

ABSTRACT
Varicella-zoster virus (VZV) maintains lifelong latency in neurons following initial infection and can subsequently be reactivated to result in herpes zoster or severe neurological manifestations such as encephalitis. Mechanisms of VZV neuropathogenesis have been challenging to study due to the strict human tropism of the virus. Although neuronal entry mediators of other herpesviruses, including herpes simplex virus, have been identified, little is known regarding how VZV enters neurons. Here, we utilize a human stem cell-based neuronal model to characterize cellular factors that mediate entry. Through transcriptional profiling of infected cells, we identify the cell adhesion molecule nectin-1 as a candidate mediator of VZV entry. Nectin-1 is highly expressed in the cell bodies and axons of neurons. Either knockdown of endogenous nectin-1 or incubation with soluble forms of nectin-1 produced in mammalian cells results in a marked decrease in infectivity of neurons. Notably, while addition of soluble nectin-1 during viral infection inhibits infectivity, addition after infection has no effect on infectivity. Ectopic expression of human nectin-1 in a cell line resistant to productive VZV infection confers susceptibility to infection. In summary, we have identified nectin-1 as a neuronal entry mediator of VZV.
IMPORTANCE Varicella-zoster virus (VZV) causes chickenpox, gains access to neurons during primary infection where it resides lifelong, and can later be reactivated. Reactivation is associated with shingles and postherpetic neuralgia, as well as with severe neurologic complications, including vasculitis and encephalitis. Although the varicella vaccine substantially decreases morbidity and mortality associated with primary infection, the vaccine cannot prevent the development of neuronal latency, and vaccinated populations are still at risk for reactivation. Furthermore, immunocompromised individuals are at higher risk for VZV reactivation and associated complications. Little is known regarding how VZV enters neurons. Here, we identify nectin-1 as an entry mediator of VZV in human neurons. Identification of nectin-1 as a neuronal VZV entry mediator could lead to improved treatments and preventative measures to reduce VZV related morbidity and mortality.
KEYWORDS: embryonic stem cells, entry mediator, nectin-1, varicella-zoster virus
INTRODUCTION
Varicella-zoster virus (VZV) is an alphaherpesvirus responsible for causing varicella (chickenpox) and zoster (shingles) exclusively in humans, making it challenging to develop experimental models (1). VZV primary infection begins with viral replication in respiratory epithelial tissue, followed by viremia in T cells, resulting in dermal and epidermal lytic infection. The virus gains access to and establishes lifelong latency in neurons of the dorsal root, cranial nerve, and autonomic ganglia via direct T-cell interactions or retrograde transport along sensory axons (2, 3). In the setting of diminished cell-mediated immunity, viral reactivation can result in severe complications, including vasculitis, myelitis, and encephalitis (4, 5). Although the use of the live attenuated varicella vaccine has significantly reduced VZV morbidity in immunocompetent individuals, viral latency occurs in vaccinated populations (6). Furthermore, the increasing use of immunosuppressive and immunomodulatory therapy for autoimmune conditions has been linked to increased VZV reactivation and associated complications (7).
Ongoing work has provided insights into the mechanisms of VZV infection in nonneuronal cells, with some parallels with the established mechanisms of infection by the other human alphaherpesviruses, herpes simplex virus 1 (HSV-1), and HSV-2. Human alphaherpesviruses attach to heparan sulfate proteoglycans (HSPGs) to initiate viral entry (8, 9), and binding of VZV glycoprotein B (gB) to HSPGs in lung fibroblasts has been shown to mediate attachment (10). In melanoma cells, the extracellular domain of insulin-degrading enzyme (IDE) binds to and elicits a conformational change in VZV gE that enhances infectivity and fusogenicity (11). Notably, however, IDE does not contribute to infection in neurons or T cells (1, 12, 13). A role for mannose 6-phosphate (Man 6-P) is highlighted by the findings that multiple VZV envelope glycoproteins contain Man 6-P groups, Man 6-P can block infection of human embryonic lung fibroblasts by cell-free VZV, and melanoma cells deficient in Man 6-P receptors are resistant to infection by cell-free VZV (9, 14). In addition, in an oligodendrocyte cell line, myelin-associated glycoprotein has been reported to associate with VZV gB, and this interaction modulates viral fusion and infectivity (15). Thus, viral entry appears to be a complex phenomenon that is governed by cell-specific mechanisms.
The mechanisms of VZV infection in neurons remain poorly understood, in part due to the strict human specificity of the virus. Over the past decade, human pluripotent stem cell-derived neurons have become increasingly utilized to study VZV neurotropism, resulting in a number of insights regarding viral gene expression, neurovirulence, intracellular viral transport, and latency and reactivation in human neurons (16–24). However, the fundamental question of how VZV enters neurons and which cellular molecules mediate VZV entry remain unsolved. Here, we identify nectin-1 as a neuronal entry mediator for VZV using a human stem cell-based neuronal model.
RESULTS
Identification of potential candidates as VZV neuronal entry mediators.
Neurons derived from human embryonic stem cells (hESCs) were infected with VZV-green fluorescent protein (GFP) and sorted by fluorescence-activated cell sorting (FACS) into infected (GFP+) and bystander (GFP–) neurons; mock-infected neurons were treated identically but without viral infection (Fig. 1A). RNA isolated from cells was analyzed on microarrays, and transcriptional differences were binned according to the function of the encoded proteins, with a focus on proteins that could influence viral entry such as adhesion molecules or plasma membrane proteins and receptors. Upon cross-referencing gene array data with relevant literature, nine candidate genes were identified as potential entry mediators in neurons.
FIG 1.

Identification of potential candidates of neuronal entry mediators for VZV infection. (A) Schematic of methodology. Neural stem cells (NSCs) were differentiated into neurons, infected for 4 days, and then sorted using FACS. RNA was extracted from sorted cells, and transcriptional profiling was performed. (B) RT-qPCR analysis of candidate genes on day 1 after infection normalized to expression of the housekeeping gene HPRT1 and relative to mock-infected cells is shown. Error bars indicate the standard errors of the mean (SEM; P < 0.001 when comparing GFP+ to either GFP– or mock samples; two-way analysis of variance with Bonferroni posttest). (C) Mock-infected (upper panel) and pOka-infected neurons immunostained with antibodies against nectin-1 (green) and beta-III tubulin (red) and counterstained with DAPI (cyan). Scale bar, 50 μm. (D) Mock-infected (upper panel) and VZV-GFP-infected neurons immunostained with antibodies against VZV gE (purple) and nectin-1 (red) and counterstained with DAPI (cyan). Scale bar, 20 μm. (E) pOka-infected neurons stained with antibodies against VZV gE (red), TGN46 (purple), and nectin-1 (green). The merged panel includes DAPI counterstain (cyan). Scale bar, 20 μm.
