

Abstract
Astrocytic processes interact with synapses throughout the brain modulating neurotransmitter signaling and synaptic communication. During conditions such as exposure to drugs of abuse and neurological diseases, astrocytes respond by altering their morphological and functional properties. Reactive astrocyte phenotypes exhibit a bushy morphology with altered soma volume and an increased number of processes compared to resting astrocytes. The reactive astrocytic phenotype also overexpresses proteins one of which can be glial fibrillary acidic protein (GFAP). Fluorescence microscopy on thin tissue sections (< 20 μm) requires reconstruction, often through multiple sections, to delineate the full astrocytic morphology. In constrast, tissue clearing methods have been developed that enable imaging of larger sections including the whole brain, providing an opportunity to see in-depth changes in single cell structure. In this unit a detailed protocol for studying astrocyte morphology using tissue clearing and subsequent imaging of whole brains as well as region-specific slices is provided. This method is ideal for understanding the effect of different physiological conditions on astrocyte morphology. A standard biochemistry laboratory has the resources to accomplish tissue clearing using this protocol and most universities have the required imaging facilities. Protocol to study brains from both genetically modified mice that contain an astrocyte-specific marker and from wild-type mice using antibody labeling steps after tissue clearing are provided. We also describe general protocols to conduct fluorescence imaging of astrocytes in cleared tissue to characterize their morphology. This protocol could be useful for researchers working in the rapidly growing field of astrocyte biology.
Keywords: Astrocyte, Tissue clearing, Brain, Microscopy, Fluorescence
INTRODUCTION:
While astrocytes were previously considered relatively nonfunctional cells (Allen & Barres, 2009), recent studies have demonstrated that astrocytes play a key role in the activity of a healthy brain and that both their morphology and function are altered under many neuropathological conditions (Khakh & Deneen, 2019; Oberheim, Goldman, & Nedergaard, 2012). With the development of a variety of genetic and optical tools, the function of astrocytes is now being explored to understand their role in conditions such as metabolism (Yi, Habegger, Chowen, Stern, & Tschöp, 2011) substance abuse, traumatic brain injury and neurological disorders (Sidoryk-Wegrzynowicz, Wegrzynowicz, Lee, Bowman, & Aschner, 2011; X. Zhang et al., 2021). Utilizing fluorescence microscopy, studies on astrocyte biology range from cell and tissue-based imaging to monitoring in vivo calcium activity, and have led to advances in morphological and functional characterization of astrocytes. For example, characterization of fine astrocytic processes has been performed in vitro, in situ and in vivo using genetically encoded calcium sensors (Lia et al., 2021).Techniques such as the neuron astrocyte proximity assay (NAPA) have characterized the proximity of astrocytes and neurons at synaptic scales (Badia-Soteras, Octeau, Verheijen, & Khakh, 2020; Octeau et al., 2018). Even though a great deal of information is obtained from cell and tissue-based studies, imaging the whole brain is especially important to monitor changes the 3-D architecture of astrocytes within the brain. Astrocytes are heterogeneous cells and brain region-specific information is important to understand astrocytic function and physiology (Rusnakova et al., 2013). Imaging deep inside the brain in either a live animal or in fixed tissue is complicated by lack of penetration of light into the tissue and of scattering of the fluorescence signal (Richardson & Lichtman, 2015). In vivo two-photon imaging is a powerful method to study live animals and especially useful to image shallow regions of the brain just below the skull through a cranial window. However, to date, this approach can not be used to access deeper regions. Additionally, the surgery to implant a cranial window followed by high intensity laser exposure increases the likelihood of an inflammatory response and thus reactive astrocytosis (Cramer et al., 2021).
