

ABSTRACT
Respiratory syncytial virus (RSV) is the main cause of acute respiratory infections in young children and also has a major impact on the elderly and immunocompromised people. In the absence of a vaccine or efficient treatment, a better understanding of RSV interactions with the host antiviral response during infection is needed. Previous studies revealed that cytoplasmic inclusion bodies (IBs), where viral replication and transcription occur, could play a major role in the control of innate immunity during infection by recruiting cellular proteins involved in the host antiviral response. We recently showed that the morphogenesis of IBs relies on a liquid-liquid-phase separation mechanism depending on the interaction between viral nucleoprotein (N) and phosphoprotein (P). These scaffold proteins are expected to play a central role in the recruitment of cellular proteins to IBs. Here, we performed a yeast two-hybrid screen using RSV N protein as bait and identified the cellular protein TAX1BP1 as a potential partner of this viral protein. This interaction was validated by pulldown and immunoprecipitation assays. We showed that TAX1BP1 suppression has only a limited impact on RSV infection in cell cultures. However, RSV replication is decreased in TAX1BP1-deficient (TAX1BP1 knockout [TAX1BP1KO]) mice, whereas the production of inflammatory and antiviral cytokines is enhanced. In vitro infection of wild-type or TAX1BP1KO alveolar macrophages confirmed that the innate immune response to RSV infection is enhanced in the absence of TAX1BP1. Altogether, our results suggest that RSV could hijack TAX1BP1 to restrain the host immune response during infection.
IMPORTANCE Respiratory syncytial virus (RSV), which is the leading cause of lower respiratory tract illness in infants, remains a medical problem in the absence of a vaccine or efficient treatment. This virus is also recognized as a main pathogen in the elderly and immunocompromised people, and the occurrence of coinfections (with other respiratory viruses and bacteria) amplifies the risks of developing respiratory distress. In this context, a better understanding of the pathogenesis associated with viral respiratory infections, which depends on both viral replication and the host immune response, is needed. The present study reveals that the cellular protein TAX1BP1, which interacts with the RSV nucleoprotein N, participates in the control of the innate immune response during RSV infection, suggesting that the N-TAX1BP1 interaction represents a new target for the development of antivirals.
KEYWORDS: RSV, TAX1BP1, nucleoprotein, innate immunity, interferons, lung, yeast two-hybrid screening
INTRODUCTION
Respiratory syncytial virus (RSV) is the main pathogen responsible for acute respiratory infections and bronchiolitis in children (1). Almost all children are infected by the age of 2 years. A systemic multisite study of the cause of infant pneumonia in hospitalized children in Asia and Africa recently revealed that RSV is the main etiological agent of severe pneumonia, accounting for over 30% of infections (2). In the United States, RSV is estimated to be responsible for the hospitalization of 86,000 children per year, with a related cost of $394 million (3). Furthermore, RSV infections in early childhood are recognized to later increase susceptibility to chronic asthma (4, 5). Reinfections occur throughout life, and if healthy adults generally present symptoms of a bad cold, RSV infections are associated with significant morbidity and mortality in the elderly and immunocompromised people (6–9). Indeed, RSV is estimated to cause over 17,000 deaths per year in the United States, 78% of which occur in adults over 65 years of age, and is responsible for 5% of total hospital admissions in the elderly (10). Although RSV has a major impact on human health and the economy, there is still no vaccine available. The development of vaccines has been hampered by the repercussions of a failed vaccine trial using a formalin-inactivated virus in the 1960s, which resulted in an exacerbation of pathology upon infection and led to two deaths (11). The current standard of care consists of prophylactic treatment of at-risk infants with a monoclonal antibody (palivizumab), but its use is limited by its moderate effectiveness and high cost (12).
The pathology associated with RSV infection results from both viral replication and the host’s immune response (13). RSV infection triggers an early immune response mediated by the production of type I interferon (IFN-I), which induces the transcription of IFN-stimulating genes (ISGs) and the production of proinflammatory mediators (14–17). On the other hand, RSV has developed multiple strategies to hijack cellular pathways controlling the IFN-I and NF-κB (nuclear factor kappa B) pathway in order to blunt the host antiviral response (17–19). In particular, the two nonstructural viral proteins NS1 and NS2 are known to suppress IFN-I production and cell signaling during infection (20). Although type I IFNs are major players in viral clearance and are essential to induce an appropriate immune response (21), they could also contribute to RSV pathogenesis with potentially different roles in infants and adults (17, 22–26). Indeed, high levels of IFN-I and inflammatory cytokines usually correlate with severity, as this reflects the inability of the immune response to control the virus. It is thus essential to better characterize the complex interactions between RSV and the host immune response to decipher pathogenesis and design effective treatments.
RSV belongs to the Mononegavirales order and the Pneumoviridae family (27). It is an enveloped virus with a nonsegmented negative-strand RNA genome containing 10 genes that encode 11 proteins. The two surface glycoproteins G and F are involved in the initial steps of infection, i.e., attachment and fusion with the cell membrane. The viral membrane, which also contains the small hydrophobic protein SH, is lined by the matrix protein M that drives virus assembly. The genome is encapsidated by the nucleoprotein N, forming a helical nucleocapsid (28). The polymerase complex composed of the large polymerase (L) and its main cofactor, phosphoprotein P, is associated with this ribonucleoprotein (RNP) complex, which serves as a template for viral transcription and replication (29). The viral transcription factor M2-1 is also present in the viral particle. After cell entry, RSV replicates in the cytoplasm of host cells within virus-induced spherical cytoplasmic granules called inclusion bodies (IBs). These structures are viral factories where all the viral proteins of the polymerase complex concentrate to perform the replication and transcription of the viral genome (30). These structures also play a role in viral escape from the innate immune system by limiting the recognition of viral RNAs by cytoplasmic pattern recognition receptors (PRRs) such as RIG-I (retinoic acid-inducible gene I) and MDA5 (melanoma differentiation-associated gene 5). Once stimulated, these PRRs activate the transcription factors NF-κB and interferon regulatory factors 3 and 7 (IRF3/7) (31). The function of IBs in the modulation of the host innate immune response was further supported by a study showing that MDA5 interacts with the RSV N protein (RSV-N). In addition, MDA5 and the downstream signaling molecule MAVS (mitochondrial antiviral signaling) both colocalize to IBs as soon as 12 h postinfection (p.i.), leading to the downregulation of IFN-β mRNA expression (32). More recently, a study also revealed the sequestration of the NF-κB subunit p65 in RSV IBs (33). It is thus now recognized that the recruitment of cellular proteins into IBs not only participates in viral replication but also is involved in the control of cellular responses (34).
We recently showed that RSV IBs display hallmarks of liquid-liquid-phase separation and that the N and P proteins are at the core of RSV IB biogenesis (35). Their role as scaffold proteins suggests that N and P are directly involved in the partitioning of cellular proteins into IBs. However, their interactions with cellular factors are still poorly characterized. Here, we report the identification of Tax1-binding protein 1 (TAX1BP1) as an interactor of RSV-N. TAX1BP1 was initially identified as a partner of the Tax protein from human T-lymphotropic virus 1 (HTLV-1) (36). Since then, TAX1BP1 was shown to interact with viral proteins from papillomaviruses (37), measles virus (MeV) (38), and mammarenaviruses (39). Among the described activities of TAX1BP1, this protein was involved in the negative regulation of NF-κB and IRF3 signaling by editing the ubiquitylation of its catalytic partner, the A20 protein (40, 41). We thus investigated the role of TAX1BP1 in both RSV replication and the control of the host antiviral response using in vitro and in vivo infection models. Altogether, our results suggest that TAX1BP1 is recruited by RSV to inhibit the host antiviral response.
RESULTS
Identification of TAX1BP1 interaction with the viral nucleoprotein N.
To identify cellular interactors of the RSV-N protein, we first performed a yeast two-hybrid (Y2H) screen. Yeast cells were transformed with a vector encoding the RSV-N protein fused to the GAL4 DNA-binding domain (GAL-BD) in order to use it as bait in the Y2H system. Surprisingly, no yeast clones were obtained, suggesting that RSV-N is toxic. This could be due to the nonspecific RNA-binding properties of N (42). We thus decided to use as a substitute the N protein harboring the K170A/R185A mutations that were previously shown to impair the interaction of N with RNA. This mutant is expressed as a monomeric RNA-free N, named Nmono, which can mimic the natural N0 form (42). When yeast cells were transformed with a vector encoding Nmono fused to the GAL4-BD, growing colonies were obtained on selective medium, as expected. Yeast cells expressing Nmono were then mated with yeast cells transformed with a human spleen cDNA library or a normalized library containing 12,000 human open reading frames (ORFs) fused to the GAL4 activation domain (GAL4-AD) (prey libraries). Yeast diploids were grown on the appropriate medium for the selection of bait-prey interactions, and positive colonies were analyzed by PCR and sequencing to identify human proteins captured by Nmono in the Y2H system. This screen allowed us to identify, among others, the protein TAX1BP1 as an interactor of the Nmono protein (Table 1). For this specific interaction, 40 positive yeast colonies were obtained, and the alignments of the reads from the PCR products showed that the C-terminal part of TAX1BP1 (residues 401 to 789), including half of the central coiled-coil domain involved in TAXBP1 dimerization and the C-terminal zinc fingers (ZFs), is involved in the interaction with N (Fig. 1A). None of the cDNA clones expressed full-length TAX1BP1. This probably reflects the fact that isolated domains often perform better than full-length proteins in the Y2H system as the reconstitution of a functional GAL4 transcription factor is usually facilitated (43).