Transcription of these potential candidates was analyzed by reverse transcription-quantitative PCR (RT-qPCR) shortly after infection (Fig. 1B). One day postinfection, levels of nectin-1 mRNA were markedly higher in GFP+ infected neurons compared to GFP– bystander and mock-infected neurons. The marked elevation in nectin-1 mRNA expression early on during infection, along with its known role in HSV infection, suggested a potential role for this molecule in mediating entry; thus, subsequent experiments focused on nectin-1.
We next examined the localization of nectin-1 in human neurons. In mock-infected neurons, nectin-1 expression is present diffusely in neuronal cell bodies and axons, as evidenced by colocalization of nectin-1 and beta III tubulin (Fig. 1C, upper panel). After infection, nectin-1 accumulates in foci in cell bodies (Fig. 1C, lower panel by pOka infection; and Fig. 1D, lower panel by VZV-GFP infection). Notably, in infected cells, nectin-1 and VZV gE exhibit areas of colocalization (Fig. 1D, arrows), potentially providing support for an interaction between these two proteins. Moreover, colocalization of nectin-1, VZV glycoprotein E (gE), and the trans-Golgi network marker TGN46 was observed (Fig. 1E), suggestive of involvement of nectin-1 in entry and/or intracellular trafficking of VZV.
Knockdown of nectin-1 inhibits VZV infection in human neurons.
While the data presented above suggest a role for nectin-1 in VZV infection, we next sought to directly determine whether and how nectin-1 may impact VZV infection of neurons. We began with experiments involving knockdown of nectin-1 in neurons. RT-qPCR was performed to assess the level of mRNAs 3 days after transfection with selected siRNAs (Fig. 2A). As expected, siRNA directed against GAPDH reduced GAPDH mRNA expression by 78% (Fig. 2A). Notably, siRNA directed against nectin-1 reduced nectin-1 mRNA expression by 71% (Fig. 2A). Despite efficient knockdown of mRNA level at 3 days posttransfection, siRNA targeting NECTIN1 or GAPDH did not cause significant reduction of targeted protein expression; as a result, we serially transfected siRNAs every 4 days.
FIG 2.

Effect of nectin-1 siRNA knockdown on VZV infection of human neurons. (A) RNA fold changes normalized to HRPT1 expression. The error bars indicate the SEM (P values refer to two-way analysis of variance with Bonferroni posttest). (B) Effect of siRNA-mediated knockdown against GAPDH or nectin-1 (50 μM) at 7 and 10 days by Western blotting with whole-cell lysates. (C) Effect of siRNA-mediated knockdown on cell surface level of nectin-1 by flow cytometry at 7 days posttransfection. (D) Neurons transfected with siRNA against nectin-1 (7 days) were infected with VZV, and the effect of nectin-1 knockdown on VZV infection was analyzed by Western blotting for VZV gE expression at 4 days after infection. (E) Dose-dependent effect of nectin-1 siRNA (0, 10, 25, and 50 μM) on VZV infection was analyzed by Western blotting with anti-VZV gE and pORF63 antibodies.
At 7 days after initial transfection (i.e., transfection on day 0 and day 4), nectin-1 and GAPDH protein levels were reduced by 61 and 80%, respectively (Fig. 2B). At 10 days after the initial siRNA transfection (i.e., transfection on days 0, 4, and 8), protein expression was still reduced, though it had begun to recover (34% reduction in GAPDH and 53% reduction in nectin-1; Fig. 2B). Live cell immunostaining with antibody against nectin-1, followed by flow cytometry, confirmed that knockdown of nectin-1 resulted in decreased protein levels at the neuronal cell surface (Fig. 2C).
To examine whether downregulation of nectin-1 impacted VZV infection, neurons were infected at day 7 after siRNA treatment, a time point when nectin-1 protein expression was observed to be lowest in silenced neurons. Nectin-1 downregulation blocked VZV infection, as evidenced by the absence of gE in the cells (Fig. 2D). Neither expression of housekeeping genes (GAPDH or beta-actin; Fig. 2D and E) nor cell viability (104% ± 2% with 25 μM siRNA treatment and 106% ± 5% for 50 μM siRNA treatment when normalized to mock siRNA treatment) were affected, suggesting that the observed decrease in viral infectivity was not explained by cell death in the setting of nectin-1 knockdown. In separate experiments in which we added increasing concentrations of nectin-1 siRNA, decreasing levels of gE and the VZV immediate early ORF63 protein (pORF63) were observed (Fig. 2E), thus demonstrating a dose-dependent effect of nectin-1 knockdown on VZV infection of neurons.
Recombinant nectin-1 modulates VZV infectivity in human neurons.
Since nectin-1 functions as an entry receptor for several other alphaherpesviruses (25), we assessed its role as an entry mediator for VZV infection in neurons by utilizing soluble recombinant forms of the protein. While such methods have successfully been used to block entry of HSV into cells, the prominent cell-to-cell spread of VZV in cell culture could potentially confound such assays. Thus, we first sought to determine whether a soluble recombinant gE (sgE) could inhibit VZV infection in human neurons. Preincubation of various amounts of sgE, but not bovine serum albumin (BSA), with human neurons, followed by VZV infection, resulted in a dose-dependent decrease in VZV infection, as evidenced by reduced de novo gE synthesis and the nearly complete absence of de novo gE upon addition of 25 μg of sgE (Fig. 3A). These data indicate that sgE can effectively compete with VZV at the neuronal cell surface and block VZV entry into neurons.
FIG 3.

