

Short abstract
Content available: Author Interview and Audio Recording
Abbreviations
- ASL
Argininosuccinate lyase
- ASS
Argininosuccinate synthase
- CPSI
carbamoylphosphate synthetase‐1
- HHH
hyperammonemia, hyperornithemia, homocitrullinemia
- IV
intravenous
- MCT
medium‐chain triglycerides
- NAGS
N‐acetylglutamate synthase
- OTC
ornithine transcarbamylase
- UC
urea cycle
- UCD
urea cycle disorder
Overview
Watch the interview with the author.
Listen to an audio presentation of this article.
All tissues produce the neurotoxin ammonia as a by‐product of amino acid metabolism. The primary route of ammonia removal in mammals is the hepatic urea cycle (UC), a series of biochemical reactions that enables urinary excretion of waste ammonia as urea. The urea cycle disorders (UCDs) are monogenic disorders caused by a deficiency of an enzyme or cellular transporter essential to ureagenesis (Fig. 1). The primary acute manifestations of UCDs are hyperammonemia, liver dysfunction, and encephalopathy. Common long‐term sequelae are neurological deficits, cognitive impairments, and hepatic disease. 1 Prompt diagnosis and initiation of treatment are critical to survival and optimal neurocognitive outcome. 2
FIG 1.
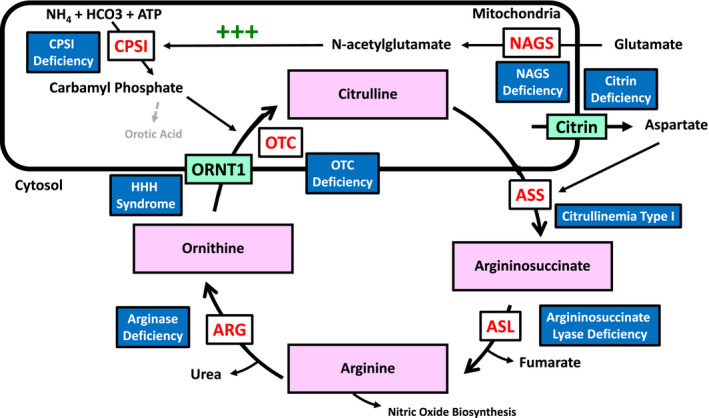
Schematic of the urea cycle. CPSI catalyzes the reaction of ammonium, bicarbonate, and ATP to form carbamyl phosphate, which combines with mitochondrial ornithine to generated citrulline in a reaction catalyzed by OTC. Aspartate is transported to the cytosol through the citrin carrier and combines with citrulline to form argininosuccinate in a reaction catalyzed by argininosuccinate synthetase. Argininosuccinate is converted to arginine by argininosuccinate lyase. Arginine can be used in nitric oxide biosynthesis or can be converted to urea and ornithine via arginase. Generated ornithine is transported into the mitochondria by the ornithine transporter to combine with carbamyl phosphate and generate citrulline and replenish the cycle. CPSI is the rate‐limiting step in the urea cycle, and is allosterically activated by N‐acetylglutamate, which is generated from glutamate by NAGS. Abbreviations: ARG: arginase; ASL: argininosuccinate lyase; ASS: argininosuccinate synthetase; CPSI: carbamylphosphate synthetase‐1; HHH: Hyperornithinemia‐hyperammonemia‐homocitrullinuria; NAGS: N‐acetylglutamate synthetase; ORNT1: ornithine transporter; OTC: ornithine transcarbamylase.