Recent advances in tissue clearing that render brain tissue optically transparent have enabled fluorescence imaging of the whole brain as well as region-specific slices to obtain 3D information of structural and functional features of neurons, vasculature, as well as glial cells such as astrocytes and microglia (Ueda et al., 2020a). Even though there are various methods to clear whole brains and region-specific slices, many of these methods require sophisticated techniques and expensive equipment (Richardson & Lichtman, 2015). Some techniques use methods to actively remove lipids by performing electrophoresis which can affect the underlying cellular and subcellular tissue structure, alter genetically encoded labels, and diminish immunostaining efficiency (Tomer, Ye, Hsueh, & Deisseroth, 2014). Other techniques utilize a passive clearing approach and rely on chemical clearing of tissue that often contains reagents that can alter fluorescent labels and/or damage tissue (Becker, Jährling, Saghafi, Weiler, & Dodt, 2012). Commercially available clearing kits are also available, but can be costly and are not compatible with all cell types or antibodies. Here, we detail a protocol for tissue clearing using a modified version of fast free-of-acrylamide clearing tissue (FACT) reagents that is compatible with the study of astrocyte morphology.(S. P. Aryal et al., 2021; Wang, Khoradmehr, & Tamadon, 2019; Xu et al., 2017)
This tissue clearing method uses readily available reagents and leaves astrocyte structure intact allowing researchers to perform quantitative analysis of astrocytic morphology, correlating structural changes and function. For example, the brain slices which are used for electrophysiology or calcium imaging can be cleared and used for assessment of morphological changes. This protocol describes how to characterize astrocyte morphology. However, detailed steps should be followed based on standard operating procedures for the specific microscopes and software analysis packages utilized for the experiments. The described approach can be used to clear and analyze tissue from both fluorescent protein knock-in animals as well as wild-type animals which have been labeled using fluorescent antibodies. This method is simple, fast, and reliable, applicable to astrocytes, and is compatible with astrocyte-specific antibodies. The first part of this protocol discusses how to prepare and extract a rodent brain for clearing. This results in optically transparent brain tissue which can be confirmed by simple visual inspection. The second part of this protocol discusses some of the key steps of the process which includes successive labeling of antibody and refractive index matching. The third part of this protocol focuses on general information regarding fluorescence imaging of a whole brain or thick slice and the characterization of astrocyte morphology.
BASIC PROTOCOL 1: Brain perfusion, fixation, and tissue clearing
Introductory Paragraph:
This protocol details perfusion, fixation, and clearing of mouse brain tissue for fluorescence imaging of labeled astrocytes (Figure 1). A similar procedure can also be applied to other rodents such as rats. Complete perfusion of the mouse brain will result in a white brain tissue with no observable blood in the vasculature of the brain. The presence of reddish color indicates that blood was not perfused properly which can affect the tissue clearing process and create difficulties in imaging of astrocytes. The protocol outlines post-extraction fixation of the brain tissue, slicing of brain region-specific imaging, and tissue clearing. Post-extraction fixation is necessary for tissue clearing and can be achieved by fixing the brain in 4 % PFA (in PBS) for 24-48 hours. Clearing is performed by incubating the brain in an 8% Sodium Dodecyl Sulfate (SDS) solution at 37°C. The clearing solution should be changed every 2 days. If implemented successfully, this will result in a near-transparent whole brain or slice which can be imaged using a fluorescence microscope (S. P. Aryal et al., 2021; Xu et al., 2017). PFA is shown to be carcinogenic so it should be handled carefully. A chemical fume hood must be used while preparing the reagents. The protocol must be approved by respective Institutional Animal Care and Use Committees (IACUC).
FIGURE 1:

Schematic of FACT protocol for astrocyte imaging. The mouse brain is transcardially perfused with fixative and then the brain is extracted and subjected to post-extraction fixation. The fixed brain is cleared by using FACT reagents and antibody labeling is performed. Finally, the brain is cleared by TDE and imaged using a fluorescence microscope.