TABLE 1.
Cellular proteins interacting with RSV Nmono identified by Y2H screeninga
| Gene | Gene ID | No. of hits | Functional annotation (GO biological process) |
|---|---|---|---|
| MAGEA11 | 4110 | 2/67 | Undetermined |
| TAX1BP1 | 8887 | 40/0 | Negative regulation of NF-κB transcription factor activity |
| Negative regulation of type I interferon production | |||
| Negative regulation of apoptotic process | |||
| TMCC3 | 57458 | 11/0 | Undetermined |
| IHO1 | 339834 | 5/0 | Synapsis |
| Regulation of homologous chromosome segregation | |||
| DNA recombination | |||
| Spermatogenesis | |||
| Oogenesis | |||
| Meiotic DNA double-strand break formation | |||
| BEND7 | 222389 | 0/4 | Undetermined |
| CCDC102B | 79839 | 4/0 | Undetermined |
The first and second columns correspond to the canonical gene names and gene identifiers of interacting cellular proteins, respectively. The third column shows the number of positive yeast colonies (hits) obtained for each cellular protein when screening the human spleen cDNA or the human ORFeome library. The fourth column provides information on the roles of the corresponding proteins using the Gene Ontology (GO) annotation (79, 80).
FIG 1.

Identification and validation of the TAX1BP1-N interaction. (A) Multiple-sequence alignment of sequencing reads obtained from the 40 yeast colonies matching TAXBP1. As the cDNA library used in the screen was built by oligo(dT) priming, TAX1BP1 fragments captured in the screen extend from the beginning of the sequencing reads (thick green line) to the end of the TAX1BP1 sequence. The shortest TAX1BP1 fragment captured with Nmono is depicted in blue. Below the alignment, a scheme of the TAX1BP1 structural organization is presented, with numbers indicating residues of TAX1B1P1: the SKIP carboxyl homology domain (SKICH), the LC3-interacting region (LIR), central coiled coils constituting the oligomerization domain, and the two C-terminal zinc fingers (ZF). (B) Validation of the N-TAX1BP1 interaction by GST pulldown with recombinant proteins. GST and GST-TAX1BP1 proteins were purified on glutathione-Sepharose beads and incubated in the presence of recombinant N protein, and interactions were analyzed by SDS-PAGE and Coomassie blue staining. The asterisks indicate the product of degradation of GST-TAX1BP1 corresponding to the deletion of the C-terminal domain. Molecular masses (MW) corresponding to the ladder’s bands are indicated. (C) Western blot analysis of the TAX1BP1-N interaction after an immunoprecipitation assay. Cells were transiently transfected with constructs allowing the expression of the Flag tag alone or the Flag-TAX1BP1 fusion protein with N protein. Immunoprecipitations (IP) were performed with an anti-Flag antibody.
To validate the interaction between TAX1BP1 and the RSV-N protein, we then performed pulldown assays using recombinant proteins. Analysis of purified glutathione S-transferase (GST)-TAX1BP1 by SDS-PAGE and staining with Coomassie blue revealed two main bands of equivalent intensities, with an apparent molecular weight (MW) close to 120 kDa (Fig. 1B). Mass spectrometry analysis of these products allowed us to identify the higher-migrating band as full-length GST-TAX1BP1 (theoretical mass, 112 kDa). The lower band corresponds to GST-TAX1BP1 deleted from the last 77 residues of TAX1BP1 (data not shown), which include the two C-terminal ZFs of the protein (Fig. 1A). This analysis revealed the strong instability of the TAX1BP1 C-terminal domain when expressed alone in bacteria. When coincubated with Sepharose-glutathione beads bound to either GST or GST-TAX1BP1, recombinant N protein was specifically captured in the presence of GST-TAX1BP1 (Fig. 1B). This result confirmed that RSV-N and TAX1BP1 can directly interact. Finally, we investigated the capacity of RSV-N protein to interact with TAX1BP1 in mammalian cells. Cells were cotransfected with plasmids encoding RSV-N and Flag-tagged TAX1BP1 or the Flag tag alone as a control, and an immunoprecipitation assay was performed using an anti-Flag antibody. As shown in Fig. 1C, the RSV-N protein coprecipitated specifically with Flag-TAX1BP1. Altogether, if our results indicate that the RSV-N protein can interact directly with TAX1BP1, further characterization of the domain of TAX1BP1 involved in the interaction would be required to validate the potential role of the oligomerization and ZF domains in N binding.
Downregulation of TAX1BP1 expression has a limited impact on RSV replication in human cells.
TAX1BP1 was recently shown to control the cellular antiviral response during RSV infection (44). We thus determined whether the downregulation of TAX1BP1 expression has an impact on RSV replication in cell culture (45). Human epithelial A549 cells were transfected with control small interfering RNA (siRNA) (siCT) or siRNA targeting TAX1BP1 (siTAX1BP1). After 24 h of culture, cells were infected with recombinant strains of human RSV (HRSV) expressing either the fluorescent protein mCherry (rHRSV-mCherry) or the bioluminescent enzyme firefly luciferase (rHRSV-Luc). After 48 h of culture, mCherry and luciferase expression levels were determined as a proxy for viral infection. Lower signals were observed in siTAX1BP1-treated cells, thus suggesting a role of TAX1BP1 in RSV replication (Fig. 2A). Western blot analysis of cell lysates confirmed that TAX1BP1 expression is suppressed at this time point (Fig. 2B). Somewhat unexpectedly, RSV-N expression in siTAX1BP1-treated cells was similar to that in control cells (Fig. 2B), suggesting that TAX1BP1 has no major impact on viral replication in this cell culture system. We then further assessed the consequence of TAX1BP1 downregulation on viral shedding by quantifying virions in culture supernatants of infected cells. As shown in Fig. 2C, viral titers in the supernatants of siTAX1BP1-treated cells were similar to those in the siCT-treated controls. These results corroborate those of Martin-Vicente et al. (44) showing only a weak reduction of virus titers upon the downregulation of TAX1BP1 expression. Altogether, these results led to the conclusion that although a slight decrease of RSV replication was detected using quantitative approaches based on fluorescent or luminescent reporter proteins, TAX1BP1 does not have a strong impact on RSV replication. This suggested a more indirect effect of TAX1BP1 on RSV replication that could depend on its regulatory role in the innate immune response.
FIG 2.
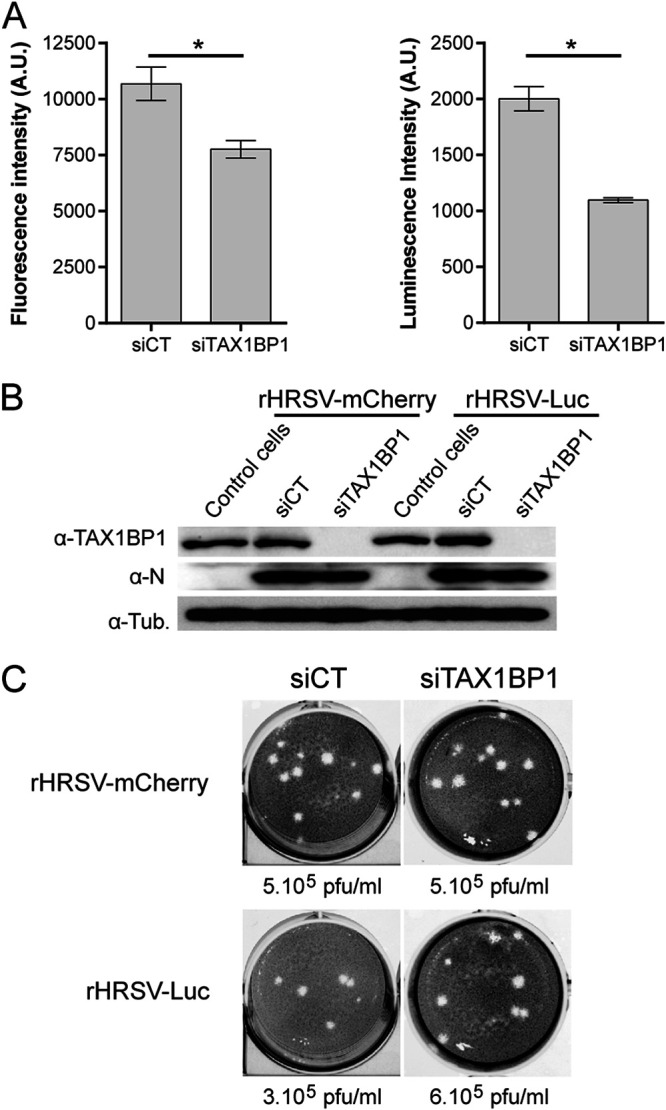
Impact of TAX1BP1 depletion on RSV replication in cells. A549 cells were transfected with control siRNA (siCT) or siRNA targeting TAX1BP1 (siTAX1BP1) and then infected 24 h later with either rHRSV-mCherry or rHRSV-Luc at an MOI of 0.5. (A) RSV replication was quantified at 48 h postinfection by measurement of fluorescence (left) and luminescence (right), expressed in arbitrary units (A.U.), in cell lysates. Data are representative of results from three experiments made in quadruplicates. Data are means ± standard errors of the means (SEM). *, P < 0.05. (B) Western blot analysis of TAX1BP1 silencing and RSV-N expression in cells infected with either rHRSV-mCherry or rHRSV-Luc, at 48 h postinfection. (C) Titration of virions released into the culture media of cells treated with siCT (left) and siTAX1BP1 (right) and infected with rHRSV-mCherry (top) or rHRSV-Luc (bottom). Calculated viral titers in PFU per milliliter are indicated.