Epidemiology and Genetics
UCDs affect all ethnicities and have an estimated US incidence of 1/35,000 births. Ornithine transcarbamylase (OTC) deficiency accounts for ~60% of cases. 3 UCDs are predominantly inherited in an autosomal recessive fashion with the notable exception of OTC, which is X linked. Null genetic variants, typically gene deletions, nonsense mutations, and frameshift mutations, result in near‐complete enzyme deficiency and severe disease, whereas missense mutations are associated with partial enzyme deficiency and attenuated disease. 4
Diagnosis
UCDs may present at any age, even in adult life, but two‐thirds occur in the neonatal period (Table 1). A major diagnostic advance is newborn screening for UCDs, which uses mass spectrometry to measure amino acids in presymptomatic neonates. Regrettably, the lack of a reproducible amino acid marker for OTC has confounded reliable presymptomatic screening for this disorder. 5 Symptomatic infants typically present with lethargy, seizures, hypotonia, poor feeding, and tachypnea in the first days to weeks of life. 1 These signs often prompt an erroneous diagnosis of neonatal sepsis, a confusion that may be compounded by the absence in UCDs of other stigmata of metabolic disease, such as anion gap acidosis, lacticemia, and hypoglycemia. In fact, many symptomatic neonates demonstrate a respiratory alkalosis. The biochemical hallmark of UCDs is hyperammonemia, which is often extreme. Ammonia levels less than 100 μmol/L are considered normal in neonates, whereas 60 μmol/L is considered the upper limit of normal after 1 month of age. Plasma ammonia samples preferably should be collected from a free‐flowing puncture with no tourniquet, stored on ice, and run immediately on receipt by the laboratory. Improper sample collection or handling may lead to falsely elevated values.
TABLE 1.
UCDS
| Disorder Type | Disease | Causal Gene | Inheritance | Classical Phenotype | Mild/Late‐Onset Disease | Liver Disease |
|---|---|---|---|---|---|---|
| Proximal UC defect | NAGS deficiency | NAGS | Autosomal recessive | Hyperammonemia, encephalopathy, seizures, developmental delay, regression | Developmental delay, episodic hyperammonemia | Steatosis |
| CPSI deficiency | CPSI | Autosomal recessive | Hyperammonemia, encephalopathy, seizures, developmental delay, regression | Episodic hyperammonemia | Acute liver failure, steatosis, fibrosis | |
| Distal UC defect | OTC deficiency | OTC | X‐linked recessive | Hyperammonemia, encephalopathy, seizures, neuropsychiatric symptoms | Neuropsychiatric disturbances, neurocognitive deficits, seizures, encephalopathy | Hepatitis, synthetic liver dysfunction |
| Citrullinemia type I | ASS | Autosomal recessive | Hyperammonemia, encephalopathy, seizures, developmental delay, regression | Pregnancy‐induced acute liver failure, neurocognitive disturbances, episodic hyperammonemia | Acute liver failure, steatosis, fibrosis, hepatocellular carcinoma | |
| Argininosuccinate lyase deficiency | ASL | Autosomal recessive | Hyperammonemia, encephalopathy, seizures, developmental delay, regression, trichorrhexis nodosa, brittle hair, hypertension | Neurocognitive disturbances, episodic hyperammonemia | Hepatomegaly, hepatic fibrosis, cirrhosis, portal hypertension, hepatocellular carcinoma | |
| Arginase deficiency | ARGI | Autosomal recessive | Regression, progressive spasticity, seizures, hyperammonemia (rarer) | Developmental delay, regression, spasticity, episodic hyperammonemia | Hepatomegaly, hepatitis, steatosis, fibrosis, hepatocellular carcinoma | |
| Transporter defect | HHH syndrome | SLC25A15 | Autosomal recessive | Hyperammonemia, encephalopathy, seizures, spasticity, regression | Neurocognitive deficits, ataxia, spasticity, seizures | Liver dysfunction, coagulopathy, steatosis, fibrosis (mild) |
| Citrin deficiency (citrullinemia type II) | SLC25A13 | Autosomal recessive | Liver disease, poor growth | Neuropsychiatric disturbances, neurocognitive deficits, pancreatitis | Cholestasis, hepatomegaly, steatosis, fibrosis |
Individuals with partial enzyme deficiencies may present more mildly, with their first symptoms emerging after the neonatal period. Common childhood manifestations include global developmental delay, neuroregression, spasticity, epilepsy, psychiatric symptoms, growth failure, and aminotransferase elevation. Many children and adults self‐restrict intake of high‐protein foods, such as meat and dairy, as a learned response to feeling unwell after protein‐rich meals. Children and adults with UCDs are at risk for acute‐onset encephalopathy, seizure, and coma in the setting of critical illness or exposure to certain medications, such as systemic steroids or valproate. 6
Adult women are at particular risk for postpartum hyperammonemic crises due to the increased protein load from uterine remodeling and red blood cell turnover. 7 , 8 Gastric bypass surgery can also unmask an underlying UCD by inducing a catabolic state and altering protein absorption. Other triggers of hyperammonemia in adults with UCDs include short bowel disease and gastrointestinal bleeding. 8
Diagnosis of UCDs in children and adults is established using complementary biochemical testing (measurement of plasma ammonia, plasma amino acids, urine organic acids, and urine orotic acid testing), as well as gene sequencing (Fig. 2). 9
FIG 2.