Soluble nectin-1 influences VZV infectivity of neurons. (A) Neurons were preincubated with soluble VZV gE (sgE) and infected with VZV for 4 days. Dose-dependent effect of sgE treatment on VZV infection was analyzed by Western blotting with anti-VZV gE antibody. BSA was used as an experimental control. (B) Neurons preincubated without (lane 2) or with 100 μg of nectin-1(Bac) produced in Sf-9 cells (lane 3) were infected with VZV, and whole-cell lysate collected 4 days after infection was subjected to Western blotting with anti-VZV gE antibody. (C) Effect of preincubation of soluble nectin-1 (sNectin-1) produced in NS0 cells with HSV-1 was analyzed by Western blotting with anti-HSV gD antibody. (D) VZV was preincubated with increasing doses of sNectin-1 prior to infection of neurons. Four days later, whole-cell lysates were collected and subjected to Western blotting with anti-VZV gE antibody. Anti-GAPDH antibody was used as an internal control. (E) VZV was preincubated with 20 μg of Nectin-1(FL) produced in HEK293T cells prior to infection of neurons. Four days later, whole-cell lysates were collected and subjected to Western blotting using anti-VZV gE antibody. Anti-GAPDH antibody was used as internal control.
Having established that sgE can interfere with VZV infection of human neurons, we performed a VZV entry competition assay using recombinant human nectin-1 proteins. The extracellular domain of nectin-1 produced in a baculovirus expression system, nectin-1(Bac), has been shown to block HSV-1 entry into mouse and rat neurons (26) and human fetal neurons (27). Surprisingly, nectin-1(Bac) enhanced VZV gE expression in human ESC-derived neurons (Fig. 3B). Next, another soluble nectin-1, sNectin-1, produced in mouse myeloma cells in which posttranslational modifications are expected to be more similar to those in human endogenous nectin-1 than those of nectin-1(Bac) was used. sNectin-1 was able to effectively diminish neuronal infection by HSV-1 (Fig. 3C). Notably, incubation with sNectin-1 resulted in a dose-dependent decrease in VZV infection, as evidenced by reduced gE, such that treatment with 25 μg of sNectin-1 reduced VZV infection by 97% (Fig. 3D). Similar results were seen with addition of full-length nectin-1 protein, Nectin-1(FL) produced in HEK293 cells, which reduced VZV infection by 86% compared to control (Fig. 3E). Taken together, recombinant nectin-1 proteins dramatically modulate VZV infection of human neurons, and posttranslational modifications of the protein may be critical for its entry blocking activity against VZV.
Soluble nectin-1 protein blocks VZV infection during, but not after, virus entry.
To determine whether sNectin-1 inhibits VZV entry, spread, or both, we conducted experiments in which we added sNectin-1 either at the time of VZV infection or 2 h postinfection. When added to neurons at the time of infection, sNectin-1 inhibited VZV infection in a dose-dependent manner, as assessed by VZV gE and pORF63 (Fig. 4A, left; quantification of the density of viral protein bands normalized to GAPDH demonstrated that addition of 5 μg/ml of sNectin-1 resulted in a 24% reduction in gE and 34% reduction in pORF63, while addition of 10 μg/ml of sNectin-1 resulted in a 64% reduction in gE and 76% reduction in pORF63). In contrast, when sNectin-1 was added to neurons after infection and extracellular virus had been inactivated with sodium citrate buffer, VZV infectivity was unaffected (Fig. 4A, right). The addition of the control protein BSA either during or after infection did not affect VZV infectivity (Fig. 4B). These data support a role for sNectin-1 in blocking the entry, but not the subsequent spread, of VZV in neurons.
FIG 4.
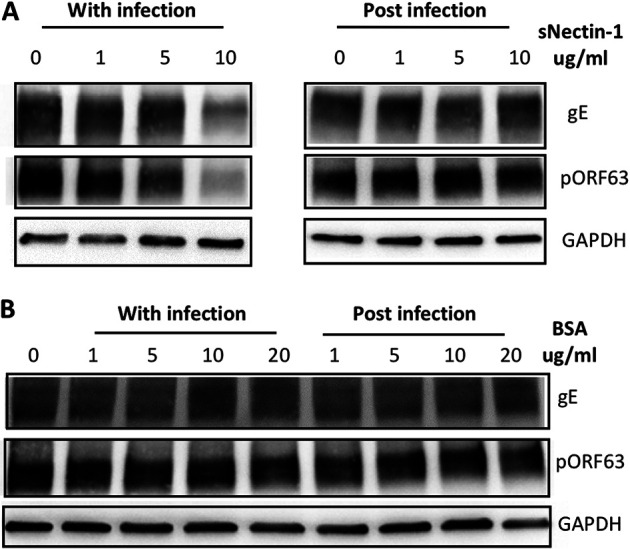
Addition of soluble nectin-1 during, but not after, infection inhibits VZV infectivity of neurons. (A) Neurons were infected with VZV in the presence of increasing concentrations of sNectin-1 during infection (left) or at 2 h postinfection (right). Whole-cell lysates were collected at 4 days after infection and subjected to Western blotting with anti-VZV gE and pORF63 antibody. Anti-GAPDH antibody was used as an internal control. (B) BSA was used as an experimental control.
Expression of exogenous human nectin-1 increases VZV infectivity.
Finally, in a gain-of function analysis, human nectin-1 was ectopically expressed in CHO-K1 cells, followed by infection with cell-free VZV pOka. CHO-K1 is a Chinese hamster ovary epithelial cell line that is broadly resistant to productive infection by human alphaherpesviruses and thus has been widely used to confirm the role of candidate alphaherpesvirus entry mediators (8, 12, 28, 29). While little to no endogenous nectin-1 was detected in CHO-K1 cells, upon transfection of a plasmid encoding human nectin-1 CHO-K1 cells were found to abundantly express human nectin-1 (Fig. 5A). Expression of human nectin-1 was readily detected at the cell surface, increasing from 39% of cells at 24 h posttransfection to 83% at 48 h posttransfection (Fig. 5B). Upon infection with VZV, only CHO-K1 cells that had been transfected with the nectin-1 plasmid expressed substantial amounts of gE (Fig. 5C, compare lanes 2 and 3). In separate experiments, ectopic expression of human nectin-1 in CHO-K1 cells, followed by VZV infection, resulted in a dose-dependent increase in expression of VZV gE and pORF63, indicating that human nectin-1 functions as an entry mediator for VZV (Fig. 5D).
FIG 5.