Depletion of TAX1BP1 impairs RSV replication in mice.
Given the complexity of the immune response triggered upon RSV infection, we assessed the impact of TAX1BP1 depletion directly in vivo using TAX1BP1-deficient (TAX1BP1 knockout [TAX1BP1KO]) mice. These mice being generated in 129 strain mice (40), we first investigated the kinetics of rHRSV-Luc replication in this genetic background. Although luminescence was shown to be correlated with viral replication by direct measurement in live BALB/c mice using the in vitro imaging system (IVIS) (45, 46), the skin pigmentation of 129 mice impaired luminescence detection. We thus decided to monitor viral replication in infected animals by measuring the luciferase activity in lung homogenates. Wild-type (WT) 129 mice were either instilled with the mock control (mock) consisting of a HEp-2 cell culture supernatant or infected with 1.87 × 105 PFU of rHRSV-Luc via intranasal (i.n.) inoculation. Viral replication was quantified in the first 4 days postinfection (dpi). The bioluminescence in lung homogenates was detected at day 1 p.i., and viral replication in the lungs increased from day 2 to day 4 p.i. (Fig. 3A, left). In parallel, the expression of the N-RSV gene in the lung lysates was quantified by reverse transcription-quantitative PCR (RT-qPCR) (Fig. 3A, right). The data showed that N-RSV mRNA could be detected from day 2 p.i. and that the peak of infection was reached at days 3 and 4 p.i. These results revealed a correlation between bioluminescence intensity and N-RSV mRNA expression, in line with previous reports (45), with clear detection of RSV replication at days 3 and 4 p.i. Of note, this kinetics of replication is similar to the one described in BALB/c mice, a reference mouse strain used to study RSV infection (45, 46).
FIG 3.

TAX1BP1-deficient mice infected with RSV present reduced virus replication in the lungs. (A) Kinetics of RSV infection in 129 mice. Wild-type (WT) strain 129 mice were infected with the HEp-2 supernatant (mock) (n = 1) or rHRSV-Luc (n = 4). (Left) Luciferase activity associated with viral replication was measured at different days postinfection (dpi) in lung lysates by quantification of photon emission (radiance in photons per second per square centimeter per steradian) and normalized to the amount of lysed tissue. (Right) In parallel, N-RSV gene expression was measured in the lung lysates by RT-qPCR and calculated by the formula 2−ΔCT, where ΔCT = CTN-RSV − CTHPRT. Data are means ± SEM. *, P < 0.05; ns, not significant. (B) WT or TAX1BP1KO 129 mice were infected with the HEp-2 supernatant (mock) or rHRSV-Luc. Luciferase activity associated with viral replication was measured at 2 or 4 dpi (left and right, respectively) in lung lysates by quantification of photon emission (radiance in photons per second per square centimeter per steradian) and normalized to the amount of lysed tissue. Data are means ± SEM from two independent experiments with 7 RSV-infected WT mice and 11 RSV-infected TAX1BP1KO mice. (C) Quantification of N-RSV gene expression at 4 dpi in RSV-infected WT or TAX1BP1KO mice (n = 4). N-RSV gene expression was measured in the lung lysates by RT-qPCR and calculated by the formula 2−ΔCT, where ΔCT = CTN-RSV − CTHPRT (right). Data are means ± SEM. *, P < 0.05.
Based on these results, we decided to compare rHRSV-Luc replication in WT and TAX1BP1KO 129 mice. We chose to quantify bioluminescence in the lungs of mock-treated and HRSV-infected animals at day 2 and day 4 p.i. in order to compare viral replication at an early time point and at the peak of infection. Our results showed a strong reduction in RSV replication in TAX1BP1KO mice compared to WT mice, at both days 2 and 4 p.i. (Fig. 3B). In order to confirm these results, viral replication in the lungs of infected mice at day 4 p.i. was assessed by quantification of N-RSV gene expression in the lungs by RT-qPCR and virion production in the lungs using a plaque assay approach. As shown in Fig. 3C, the level of N-RSV mRNA was significantly lower in TAX1BP1KO mice than in wild-type mice. Once again, these results reveal that in vivo quantification of viral replication by bioluminescence correlates with the viral load, as previously reported (45). However, we did not manage to recover virus from lung lysates to quantify virion production. Altogether, our results revealed a supportive role of TAX1BP1 in RSV replication in vivo.
Depletion of TAX1BP1 favors antiviral and inflammatory responses during RSV infection.
As mentioned above, among the various functions of TAX1BP1, this cellular protein acts as a cofactor of the A20 protein, which is a negative regulator of the NF-κB and IRF3/7 pathways that are involved in inflammatory and antiviral responses, respectively. In the mouse model of RSV infection, the induction of inflammatory cytokines and IFN-I in the first hours after exposure to the virus is well documented (47–50). We thus assessed if the inhibition of RSV replication upon TAX1BP1 depletion could be associated with a modulation of the antiviral and inflammatory responses in the lungs of infected mice at early time points postinfection. Mice were mock treated or infected with 1.87 × 105 PFU of rHRSV-Luc, and at day 1 p.i., the expression levels of IFN-I (IFN-α and IFN-β) and the inflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) were determined from lung lysates of WT or TAX1BP1KO mice. As shown in Fig. 4, RSV infection of WT mice induced the production of IFN-α and IFN-β in all the animals. Of note, one of the infected WT mice that presented a strong induction of IFN-α and IFN-β also displayed induction of IL-6 and TNF-α. TAX1BP1KO mice were infected in parallel, and higher levels of IFN-α and TNF-α were detected in the lungs of TAX1BP1KO mice than in WT mice (Fig. 4A and D). On the contrary, IFN-β induction by RSV was unchanged (Fig. 4B). Although IL-6 was induced in only one of the infected WT animals, this cytokine was induced in all infected TAX1BP1KO mice (Fig. 4C). However, IL-6 expression levels were not statistically significant when comparing TAX1BP1KO to WT mice. Of note, all groups of animals showed comparable levels of RSV infection at this early time point (1 dpi) as assessed by bioluminescence quantification in the lung homogenates (not shown). Because the measurements were performed in whole-lung lysates, the quantified cytokines and chemokines are probably produced by several cell populations (i.e., both epithelial and immune cells). We thus decided to specifically focus on alveolar macrophages (AMs), which are major actors in the antiviral response to RSV (48). AMs were isolated from WT and TAX1BP1KO mice after repeated bronchoalveolar lavages (BALs) and cultured for 24 h before incubation for another 24 h in the presence of either rHRSV-mCherry or UV-inactivated rHRSV-mCherry (multiplicity of infection [MOI] = 5). Culture supernatants were collected, and IFN-α, IFN-β, IL-6, and TNF-α were quantified by immunoassays. Strong induction of both antiviral (Fig. 5A and B) and inflammatory (Fig. 5C and D) cytokines was detected in the supernatant of AMs exposed to RSV, whereas much weaker induction of these molecules was observed for AMs exposed to inactivated RSV, thus validating the efficient infection of AMs. Of note, although AMs can be infected by RSV, these cells do not productively replicate the virus (51). Most interestingly, the production of IFN-α, IFN-β, IL-6, and TNF-α was enhanced in AMs derived from TAX1BP1KO mice compared to AMs isolated from WT mice (Fig. 5). Altogether, these results demonstrate that TAX1BP1 is a key factor involved in the inhibition of the antiviral and inflammatory responses in the lungs of RSV-infected animals and in isolated AMs.
FIG 4.

Study of antiviral/inflammatory immune responses in the lungs of infected TAX1BP1KO mice. WT or TAX1BP1KO mice were infected with the HEp-2 supernatant (mock) or rHRSV-Luc. (A and B) The levels of production of IFN-α and IFN-β were measured at 24 h postinfection in lung lysates using a ProcartaPlex immunoassay. (C and D) The levels of production of IL-6 and TNF-α were measured at 24 h postinfection in lung lysates using a MilliPlex Map immunoassay. The concentrations were normalized to lung weights. Data are means ± SEM (*, P < 0.05; **, P < 0.01) and are representative of results from two independent experiments with 5 to 6 mice per group.