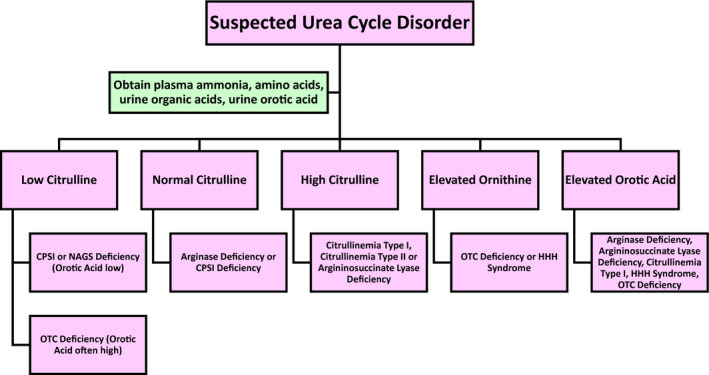
Diagnostic algorithm for UC diagnosis. Assessment should begin with evaluation of plasma amino acids, urine organic acids, and urine orotic acid. Citrulline level can help distinguish CPSI, OTC, citrullinemia I, citrullinemia II, and argininosuccinate lyase deficiency. Ornithine level can help diagnose OTC deficiency and HHH syndrome. Orotic acid levels can help diagnose arginase deficiency, argininosuccinate lyase deficiency, citrullinemia type I, HHH syndrome, and OTC deficiency.
Treatment
Acute hyperammonemia causes cerebral accumulation of glutamate and glutamine, leading to both excessive activation of glutamatergic receptors and increased osmotic pressure. The result is encephalopathy, often with seizures, brain swelling, hypoxic injury, and ultimately, neuronal death. Chronic hyperammonemia leads to down‐regulation of glutamate receptors and increased GABAergic signaling, causing neurocognitive decline. 10 Management of patients with UCDs focuses on the prevention of both acute and chronic hyperammonemia.
The mainstay of treatment is a protein‐restricted diet to decrease the nitrogen load. Most patients also use oral nitrogen scavengers (sodium benzoate and glycerol phenylbutyrate), which provide a route other than the defective UC for the elimination of waste nitrogen, thereby lessening the risk for development of hyperammonemia. An accessory intervention is supplementation of deficient UC intermediates, such as arginine and citrulline. For individuals with a deficiency of N‐acetylglutamate synthase, one of the proximal UC defects, dramatic improvement is realized by treatment with N‐carbamylglutamate (Carbaglu), a synthetic analogue of N‐acetylglutamate, which is an obligatory effector of the hepatic UC. Rarely, lactulose is given to decrease the nitrogen load from gut flora (Table 2 and Fig. 3). 9 Routine surveillance includes blood pressure measurements; assessment of nutritional status through measurement of the level of blood albumin, amino acids, and vitamins; and surveillance of liver function and bone health. Even with careful management and adherence to medical protocols, some patients may progress to liver steatosis, fibrosis, cirrhosis, portal hypertension, and hepatocellular carcinoma. Liver fibrosis and malignancy are of particular concern for individuals with argininosuccinate lyase deficiency, likely caused by the hepatotoxicity of the accumulating intermediate argininosuccinate. 9
TABLE 2.