CHO-K1 cells expressing human nectin-1 permits efficient VZV infection. (A) Total nectin-1 expression in CHO-K1 cells transfected with control plasmid (TdT, 1.5 μg of plasmid; lane 1), mock transfected (lane 2), infected with VZV (lane 3), and transfected with human nectin-1 expressing plasmid, followed by VZV infection (1.5 μg; lane 4). (B) Cell surface expression of human nectin-1 in CHO-K1 cells after nectin-1 plasmid transfection at 24 and 48 h after transfection. (C) CHO-K1 cells (lane 1), transfected with control plasmid followed by VZV infection (lane 2) and transfected with human nectin-1 plasmid followed by VZV infection (lane 3), were analyzed for VZV gE expression (arrow) at 4 days postinfection. CHO-K1 cells were infected with VZV 48 h after transfection. (D) CHO-K1 cells were transfected with human nectin-1 plasmid (0, 0.25, 0.5, and 0.75 μg) and infected with VZV at 48 h after transfection. Whole-cell lysates at 4 days after infection were subjected to Western blotting with anti-nectin-1 antibody, anti-VZV pORF63 antibody, anti-VZV gE antibody, and anti-GAPDH antibody.
DISCUSSION
Despite the key role of neurons in the life cycle of VZV in humans, the mechanisms by which the virus enters human neurons remains enigmatic. Here, we show that the cellular adhesion molecule nectin-1 plays an important role in VZV neurotropism using hESC-derived neurons. Loss-of-function and gain-of-function analyses, together with a virus entry competition assay using soluble nectin-1 proteins, provide evidence that nectin-1 mediates efficient VZV entry into human neurons.
Nectin-1 has been identified as an entry receptor for a number of Alphaherpesvirinae, including HSV-1, HSV-2, pseudorabies virus (PRV), and bovine herpesvirus 1 (BHV-1), each of which utilizes gD as a viral ligand for nectin-1 (25). In addition, cercopithecine herpesvirus-1 (CeHV-1, also known as herpes B virus) and CeHV-2 also use nectin-1 as an entry receptor and for gD/gH/gL/gB-mediated cell-to-cell fusion (30), although it has not been determined whether their gDs directly bind to nectin-1. Equine herpesvirus 1 (EHV-1) is a well-studied member of the Varicellovirus genus, along with PRV and BHV-1, and the interaction between EHV-1 gD and equine major histocompatibility complex class I mediates its entry into multiple equine cells (31). EHV-1 also enters CHO-K1 cells using gD and replicates well, but the entry process does not require nectin-1 (32). VZV and simian varicella virus (SVV or CeHV-9) also belong to the Varicellovirus genus. However, unlike the other members of Alphaherpesvirinae noted above, they lack a gD ortholog, possess an N-terminal extended gE, and are highly contagious via airborne transmission in vivo while exhibiting marked cell association in vitro (33–36). The genes encoding Alphaherpesvirinae gD and gE share several features and are thought to have evolved via duplication and subsequent divergence (37). Thus, gE in VZV or SVV has been posited to play an analogous role to gD as a viral ligand for cellular entry. While we were unable to detect a direct interaction between VZV gE and nectin-1 via immunoprecipitation (data not shown), a more transient or lower affinity interaction between these two proteins is suggested by our immunohistochemical data demonstrating colocalization in infected neurons. Further investigations into interactions between human nectin-1 and VZV gE or other glycoproteins, along with evaluation of the potential role of nectin-1 in SVV infection, may serve to highlight both conserved mechanisms of infection in Alphaherpesvirinae, along with evolutionarily acquired strategies specific to Varicellovirus infection of humans and nonhuman primates by VZV and SVV, respectively.
Interestingly, nectin-1 mRNA was markedly increased in VZV-infected cells compared to bystander or mock-infected cells. This finding may seem counterintuitive since many viruses downregulate expression of cell surface receptors upon infection, perhaps in an effort to avoid superinfection (38–40). Most studies, however, have not addressed the level of virus receptor mRNA after infection, only the level of receptor protein on the surface of the cell. For example, nectin-1 is removed from the surface of HSV-1 infected cells upon binding to HSV gD (41), a process recently shown to be mediated by the Cbl E3 ligase (42); however, the kinetics of nectin-1 mRNA after infection are unknown. After VZV infection, there were areas of nectin-1 accumulation within cell bodies. It is possible that nectin-1 is recycled after internalizing with its viral ligand(s); however, it is also possible that nectin-1 is degraded after mediating VZV entry, and its mRNA is upregulated in a compensatory manner with de novo-synthesized nectin-1 localizing to the nucleus. The specific mechanisms and contribution of intracellular accumulation of nectin-1 after VZV neuronal infection will need to be clarified in future studies.
In contrast to the effects on HSV-1 infection, a soluble form of nectin-1 produced in a baculovirus expression system did not block, but rather enhanced, VZV infection of human neurons. A secreted form of human nectin-1 produced in mouse cells, however, efficiently blocked neuronal entry of both VZV and HSV-1, and a full-length form of human nectin-1 produced in human cells also blocked VZV entry in neurons. Although the cause of the differential effects of the soluble proteins on VZV infectivity remains unclear, there are several possible explanations. The ectodomain of the human nectin-1 protein consists of amino acid residues Gln31-Thr334 (43). While the nectin-1 produced in the mouse cells contains only this ectodomain, the nectin-1 produced in insect cells contains residues Gln31-His346 and an additional N-terminal aspartic acid not normally found in nectin-1 (44). The additional 13 amino acids could contribute to the differential effects of the two proteins. Another possible—and perhaps more likely—explanation lies in potential differences in posttranslational modifications of nectin-1 produced in mammalian versus insect cells. Indeed, human nectin-1 contains several known posttranslational modifications, including N-linked glycosylation and multiple phosphorylation sites (45). In addition, the nectin-1(Bac) used in our experiments contains an additional N-linked glycosylation site at Asn202. The nectin family of proteins, including nectin-1, are known to form homo- and heterodimers in cis and in trans with each other to produce cell-cell adhesions (46). They can also form heterophilic complexes with other immunoglobulin-like cell adhesion molecules and can interact with matrix metalloproteinases, thus resulting in a wide range of multiprotein complexes that can vary between cell types (47). It is possible that the posttranslational modifications specific to insect cell-produced nectin-1 alter such protein-protein interactions at the neuronal surface in a manner that enables the virus to either attach or enter more readily. Notably, contrasting effects of mammalian and insect cell-produced soluble entry mediator proteins in the entry of VZV have previously been observed. Preincubation with a soluble form of mammalian cell-produced IDE reduced VZV infectivity by approximately 70% in MRC-5 cells, while a form of IDE produces in a baculovirus expression system enhanced infectivity by increasing stability of the virus (11, 12). Interestingly, cocrystallization of nectin-1 and HSV gD was achieved using soluble nectin-1 produced in a baculovirus expression system, and a recombinant nectin-1 ectodomain produced in an Escherichia coli expression system bound gD and blocked HSV entry (43). Thus, mammalian glycosylation or other posttranslational modifications of nectin-1 may play a critical role in VZV entry.
