion form of gB have been visualized for multiple herpesviruses (3–10). gB is a trimer that consists of five domains (Fig. 1A). Domain I includes hydrophobic fusion loops that insert into the cell membrane at the base of the structure, where the transmembrane region would also be positioned (6, 11, 12). Domain III forms a trimeric coiled coil at the core of the structure, against which a domain V arm packs in an antiparallel orientation. More recently, the structures of part of the membrane proximal region (MPR), the transmembrane domain (TMD), and the cytoplasmic tail domain (CTD) of postfusion HSV-1 gB were visualized (3). The gB CTD is proposed to act as a clamp that regulates fusion, and mutations in this region are hyperfusogenic (3, 13).
FIG 1.

gB structures. (A) Postfusion HSV-1 gB trimer (PDB accession number 2GUM) (5). Domain I (DI) (blue), including the fusion loops (magenta), sits closest to the membrane. Domain III (DIII) (yellow) includes a large central trimer of helices, with the N termini down (yellow N) and the C termini up (yellow C). Domain V (DV) (red) packs against the domain III helices in an antiparallel orientation down the length of the structure, with domain V from one protomer contacting domain III from the other two protomers. The residues mutated in gB3A (red space-filled residues I671, H681, and F683) form extensive contacts with domain III. Domain II (DII) (green) and domain IV (DIV) (orange) are also shown. (B) Prefusion model of HSV-1 gB (PDB accession number 6Z9M) (15). Domains I to V and the fusion loops are colored as described above for panel A. A transparent surface rendering of the gB trimer is shown, with a single protomer shown in ribbon format. Domain I comprises the legs of the trimer with fusion loops facing the membrane. Domain III includes a long helix at the center of the trimer with its N terminus up (yellow N label) and its C terminus down (yellow C label), the opposite orientation compared to the postfusion structure. Domain V includes an extended strand at its N terminus (red N label) and a central helix at its C terminus (red C label) that descends toward the membrane. The domain V extended strand is packed between the domains of a neighboring protomer. I671, H681, and F683 (red space-filled residues) are positioned near the top and bottom of this strand. (C) Ribbon model of prefusion HSV-1 gB with an inset. The prefusion trimer from panel B is shown in ribbon format with the same coloring. The extended strand of domain V packs between domains I (blue), II (green), and III (yellow) of a neighboring protomer. The inset shows I671, H681, and F683 in one protomer (red sticks) and their contacts in the neighboring protomer (space-filled residues). I671 contacts domain II residue K151 (green) and domain III residue R557 (yellow). H681 contacts domain III at residue T120 (yellow) and domain I at two sites, Y165 (blue) and T291 (blue). F683 contacts domain I at R189 (blue) as well as at G292 and D293 (blue). A contact is defined as a location within a 4-Å proximity.
The prefusion form of gB is unstable in a secreted form of the protein (14) and has not been crystallized. Structural models of prefusion gB have been proposed based on cryo-electron microscopy (cryoEM) (15–18) and homology to the fusion protein of vesicular stomatitis virus (19, 20). Most recently, a model of prefusion HSV-1 gB, including nearly the complete ectodomain, was visualized (15) (Fig. 1B). Comparison of this prefusion model with the postfusion HSV-1 gB structure reveals extensive conformational changes due to rotations at interdomain connections. In the prefusion form, the central helices of domain III are inverted with respect to the membrane, and the domain V arm does not pack against domain III.
The packing of domain V against domain III to form a “coil-arm” complex in postfusion gB is reminiscent of a stable six-helix bundle that is characteristic of class I viral fusion proteins (21). In class I fusion, the formation of this six-helix bundle is critical for fusion, and disruption of the six-helix bundle inhibits fusion. Using mutagenesis, we previously examined the HSV-1 gB coil-arm interface and identified three residues in the domain V arm (I671, H681, and F683) that are important for fusion function (22). Replacing these three residues with alanine (called gB3A) resulted in greatly reduced fusion in a cell-cell fusion assay. Fusion could be restored by introducing a hyperfusogenic mutation into the gB CTD, suggesting that the protein is properly folded. We proposed that the coil-arm interaction drives the gB transition to a postfusion conformation and provides energy for fusion.
Using a bacterial artificial chromosome (BAC), we generated a virus carrying the gB3A mutations (gB-I671A/H681A/F683A) (23). gB3A BAC DNA was transfected into Vero cells; after 3 weeks, the cells exhibited cytopathic effect (CPE), and gB3A virus was harvested. For comparison, transfection of cells with wild-type (WT) BAC DNA resulted in CPE after only 5 to 7 days. The gB3A virus formed significantly smaller plaques on Vero cells than the WT virus and demonstrated a substantial delay in entry into cells. We proposed that the gB3A mutations hindered gB refolding from a prefusion to a postfusion form. The small-plaque phenotype of the gB3A virus provided an opportunity to select for mutations that would reverse the gB3A defects, potentially offering insight into the structural basis for gB refolding. Using serial passage of gB3A virus, we previously reported several mutations in gB3A that partially restored fusion function (24). In this study, we report additional novel second-site mutations in gB3A that partially restore the entry kinetics of gB3A.
RESULTS
Isolation of gB3A revertant viruses.
To select for second-site mutations that could rescue the gB3A entry defect, gB3A viruses were passaged serially on Vero cells to select for virus isolates with larger plaque sizes. The gB3A virus stock was passaged at a low multiplicity of infection (MOI) in three independent lineages on Vero cells for multiple passages. After larger plaques became apparent, a single large plaque was picked and subjected to plaque purification. These large plaque isolates are designated “revertant viruses” going forward.
To identify mutations that can enhance plaque size in the gB3A virus background, DNA was extracted from the revertant virus isolates, and whole-genome sequencing was performed. For comparison, the BAC genomes of the parental WT HSV-1 (GS3217) and gB3A were sequenced.
Revertant viruses with larger plaques from three independent lineages were sequenced. The first revertant virus acquired the mutation gB-E187A. This virus was sequenced after 18 passages, and an additional mutation, gB-A417T, was also found at a low frequency (29% of sequencing reads). To retrospectively determine the acquisition of gB mutations over time, the gB gene from earlier passages was PCR amplified and sequenced. The results indicated that the gB-E187A mutation arose first, prior to passage 5, and the gB-E187A mutation alone was associated with large plaques in that early passage. Thus, further analysis focused on gB-E187A. The second revertant virus was sequenced after 39 passages, and it acquired the mutation gB-M742T. Sequencing revealed an additional mutation in virion host shutoff (vhs-L225W) and in gH (gH-Y41S). Further analysis focused on gB-M742T because vhs does not have a known role in fusion, and gH residue 41 lies in an N-terminal region that is dispensable for fusion (25). The third revertant virus acquired four mutations in gB3A (S383F, G645R, V705I, and V880G) after 25 passages. This virus also acquired the mutation gC-L204F. In our previous study using the same approach, a gB3A revertant virus carrying three of these gB mutations (S383F, G645R, and V705I) was selected, concurrent with a different mutation in the gB CTD (A855V) (24). Thus, further analysis focused on the new mutation gB-V880G.