Treatments for UCDs
| Disorder Type | Disease Name | Acute Treatment | Chronic Treatment | Surveillance |
|---|---|---|---|---|
| Proximal UC defects | NAGS deficiency | Halt protein intake, nitrogen scavengers, IV dextrose, carglumic acid | Protein restriction, carglumic acid | Monitor liver function, nutritional status, and vitamin deficiencies |
| CPSI deficiency | Halt protein intake, IV scavengers, IV arginine, dextrose‐containing fluids | Protein restriction, arginine or citrulline supplementation, hospitalization for dextrose‐containing fluids with illness | Monitor liver function, nutritional status, and vitamin deficiencies | |
| Distal UC defects | OTC deficiency | Halt protein intake, IV scavengers, IV arginine, dextrose‐containing fluids | Protein restriction, arginine or citrulline supplementation, nitrogen scavengers, liver transplant | Monitor liver function, nutritional status, and vitamin deficiencies |
| Citrullinemia type I | Halt protein intake, IV scavengers, IV arginine, dextrose‐containing fluids | Protein restriction, nitrogen scavengers, arginine supplementation, hospitalization for dextrose‐containing fluids with illness, citrulline is contraindicated | Monitor liver function, nutritional status, and vitamin deficiencies | |
| Argininosuccinate lyase deficiency | Halt protein intake, IV scavengers, IV arginine, dextrose‐containing fluids | Protein restriction, nitrogen scavengers, arginine supplementation, hospitalization for dextrose‐containing fluids with illness | Monitor liver function, fibrosis, hepatocellular carcinoma, vitamin deficiencies, nutritional status | |
| Arginase deficiency | Arginine and citrulline are contraindicated | Protein restriction, nitrogen scavengers, arginine and citrulline are contraindicated | Monitor for movement disorder, liver function, liver fibrosis, nutritional status, and vitamin deficiencies | |
| Transporter defect | HHH syndrome | Halt protein intake, IV scavengers, IV arginine, dextrose‐containing fluids | Protein restriction, nitrogen scavengers, ornithine supplementation | Monitor liver function, fibrosis, hepatocellular carcinoma, vitamin deficiencies, nutritional status |
| Citrin deficiency (citrullinemia type II) | Protein‐ and lipid‐rich diet, IV nitrogen scavengers, arginine supplementation | Lipid‐ and protein‐rich diet, MCT oil, galactose‐free diet, nitrogen scavengers, arginine supplementation, sodium pyruvate supplementation | Monitor liver function, screen for steatosis, monitor for zinc deficiency |
FIG 3.
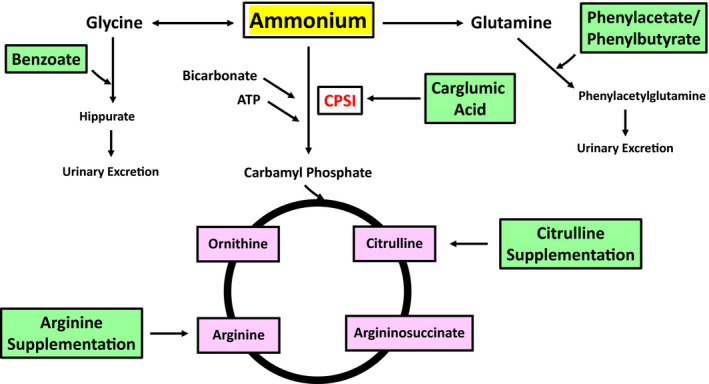
Treatments modalities for UCDs. Benzoate complexes with glycine and is converted to Hippurate, which can be excreted in urine, bypassing the urea cycle. Phenylacetate and its precursor phenylbutyrate complex to glutamine, forming phenylacetylglutamine, which is excreted in urine. Carglumic acid, like NAGS, is an allosteric activator of CPSI, increasing flux through the urea cycle, and is useful in proximal urea cycle disorders. Arginine and citrulline supplementation replenishes deficient urea cycle substrates. Importantly, arginine supplementation is contraindicated in arginase deficiency. Abbreviation: CPSI, arbomylphosophate synthetase I.
Acute hyperammonemic crises are treated with elimination of dietary protein and infusion of intravenous (IV) dextrose (most commonly 10% dextrose). Importantly, complete protein restriction should last no longer than 24 to 48 hours to avoid paradoxical hyperammonemia from essential amino acid deficiency and consequent muscle catabolism. In addition, patients with neurological symptoms of hyperammonemia may require IV sodium phenylacetate as a nitrogen scavenger and IV (or enteric) administration of citrulline and/or arginine. Severe and refractory hyperammonemia may require hemodialysis. 1 , 9
The only “curative” therapy for UCDs is liver transplantation, which replaces the impaired UC; however, this does not reverse past neurological damage.