CHO-K1 cells are resistant to HSV-1 entry and have been used to identify viral entry mediators (29, 48). Finnen et al. used CHO-K1 cells to study VZV infection and reported evidence for viral entry via the use of sensitive reporter assays and by assessing the presence of several viral proteins, including pORF62 and gE by immunohistochemistry (49). In contrast, we detected little or no viral protein production in CHO-K1 cells unless exogenous nectin-1 was expressed. Although the ORF68 mRNA, RNA 68, which encodes gE has recently been recognized to be transcribed with early-late kinetics (22), substantial gE protein production does not occur until late in infection and thus sole reliance on the presence of gE as a measure of infectivity in CHO-K1 cells, as well as in neurons, might reflect both entry and postentry mechanisms. As a result, we also assessed the presence of the canonical VZV immediate early protein pORF63. Upon attempted infection of CHO-K1 cells, we found little or no pORF63 expression in the absence of exogenous nectin-1, and pORF63 expression increased in parallel with increased nectin-1 expression. It is possible that multiple routes of VZV entry can occur in CHO-K1 cells and that nectin-1 independent mechanisms may allow for limited VZV entry; however, ectopic human nectin-1 expression mediates efficient VZV entry in CHO-K1 cells.
Finally, while our data highlight the importance of nectin-1 in mediating entry of VZV into neurons, the process of VZV attachment followed by entry is likely to involve multiple factors and may occur via several nonexclusive mechanisms. Future work will need to address viral binding partners of nectin-1 and the potential role of other cell surface proteins in mediating VZV entry into neurons and other cell types.
MATERIALS AND METHODS
Human neurons.
Human neurons were differentiated from human embryonic stem cell (hESC; H9 line)-derived neural stem cells (Thermo Fisher Scientific), as described previously (20). Briefly, neural stem cells (passages 4 to 10) were seeded in 12-well plates coated with poly-d-lysine (100 μg/ml) and Matrigel (100 μg/ml) at a density of 1 × 105 cells/cm2. Cells proliferated for 2 days in knockout DMEM/F-12 medium containing 2% (vol/vol) StemPro neural supplement, 2 mM GlutaMAX-I, epidermal growth factor, and basic fibroblast growth factor (20 ng/ml; Thermo Fisher Scientific). Cells were then differentiated into neurons in differentiation medium consisting of 2% (vol/vol) B27 neurobasal medium, 2 mM GlutaMAX-I (Thermo Fisher Scientific), and 200 μM ascorbic acid (Sigma-Aldrich) for a minimum of 14 days.
Virus infection.
The VZV strain pOka (parental Oka) was a generous gift from Michiaki Takahashi (Osaka University, Osaka, Japan). A recombinant pOka-based VZV expressing GFP, VZV-GFP, was reconstituted in MRC-5 cells by transfection of VZVLUCBAC DNA (50; a gift from Hua Zhu, New Jersey Medical School, Rutgers University, Newark, NJ) as described previously (51). Cell-free virus was prepared in MRC-5 cells (52) and used to infect cells in 400 μl of the differentiation medium for 2 h in 12-well plates, followed by the addition of sodium citrate buffer (40 mM sodium citrate, 10 mM potassium chloride, 135 mM sodium chloride [pH 3.2], <30 s) to inactivate noninternalized virus. Cells were washed and cultured in differentiation medium for 4 days before harvesting for flow cytometry, protein, or RNA analyses.
Fluorescence-activated cell sorting.
Following infection, cells were harvested using Accutase (A6964; Sigma-Aldrich) treatment for 10 min at 37°C. Samples were collected and resuspended in 500 μl of cold phosphate-buffered saline (PBS) and transferred into 5 ml polystyrene round bottom flow tube with cell strainer cap (catalog no. 352235; Corning) to prepare single-cell suspensions. Cells were gated based on GFP positivity, and a minimum of 10,000 cells was collected for each condition.
Microarray data analysis.
RNA was isolated following FACS using an RNeasy Plus minikit (Qiagen) and amplified using the whole transcript sense target labeling protocol according to the manufacturer’s directions (Affymetrix). First-strand cDNA was synthesized using 100 ng of total RNA and random oligonucleotides with a T7 promoter sequence as a primer at the 5′ end and using the SuperScript Choice system (Invitrogen). cDNA was hybridized using Affymetrix Human Genome GeneChip array 2.0 for 16 h at 45°C with constant rotation at 60 rpm. An Affymetrix Fluidics Station 450 was then used to wash and stain the chips, removing the nonhybridized target and incubating with a streptavidin-phycoerythrin (PE) conjugate to stain the biotinylated cRNA. Goat IgG blocking reagent and biotinylated anti-streptavidin goat antibody was added, followed by streptavidin-phycoerythrin conjugate to amplify the signal. An Affymetrix G3000 GeneArray scanner was then used for fluorescence detection, and image analysis of each GeneChip was performed using GeneChip operating system software (version 2.0; AGCC) from Affymetrix. Differential gene expression was detected using analysis of variance, and Spotfire Functional Genomic DecisionSite was used for visualization and further functional analysis.
cDNA synthesis and RT-qPCR.
Fifty nanograms of each RNA sample were converted to cDNA using a high-capacity RNA-to-cDNA kit (Applied Biosystems) in a T100 thermal cycler (Bio-Rad). cDNA was subjected to quantitative PCR using iQ-SYBR green master mix (Bio-Rad) containing primers (Integrated DNA Technologies) to genes of interest, along with hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The reaction mix was run on a CFX384 real-time system thermal cycler (Bio-Rad) and analyzed according to the manufacturer’s protocol.
Antibodies.