Plaque morphology of the gB3A revertant viruses.
To compare plaque phenotypes, Vero cells were infected with WT HSV-1, gB3A virus, or revertant viruses. Plaques were stained after 3 days. The gB3A virus showed much smaller plaques than the WT virus, as seen previously (23). The plaques of all three revertant viruses were larger than the gB3A plaques (Fig. 2A). Plaques from the gB3A-S383F/G645R/V705I/V880G revertant virus showed a distinct syncytial plaque morphology.
FIG 2.

Revertant virus plaques. (A) Plaque morphology. Vero cells were infected with BAC-derived WT HSV-1 (GS3217), gB3A virus, or gB3A revertant viruses at an MOI of 0.01. Three days after infection, cells were stained, and plaques were imaged at a ×40 total magnification. Arrows indicate small plaques formed by gB3A virus. Squares outlined by dotted lines were magnified and are shown as insets at the top right of each panel to highlight the syncytial plaque phenotype of gB3A-S383F/G645R/V705I/V880G revertant virus. The circles at the top right serve as a reference to show a 10-fold increase in the average plaque area. (B) Plaque size. Vero cells were infected as described above for panel A. The diameters of at least 50 randomly selected plaques from each virus were measured using Evos cell imaging. The average plaque area was calculated and expressed as a percentage of the WT HSV-1 plaque area. The error bars represent standard deviations. Asterisks indicate that the difference in plaque sizes between the mutant and gB3A viruses was statistically significant (Mann-Whitney U test). (C) Cell surface expression. Revertant virus gB genes were cloned into an expression construct with an N-terminal FLAG tag. The expression of gB on the surface of Vero cells was detected by a CELISA using anti-gB MAb II-105. The averages from three experiments are shown, and the error bars represent standard deviations. Asterisks indicate that the difference in expression between one mutant and WT gB was statistically significant (P < 0.05 using a t test).
The average plaque area for each virus was determined by measuring the plaque diameters of at least 50 plaques at a ×40 magnification. Although plaque sizes were variable, the average plaque area for the WT virus was 90 times larger than that of the gB3A virus (Fig. 2B). The gB3A-E187A and gB3A-M472T revertant viruses had plaques that were 7 to 10 times larger than gB3A plaques but still much smaller than WT virus plaques. The gB3A-S383F/G645R/V705I/V880G revertant virus had larger plaques than the other revertant viruses, but the plaques were still smaller than those of the WT virus on average.
Expression of gB from the revertant viruses.
To determine whether the mutations in gB that arose in the revertant viruses affect gB expression, the gB genes from the revertant viruses were cloned into expression constructs, and cell surface expression in Vero cells was assessed by a cell-based enzyme-linked immunosorbent assay (CELISA). gB3A-E187A and gB3A-M472T were expressed at levels similar to those of WT gB. Unexpectedly, the surface expression of gB3A-S383F/G645R/V705I/V880G was reduced despite this revertant virus showing the largest plaque size (Fig. 2C).
Fusion function of gB from the revertant viruses.
To examine whether the mutations in gB that arose in the revertant viruses can restore fusion function to gB3A, the mutants were assessed using a cell-cell fusion assay performed in CHO-K1 cells. Since the S383F, G645R, and V705I mutations were examined previously (24), only the V880G mutation from the S383F/G645R/V705I/V880G revertant virus was investigated in the fusion assay. The cell surface expression of the gB mutants in CHO-K1 cells was determined by a CELISA (Fig. 3A). All of the mutants were expressed at near-WT gB levels. When examined by SDS-PAGE and Western blotting of transfected CHO-K1 cell lysates, all of the gB mutants migrated as expected, similar to gB3A (Fig. 3B). The mutants were examined for fusion function using a quantitative cell-cell fusion assay (Fig. 3A). Adding V880G to gB3A restored fusion function, suggesting that this mutation in the gB CTD is hyperfusogenic. V880G may restore fusion through a mechanism similar to that of the A855V mutation that we identified previously (24). This enhanced fusion may account for the enhanced plaque size of the revertant virus. Unexpectedly, adding E187A or M742T to gB3A did not enhance fusion (Fig. 3A). Fusion results with cells expressing the HSV entry receptor nectin-1 or herpesvirus entry mediator (HVEM) were similar (Fig. 3A).
FIG 3.

Expression and fusion mediated by mutant gB constructs in a gB3A background. (A) Cell surface expression and fusion function. Mutations identified in the revertant viruses were cloned into a gB3A expression construct. One set of CHO-K1 cells (effector cells) was transfected with plasmids encoding T7 polymerase, gD, gH, gL, and either a version of gB or an empty vector. Another set of CHO-K1 cells (target cells) was transfected with plasmids carrying the luciferase gene under the control of a T7 promoter and either the nectin-1 or HVEM receptor. The expression of gB on the surface of effector cells was detected by a CELISA using anti-gB PAb R74. To assess fusion function, effector cells were cocultured with target cells for 18 h, and luciferase expression was assayed. Fusion with target cells expressing nectin-1 or HVEM is shown. Results are expressed as a percentage of the expression of fusion mediated by WT gB, after subtracting background values measured in the absence of gB (vector control). The means and standard deviations from at least three independent experiments are shown. Asterisks indicate that the difference in fusion levels between the mutants and WT gB was statistically significant (P < 0.05 using a t test). Differences in cell surface expression were not statistically significant. (B) Western blots. CHO-K1 cells were transfected with plasmids encoding gB constructs or an empty vector. Cell lysates were subjected to SDS-PAGE followed by Western blotting using anti-gB PAb R74. The location of the 100-kDa molecular weight marker is shown.
Impact of mutations on WT gB expression and fusion function.
To examine how the mutations from the revertant viruses would affect the expression and fusion activity of WT gB, the mutations were introduced into a FLAG-tagged WT gB expression construct using site-specific mutagenesis. All three mutants showed near-WT levels of expression (Fig. 4A). Western blotting of transfected cell lysates showed that the mutants migrated similarly to WT gB (Fig. 4B). Consistent with a hyperfusogenic property, adding V880G to WT gB enhanced fusion with both nectin-1- and HVEM-expressing cells (Fig. 4A). The E187A mutation impacted WT gB fusion function only modestly, consistent with this mutation also failing to enhance gB3A-mediated fusion (Fig. 4A). Unexpectedly, the M742T mutation greatly decreased fusion function in this WT gB background (Fig. 4A), suggesting that this mutation paradoxically inhibits fusion in the absence of the gB3A mutations.