FIG 5.

Deletion of TAX1BP1 enhances the production of type I IFN and inflammatory cytokines in AMs following RSV infection. AMs from WT or TAX1BP1KO mice were either not infected (mock) (black triangles) or exposed to rHRSV-mCherry (RSV) (inverted black triangles) or UV-inactivated rHRSV-mCherry (UV-RSV) (white circles) at an MOI of 5 for 2 h. (A and B) The levels of production of IFN-α and IFN-β were measured at 24 h postinfection in the supernatants using a ProcartaPlex immunoassay. (C and D) The levels of production of IL-6 and TNF-α were measured at 24 h postinfection in supernatants using a MilliPlex Map immunoassay. Data are means ± SEM from two independent experiments. ***, P < 0.001.
DISCUSSION
Previous studies using microarray and proteomic approaches have provided key information on RSV-host interactions (52, 53), but the interactome of RSV proteins remains poorly characterized. Due to their pivotal role during virus entry, replication, and assembly, it is expected that components of the viral polymerase complex, and especially the N protein, are involved in various interactions with cellular factors. The objective of this study was to find new cellular partners of RSV-N by performing a yeast two-hybrid screen. Using this approach, we captured 6 cellular proteins using RSV-N as bait, among which TAX1BP1 was overrepresented. We thus focused on TAX1BP1 as TAX1BP1 depletion has recently been shown to favor the innate immune response to RSV infection and to impair viral replication in cell culture (44). In addition, TAX1BP1 is already known to interact with different viral proteins, including the N protein of measles virus, which belongs to the Mononegavirales order (36–39), like RSV, suggesting that this protein is often hijacked by viruses. TAX1BP1 is a homodimer of about 90 kDa and is organized into three main structural domains. The N-terminal SKIP carboxyl homology (SKICH) domain (54) was recently shown to interact with the adaptor protein NAP1, allowing the recruitment of the TANK-binding kinase 1 (TBK1), which is involved in selective autophagy of invading pathogens and damaged mitochondria but is also critical for the induction of IFN-I by RIG-I, MDA5, and STING (55–59). It is followed by an LC3-interacting region (LIR) that can bind different LC3/GABARAP orthologs (60) involved in the recruitment of TAX1BP1 to autophagosomes. The central part of TAX1BP1 exhibits coiled coils forming the oligomerization domain that interacts with TRAF6 protein (61) and is followed by two C-terminal zinc fingers (UBZ1 and UBZ2) (62). These zinc fingers were shown to interact with ubiquitinylated proteins, myosin VI, and the A20 protein (63–65).
Here, the alignment of the PCR reads obtained from the 40 yeast clones that expressed TAX1BP1 in the two-hybrid screen revealed that the C-terminal part of this protein is involved in the interaction with RSV-N. Based on our results, it is expected that the site of TAX1BP1 binding to RSV-N is located within the oligomerization domain and/or the C-terminal zinc finger domains. The N-TAX1BP1 interaction was validated first by pulldown using recombinant TAX1BP1 and RSV-N proteins and then by immunoprecipitation when coexpressing the two proteins in human cells. Noteworthy, we managed to purify the recombinant TAX1BP1 protein to validate the direct interaction with the RSV-N protein. However, the purification of this protein was challenging as TAX1BP1 tends to be cleaved at its C terminus, and this hampered affinity studies with RSV-N by biophysical approaches. To gain structural and functional insights into this interaction that could represent a new therapeutic target, precise characterization of domains of TAXBP1 binding to RSV-N is required. The structure of the C-terminal UBZ domain of TAX1BP1 either alone or in complex with myosin VI has already been resolved (62, 65). The crystal structure of RSV nucleocapsid-like structures consisting of rings containing 10 N protomers and RNA of 70 nucleotides has been determined (66). Recently, a recombinant RSV N0-P complex has also been characterized (67). The reconstitution of a recombinant complex of RSV-N (monomeric or oligomeric form) bound to the C-terminal fragment of TAX1BP1 could thus provide key structural information on this interaction. Finally, given the strong homology between the N proteins of RSV and human metapneumovirus (hMPV), another pneumovirus also responsible for acute respiratory infections, the potential interaction between hMPV-N and TAX1BP1 and its functional relevance during infection should also be investigated.
We then investigated the potential role of TAX1BP1 in RSV infection. TAX1BP1 suppression showed a limited or no impact on viral protein expression in cell culture, and the production of new viral particles was unaffected. However, a model of RSV-infected TAX1BP1KO mice revealed the critical role of TAX1BP1 in RSV infection in vivo, with the depletion of TAX1BP1 leading to a nearly 3-fold decrease in viral replication in the lungs of infected mice. We also showed that RSV-infected TAX1BP1KO mice present higher levels of IFN-α and TNF-α in the lungs than WT mice at day 1 p.i. Besides, RSV-infected AMs isolated from TAX1BP1KO mice produced higher levels of IFN-I (IFN-α and -β) and inflammatory cytokines (IL-6 and TNF-α) than those isolated from WT mice.
These results reveal that TAX1BP1 participates in the attenuation of the host antiviral and inflammatory responses during RSV infection in vivo and especially in AMs. Altogether, these results suggest that TAXBP1 recruitment by RSV-N indirectly promotes RSV growth by inhibiting the innate immune response. It is noteworthy that this interaction could compete with the interaction of TAX1BP1 with another partner. Overall, this conclusion is consistent with a recent study by Martin-Vicente et al. (44), but significant differences should be highlighted. Indeed, those authors found that the production of infectious RSV particles in A549 cells decreases when silencing TAX1BP1 or the interacting cofactors A20, ABIN1, and ITCH. In our hands, the effect of TAX1BP1 silencing on RSV infection was striking only in vivo. At this point, we do not have an explanation for this discrepancy as we both used the same in vitro model of A549-infected cells. Besides, they found in their study that A549 cells silenced for TAX1BP1 express higher levels of ISG15, IL-6, and IL-8 upon RSV infection, but IFN-β and TNF-α expression levels were not significantly affected. On the contrary, we found that TAX1BP1-deficient AMs express higher levels of TNF-α, IL-6, IFN-β, and IFN-α when infected by RSV. The use of distinct cellular models and TAX1BP1 depletion methods could account for these differences. Indeed, TAX1BP1 is directly involved in the regulation of innate immune pathways but is also an adaptor for autophagy (63), which is required for the induction of an optimal antiviral response in RSV-infected macrophages (68). Thus, the role of TAX1BP1 in the regulation of the innate immune response induced upon RSV infection could vary between epithelial and immune cells depending on the relative contribution of autophagy to the activation of the innate immune response. Finally, it should be noted that TAX1BP1 has been previously described to regulate B cell differentiation (69). It would thus be interesting to study whether TAX1BP1 could also be involved in acquired immune responses in the context of RSV infection in vivo and in particular the production of antibodies.
As TAX1BP1 works as an adaptor protein in different processes, it is essential to characterize TAX1BP1 partners in different cell lines when infected by RSV. During our study, we investigated the cellular localization of TAX1BP1 in the context of viral infection or overexpression of N in order to determine in particular if TAX1BP1 could be recruited to IBs, as previously shown for MDA5 and MAVS (32), or if TAX1BP1 could recruit RSV-N to specific cellular compartments. However, we were not able to clearly detect TAX1BP1 by immunolabeling using commercial antibodies. Furthermore, upon the overexpression of Flag- or green fluorescent protein (GFP)-tagged TAX1BP1 in cells, TAX1BP1 was shown to concentrate into cytoplasmic granules and to induce cell death, thus precluding further analysis (data not shown).
In conclusion, we have shown that TAX1BP1 suppresses the innate immune response to RSV in vivo and in AMs. The results also suggest that RSV hijacks this mechanism through a direct physical interaction with RSV-N. Although the precise role of TAX1BP1 in RSV infection needs to be further characterized, this interaction helps in understanding the pathology associated with infection and represents a new target for antiviral approaches.
MATERIALS AND METHODS
Plasmids and siRNA.
The plasmid pFlag-TAX1BP1 encoding TAX1BP1 fused with an N-terminal Flag tag was kindly provided by C. Journo (ENS, Lyon, France). The plasmid pFlag was obtained by inserting a stop codon into the pFlag-TAX1BP1 vector using the QuikChange site-directed mutagenesis kit (Stratagene). The previously described plasmid p-N (70) was used for cell transfection and immunoprecipitation assays.
The pGEX-4T-3 vector was used to produce recombinant glutathione S-transferase (GST) protein. The pGEX-TAX1BP1 plasmid expressing GST fused with the N terminus of TAX1BP1 was obtained by cloning the TAX1BP1 sequence between the BamHI and XhoI sites of the pGEX-4T-3 plasmid. For the purification of recombinant N protein, the pET-N and pGEX-PCT (P C-terminal domain) plasmids described previously (42) were used. For yeast two-hybrid screening, the DNA sequence encoding Nmono (monomeric N mutant K170A/R185A) was cloned by in vitro recombination (Gateway technology; Invitrogen) from pDONR207 into the yeast two-hybrid vector pPC97-GW for expression as a fusion downstream of the GAL4 DNA-binding domain (GAL4-BD). The control siRNA and a pool of TAX1BP1 siRNAs (Ambion) were used for TAX1BP1 silencing experiments.