Materials
70 % ethanol (v/v, in water)
Phosphate Buffer Saline (PBS, pH=7, BDH7447-1, VWR Chemicals)
Paraformaldehyde (PFA, CAS Number: 30525-89-4)
Magnetic stirrer and heater (Catalog number: HP88854100, Thermo Scientific)
Magnetic stirring bar (58948-091, VWR international)
50 ml centrifuge tubes (VWR, catalog number: 89039-658)
4% Paraformaldehyde solution (4% PFA in PBS, pH 7.0-7.4): See the recipe
Fatal Plus (Drugs.com, product number: V.P.L. 9373, NDC number: 0298-9373-68)
Fatal plus injection solution (10% Fatal plus in PBS): See the recipe
C57BL6 mice (RRID: MGI:5652902) (6 animals per condition, older than 2 months)
Aldh1l1-tdTomato mice (RRID: MMRRC_036700-UCD) (6 animals per condition, older than 2 months)
Sodium dodecyl sulfate (VWR Catalog no. 97064-496, VWR International)
FACTS clearing solution (8% Sodium Dodecyl Sulfate (SDS) in PBS): See the recipe
Milli-Q water (Fisher Scientific, catalog number: QGARD00D2)
Tygon tubing (Garinger, Item number: 9MH86)
1 ml plastic syringe (Grainger, Item number: 19G334, ThermoFisher Scientific, catalog number: NC0786233)
Syringe needles (Air-Tite, catalog number: NH252, 25g, 2”)
5 ml Serological pipettes (VWR, catalog number: 89130-896)
Ice
Smooth fine forceps (Electron Microscopy Sciences, 1209K43, catalog number: 72911-6)
Surgical scissors (Medline, catalog number: MDS0838410)
Operating scissors (Stoelting, catalog number: 52138-51)
Flat tip forceps (Fisher Scientific, catalog number: 12-000-123)
Hemostat (VWR international, catalog number: 10806-170)
Ice bucket (VWR, catalog number: 10146-202)
Adult Mouse Brain Slicer Matrix (Zivic Instruments, catalog number: BSMAS002-1)
1 ml pipettor (Gilson, catalog number: F123602)
Pipet controller (VWR, catalog number: 613-4442)
Perfusion pump (Grainger, Item No.: 2P305, Model No.: UD50-XA-LSAUXXX, catalog number: 2688)
Swing bucket rotor (Beckman Coulter, model: SW 28)
Fixed angle rotor (Beckman Coulter, model: 70 Ti)
Belly dancer or orbital shaker (Fisher Scientific, catalog number: 15-453-211)
Sonicator (Amsco Reliance Sonic 550)
Vortex (VWR, catalog number: 10153-836)
Zivic mouse brain matrix (Zivic instruments, BSMAA002-1)
Razor blades (VWR, catalog number: 10835-969)
Glass bottom dish (MatTek, catalog number: P35G-1.5-14-C)
4 °C fridge
−20 °C freezer
Shaker with incubator (VWR, catalog number: 75874-516)
Protocol steps with step annotations:
Mouse perfusion
Prepare all the necessary solutions (4% PFA in PBS, Fatal plus injection solution) as shown in the recipe.
-
Wipe all surgical tools with 70% ethanol.
This will kill most of the bacterial pathogens oin tools if present. Autoclaving or UV treatment can be performed to remove more refractory pathogens.
-
Set-up the perfusion system.
You will need a syringe, a needle (see the specifications in the Materials section), a Tygon tube, and perfusing liquid in a container. A syringe containing a needle should be inserted into one end of the Tygon tube. The other end of the Tygon tube should go inside the solution through the perfusion pump.
-
Inject 200 μl of Fatal plus solution subcutaneously or intravenously to a mouse (as shown in Figure 2).
The animal should be handled properly while performing the injection. Subcutaneous injection is the easiest way to deliver the drug for most users. Experienced users can also choose to perform intravenous injection through a tail vein or retroorbital injection. Injection should be made on the back of the mouse near the leg.
-
Wait until the mouse becomes nonresponsive by using the pinch reflex. Once it becomes nonresponsive transfer it to the perfusion area.