FIG 4.
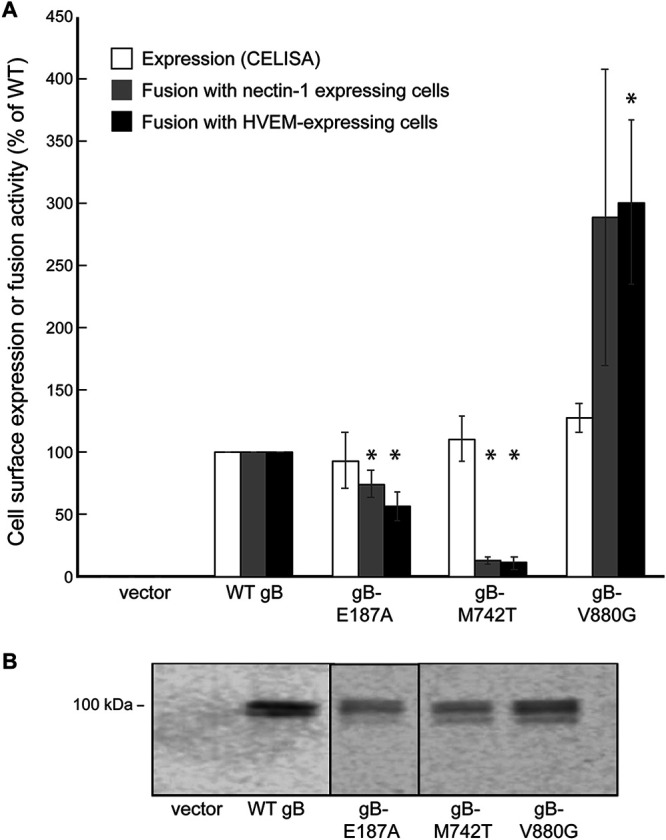
Expression and fusion mediated by mutant gB constructs in a WT gB background. (A) Cell surface expression and fusion function. Mutations identified in the revertant viruses were cloned into a WT gB expression construct with an N-terminal FLAG tag. Effector and target cells were transfected and cocultured as described in the legend of Fig. 3. The expression of gB on the surface of effector cells was detected by a CELISA using anti-FLAG MAb F1804. Fusion with target cells expressing nectin-1 or HVEM is shown. Results are expressed as a percentage of the expression of WT fusion, as described in the legend of Fig. 2. The means and standard deviations from at least three independent experiments are shown. Asterisks indicate that the difference in fusion levels between the mutants and WT gB was statistically significant (P < 0.05 using a t test). Differences in cell surface expression were not statistically significant. (B) Western blots. Cell lysates from transfected CHO-K1 cells were analyzed by Western blotting using anti-FLAG PAb F7425. The location of the 100-kDa molecular weight marker is shown.
Rate of revertant virus entry into cells.
Although the enhanced plaque size for the revertant virus carrying the hyperfusogenic V880G mutation may be explained by enhanced fusion, the large-plaque phenotype of the revertant viruses carrying E187A or M742T mutations is not explained by the cell-cell fusion assay results. We previously demonstrated that the gB3A virus has delayed entry into cells (23). To determine if the revertant viruses exhibited enhanced virus entry kinetics, a penetration assay was performed to compare virus entry after 2 h (Fig. 5). Vero cells were used because they form uniform plaques and entry occurs at the cell surface (26). Viruses were added to cell monolayers at 37°C for 2 h to allow entry. Extracellular virus particles were then inactivated with an acid wash. As a control, duplicate samples were rinsed in parallel with phosphate-buffered saline (PBS) instead of citric acid to determine the maximum number of PFU. After 3 days, plaques were counted to determine how many virus particles had entered the cells during the 2-h window. As an additional control, a gB3A revertant virus that was previously shown to have a large plaque size and enhanced cell-cell fusion, HSV-1 gB3A-A683V (24), was included. As expected, although most WT HSV-1 entered after 2 h at 37°C, very little gB3A virus entered the cells. In addition, none of the revertant viruses showed significantly enhanced entry after 2 h at 37°C.
FIG 5.

Entry of revertant viruses. BAC-derived WT HSV-1 (GS3217), gB3A virus, or gB3A revertant viruses were added to Vero cells at 37°C for 2 h and then rinsed with citric acid buffer to inactivate extracellular virus. As a positive control, duplicate samples were rinsed with PBS instead of acid. Methylcellulose was overlaid onto the cells, and plaques were counted after 3 days. For each virus, the number of plaques present after the citric acid rinse is expressed as a percentage of the number of plaques present in the duplicate control samples that received a PBS rinse only. The means and standard deviations from at least four independent experiments are shown. Asterisks indicate that the difference in the numbers of plaques formed between the mutant and WT viruses was statistically significant (P < 0.05 using a t test). The difference between the mutant and gB3A viruses was not significant.
To determine if the second-site mutations conveyed a faster rate of entry for the revertant viruses than for the gB3A virus, the penetration assay was performed at different time points (Fig. 6). Virus was bound to cells at 4°C, and the samples were then transferred to 37°C for 2 to 12 h prior to acid inactivation. As before, duplicate control samples were rinsed with PBS instead of acid, and plaques were counted after 3 days to determine the entry kinetics for each virus. As expected, most WT virus entry occurred within the first 2 h at 37°C. gB3A entry was very low after 6 h at 37°C but gradually increased after 8 to 12 h. Both of the revertant viruses that showed enhanced cell-cell fusion (HSV-1 gB3A-A683V and HSV-1 gB3A-S383F/G645R/V705I/V880G) showed enhanced entry kinetics compared to gB3A, with nearly half of the virus entering after 4 h at 37°C (Fig. 6C and F). Compared to gB3A virus, gB3A-S383F/G645R/V705I/V880G virus showed statistically significant enhanced entry at the 2- to 10-h postinfection time points (Fig. 6F).
FIG 6.