Antibodies.
The following primary antibodies were used for immunoprecipitation assays and/or immunoblotting: mouse anti-Flag and mouse anti-Flag-horseradish peroxidase (HRP) antibodies (Sigma), rabbit anti-N antiserum (71), and mouse monoclonal anti-β-tubulin antibody (Sigma). Secondary antibodies directed against mouse and rabbit IgG coupled to HRP (PARIS) were used for immunoblotting.
Cell lines.
BHK-21 cells (clone BSRT7/5), hamster kidney cells constitutively expressing the T7 RNA polymerase (72), HEp-2 cells (ATCC CCL-23), and human lung carcinoma epithelial A549 cells were grown in Dulbecco modified essential medium (DMEM; Lonza) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, and 1% penicillin-streptomycin. The transformed human bronchial epithelial cell line BEAS-2B (ATCC) was maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 1% l-glutamine, and 1% penicillin-streptomycin.
Viruses.
The recombinant RSVs rHRSV-mCherry and rHRSV-Luc corresponding to the RSV Long strain expressing either the mCherry or the luciferase protein were amplified in HEp-2 cells and titrated using a plaque assay procedure as previously described (45). Briefly, for titration, cells were infected with serial 10-fold dilutions of the viral supernatant in complete minimum essential medium (MEM). The overlay was prepared with Avicel RC581 microcrystalline cellulose (FMC Biopolymer) at a final concentration of 0.6% in complete MEM containing 1% fetal calf serum. After 6 days at 37°C with 5% CO2, plaques were revealed by staining of the cell layers with a solution containing 0.5% crystal violet and 20% ethanol, and the number of PFU per well was counted.
Yeast two-hybrid screening.
Yeast two-hybrid screens were performed according to the protocol described previously by Vidalain et al. (73). AH109 yeast cells (Clontech, TaKaRa, Mountain View, CA, USA) were transformed with pGAL4-BD-Nmono using a standard lithium-acetate protocol. Screens were performed on a synthetic medium lacking histidine (−His) and supplemented with 3-amino-1,2,4-triazole (3-AT) at 10 mM. A mating strategy was used to screen two different prey libraries with distinct characteristics: a human spleen cDNA library and a normalized library containing 12,000 human ORFs (74). All libraries were established in the yeast two-hybrid expression plasmid pPC86 to express prey proteins as fusions downstream of the GAL4 transactivation domain (GAL4-AD). After 6 days of culture, colonies were picked, replica plated, and incubated over 3 weeks on selective medium to eliminate potential contamination with false positives. Prey proteins from selected yeast colonies were identified by PCR amplification using primers that hybridize within the pPC86 regions flanking the cDNA inserts. PCR products were sequenced, and cellular interactors were identified by multiparallel BLAST analysis.
Expression and purification of recombinant proteins.
Escherichia coli BL21(DE3) bacteria (Novagen, Madison, WI) transformed with the pGEX-4T-3 and pGEX-TAX1BP1 plasmids were grown at 37°C for 2 to 3 h in 200 ml of Luria-Bertani (LB) medium containing 100 μg/ml of ampicillin until the optical density at 600 nm (OD600) reached 0.6. Protein expression was then induced by the addition of 1 mM isopropyl-β-d-thiogalactoside (IPTG) in the presence of 50 mM ZnSO4 for 4 h at 37°C before harvesting by centrifugation. Expression and purification of the recombinant N protein were performed as previously described (66, 75). Briefly, BL21 bacteria cotransformed with pET-N and pGEX-PCT plasmids were grown in LB medium containing kanamycin (50 μg/ml) and ampicillin for 8 h at 37°C. Next, the same volume of fresh LB medium was added, and protein expression was induced by the addition of IPTG at 80 μg/ml to the culture. The bacteria were incubated for 15 h at 28°C and then harvested by centrifugation. For GST fusion protein purification, bacterial pellets were resuspended in lysis buffer (50 mM Tris-HCl [pH 7.8], 60 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol [DTT], 0.2% Triton X-100, 1 mg/ml lysozyme) supplemented with a complete protease inhibitor cocktail (Roche, Mannheim, Germany), incubated for 1 h on ice, sonicated, and centrifuged at 4°C for 30 min at 10,000 × g. Glutathione-Sepharose 4B beads (GE Healthcare, Uppsala, Sweden) were added to the clarified supernatants, and the mixture was incubated at 4°C for 15 h. The beads were then washed two times in lysis buffer and three times in 1× phosphate-buffered saline (PBS) and then stored at 4°C in an equal volume of PBS. To isolate the recombinant N protein, beads containing the bound GST-PCT+N complex were incubated with thrombin (Novagen) for 16 h at 20°C. Purified recombinant N proteins were loaded onto a Superdex 200 16/30 column (GE Healthcare) and eluted in a solution containing 20 mM Tris-HCl (pH 8.5) and 150 mM NaCl.
(i) Pulldown assays.
Purified recombinant N protein was incubated in the presence of GST or the GST-TAX1BP1 fusion protein fixed on beads in a final volume of 100 μl in buffer containing 20 mM Tris (pH 8.5) and 150 mM NaCl. After 1 h under agitation at 4°C, the beads were extensively washed with 20 mM Tris (pH 8.5)–150 mM NaCl, boiled in 30 μl Laemmli buffer, and analyzed by SDS-PAGE and Coomassie blue staining.
(ii) Coimmunoprecipitation assays.
BSRT7 cells were cotransfected with pFlag or pFlag-TAX1BP1 and pN for 36 h. Transfected cells were then lysed for 30 min at 4°C in ice-cold lysis buffer (50 mM Tris-HCl [pH 7.4], 2 mM EDTA, 150 mM NaCl, 0.5% NP-40) with a complete protease inhibitor cocktail (Roche), and coimmunoprecipitation experiments were performed on cytosolic extracts. Cell lysates were incubated for 4 h at 4°C with an anti-Flag antibody coupled to agarose beads (Euromedex). The beads were then washed 3 times with lysis buffer and 1 time with PBS, and proteins were eluted in Laemmli buffer at 95°C for 5 min and then subjected to SDS-PAGE and immunoblotting.
siRNA transfection and infection.
Freshly passaged A549 cells were transfected with the indicated siRNAs at a final concentration of 10 nM by reverse transfection into 48-well plates using Lipofectamine RNAiMAX (Thermo Fisher) according to the manufacturer’s instructions. Briefly, a mixture containing Opti-MEM (Invitrogen), Lipofectamine RNAiMAX, and siRNA was incubated for 5 min at room temperature before being deposited at the bottom of the wells. The cells in DMEM without antibiotics were then added dropwise before incubation at 37°C with 5% CO2. After 24 h of transfection in the presence of siRNA, the medium was removed, and the cells were infected with recombinant rHRSV-mCherry or rHRSV-Luc at an MOI of 0.5 in DMEM without phenol red and without FCS for 2 h at 37°C. The medium was then replaced by DMEM supplemented with 2% SVF, and the cells were incubated for 48 h at 37°C. For cells infected with rHRSV-mCherry, quantification of replication was performed by measuring the mCherry fluorescence (excitation at 580 nm and emission at 620 nm) using a Tecan Infinite M200 Pro luminometer. For HRSV-Luc replication quantification, cells were lysed in luciferase lysis buffer (30 mM Tris [pH 7.9], 10 mM MgCl2, 1 mM DTT, 1% Triton X-100, and 15% glycerol). After the addition of luciferase assay reagent (Promega), luminescence was measured using a Tecan Infinite M200 Pro luminometer. Noninfected A549 cells were used as standards for fluorescence or luminescence background levels. Each experiment was performed in triplicates and repeated at least three times. For each experiment, cells treated under the same conditions were lysed, and protein expression was analyzed by Western blotting.
RSV infection of mice and luciferase measurement.
TAX1BP1-deficient (TAX1BP1KO) 129 mice were created by gene targeting, as previously described (40). TAX1BP1KO mice and wild-type 129 cohoused control animals were bred and housed under specific-pathogen-free (SPF) conditions in our animal facilities (IERP, INRAE, Jouy-en-Josas, France). Wild-type (WT) and TAX1BP1KO female and male mice at 8 weeks of age (n = 11 per group) were anesthetized with a mixture of ketamine and xylazine (1 and 0.2 mg per mouse, respectively) and infected by intranasal administration of 80 μl of recombinant RSV expressing luciferase (rHRSV-Luc) (2.34 × 106 PFU/ml) (45, 76, 77) or cell culture medium as a mock infection control. Mice were then sacrificed at different time points by intraperitoneal (i.p.) injection of pentobarbital, and lungs were frozen.
Viral N-RNA gene expression by RT-qPCR.