Considerations For Prolonged Survival in UCDs
The UCDs first were described about 50 years ago. At that time, the outlook was dire for babies with the classical, severe disease. Few survived to adulthood. The development of special low‐protein metabolic foods, nitrogen scavengers, and liver transplantation have enabled long‐term survival for most of these infants. Of course, survival is not cost‐free, and we now recognize an array of complications that afflict older children and adults, including hypertension from impaired nitric oxide generation, chronic kidney disease and electrolyte abnormalities, often profound cognitive impairment, a distinct neuropsychiatric profile, and a hepatopathy that ranges from clinically silent aminotransferase elevation to cholestasis, steatosis, fibrosis, cirrhosis, portal hypertension, and hepatocellular carcinoma (Table 2). 11
Liver disease in UCDs likely results from chronic accumulation of amino acids such as glutamine, toxic products such as argininosuccinate and guanidino compounds, ongoing steatosis and glycogen deposition, a deficiency of essential amino acids and nitric oxide, and a failure of adequate adenosine triphosphate production consequent to mitochondrial dysfunction. 11
As women with UCDs attain reproductive age, there is a concomitant need for genetic counseling and careful monitoring during pregnancy and the postpartum period.
Conclusions
Prompt diagnosis and treatment of UCDs improves outcome. These disorders should be considered in any patient presenting with unexplained developmental regression, encephalopathy (especially if recurrent), seizures, movement disorder, or liver disease. Despite advances in the diagnosis and treatment of UCDs, these disorders remain difficult to manage and are associated with significant neurological and hepatic morbidity and mortality. Liver transplantation corrects hyperammonemia but cannot reverse past neurological damage. Liver‐directed gene therapy research is actively ongoing as an alternative to transplantation.
Potential conflict of interest: Nothing to report.
References
- 1. Yudkoff M, Summar M, Haberle J. Urea cycle disorders. In: Sarafoglou K, Hoffmann GF, Roth KS, eds. Pediatric Endocrinology and Inborn Errors of Metabolism. 2nd ed. New York, NY: McGraw‐Hill; 2013:191‐208. [Google Scholar]
- 2. Posset R, Gropman AL, Nagamani SCS, et al. Impact of diagnosis and therapy on cognitive function in urea cycle disorders. Ann Neurol 2019;86:116‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab 2013;110:179‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caldovic L, Abdikarim I, Narain S, et al. Genotype‐phenotype correlations in ornithine transcarbamylase deficiency: a mutation update. J Genet Genomics 2015;42:181‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cavicchi C, Malvagia S, la Marca G, et al. Hypocitrullinemia in expanded newborn screening by LC‐MS/MS is not a reliable marker for ornithine transcarbamylase deficiency. J Pharm Biomed Anal 2009;49:1292‐1295. [DOI] [PubMed] [Google Scholar]
- 6. Batshaw ML, Brusilow SL. Valproate‐induced hyperammonemia. Ann Neurol 1982;11:319‐321. [DOI] [PubMed] [Google Scholar]
- 7. Mendez‐Figueroa H, Lamance K, Sutton VR, et al. Management of ornithine transcarbamylase deficiency in pregnancy. Am J Perinatol 2010;27:775‐784. [DOI] [PubMed] [Google Scholar]
- 8. Stepien KM, Geberhiwot T, Hendriksz CJ, et al. Challenges in diagnosing and managing adult patients with urea cycle disorders. J Inherit Metab Dis 2019;42:1136‐1146. [DOI] [PubMed] [Google Scholar]
- 9. Häberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis 2019;42:1192‐1230. [DOI] [PubMed] [Google Scholar]
- 10. Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis 2007;30:865‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ranucci G, Rigoldi M, Cotugno G, et al. Chronic liver involvement in urea cycle disorders. J Inherit Metab Dis 2019;42:1118‐1127. [DOI] [PubMed] [Google Scholar]