Entry kinetics of revertant viruses. BAC-derived WT HSV-1 (A), gB3A virus (B), or gB3A-revertant viruses (C to F) were bound to Vero cells at 4°C for 1 h and then shifted to 37°C. At specific time points (2 to 12 h), cells were rinsed with either citric acid buffer to inactivate extracellular virus or PBS as a positive control. Methylcellulose was overlaid onto the cells, and plaques were counted after 3 days. For each time point, the number of plaques present after the citric acid rinse is expressed as a percentage of the number of plaques present in the PBS control samples at that specific time point. The means and standard deviations from at least four independent experiments are shown. Asterisks indicate that the difference in the numbers of plaques formed between the mutant and gB3A viruses was statistically significant at each time point noted (P < 0.05 using a t test).
Compared to gB3A virus, both the gB3A-M742T and gB3A-E187A viruses showed statistically significant enhanced entry at the 4- to 8-h postinfection time points (Fig. 6D and E). Interestingly, approximately half of the HSV-1 gB3A-M742T virus entered by 4 h at 37°C (Fig. 6E) despite this mutation not resulting in enhanced fusion in the cell-cell fusion assay. All of the gB3A-containing revertant viruses showed some degree of enhanced entry kinetics, consistent with the large-plaque phenotype observed for these revertant viruses.
DISCUSSION
We previously demonstrated that HSV-1 carrying three mutations in the domain V arm region of gB (gB3A) showed a small-plaque phenotype and delayed entry kinetics (22, 23). We also showed previously that serial passage of the gB3A virus selected for second-site mutations in gB that restored plaque size, and these second-site mutations enhanced cell-cell fusion in the context of gB3A (24). Other studies have also reported the selection of gB mutations after serial passage of herpesviruses (27, 28). In this new study, we isolated three additional revertant gB3A viruses carrying second-site mutations in gB that conveyed a larger plaque size (Fig. 2). Unexpectedly, two of these mutations did not enhance cell-cell fusion mediated by gB3A (Fig. 3) or WT gB (Fig. 4); however, all three revertant viruses showed enhanced entry kinetics (Fig. 6).
These results reinforce the fact that cell-cell fusion and virus entry are distinct processes (29–31). The presence of accessory glycoproteins may influence gB function during virus entry (32–34). In addition, glycoprotein density, membrane curvature, or membrane composition may differ during cell-cell fusion compared to virus-cell fusion. Differences in the efficiency of egress, a process that involves gB, would also impact plaque size (35, 36). The route of entry or site of fusion also may impact how these gB mutants function. Depending on the cell type, HSV entry can occur by fusion at the cell surface or by fusion with the endosomal membrane (26). Virus fusion with Vero cells (the cells used for our plaque assay) occurs at the cell surface in a pH-independent manner, whereas virus fusion with CHO-K1 cells (the cells used for our cell-cell fusion assay) occurs after endocytosis and exposure to low pH.
Since our previous study, a pseudoatomic model of prefusion HSV-1 gB (PDB accession number 6Z9M) was determined using cryo-electron microscopy of a full-length gB mutant anchored in vesicles (15) (Fig. 1B). The prefusion model suggests that the conformational changes that permit the transition from a prefusion to a postfusion form occur as a result of rearrangements at interdomain connections. Compared to the postfusion form, the prefusion coiled coil is broken, and its N-terminal-to-C-terminal orientation is reversed with respect to the membrane. The prefusion form shows a centrally located domain V that does not pack against domain III, as it does in postfusion gB (Fig. 1A). This indicates that domain V undergoes extensive refolding during the conformational change that mediates fusion. In the prefusion model, the C-terminal portion of domain V forms a central helix that descends toward the TMD, and the N-terminal portion of domain V consists of an extended strand that packs between domains I, II, and III of a neighboring protomer.
This prefusion model allows us to examine the position of the residues mutated in gB3A in both a prefusion and a postfusion structure. The residues mutated to alanine in gB3A (I671, H681, and F683) were selected because each had multiple side chain contacts with residues in the central coiled coil in gB domain III (Fig. 1A). We hypothesized that alanine substitutions at these sites would reduce the number of contacts and weaken the coil-arm interactions, thereby favoring a prefusion gB fold. Alternatively, gB3A may exhibit reduced fusion because the mutations impact the structure of a prefusion or intermediate form of gB. The residues mutated in gB3A map to an N-terminal extended strand in prefusion domain V, near the top (I671) and bottom (H681 and F683) of the strand (Fig. 1B). I671 contacts residues K151 in domain II and R557 in domain III (Fig. 1C). H681 contacts domain III at T120 and domain I at two places, Y165 and T291. F683 also contacts domain I at two places, R189 and G292-D293. The substitution of I671, H681, and F683 in gB3A may influence the packing of this strand within prefusion gB and impact the stability of this form of the gB trimer.
The first gB3A revertant virus selected in this study acquired the second-site mutation E187A in gB. E187 maps to domain I near the base of the molecule, the domain that includes the fusion loops (Fig. 7A and B). In both the prefusion and postfusion structures, E187 participates in a β sheet and contacts V168 in the neighboring β strand. In the prefusion model, E187 also makes multiple contacts with V684 in domain V of a neighboring protomer. V684 is immediately downstream from F683, a residue that is replaced with alanine in gB3A. The proximity of F683 and E187 in the prefusion structure suggests that the E187A mutation may influence the prefusion packing of this critical region in domain V. E187A may enhance gB3A virus entry kinetics by counteracting the effects of the original F683A mutation in the prefusion structure. Accordingly, this suggests that the original F683A mutation may reduce fusion function by altering both the prefusion and postfusion conformations of gB. An interaction between these two sites in domains I and V would validate this prefusion model.
FIG 7.

Location of mutations in revertant viruses. (A) Postfusion structure of full-length HSV-1 gB (PDB accession number 5V2S) (3). Domains (DI to DV) in the ectodomain are colored as described in the legend of Fig. 1. In addition, the membrane proximal region (MPR) (dark green ribbons), transmembrane region (TMR) (purple ribbons), and cytoplasmic tail domain (CTD) (pink ribbons) are shown. The location of the membrane is indicated by dotted lines. The ribbons are partially transparent. The residues mutated in gB3A (I671, H681, and F683) and the residues mutated in the gB3A revertant viruses are shown as space-filled residues. E187 (blue spheres) was mutated in one of the gB3A revertant viruses and lies in domain I near the base of the structure. M742 was mutated in another revertant virus. M742 maps to an unresolved region (green dotted lines) between the C terminus of the ectodomain (position 725) and the N terminus of the MPR (position 753). In a third revertant virus, S383 (green spheres), G645 (orange spheres), V705 (pink spheres), and V880 were mutated. V880 maps downstream of the C terminus of the CTD depicted here. (B) Prefusion model of HSV-1 gB (PDB accession number 6Z9M) (15). Domains and residues in the trimer are colored as described above for panel A. E187 (blue spheres) in domain I contacts domain V of a neighboring protomer, adjacent to F683.