Frozen lungs were homogenized in NucleoSpin RNA XS kit lysis buffer (Macherey-Nagel) with a Precellys 24 bead grinder homogenizer (Bertin Technologies, St Quentin en Yvelines, France). Total RNA was extracted from lungs or infected cells using a NucleoSpin RNA kit (Macherey-Nagel) and reverse transcribed using the iScript reverse transcription supermix for RT-qPCR kit (Bio-Rad) according to the manufacturer’s instructions. The primers (Sigma-Aldrich) used are listed below. The qPCRs were performed with MasterCycler RealPlex (Eppendorf) and SYBR green PCR master mix (Eurogenetec), and data were analyzed with RealPlex software (Eppendorf) to determine the cycle threshold (CT) values. Results were determined with the formula 2−ΔCT, where ΔCT = CTgene − CTHPRT. The primers (Sigma-Aldrich) used are as follows: forward primer 5′-CAGGCCAGACTTTGTTGGAT-3′ and reverse primer 5′-TTGCGCTCATCTTAGGCTTT-3′ for HPRT (hypoxanthine-guanine phosphoribosyltransferase) and forward primer 5′-AGATCAACTTCTGTCATCCAGCAA-3′ and reverse primer 5′-TTCTGCACATCATAATTAGGAGTATCAAT-3′ for N-RSV.
Luciferase expression in lung lysates.
Frozen lungs were weighed and then homogenized in 300 μl of passive lysis buffer (PLB) (1 mM Tris [pH 7.9], 1 mM MgCl2, 1% Triton X-100, 2% glycerol, 1 mM DTT) with a Precellys 24 bead grinder homogenizer (Bertin Technologies, St Quentin en Yvelines, France) and a cycle of two 15-s pulses at 4 m/s. Lung homogenates were clarified by centrifugation for 5 min at 2,000 × g and distributed onto microplates (50 μl). Next, 50 μl of luciferase assay reagent (Promega) was added to each well. The detection of firefly luciferase activity was measured by photon emission using an in vivo imaging system (IVIS-200; Xenogen, Advanced Molecular Vision) and Live Imaging software (version 4.0; Caliper Life Sciences). Data were expressed in radiance (photons per second per square centimeter per steradian) and normalized to lung weights.
RSV infection of AMs.
A cannula was inserted into the tracheas of mice, and repeated bronchoalveolar lavages (BALs) were made with PBS. AMs were isolated after centrifugation of the BAL fluids of 5 mice per group and pooled, and 1 × 105 AMs were plated into 96-well cell culture plates in RPMI 1640 supplemented with 2 mM l-glutamine, 5% FCS, and antibiotics for 24 h to allow adhesion, as previously described (78). AMs were then exposed to rHRSV-mCherry or UV-inactivated rHRSV-mCherry (the same batch exposed to UV for 20 min) at an MOI of 5 or a HEp-2 cell culture supernatant (mock). After 24 h, the supernatants were collected and frozen for cytokine quantification.
Cytokine quantification.
IFN-α and IFN-β or IL-6 and TNF-α were measured in the supernatants of AMs or lung lysates using an IFN alpha/IFN beta 2-plex mouse ProcartaPlex immunoassay (eBioscience) or a MilliPlex Map mouse assay (Merck), respectively. Data were acquired using a Magpix multiplex system (Merck) in order to determine the mean fluorescence intensities (MFIs), and the results were analyzed using Bio-Plex Manager software. The concentrations were normalized to lung weights.
Ethics statement.
The in vivo work of this study was carried out in accordance with INRAE guidelines in compliance with European animal welfare regulation. The protocols were approved by the Animal Care and Use Committee at the Centre de Recherche de Jouy-en-Josas (COMETHEA) under relevant institutional authorization (Ministère de l’Éducation Nationale, de l’Enseignement Supérieur et de la Recherche) under authorization number 2015060414241349_v1 (APAFIS number 600). All experimental procedures were performed in a biosafety level 2 facility.
Statistical analysis.
A nonparametric Mann-Whitney test (comparison of two groups) (n ≥ 4) was used to compare unpaired values (GraphPad Prism software). Significance is represented in the figures (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
ACKNOWLEDGMENTS
We thank Sabine Riffault (INRAE, Jouy-en-Josas, France) for helpful discussion and critical reading of the manuscript. We are grateful to Chloé Journo (ENS-Lyon, France) for providing the pFlag-TAX1BP1 plasmid, Céline Urien (INRAE, Jouy-en-Josas) for mouse genotyping, Fortune Bidossessi (INRAE, Jouy-en-Josas) for qPCR, and the Infectiology of Fishes and Rodent Facility (IERP, INRAE [https://doi.org/10.15454/1.5572427140471238E12]) for animal facilities and birth management.
We thank the Emerg’in platform for access to the IVIS-200 instrument, which was financed by the Région Ile de France (SESAME and DIM-OneHealth), and the Plateforme d’Analyse Protéomique de Paris Sud-Ouest (PAPPSO, INRAE) for mass spectrometry analysis. C.D. and Q.M. were recipients of Ph.D. and postdoctoral fellowships of the Région Ile de France (DIM-Malinf and DIM-OneHealth, respectively), A.P.D.O. was the recipient of a postdoctoral fellowship (CAPES-Brazil 14809-13-3/CAPES-COFECUB 769-13). This study was supported in part by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, to H.I. and with the financial support of the French Agence Nationale de la Recherche, specific program ANR Blanc 2013 Respisyncycell (ANR-13-IVS3-0007 and FAPESP-Brazil/ANR-Blanc-Respisyncycell 2013/50299-2).
We declare that we have no conflicts of interest with the contents of this article.
D.D., A.M.V., J.-F.E., P.-O.V., and M.G. designed experiments. A.P.D.O., S.M., L.G., J.F., E.B., and M.G. performed molecular and cellular assays. S.M., C.D., V.P., Q.M., E.B., H.I., and D.D. performed mouse experiments, sample treatments, and analysis of in vivo experiments. A.P.D.O., F.T., and P.-O.V. performed two-hybrid screens. M.G., D.D., P.-O.V., and J.-F.E. wrote the paper. M.G. edited the manuscript. All authors commented on the manuscript.
Contributor Information
Delphyne Descamps, Email: delphyne.descamps@inrae.fr.
Marie Galloux, Email: marie.galloux@inrae.fr.
Stacey Schultz-Cherry, St. Jude Children’s Research Hospital.
REFERENCES
- 1.Shi T, McAllister DA, O’Brien KL, Simoes EAF, Madhi SA, Gessner BD, Polack FP, Balsells E, Acacio S, Aguayo C, Alassani I, Ali A, Antonio M, Awasthi S, Awori JO, Azziz-Baumgartner E, Baggett HC, Baillie VL, Balmaseda A, Barahona A, Basnet S, Bassat Q, Basualdo W, Bigogo G, Bont L, Breiman RF, Brooks WA, Broor S, Bruce N, Bruden D, Buchy P, Campbell S, Carosone-Link P, Chadha M, Chipeta J, Chou M, Clara W, Cohen C, de Cuellar E, Dang D-A, Dash-Yandag B, Deloria-Knoll M, Dherani M, Eap T, Ebruke BE, Echavarria M, de Freitas Lazaro Emediato CC, Fasce RA, Feikin DR, Feng L, et al. 2017. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390:946–958. 10.1016/S0140-6736(17)30938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pneumonia Etiology Research for Child Health Study Group. 2019. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: the PERCH multi-country case-control study. Lancet 394:757–779. 10.1016/S0140-6736(19)30721-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olszewska W, Openshaw P. 2009. Emerging drugs for respiratory syncytial virus infection. Expert Opin Emerg Drugs 14:207–217. 10.1517/14728210902946399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Backman K, Piippo-Savolainen E, Ollikainen H, Koskela H, Korppi M. 2014. Adults face increased asthma risk after infant RSV bronchiolitis and reduced respiratory health-related quality of life after RSV pneumonia. Acta Paediatr 103:850–855. 10.1111/apa.12662. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths C, Drews SJ, Marchant DJ. 2017. Respiratory syncytial virus: infection, detection, and new options for prevention and treatment. Clin Microbiol Rev 30:277–319. 10.1128/CMR.00010-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asner S, Stephens D, Pedulla P, Richardson SE, Robinson J, Allen U. 2013. Risk factors and outcomes for respiratory syncytial virus-related infections in immunocompromised children. Pediatr Infect Dis J 32:1073–1076. 10.1097/INF.0b013e31829dff4d. [DOI] [PubMed] [Google Scholar]
- 7.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 8.Fleming DM, Taylor RJ, Lustig RL, Schuck-Paim C, Haguinet F, Webb DJ, Logie J, Matias G, Taylor S. 2015. Modelling estimates of the burden of respiratory syncytial virus infection in adults and the elderly in the United Kingdom. BMC Infect Dis 15:443. 10.1186/s12879-015-1218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah JN, Chemaly RF. 2011. Management of RSV infections in adult recipients of hematopoietic stem cell transplantation. Blood 117:2755–2763. 10.1182/blood-2010-08-263400. [DOI] [PubMed] [Google Scholar]
- 10.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186. 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 11.Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol 89:422–434. 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 12.Mac S, Sumner A, Duchesne-Belanger S, Stirling R, Tunis M, Sander B. 2019. Cost-effectiveness of palivizumab for respiratory syncytial virus: a systematic review. Pediatrics 143:e20184064. 10.1542/peds.2018-4064. [DOI] [PubMed] [Google Scholar]