Rabbit anti-VZV pORF63 antibody was generated and characterized previously (53). Mouse anti-VZV glycoprotein E (gE) antibody (MAB612; EMD Millipore), mouse anti-beta-III tubulin antibody (SDL.3D10; Sigma-Aldrich), mouse anti-beta actin antibody (3700S; Cell Signaling Technology), rabbit anti-nectin-1 antibody (ab66985 and ab229464; Abcam), sheep anti-TGN46 antibody (AbD Serotech), PE-conjugated mouse anti-human nectin-1 antibody (catalog no. 565766; BD Biosciences), and rabbit anti-GAPDH antibody (catalog no. 5174; Cell Signaling Technology) were used as primary antibodies. Alexa Fluor 594/647-conjugated donkey anti-mouse IgG, Alexa Fluor 488/555-conjugated donkey anti-rabbit IgG, and Alexa Fluor 647-conjugated donkey anti-sheep IgG (Thermo Fisher Scientific) were used as secondary antibodies for immunostaining (1:250 to 1:300). Anti-mouse IgG HRP-linked sheep or anti-rabbit IgG HRP-linked donkey antibodies (GE Healthcare Bio-Sciences) were used as secondary antibodies for Western blotting (1:5,000).
Immunostaining and confocal imaging.
Human neurons infected with VZV pOka or VZV-GFP were washed once with PBS and fixed with 4% paraformaldehyde/PBS for 20 min at room temperature. Cells were permeabilized and blocked using 0.25% Triton X-100/5% normal goat serum/PBS for 1 h and then stained with primary antibodies overnight at 4°C (1:300 for mouse anti-VZV gE antibody, 1:100 for rabbit anti-nectin-1 antibodies and sheep anti-TGN46 antibody, and 1:200 for mouse anti-beta-III tubulin antibody). The following day, the samples were incubated in the secondary antibodies in 5% normal goat serum/PBS for 1.5 h at room temperature. Cells were then counterstained with 1 μM DAPI (4′,6′-diamidino-2-phenylindole; Thermo Fisher Scientific) and imaged using an LSM 800/900 confocal microscope (Zeiss).
siRNA treatment.
siRNA targeting human NECTIN1 (catalog no. 4392420) or GAPDH (catalog no. 4390849) was used to knock down each gene in human neurons, along with a nontargeting siRNA (negative control) not known to be complementary to human mRNA (catalog no. 4390843; Silencer Select siRNAs; Thermo Fisher Scientific). Cells were transfected with siRNA (at a final concentration of 50 nM) using Lipofectamine RNAiMAX (catalog no. 56531; Thermo Fisher Scientific) in Opti-MEM reduced serum media (Thermo Fisher Scientific) every 4 days to ensure a sustained reduction in target mRNAs/proteins. Live/Dead assay (catalog no. L3224; Thermo Fisher Scientific) was performed according to the manufacturer’s protocol.
Competition assays.
Cell-free VZV pOka was preincubated with recombinant nectin-1 protein for 15 min at room temperature, followed by infection of human neurons. After infection, the cells were washed, treated with sodium citrate buffer, and cultured for 4 days before protein was collected for Western blot analysis. Three recombinant human nectin-1 proteins were used. sNectin-1, a soluble form of nectin-1 (approximate molecular weight, 35 kDa; Gln31-Thr334 of 517 amino acids), was produced in NS0 mouse myeloma cells (2880-N1; R&D Systems). Nectin-1(FL) contains full-length nectin-1 produced in HEK293 cells (TP311214; OriGene). Nectin-1(Bac) contains Gln31-His346 of nectin-1 and was produced using a baculovirus expression system in Spodoptera frugiperda Sf9 cells.
VZV gE (Shingrix zoster vaccine; approximate molecular weight, 77 kDa) and BSA (Thermo Fischer Scientific; approximate molecular weight, 67 kDa) were added directly to cells after cooling the plate for 5 min on ice, followed by incubation for 1 h; ice was utilized to prevent protein internalization. Subsequently, cell-free VZV pOka was added to the cells, and the infection was performed as described above. Protein lysates were harvested at 4 days postinfection for Western blotting.
Flow cytometry.
Chinese hamster ovary (CHO-K1) cells were transfected with up to 1 μg of human nectin-1 plasmid (25) or TdTomato plasmid (used as a control; constructed in Addgene plasmid 14883 by replacing GFP with TdTomato) using Lipofectamine 2000 (Thermo Fisher Scientific). A single-cell suspension was obtained using Accutase at the indicated time points, and the cells were stained with PE-conjugated mouse anti-human nectin-1 antibody and subjected to flow cytometric analysis. At least 10,000 events were analyzed for each condition.
Western blotting.
Proteins were extracted using radioimmunoprecipitation assay buffer (BP-115; Boston Bio) containing protease and phosphatase inhibitor. BCA assay kit (catalog no. 23227; Thermo Fisher Scientific) was used to determine protein concentrations per the manufacturer’s protocol. Proteins (20 μg/lane) were separated on a 4 to 15% Mini-PROTEAN TGX gel (catalog no. 456-1084; Bio-Rad) and transferred onto a 0.2-μm nitrocellulose membrane using a Trans-Blot Turbo (Bio-Rad), followed by blocking for 30 min with 5% milk in PBS and 0.05% Tween 20. The membrane was probed with primary antibodies overnight at 4°C (1:1,000 for anti-nectin 1 antibody [ab66985], 1:3,000 for mouse anti-VZV gE antibody, 1:5,000 for mouse anti-beta actin antibody and rabbit anti-GAPDH antibody, and 1:30,000 for rabbit anti-VZV pORF63 antibody). After several washes with PBS, the membrane was incubated with the secondary antibody for 45 min at room temperature following multiple washings with PBS. Antibody binding was visualized using SuperSignal West Femto maximum sensitivity substrate (catalog no. 34095; Thermo Fisher Scientific) and captured using a ChemiDoc imaging system (Bio-Rad). Western blot data were analyzed using ImageJ to quantify the intensity of given bands. Intensity data were normalized to controls after the background intensity was subtracted. All experiments were performed independently at least three times, and representative blots are shown in the figures.
ACKNOWLEDGMENTS
This study was supported by NIH R21NS107991 (A.V.) and the intramural research program of the National Institute of Allergy and Infectious Diseases. T.S. received funding from the Tokyo Biochemical Research Foundation and the Ministry of Education, Culture, Sports, Science, and Technology (MEXT; KAKENHI JP21H02741).
This paper is dedicated to the memory of Qingxue Li, who devoted her research career to the study of herpesvirus entry.