In the second gB3A revertant virus, the second-site mutation M742T arose in gB. M742 maps to the N-terminal region of the MPR of the gB ectodomain. Although the N terminus of the MPR, including M742, is not present in either the prefusion or postfusion models, the structure of the C terminus of the MPR has been visualized (3) (Fig. 7A). The MPR C-terminal region does not make substantial contact with the ectodomain, meaning that the MPR N-terminal region tethers the ectodomain to the TMD and CTD. Mutations in the gB CTD influence fusion (13, 37), indicating that the gB CTD communicates with the ectodomain, possibly through the MPR N-terminal region. To facilitate the conformational change required during fusion, the MPR may undergo significant rearrangement. The M742T mutation may promote MPR rearrangements in the context of gB3A and virus-cell fusion. Interestingly, in prefusion models of gB, the fusion loops are oriented toward the membrane (15, 18). The MPR N-terminal region might interact with the fusion loops, influencing the stability of the prefusion conformation.
Unexpectedly, adding M742T to the WT gB sequence reduced cell-cell fusion, suggesting that enhanced plaque size and virus-cell fusion occur only in the presence of the gB3A mutation. This finding suggests that the prefusion form of gB3A may have a structure that is distinct from that of WT gB in the regions that interact with the MPR N-terminal region.
In the third revertant virus, multiple concurrent second-site mutations in gB were selected, including S383F, G645R, V705I, and V880G. The first three of these mutations were examined in our previous study (24) when they arose in combination with the gB-A855V hyperfusogenic mutation in the CTD of gB. We previously reported that V705I alone boosted gB3A fusion function but did not impact WT gB fusion. Mapping V705 to the recent prefusion model shows that this residue lies in domain V in the central helix that links the ectodomain to the MPR and TMD (Fig. 7B). Although V705 contacts in prefusion gB are restricted to domain V residues in the same protomer, mutation of V705 may impact the refolding of domain V during the transition from a prefusion to a postfusion form.
This study selected a new CTD mutation, V880G, that enhances cell-cell fusion, in both the gB3A and WT gB backgrounds. Residue V880 lies C terminally to the resolved crystal structure of the gB CTD (Fig. 7A), in a region analyzed by electron spin resonance (ESR) (Fig. 8) (3). The CTD is proposed to act as a clamp to stabilize gB in a prefusion state, and multiple mutations in the CTD have been shown to enhance fusion (13, 37). The hyperfusogenic mutation V880G likely accounts for the large plaque size of this revertant virus. Substitution of cysteine at residue 880 was reported previously to result in enhanced fusion (3). V880 lies in a putative C-terminal amphipathic helix of the CTD (helix 3) that may stabilize the attachment of the CTD to the membrane (3, 38). A glycine at position 880 may have a weaker interaction with the membrane than the valine, or a substitution at this site may influence the membrane interaction of neighboring residues. Indeed, the adjacent residue gB-M879 was modeled to penetrate deepest into the membrane for this region (3).
FIG 8.

Sequence alignment of alphaherpesvirus gB homologue cytoplasmic tails. Species and their corresponding GenBank accession numbers (in parentheses) include HSV-1 (KT899744), HSV-2 (NC_001798), varicella-zoster virus (VZV) (X04370), saimiriine herpesvirus 1 (SaHV) (NC_014567), pseudorabies virus (PRV) (JF797218), bovine herpesvirus 1 (BHV1) (AJ004801), equine herpesvirus 1 (EHV1) (AY665713), feline herpesvirus 1 (FHV1) (S49775), canid herpesvirus 1 (CHV1) (NC_030117), and gallid herpesvirus 2/Marek’s disease virus (GHV2) (AF147806). The locations of helices h1a, h1b, h2, and h3 are indicated by the letter “h,” as defined previously by Cooper et al. (3). Regions resolved in the HSV-1 gB crystal structure (3) are underlined with a thick line. The region analyzed using electron spin resonance (ESR) is underlined with a thin line. Conserved residues at each position are highlighted in blue when identical or gray when similar. The position of HSV-1 gB-V880 is highlighted in yellow.
Sequence alignment of multiple alphaherpesviruses shows substantial conservation in the helical regions of the CTD, suggesting a conserved structure. The regions between these helices have variable lengths and amino acid compositions. Varicella-zoster virus (VZV) has a long insertion between the h1b and h2 helices. The residue corresponding to HSV-1 gB-V880 is a valine or an alanine in most species. Notably, canid herpesvirus 1 (CHV1) and gallid herpesvirus 2 (GHV2) both have polar residues at this position (serine and arginine, respectively), suggesting that the h3 helix structures in these two species might differ.
This study demonstrated that mutations selected in three different domains of gB (domain I, MPR, and CTD) can compensate for the reduced entry kinetics of gB3A virus. The results suggest that the gB3A mutations may slow entry due to intramolecular effects on both the prefusion and postfusion forms of gB. Altering the interaction between domains I and V in prefusion gB may affect gB stability and fusion function. The N terminus of the MPR and the C terminus of the CTD also impact entry kinetics, partially counteracting the entry impairment imparted by the gB3A mutations.
MATERIALS AND METHODS
Cells and antibodies.
Chinese hamster ovary (CHO-K1; American Type Culture Collection [ATCC], USA) cells were grown in Ham’s F-12 medium supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, USA) and penicillin and streptomycin (PS). Vero cells (ATCC, USA) and Vero-Cre cells (kindly provided by Gregory Smith at Northwestern University) expressing Cre recombinase were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS and PS. Antibodies used in this study include rabbit anti-gB polyclonal antibody (PAb) serum R74 (39), mouse anti-gB monoclonal antibody (MAb) II-105 (40) (both kindly provided by Patricia Spear at Northwestern University), anti-FLAG MAb F1804 (Sigma), and rabbit anti-FLAG PAb F7425 (Sigma).
Plasmids.
Plasmids encoding HSV-1 KOS strain gB (pPEP98), gD (pPEP99), gH (pPEP100), and gL (pPEP101) were kindly provided by Patricia Spear at Northwestern University and were previously described (41). Plasmids encoding human nectin-1 (pBG38) (49) and HVEM (pBEC10) (42) were also provided by Patricia Spear.