- 13.Walsh EE, McConnochie KM, Long CE, Hall CB. 1997. Severity of respiratory syncytial virus infection is related to virus strain. J Infect Dis 175:814–820. 10.1086/513976. [DOI] [PubMed] [Google Scholar]
- 14.Durbin RK, Kotenko SV, Durbin JE. 2013. Interferon induction and function at the mucosal surface. Immunol Rev 255:25–39. 10.1111/imr.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbert K, Schlaak JF, Yang D, Dittmer U. 2013. IFN-alpha subtypes: distinct biological activities in anti-viral therapy. Br J Pharmacol 168:1048–1058. 10.1111/bph.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell CD, Unger SA, Walton M, Schwarze J. 2017. The human immune response to respiratory syncytial virus infection. Clin Microbiol Rev 30:481–502. 10.1128/CMR.00090-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hijano DR, Vu LD, Kauvar LM, Tripp RA, Polack FP, Cormier SA. 2019. Role of type I interferon (IFN) in the respiratory syncytial virus (RSV) immune response and disease severity. Front Immunol 10:566. 10.3389/fimmu.2019.00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isaacs D. 1989. Production of interferon in respiratory syncytial virus bronchiolitis. Arch Dis Child 64:92–95. 10.1136/adc.64.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor CE, Webb MS, Milner AD, Milner PD, Morgan LA, Scott R, Stokes GM, Swarbrick AS, Toms GL. 1989. Interferon alfa, infectious virus, and virus antigen secretion in respiratory syncytial virus infections of graded severity. Arch Dis Child 64:1656–1660. 10.1136/adc.64.12.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sedeyn K, Schepens B, Saelens X. 2019. Respiratory syncytial virus nonstructural proteins 1 and 2: exceptional disrupters of innate immune responses. PLoS Pathog 15:e1007984. 10.1371/journal.ppat.1007984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephens LM, Varga SM. 2020. Function and modulation of type I interferons during respiratory syncytial virus infection. Vaccines (Basel) 8:177. 10.3390/vaccines8020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drajac C, Laubreton D, Riffault S, Descamps D. 2017. Pulmonary susceptibility of neonates to respiratory syncytial virus infection: a problem of innate immunity? J Immunol Res 2017:8734504. 10.1155/2017/8734504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIntosh K. 1978. Interferon in nasal secretions from infants with viral respiratory tract infections. J Pediatr 93:33–36. 10.1016/s0022-3476(78)80595-2. [DOI] [PubMed] [Google Scholar]
- 24.Remot A, Descamps D, Jouneau L, Laubreton D, Dubuquoy C, Bouet S, Lecardonnel J, Rebours E, Petit-Camurdan A, Riffault S. 2016. Flt3 ligand improves the innate response to respiratory syncytial virus and limits lung disease upon RSV reexposure in neonate mice. Eur J Immunol 46:874–884. 10.1002/eji.201545929. [DOI] [PubMed] [Google Scholar]
- 25.Cormier SA, Shrestha B, Saravia J, Lee GI, Shen L, DeVincenzo JP, Kim YI, You D. 2014. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J Virol 88:9350–9360. 10.1128/JVI.00818-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall CB, Douglas RG, Jr, Simons RL, Geiman JM. 1978. Interferon production in children with respiratory syncytial, influenza, and parainfluenza virus infections. J Pediatr 93:28–32. 10.1016/S0022-3476(78)80594-0. [DOI] [PubMed] [Google Scholar]
- 27.Afonso CL, Amarasinghe GK, Bányai K, Bào Y, Basler CF, Bavari S, Bejerman N, Blasdell KR, Briand F-X, Briese T, Bukreyev A, Calisher CH, Chandran K, Chéng J, Clawson AN, Collins PL, Dietzgen RG, Dolnik O, Domier LL, Dürrwald R, Dye JM, Easton AJ, Ebihara H, Farkas SL, Freitas-Astúa J, Formenty P, Fouchier RAM, Fù Y, Ghedin E, Goodin MM, Hewson R, Horie M, Hyndman TH, Jiāng D, Kitajima EW, Kobinger GP, Kondo H, Kurath G, Lamb RA, Lenardon S, Leroy EM, Li C-X, Lin X-D, Liú L, Longdon B, Marton S, Maisner A, Mühlberger E, Netesov SV, Nowotny N, et al. 2016. Taxonomy of the order Mononegavirales: update 2016. Arch Virol 161:2351–2360. 10.1007/s00705-016-2880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bakker SE, Duquerroy S, Galloux M, Loney C, Conner E, Eleouet JF, Rey FA, Bhella D. 2013. The respiratory syncytial virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J Gen Virol 94:1734–1738. 10.1099/vir.0.053025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res 162:80–99. 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rincheval V, Lelek M, Gault E, Bouillier C, Sitterlin D, Blouquit-Laye S, Galloux M, Zimmer C, Eleouet J-F, Rameix-Welti M-A. 2017. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat Commun 8:563. 10.1038/s41467-017-00655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mogensen TH. 2009. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22:240–273. 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE, Jr, Santangelo PJ. 2012. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258. 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jobe F, Simpson J, Hawes P, Guzman E, Bailey D. 2020. Respiratory syncytial virus sequesters NF-kappaB subunit p65 to cytoplasmic inclusion bodies to inhibit innate immune signaling. J Virol 94:e01380-20. 10.1128/JVI.01380-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dolnik O, Gerresheim GK, Biedenkopf N. 2021. New perspectives on the biogenesis of viral inclusion bodies in negative-sense RNA virus infections. Cells 10:1460. 10.3390/cells10061460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galloux M, Risso-Ballester J, Richard CA, Fix J, Rameix-Welti MA, Eleouet JF. 2020. Minimal elements required for the formation of respiratory syncytial virus cytoplasmic inclusion bodies in vivo and in vitro. mBio 11:e01202-20. 10.1128/mBio.01202-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gachon F, Peleraux A, Thebault S, Dick J, Lemasson I, Devaux C, Mesnard JM. 1998. CREB-2, a cellular CRE-dependent transcription repressor, functions in association with Tax as an activator of the human T-cell leukemia virus type 1 promoter. J Virol 72:8332–8337. 10.1128/JVI.72.10.8332-8337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Naidu SR, Sverdrup F, Androphy EJ. 2009. Tax1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. J Virol 83:2274–2284. 10.1128/JVI.01791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petkova DS, Verlhac P, Rozieres A, Baguet J, Claviere M, Kretz-Remy C, Mahieux R, Viret C, Faure M. 2017. Distinct contributions of autophagy receptors in measles virus replication. Viruses 9:123. 10.3390/v9050123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baillet N, Krieger S, Journeaux A, Caro V, Tangy F, Vidalain PO, Baize S. 2019. Autophagy promotes infectious particle production of Mopeia and Lassa viruses. Viruses 11:293. 10.3390/v11030293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, Smith CD, Starost MF, Yedavalli V, Heyninck K, Dikic I, Beyaert R, Jeang KT. 2008. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J 27:629–641. 10.1038/emboj.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verstrepen L, Verhelst K, Carpentier I, Beyaert R. 2011. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem Sci 36:347–354. 10.1016/j.tibs.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 42.Galloux M, Gabiane G, Sourimant J, Richard CA, England P, Moudjou M, Aumont-Nicaise M, Fix J, Rameix-Welti MA, Eleouet JF. 2015. Identification and characterization of the binding site of the respiratory syncytial virus phosphoprotein to RNA-free nucleoprotein. J Virol 89:3484–3496. 10.1128/JVI.03666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boxem M, Maliga Z, Klitgord N, Li N, Lemmens I, Mana M, de Lichtervelde L, Mul JD, van de Peut D, Devos M, Simonis N, Yildirim MA, Cokol M, Kao HL, de Smet AS, Wang H, Schlaitz AL, Hao T, Milstein S, Fan C, Tipsword M, Drew K, Galli M, Rhrissorrakrai K, Drechsel D, Koller D, Roth FP, Iakoucheva LM, Dunker AK, Bonneau R, Gunsalus KC, Hill DE, Piano F, Tavernier J, van den Heuvel S, Hyman AA, Vidal M. 2008. A protein domain-based interactome network for C. elegans early embryogenesis. Cell 134:534–545. 10.1016/j.cell.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin-Vicente M, Gonzalez-Sanz R, Cuesta I, Monzon S, Resino S, Martinez I. 2020. Downregulation of A20 expression increases the immune response and apoptosis and reduces virus production in cells infected by the human respiratory syncytial virus. Vaccines (Basel) 8:100. 10.3390/vaccines8010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rameix-Welti M-A, Le Goffic R, Herve P-L, Sourimant J, Remot A, Riffault S, Yu Q, Galloux M, Gault E, Eleouet J-F. 2014. Visualizing the replication of respiratory syncytial virus in cells and in living mice. Nat Commun 5:5104. 