Usually, a mouse becomes nonresponsive after 2-3 minutes of Fatal plus injection. If the animal is still active after 3-4 minutes, inject another 200 μl of Fatal plus solution and wait for 2-3 minutes.
Lay the nonresponsive mouse keeping the ventral side on top of a foam piece and immobilize it with tape or thumb pins. Using small scissors, snip a piece of skin on the lower thorax and make a parabolic, sagittal cut. This cut should run from one side of the abdomen to the other side. Pull out the loosened tissue and make a transverse cut through the pectoral region and tear through the diaphragm. Hold the loose tissue by using a hemostat to make sure it is out of the way. (Aryal, Fu, Masud, Neupane, & Richards, 2021)
-
Set the perfusion pump to a modest speed (5-10 ml/minute for mice) but do not start the perfusion yet. Insert the needle connected to the Tygon tube to the left ventricle (LV) of the mouse, make a small hole in the right atrium (RA) and start perfusion. Continue perfusion until the liver turns tan-colored and the solution coming out is colorless. 40 ml of PBS is enough for perfusing a mouse brain. Hoffman et al. 2016 have nicely demonstrated the dissection process during perfusion (Hoffman, Murphy, & Sita, 2016)
It is strongly recommended to ensure liquid flow through the needle before inserting it into the left ventricle. This also prevents bubble formation during the perfusion process.
Make sure one end of the Tygon tube is inside the buffer and the other end goes inside the syringe before inserting the needle (as shown in the Figure 1). The needle must be inserted into the left ventricle (LV) and can be clamped by using a hemostat. PBS perfusion and a small incision at the right atrium can be started at the same time. Good perfusions result in colorless liquid coming out from the hole at the right atrium otherwise blood vasculature will be observed during imaging of astrocytes.
-
Switch to 4% PFA fixative solution once the PBS perfusion is complete (evidenced by colorless liquid coming from the right atrium, discoloration of lungs, and tan color of the liver) and continue perfusion with 30 ml of the solution.
Fixation tremors can occur during fixation, causing the movement of limbs and parts of the body and makes the legs and other body parts stiffer.
-
Turn off the perfusion system, remove the needle from the heart, decapitate the mouse and remove the brain.
Use 6” scissors for decapitation. With the help of fine forceps locate the spinal column and remove excessive cervical vertebrae. Find the base of the magnum foramen (MF) and make a cut from its base towards the nose and another from the MF laterally towards the ears. Peel back the loosened tissue with the help of regular forceps (Moore & Alejandro, 2021).. Perform a cut in a midsagittal manner from the base of the skull up to the nose. Use blunt edges of the scissors to drag on the underside part of the brain and make the cut. Perform a transverse cut from one eye to the other eye. Peel off the loosened skull tissue with the help of tweezers. A nice illustration is shown by Moore et al. 2021.(Moore & Alejandro, 2021).
Take out the brain and put it in a tube containing 4% PFA on ice.
FIGURE 2:

Illustration of various injection methods for drug delivery in animals
Post extraction fixation and clearing
-
11.
Perform post-extraction fixation of the brain by incubating the brain in 4% PFA for 48 hours at 4°C.
-
12.
Wash the post-extraction fixed brain tissue with PBS three times and incubate in PBS for 24 hours at 4°C.
Note: whole brains and slices can be kept in PBS or PBST at 4 °C for up to 6 months.
-
13.
For making slices, place the fixed brain on the Zivic brain matrix and slice with the help of blades. Collect the slices separately in vials and incubate them in PBS for 24 hours at 4 °C if you have not done it already.
Before making slices, it is very important to locate the brain regions which you want to image with a multiphoton or confocal microscope (Allen Brain Atlas is a great resource) if you did stereotactic surgery, you could use co-ordinates to identify regions as well. For orientation, marks could be made on the sides by inserting a small pin as well.
-
14.
For whole brain tissue clearing or slices, transfer to the FACT clearing solution. Use 5 ml clearing solution for a whole-brain and 2 ml for slices.
-
15.