A plasmid encoding an N-terminally FLAG-tagged version of WT gB (pQF112) in the pFLAG-myc-CMV-21 expression vector (catalog number E5776; Sigma) was described previously (43). FLAG-tagged gB3A was also cloned into this vector (pQF302) previously (24). gB genes from the revertant viruses were cloned into this vector to create the following new FLAG-tagged gB constructs: F-gB3A-E187A (pQF346), F-gB3A-M742T (pQF348), and F-gB3A-S383F/G645R/V705I/V880G (pQF344). Individual gB mutations were introduced into pQF112 using QuikChange site-directed mutagenesis (Thermo Fisher Scientific, USA) to create the following new FLAG-tagged gB constructs: F-gB-E187A (pQF364), F-gB-M742T (pQF370), and F-gB-V880G (pQF371). Plasmids encoding untagged WT HSV-1 gB (pSG5-HSVgB, KOS strain) or gB carrying three alanine mutations (pSG5-HSVgB-I671A/H681A/F683A or pSG5-gB3A) were described previously (22). Individual mutations were introduced into this gB3A plasmid using QuikChange mutagenesis to create the new constructs gB3A-E187A (pQF387), gB3A-M742T (pQF393), and gB3A-V880G (pQF394).
Plasmids made for this study were sequenced by the Northwestern Genomic Core Facility and ACTG, Inc. A BAC encoding gB3A (pQF297) was previously described (23). This BAC was generated previously using a parental WT HSV-1 strain F BAC (GS3217) kindly provided by Gregory Smith, Northwestern University. Both BACs carry the red fluorescent protein (RFP) under the control of a cytomegalovirus promoter, inserted into US5 (gJ).
Virus stocks created from BAC DNA.
The BAC-derived virus stocks were generated as described previously (24). Briefly, WT HSV-1 BAC (GS3217) DNA and multiple independent stocks of the gB3A virus BAC DNA (pQF297) were purified. The BAC DNA was transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) into Vero cells expressing Cre recombinase to excise the LoxP-flanked BAC backbone, as previously described (23). When RFP expression was detected in the majority of cells and full cytopathic effect was observed, cells were collected and sonicated to harvest virus. For the WT BAC control, cells were harvested 5 to 7 days after transfection. For the gB3A BAC, cells were harvested 3 weeks after transfection. The gB3A-A683V virus used as a control in this study was isolated previously (24).
Selection of gB3A revertant viruses.
gB3A viruses were passaged on Vero cells as described previously (24). Briefly, Vero cells were infected with the gB3A virus stocks at a multiplicity of infection (MOI) of 0.01. After complete CPE was observed, cells were harvested and sonicated to create a virus stock. Virus was reseeded onto Vero cells for serial passages. When larger plaques were observed during the determination of the titers of stocks between passages, a single large plaque was isolated and purified through three rounds of plaque purification using limiting dilution in 96-well plates. Initially, the time required for gB3A infection to induce full CPE was 14 to 21 days. After larger plaques appeared, full CPE was achieved in 3 to 4 days, similarly to WT HSV-1.
Sequence analysis.
Unipro UGene (44) was used to create a trimmed reference sequence composed of the HSV-1 F strain genome (GenBank accession number GU734771.1) with the long and short terminal repeat regions deleted (45). The gB (UL27) gene in the reference sequence was replaced with the KOS strain gB sequence (GenBank accession number KT899744.1), and the gB3A substitutions were added. The BAC DNA used to generate the WT and gB3A viruses and DNA from the passaged gB3A viruses were submitted for genomic sequencing using HiSeq (Illumina) next-generation sequencing by the Northwestern University NUSeq Core. Using Geneious 8.1.9, sequencing reads were aligned to the reference genome, and variants were identified. Variants present in both the revertant viruses and the original BACs were ignored, as were variants in the gJ gene due to the RFP insertion. To track the acquisition of mutations, the gB gene was PCR amplified and subjected to traditional sequencing for some intermediate passages. Sequence alignments were performed using Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo/).
Determination of plaque size.
Vero cells were infected with virus and overlaid with 0.5% methylcellulose in DMEM with 1% heat-inactivated serum (Sigma-Aldrich, USA). After 3 days, plaques were visualized using Giemsa stain (Sigma-Aldrich, USA). Plaques were imaged by transmitted light microscopy using Evos cell imaging systems at a ×40 total magnification. To calculate the relative plaque area, the diameters of at least 50 plaques from each virus were measured, and the area of each plaque was calculated.
Cell-based ELISA.
To evaluate the cell surface expression of WT gB or gB mutants, CHO-K1 cells seeded into 96-well plates were transfected with 60 ng of an empty vector or a plasmid encoding a gB construct using 0.15 μl of Lipofectamine 2000 (Invitrogen) diluted in Opti-MEM (Invitrogen). After 24 h, the cells were rinsed with PBS, and a cell-based enzyme-linked immunosorbent assay (CELISA) was performed as previously described (46), using anti-gB MAb II-105, anti-gB PAb R74, or anti-FLAG MAb F1804 as the primary antibody. After incubation with primary antibody, the cells were washed, fixed, and incubated with biotinylated goat anti-mouse or anti-rabbit IgG (Sigma), followed by streptavidin-horseradish peroxidase (HRP) (GE Healthcare) and an HRP substrate (BioFX).
Cell-cell fusion assay.
The cell-cell fusion assay was performed as previously described (41). Briefly, CHO-K1 cells were seeded into 6-well plates overnight. One set of cells (effector cells) was transfected using 5 μl of Lipofectamine 2000 (Invitrogen, USA) with 400 ng each of plasmids encoding T7 RNA polymerase, gD, gH, gL, and either a gB construct or an empty vector. A second set of cells (target cells) was transfected using 5 μl of Lipofectamine 2000 with 400 ng of a plasmid carrying the firefly luciferase gene under the control of the T7 promoter and 1.5 μg of a plasmid encoding HVEM, nectin-1, or the empty vector. Six hours after transfection, the cells were detached with EDTA and resuspended in 1.5 ml of F-12 medium supplemented with 10% FBS. Effector and target cells were mixed in a 1:1 ratio and replated in 96-well plates for 18 h. Luciferase activity was quantified using a luciferase reporter assay system (Promega) in a Wallac-Victor luminometer (Perkin Elmer).
Western blotting.
Western blot assays were performed to examine the expression of HSV-1 gB (47). CHO-K1 cells were transfected with a gB construct or an empty vector in 6-well plates using 1.5 μg/μl DNA per well. After incubation overnight, the cells were lysed in 200 μl lysis buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1 mM Na3VO3, 1% Nonidet P-40) containing protease inhibitors (Roche Diagnostics, Indianapolis, IN). Proteins were boiled for 5 min under reducing conditions, separated by SDS-PAGE on 4 to 20% gels, and transferred to nitrocellulose. Blots were probed with anti-gB PAb R74 or anti-FLAG PAb F7425 at a 1:10,000 dilution for 1 h at room temperature, followed by IRDye 800CW goat anti-rabbit IgG (Li-Cor, Lincoln, NE, USA) at 1:10,000. The bands were visualized using Odyssey imaging (Li-Cor, USA).