We thank Hua Zhu for VZVLUCBAC-harboring E. coli.
Contributor Information
Arun Venkatesan, Email: avenkat2@jhmi.edu.
Felicia Goodrum, University of Arizona.
REFERENCES
- 1.Zerboni L, Sen N, Oliver SL, Arvin AM. 2014. Molecular mechanisms of varicella zoster virus pathogenesis. Nat Rev Microbiol 12:197–210. 10.1038/nrmicro3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Depledge DP, Sadaoka T, Ouwendijk WJD. 2018. Molecular aspects of varicella-zoster virus latency. Viruses 10. 10.3390/v10070349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zerboni L, Arvin A. 2011. Investigation of varicella-zoster virus neurotropism and neurovirulence using SCID mouse-human DRG xenografts. J Neurovirol 17:570–577. 10.1007/s13365-011-0066-x. [DOI] [PubMed] [Google Scholar]
- 4.Gilden D, Nagel MA, Mahalingam R, Mueller NH, Brazeau EA, Pugazhenthi S, Cohrs RJ. 2009. Clinical and molecular aspects of varicella-zoster virus infection. Future Neurol 4:103–117. 10.2217/14796708.4.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilden DH, Kleinschmidt-DeMasters BK, LaGuardia JJ, Mahalingam R, Cohrs RJ. 2000. Neurologic complications of the reactivation of varicella-zoster virus. N Engl J Med 342:635–645. 10.1056/NEJM200003023420906. [DOI] [PubMed] [Google Scholar]
- 6.Gershon AA, Chen J, Davis L, Krinsky C, Cowles R, Reichard R, Gershon M. 2012. Latency of varicella zoster virus in dorsal root, cranial, and enteric ganglia in vaccinated children. Trans Am Clin Climatol Assoc 123:17–35. [PMC free article] [PubMed] [Google Scholar]
- 7.Saylor D, Thakur K, Venkatesan A. 2015. Acute encephalitis in the immunocompromised individual. Curr Opin Infect Dis 28:330–336. 10.1097/QCO.0000000000000175. [DOI] [PubMed] [Google Scholar]
- 8.Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest 108:503–510. 10.1172/JCI13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Z, Gershon MD, Ambron R, Gabel C, Gershon AA. 1995. Infection of cells by varicella zoster virus: inhibition of viral entry by mannose 6-phosphate and heparin. Proc Natl Acad Sci USA 92:3546–3550. 10.1073/pnas.92.8.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacquet A, Haumont M, Chellun D, Massaer M, Tufaro F, Bollen A, Jacobs P. 1998. The varicella zoster virus glycoprotein B (gB) plays a role in virus binding to cell surface heparan sulfate proteoglycans. Virus Res 53:197–207. 10.1016/S0168-1702(97)00149-4. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Ali MA, Wang K, Sayre D, Hamel FG, Fischer ER, Bennett RG, Cohen JI. 2010. Insulin degrading enzyme induces a conformational change in varicella-zoster virus gE, and enhances virus infectivity and stability. PLoS One 5:e11327. 10.1371/journal.pone.0011327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Q, Ali MA, Cohen JI. 2006. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 127:305–316. 10.1016/j.cell.2006.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Krogmann T, Ali MA, Tang WJ, Cohen JI. 2007. The amino terminus of varicella-zoster virus (VZV) glycoprotein E is required for binding to insulin-degrading enzyme, a VZV receptor. J Virol 81:8525–8532. 10.1128/JVI.00286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen JJ, Zhu Z, Gershon AA, Gershon MD. 2004. Mannose 6-phosphate receptor dependence of varicella-zoster virus infection in vitro and in the epidermis during varicella and zoster. Cell 119:915–926. 10.1016/j.cell.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci USA 107:866–871. 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pugazhenthi S, Nair S, Velmurugan K, Liang Q, Mahalingam R, Cohrs RJ, Nagel MA, Gilden D. 2011. Varicella-zoster virus infection of differentiated human neural stem cells. J Virol 85:6678–6686. 10.1128/JVI.00445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markus A, Grigoryan S, Sloutskin A, Yee MB, Zhu H, Yang IH, Thakor NV, Sarid R, Kinchington PR, Goldstein RS. 2011. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J Virol 85:6220–6233. 10.1128/JVI.02396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sloutskin A, Yee MB, Kinchington PR, Goldstein RS. 2014. Varicella-zoster virus and herpes simplex virus 1 can infect and replicate in the same neurons whether co- or superinfected. J Virol 88:5079–5086. 10.1128/JVI.00252-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markus A, Lebenthal-Loinger I, Yang IH, Kinchington PR, Goldstein RS. 2015. An in vitro model of latency and reactivation of varicella-zoster virus in human stem cell-derived neurons. PLoS Pathog 11:e1004885. 10.1371/journal.ppat.1004885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadaoka T, Depledge DP, Rajbhandari L, Venkatesan A, Breuer J, Cohen JI. 2016. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc Natl Acad Sci USA 113:E2403–E2412. 10.1073/pnas.1522575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadaoka T, Schwartz CL, Rajbhandari L, Venkatesan A, Cohen JI. 2018. Human embryonic stem cell-derived neurons are highly permissive for varicella-zoster virus lytic infection. J Virol 92:e01108-17. 10.1128/JVI.01108-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braspenning SE, Sadaoka T, Breuer J, Verjans GMGM, Ouwendijk WJD, Depledge DP. 2020. Decoding the architecture of the varicella-zoster virus transcriptome. mBio 11:e01568-20. 10.1128/mBio.01568-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouwendijk WJD, Depledge DP, Rajbhandari L, Lenac Rovis T, Jonjic S, Breuer J, Venkatesan A, Verjans GMGM, Sadaoka T. 2020. Varicella-zoster virus VLT-ORF63 fusion transcript induces broad viral gene expression during reactivation from neuronal latency. Nat Commun 11:6324. 10.1038/s41467-020-20031-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grigoryan S, Kinchington PR, Yang IH, Selariu A, Zhu H, Yee M, Goldstein RS. 2012. Retrograde axonal transport of VZV: kinetic studies in hESC-derived neurons. J Neurovirol 18:462–470. 10.1007/s13365-012-0124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 26.Richart SM, Simpson SA, Krummenacher C, Whitbeck JC, Pizer LI, Cohen GH, Eisenberg RJ, Wilcox CL. 2003. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J Virol 77:3307–3311. 10.1128/jvi.77.5.3307-3311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simpson SA, Manchak MD, Hager EJ, Krummenacher C, Whitbeck JC, Levin MJ, Freed CR, Wilcox CL, Cohen GH, Eisenberg RJ, Pizer LI. 2005. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J Neurovirol 11:208–218. 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- 28.Kwon H, Bai Q, Baek HJ, Felmet K, Burton EA, Goins WF, Cohen JB, Glorioso JC. 2006. Soluble V domain of Nectin-1/HveC enables entry of herpes simplex virus 1 (HSV-1) into HSV-resistant cells by binding to viral glycoprotein D. J Virol 80:138–148. 10.1128/JVI.80.1.138-148.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 30.Fan Q, Amen M, Harden M, Severini A, Griffiths A, Longnecker R. 2012. Herpes B virus utilizes human nectin-1 but not HVEM or PILRα for cell-cell fusion and virus entry. J Virol 86:4468–4476. 10.1128/JVI.00041-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasaki M, Hasebe R, Makino Y, Suzuki T, Fukushi H, Okamoto M, Matsuda K, Taniyama H, Sawa H, Kimura T. 2011. Equine major histocompatibility complex class I molecules act as entry receptors that bind to equine herpesvirus-1 glycoprotein D. Genes Cells 16:343–357. 10.1111/j.1365-2443.2011.01491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frampton AR, Goins WF, Cohen JB, von Einem J, Osterrieder N, O’Callaghan DJ, Glorioso JC. 2005. Equine herpesvirus 1 utilizes a novel herpesvirus entry receptor. J Virol 79:3169–3173. 10.1128/JVI.79.5.3169-3173.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davison AJ, Scott JE. 1986. The complete DNA sequence of varicella-zoster virus. J Gen Virol 67(Pt 9):1759–1816. 10.1099/0022-1317-67-9-1759. [DOI] [PubMed] [Google Scholar]
- 34.Gray WL, Starnes B, White MW, Mahalingam R. 2001. The DNA sequence of the simian varicella virus genome. Virology 284:123–130. 10.1006/viro.2001.0912. [DOI] [PubMed] [Google Scholar]
- 35.Grose C. 2012. Pangaea and the Out-of-Africa model of varicella-zoster virus evolution and phylogeography. J Virol 86:9558–9565. 10.1128/JVI.00357-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berarducci B, Ikoma M, Stamatis S, Sommer M, Grose C, Arvin AM. 2006. Essential functions of the unique N-terminal region of the varicella-zoster virus glycoprotein E ectodomain in viral replication and in the pathogenesis of skin infection. J Virol 80:9481–9496. 10.1128/JVI.00533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGeoch DJ. 1990. Evolutionary relationships of virion glycoprotein genes in the S regions of alphaherpesvirus genomes. J Gen Virol 71(Pt 10):2361–2367. 10.1099/0022-1317-71-10-2361. [DOI] [PubMed] [Google Scholar]
- 38.Schneider-Schaulies S, Schneider-Schaulies J, Niewiesk S, Ter Meulen V. 2002. Measles virus: immunomodulation and cell tropism as pathogenicity determinants. Med Microbiol Immunol 191:83–87. 10.1007/s00430-002-0121-6. [DOI] [PubMed] [Google Scholar]
- 39.Geleziunas R, Bour S, Wainberg MA. 1994. Cell surface downmodulation of CD4 after infection by HIV-1. FASEB J 8:593–600. 10.1096/fasebj.8.9.8005387. [DOI] [PubMed] [Google Scholar]
- 40.Simmons G. 2013. Filovirus entry. Adv Exp Med Biol 790:83–94. 10.1007/978-1-4614-7651-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stiles KM, Milne RS, Cohen GH, Eisenberg RJ, Krummenacher C. 2008. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology 373:98–111. 10.1016/j.virol.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deschamps T, Dogrammatzis C, Mullick R, Kalamvoki M. 2017. Cbl E3 ligase mediates the removal of Nectin-1 from the surface of herpes simplex virus 1-infected cells. J Virol 91:e00393-17. 10.1128/JVI.00393-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog 7:e1002277. 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol 72:7064–7074. 10.1128/JVI.72.9.7064-7074.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harrison OJ, Vendome J, Brasch J, Jin X, Hong S, Katsamba PS, Ahlsen G, Troyanovsky RB, Troyanovsky SM, Honig B, Shapiro L. 2012. Nectin ectodomain structures reveal a canonical adhesive interface. Nat Struct Mol Biol 19:906–915. 10.1038/nsmb.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakisaka T, Takai Y. 2004. Biology and pathology of nectins and nectin-like molecules. Curr Opin Cell Biol 16:513–521. 10.1016/j.ceb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 47.Takai Y, Ikeda W, Ogita H, Rikitake Y. 2008. The immunoglobulin-like cell adhesion molecule nectin and its associated protein Afadin. Annu Rev Cell Dev Biol 24:309–342. 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- 48.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189. 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 49.Finnen RL, Mizokami KR, Banfield BW, Cai GY, Simpson SA, Pizer LI, Levin MJ. 2006. Postentry events are responsible for restriction of productive varicella-zoster virus infection in Chinese hamster ovary cells. J Virol 80:10325–10334. 10.1128/JVI.00939-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z, Rowe J, Wang W, Sommer M, Arvin A, Moffat J, Zhu H. 2007. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J Virol 81:9024–9033. 10.1128/JVI.02666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sadaoka T, Serada S, Kato J, Hayashi M, Gomi Y, Naka T, Yamanishi K, Mori Y. 2014. Varicella-zoster virus ORF49 functions in the efficient production of progeny virus through its interaction with essential tegument protein ORF44. J Virol 88:188–201. 10.1128/JVI.02245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurapati S, Sadaoka T, Rajbhandari L, Jagdish B, Shukla P, Ali MA, Kim YJ, Lee G, Cohen JI, Venkatesan A. 2017. Role of the JNK pathway in varicella-zoster virus lytic infection and reactivation. J Virol 91:e00640-17. 10.1128/JVI.00640-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Depledge DP, Ouwendijk WJD, Sadaoka T, Braspenning SE, Mori Y, Cohrs RJ, Verjans GMGM, Breuer J. 2018. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat Commun 9:1167. 10.1038/s41467-018-03569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]