10.1038/ncomms6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prince GA, Horswood RL, Berndt J, Suffin SC, Chanock RM. 1979. Respiratory syncytial virus infection in inbred mice. Infect Immun 26:764–766. 10.1128/iai.26.2.764-766.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goritzka M, Durant LR, Pereira C, Salek-Ardakani S, Openshaw PJ, Johansson C. 2014. Alpha/beta interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial virus infection. J Virol 88:6128–6136. 10.1128/JVI.00333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, Culley FJ, Mack M, Akira S, Johansson C. 2015. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med 212:699–714. 10.1084/jem.20140825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harker JA, Yamaguchi Y, Culley FJ, Tregoning JS, Openshaw PJ. 2014. Delayed sequelae of neonatal respiratory syncytial virus infection are dependent on cells of the innate immune system. J Virol 88:604–611. 10.1128/JVI.02620-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pribul PK, Harker J, Wang B, Wang H, Tregoning JS, Schwarze J, Openshaw PJ. 2008. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol 82:4441–4448. 10.1128/JVI.02541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Makris S, Bajorek M, Culley FJ, Goritzka M, Johansson C. 2016. Alveolar macrophages can control respiratory syncytial virus infection in the absence of type I interferons. J Innate Immun 8:452–463. 10.1159/000446824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rouka E, Hatzoglou C, Gourgoulianis KI, Zarogiannis SG. 2020. Interactome networks between the human respiratory syncytial virus (HRSV), the human metapneumovirus (EtaMPV), and their host: in silico investigation and comparative functional enrichment analysis. Microb Pathog 141:104000. 10.1016/j.micpath.2020.104000. [DOI] [PubMed] [Google Scholar]
- 53.Dapat C, Oshitani H. 2016. Novel insights into human respiratory syncytial virus-host factor interactions through integrated proteomics and transcriptomics analysis. Expert Rev Anti Infect Ther 14:285–297. 10.1586/14787210.2016.1141676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Y, Wang G, Huang X, Du Z. 2014. Expression, purification and crystallization of the SKICH domain of human TAX1BP1. Acta Crystallogr F Struct Biol Commun 70:619–623. 10.1107/S2053230X14006396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shembade N, Pujari R, Harhaj NS, Abbott DW, Harhaj EW. 2011. The kinase IKKalpha inhibits activation of the transcription factor NF-kappaB by phosphorylating the regulatory molecule TAX1BP1. Nat Immunol 12:834–843. 10.1038/ni.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fu T, Liu J, Wang Y, Xie X, Hu S, Pan L. 2018. Mechanistic insights into the interactions of NAP1 with the SKICH domains of NDP52 and TAX1BP1. Proc Natl Acad Sci USA 115:E11651–E11660. 10.1073/pnas.1811421115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. 2015. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moore AS, Holzbaur EL. 2016. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA 113:E3349–E3358. 10.1073/pnas.1523810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thurston TL, Boyle KB, Allen M, Ravenhill BJ, Karpiyevich M, Bloor S, Kaul A, Noad J, Foeglein A, Matthews SA, Komander D, Bycroft M, Randow F. 2016. Recruitment of TBK1 to cytosol-invading Salmonella induces WIPI2-dependent antibacterial autophagy. EMBO J 35:1779–1792. 10.15252/embj.201694491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, Buss F. 2015. The autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of Salmonella Typhimurium by autophagy. PLoS Pathog 11:e1005174. 10.1371/journal.ppat.1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ling L, Goeddel DV. 2000. T6BP, a TRAF6-interacting protein involved in IL-1 signaling. Proc Natl Acad Sci USA 97:9567–9572. 10.1073/pnas.170279097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ceregido MA, Spinola Amilibia M, Buts L, Rivera-Torres J, Garcia-Pino A, Bravo J, van Nuland NA. 2014. The structure of TAX1BP1 UBZ1+2 provides insight into target specificity and adaptability. J Mol Biol 426:674–690. 10.1016/j.jmb.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 63.Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. 2012. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol 14:1024–1035. 10.1038/ncb2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie X, Li F, Wang Y, Wang Y, Lin Z, Cheng X, Liu J, Chen C, Pan L. 2015. Molecular basis of ubiquitin recognition by the autophagy receptor CALCOCO2. Autophagy 11:1775–1789. 10.1080/15548627.2015.1082025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu S, Wang Y, Gong Y, Liu J, Li Y, Pan L. 2018. Mechanistic insights into recognitions of ubiquitin and myosin VI by autophagy receptor TAX1BP1. J Mol Biol 430:3283–3296. 10.1016/j.jmb.2018.06.030. [DOI] [PubMed] [Google Scholar]
- 66.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 67.Esneau C, Raynal B, Roblin P, Brule S, Richard CA, Fix J, Eleouet JF, Galloux M. 2019. Biochemical characterization of the respiratory syncytial virus N(0)-P complex in solution. J Biol Chem 294:3647–3660. 10.1074/jbc.RA118.006453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pokharel SM, Shil NK, Bose S. 2016. Autophagy, TGF-beta, and SMAD-2/3 signaling regulates interferon-beta response in respiratory syncytial virus infected macrophages. Front Cell Infect Microbiol 6:174. 10.3389/fcimb.2016.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsushita N, Suzuki M, Ikebe E, Nagashima S, Inatome R, Asano K, Tanaka M, Matsushita M, Kondo E, Iha H, Yanagi S. 2016. Regulation of B cell differentiation by the ubiquitin-binding protein TAX1BP1. Sci Rep 6:31266. 10.1038/srep31266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tran TL, Castagne N, Bhella D, Varela PF, Bernard J, Chilmonczyk S, Berkenkamp S, Benhamo V, Grznarova K, Grosclaude J, Nespoulos C, Rey FA, Eleouet JF. 2007. The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J Gen Virol 88:196–206. 10.1099/vir.0.82282-0. [DOI] [PubMed] [Google Scholar]
- 71.Castagne N, Barbier A, Bernard J, Rezaei H, Huet JC, Henry C, Da Costa B, Eleouet JF. 2004. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J Gen Virol 85:1643–1653. 10.1099/vir.0.79830-0. [DOI] [PubMed] [Google Scholar]
- 72.Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. 10.1128/JVI.73.1.251-259.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vidalain PO, Jacob Y, Hagemeijer MC, Jones LM, Neveu G, Roussarie JP, Rottier PJ, Tangy F, de Haan CA. 2015. A field-proven yeast two-hybrid protocol used to identify coronavirus-host protein-protein interactions. Methods Mol Biol 1282:213–229. 10.1007/978-1-4939-2438-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bourai M, Lucas-Hourani M, Gad HH, Drosten C, Jacob Y, Tafforeau L, Cassonnet P, Jones LM, Judith D, Couderc T, Lecuit M, Andre P, Kummerer BM, Lotteau V, Despres P, Tangy F, Vidalain PO. 2012. Mapping of chikungunya virus interactions with host proteins identified nsP2 as a highly connected viral component. J Virol 86:3121–3134. 10.1128/JVI.06390-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Galloux M, Tarus B, Blazevic I, Fix J, Duquerroy S, Eleouet J-F. 2012. Characterization of a viral phosphoprotein binding site on the surface of the respiratory syncytial nucleoprotein. J Virol 86:8375–8387. 10.1128/JVI.00058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cagno V, Andreozzi P, D’Alicarnasso M, Jacob Silva P, Mueller M, Galloux M, Le Goffic R, Jones ST, Vallino M, Hodek J, Weber J, Sen S, Janecek E-R, Bekdemir A, Sanavio B, Martinelli C, Donalisio M, Rameix Welti M-A, Eleouet J-F, Han Y, Kaiser L, Vukovic L, Tapparel C, Kral P, Krol S, Lembo D, Stellacci F. 2018. Broad-spectrum non-toxic antiviral nanoparticles with a virucidal inhibition mechanism. Nat Mater 17:195–203. 10.1038/nmat5053. [DOI] [PubMed] [Google Scholar]
- 77.Gaillard V, Galloux M, Garcin D, Eleouet JF, Le Goffic R, Larcher T, Rameix-Welti MA, Boukadiri A, Heritier J, Segura JM, Baechler E, Arrell M, Mottet-Osman G, Nyanguile O. 2017. A short double-stapled peptide inhibits respiratory syncytial virus entry and spreading. Antimicrob Agents Chemother 61:e02241-16. 10.1128/AAC.02241-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Descamps D, Le Gars M, Balloy V, Barbier D, Maschalidi S, Tohme M, Chignard M, Ramphal R, Manoury B, Sallenave JM. 2012. Toll-like receptor 5 (TLR5), IL-1beta secretion, and asparagine endopeptidase are critical factors for alveolar macrophage phagocytosis and bacterial killing. Proc Natl Acad Sci USA 109:1619–1624. 10.1073/pnas.1108464109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gene Ontology Consortium. 2019. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47:D330–D338. 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]