Virus penetration kinetics.
A penetration experiment was performed as described previously (23, 48) to assay the entry kinetics of the revertant viruses. WT (GS3217) virus, gB3A virus, or revertant viruses were diluted in PBS and incubated with Vero cell monolayers in 6-well plates at 37°C. Due to different plaque sizes, 150 PFU/well of WT and gB3A-S383F/G645R/V705I/V880G or 300 to 400 PFU/well of the other revertant viruses were added to the cells. After 2 h, cells were treated for 1 min with either citrate buffer (135 mM NaCl, 10 mM KCl, 40 mM citric acid [pH 3.0]) to inactivate extracellular virus or PBS as a control. For the time course experiments, viruses were adsorbed to Vero cell monolayers in 6-well plates for 1 h on ice. Cells were washed 3 times with cold PBS, and 1 ml/well of medium prewarmed to 37°C was then added (time zero). Cells were incubated at 37°C for up to 12 h. At the indicated time points, cells were treated for 1 min with either citrate buffer or PBS as a control. After the treatment, the cells were rinsed carefully five times with PBS. The monolayers were overlaid with 0.5% methylcellulose in DMEM with 1% heat-inactivated serum (Sigma-Aldrich, USA) and incubated at 37°C for 3 days. Plaques were visualized by Giemsa staining and counted under a light microscope.
Statistical analysis.
Statistical comparison of the plaque areas was performed with a Mann-Whitney U test using SPSS (version 25). Analyses were performed using IBM SPSS statistics version 25 for Windows (IBM Corp., Armonk, NY) and SAS 9.4 (SAS Institute, Cary, NC). The rest of the statistical tests were two-tailed t tests, performed using Microsoft Excel software.
ACKNOWLEDGMENTS
We thank Gregory Smith for providing the HSV-1 BAC GS3217 as well as Yasushi Kawaguchi for providing the parental BAC. We thank Kevin Bohannon for generating the HSV-1 BAC GS3217. We thank Yi Li at IQVIA for statistical analysis. We thank Sarah Kopp, Nan Susmarski, and Daniel Hippler for timely and excellent technical assistance and members of the Longnecker laboratory for their help in these studies. Sequencing services were performed at the Northwestern University Genomics Core Facility.
R.L. is the Dan and Bertha Spear Research Professor in Microbiology-Immunology. This work was supported by NIH grant AI148478.
Contributor Information
Sarah A. Connolly, Email: sarah.connolly@depaul.edu.
Rozanne M. Sandri-Goldin, University of California, Irvine
REFERENCES
- 1.Connolly SA, Jardetzky TS, Longnecker R. 2021. The structural basis of herpesvirus entry. Nat Rev Microbiol 19:110–121. 10.1038/s41579-020-00448-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vollmer B, Grunewald K. 2020. Herpesvirus membrane fusion—a team effort. Curr Opin Struct Biol 62:112–120. 10.1016/j.sbi.2019.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Cooper RS, Georgieva ER, Borbat PP, Freed JH, Heldwein EE. 2018. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat Struct Mol Biol 25:416–424. 10.1038/s41594-018-0060-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke HG, Heldwein EE. 2015. Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog 11:e1005227. 10.1371/journal.ppat.1005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 6.Vallbracht M, Brun D, Tassinari M, Vaney MC, Pehau-Arnaudet G, Guardado-Calvo P, Haouz A, Klupp BG, Mettenleiter TC, Rey FA, Backovic M. 2018. Structure-function dissection of the pseudorabies virus glycoprotein B fusion loops. J Virol 92:e01203-17. 10.1128/JVI.01203-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandramouli S, Ciferri C, Nikitin PA, Calo S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, Carfi A. 2015. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat Commun 6:8176. 10.1038/ncomms9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliver SL, Xing Y, Chen D-X, Roh SH, Pintilie GD, Bushnell DA, Sommer MH, Yang E, Carfi A, Chiu W, Arvin AM. 2020. A glycoprotein B-neutralizing antibody structure at 2.8 Å uncovers a critical domain for herpesvirus fusion initiation. Nat Commun 11:4141. 10.1038/s41467-020-17911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Yang F, Hu X, Tan F, Qi J, Peng R, Wang M, Chai Y, Hao L, Deng J, Bai C, Wang J, Song H, Tan S, Lu G, Gao GF, Shi Y, Tian K. 2017. Two classes of protective antibodies against pseudorabies virus variant glycoprotein B: implications for vaccine design. PLoS Pathog 13:e1006777. 10.1371/journal.ppat.1006777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oliver SL, Xing Y, Chen DH, Roh SH, Pintilie GD, Bushnell DA, Sommer MH, Yang E, Carfi A, Chiu W, Arvin AM. 2021. The N-terminus of varicella-zoster virus glycoprotein B has a functional role in fusion. PLoS Pathog 17:e1008961. 10.1371/journal.ppat.1008961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J Virol 83:6825–6836. 10.1128/JVI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver SL, Sommer M, Zerboni L, Rajamani J, Grose C, Arvin AM. 2009. Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol 83:7495–7506. 10.1128/JVI.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J Virol 86:8171–8184. 10.1128/JVI.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitu E, Sharma S, Stampfer SD, Heldwein EE. 2013. Extensive mutagenesis of the HSV-1 gB ectodomain reveals remarkable stability of its postfusion form. J Mol Biol 425:2056–2071. 10.1016/j.jmb.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vollmer B, Prazak V, Vasishtan D, Jefferys EE, Hernandez-Duran A, Vallbracht M, Klupp BG, Mettenleiter TC, Backovic M, Rey FA, Topf M, Grunewald K. 2020. The prefusion structure of herpes simplex virus glycoprotein B. Sci Adv 6:eabc1726. 10.1126/sciadv.abc1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeev-Ben-Mordehai T, Vasishtan D, Hernandez Duran A, Vollmer B, White P, Prasad Pandurangan A, Siebert CA, Topf M, Grunewald K. 2016. Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein B. Proc Natl Acad Sci USA 113:4176–4181. 10.1073/pnas.1523234113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Si Z, Zhang J, Shivakoti S, Atanasov I, Tao CL, Hui WH, Zhou K, Yu X, Li W, Luo M, Bi GQ, Zhou ZH. 2018. Different functional states of fusion protein gB revealed on human cytomegalovirus by cryo electron tomography with Volta phase plate. PLoS Pathog 14:e1007452. 10.1371/journal.ppat.1007452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fontana J, Atanasiu D, Saw WT, Gallagher JR, Cox RG, Whitbeck JC, Brown LM, Eisenberg RJ, Cohen GH. 2017. The fusion loops of the initial prefusion conformation of herpes simplex virus 1 fusion protein point toward the membrane. mBio 8:e01268-17. 10.1128/mBio.01268-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci USA 106:2880–2885. 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher JR, Atanasiu D, Saw WT, Paradisgarten MJ, Whitbeck JC, Eisenberg RJ, Cohen GH. 2014. Functional fluorescent protein insertions in herpes simplex virus gB report on gB conformation before and after execution of membrane fusion. PLoS Pathog 10:e1004373. 10.1371/journal.ppat.1004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison SC. 2015. Viral membrane fusion. Virology 479–480:498–507. 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connolly SA, Longnecker R. 2012. Residues within the C-terminal arm of the herpes simplex virus 1 glycoprotein B ectodomain contribute to its refolding during the fusion step of virus entry. J Virol 86:6386–6393. 10.1128/JVI.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan Q, Kopp SJ, Connolly SA, Longnecker R. 2017. Structure-based mutations in the herpes simplex virus 1 glycoprotein B ectodomain arm impart a slow-entry phenotype. mBio 8:e00614-17. 10.1128/mBio.00614-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan Q, Kopp SJ, Byskosh NC, Connolly SA, Longnecker R. 2018. Natural selection of glycoprotein B mutations that rescue the small-plaque phenotype of a fusion-impaired herpes simplex virus mutant. mBio 9:e01948-18. 10.1128/mBio.01948-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cairns TM, Friedman LS, Lou H, Whitbeck JC, Shaner MS, Cohen GH, Eisenberg RJ. 2007. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J Virol 81:5102–5111. 10.1128/JVI.00097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicola AV. 2016. Herpesvirus entry into host cells mediated by endosomal low pH. Traffic 17:965–975. 10.1111/tra.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vallbracht M, Lotzsch H, Klupp BG, Fuchs W, Vollmer B, Grunewald K, Backovic M, Rey FA, Mettenleiter TC. 2021. In vitro viral evolution identifies a critical residue in the alphaherpesvirus fusion glycoprotein B ectodomain that controls gH/gL-independent entry. mBio 12:e00557-21. 10.1128/mBio.00557-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuny CV, Bowen CD, Renner DW, Johnston CM, Szpara ML. 2020. In vitro evolution of herpes simplex virus 1 (HSV-1) reveals selection for syncytia and other minor variants in cell culture. Virus Evol 6:veaa013. 10.1093/ve/veaa013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cairns TM, Milne RS, Ponce-de-Leon M, Tobin DK, Cohen GH, Eisenberg RJ. 2003. Structure-function analysis of herpes simplex virus type 1 gD and gH-gL: clues from gDgH chimeras. J Virol 77:6731–6742. 10.1128/jvi.77.12.6731-6742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novotny MJ, Parish ML, Spear PG. 1996. Variability of herpes simplex virus 1 gL and anti-gL antibodies that inhibit cell fusion but not viral infectivity. Virology 221:1–13. 10.1006/viro.1996.0347. [DOI] [PubMed] [Google Scholar]
- 31.Weed DJ, Nicola AV. 2017. Herpes simplex virus membrane fusion. Adv Anat Embryol Cell Biol 223:29–47. 10.1007/978-3-319-53168-7_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim IJ, Chouljenko VN, Walker JD, Kousoulas KG. 2013. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J Virol 87:8029–8037. 10.1128/JVI.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dogrammatzis C, Waisner H, Kalamvoki M. 2020. “Non-essential” proteins of HSV-1 with essential roles in vivo: a comprehensive review. Viruses 13:17. 10.3390/v13010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hilterbrand AT, Daly RE, Heldwein EE. 2021. Contributions of the four essential entry glycoproteins to HSV-1 tropism and the selection of entry routes. mBio 12:e00143-21. 10.1128/mBio.00143-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, Johnson DC. 2009. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J Virol 83:11847–11856. 10.1128/JVI.01397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson DC, Wisner TW, Wright CC. 2011. Herpes simplex virus glycoproteins gB and gD function in a redundant fashion to promote secondary envelopment. J Virol 85:4910–4926. 10.1128/JVI.00011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan Z, Grantham ML, Smith MS, Anderson ES, Cardelli JA, Muggeridge MI. 2002. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell-cell fusion, but these properties are unrelated. J Virol 76:9271–9283. 10.1128/jvi.76.18.9271-9283.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chowdary TK, Heldwein EE. 2010. Syncytial phenotype of C-terminally truncated herpes simplex virus type 1 gB is associated with diminished membrane interactions. J Virol 84:4923–4935. 10.1128/JVI.00206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herold BC, Visalli RJ, Susmarski N, Brandt CR, Spear PG. 1994. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulfate and glycoprotein B. J Gen Virol 75(Part 6):1211–1222. 10.1099/0022-1317-75-6-1211. [DOI] [PubMed] [Google Scholar]
- 40.Para MF, Parish ML, Noble AG, Spear PG. 1985. Potent neutralizing activity associated with anti-glycoprotein D specificity among monoclonal antibodies selected for binding to herpes simplex virions. J Virol 55:483–488. 10.1128/JVI.55.2.483-488.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 42.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 43.Fan Q, Longnecker R, Connolly SA. 2014. Substitution of herpes simplex virus 1 entry glycoproteins with those of saimiriine herpesvirus 1 reveals a gD-gH/gL functional interaction and a region within the gD profusion domain that is critical for fusion. J Virol 88:6470–6482. 10.1128/JVI.00465-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okonechnikov K, Golosova O, Fursov M, UGENE Team. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167. 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- 45.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. 2014. Evolution and diversity in human herpes simplex virus genomes. J Virol 88:1209–1227. 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin E, Spear PG. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc Natl Acad Sci USA 104:13140–13145. 10.1073/pnas.0705926104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan Q, Lin E, Satoh T, Arase H, Spear PG. 2009. Differential effects on cell fusion activity of mutations in herpes simplex virus 1 glycoprotein B (gB) dependent on whether a gD receptor or a gB receptor is overexpressed. J Virol 83:7384–7390. 10.1128/JVI.00087-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwartz JA, Lium EK, Silverstein SJ. 2001. Herpes simplex virus type 1 entry is inhibited by the cobalt chelate complex CTC-96. J Virol 75:4117–4128. 10.1128/JVI.75.9.4117-4128.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280(5369):1618–1620. 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]