Incubate at 37 °C with 70 RPM. Change the solution every 2 days.
-
16.
Perform a visual inspection of clearing by checking every time while changing the solution. Take pictures for your record. A cleared brain will be near transparent.
Alternate Protocol 1: Clearing brain tissue with passive clarity
Introductory Paragraph:
The FACT protocol is relatively fast, reproducible, and easier to implement than many other tissue clearing techniques (Xu et al., 2017). It has also been shown to work with cellular, subcellular imaging of the brain as well as blood vasculature (Khoradmehr, Mazaheri, Anvari, & Tamadon, 2019; Mohammad Rezazadeh et al., 2018). However, this protocol might not be the best option while imaging DNA or RNA in some cells in the brain (Ueda et al., 2020a). Another option is to use passive clarity which utilizes hydrogel polymerization with a slightly different composition of reagents to perform tissue clearing. This causes an increase in the size of the brain and takes longer clearing time but helps to preserve DNA and RNA composition better(Yang et al., 2014). The major component of passive tissue clearing is still SDS, but the tissue is subjected to hydrogel polymerization prior to the SDS solution. This causes the increased swelling, but it is better suited for applications where preserving RNA/DNA is needed (Mortazavi, Stankiewicz, & Zhdanova, 2019).
The first step of the protocol discusses how to perform hydrogel prefusion, fixation and polymerization. In contrast to the the FACT protocol, where only 4% PFA is used for perfusion and fixation, the hydrogel fixation solution consists of acrylamide, bisacrylamide, polymerization initiator, and 4% PFA. The next step discusses the initiation of polymerization and tissue clearing by using a passive clarity clearing solution.
Materials
50 ml centrifuge tubes (VWR, catalog number: 89039-658)
Acrylamide (Catalog number: 97063-560, VWR international)
Bisacrylamide (Catalog number: IC800172, VWR international)
40 % acrylamide solution
2 % bisacrylamide solution
Phosphate Buffer Saline (PBS, pH=7, BDH7447-1, VWR Chemicals)
ddH2O
Polymerization thermal initiator VA044 (Novachem cat. No. 017-19362 or Wako, cat. No. VA-044)
Hydrogel fixation solution (see recipe)
Polymerization initiator device (Catalog number: C20001, X-clarity polymerization system
Boric acid (Catalog number: 470300-412, VWR international)
1 M Boric acid
Sodium dodecyl sulfate (VWR Catalog no. 97064-496, VWR International)
Sodium hydroxide (Catalog number: 97064-476, VWR international)
Passive clarity clearing solution stock (see the recipe)
Passive clarity clearing solution (see the recipe)
PBST
Refrigerator
Incubator
Freezer
Nitrogen
Polymerization initiator (Logos biosystem, catalog no.: C20001)
X-clarity polymerization system or any other polymerization instrument
Protocol steps with step annotations:
Hydrogel polymerization
All the hydrogel polymerization reagents must be kept on ice before starting polymerization.
- Prepare (see the recipe)
- Hydrogel fixation solution
- Passive clarity clearing solution stock
- Passive clarity clearing solution
-
Perform steps 1-7 as described in the FACT clearing protocol.
PBS perfusion process is exactly the same for FACT and passive clarity clearing protocol.
-
Thaw the frozen hydrogel fixation solution completely, gently mix by inverting the tube, and perfuse the mouse brain with 30 ml of hydrogel fixation solution instead of 4 % PFA.
The hydrogel fixation solution must be completely thawed, it should look transparent, but it should still be kept in cold (put it in ice). Make sure there are no air bubbles and precipitate in the solution.
Harvest the brain as described in protocol 1 and transfer the brain tissue to a vial containing 20 ml of ice-cold hydrogel fixation solution. Keep it in ice and transfer it to a 4 °C refrigerator for 48 hours.
-
Perform degassing of hydrogel-fixed brain tissue with nitrogen for 3-5 minutes.
This is important because oxygen will slow down hydrogel polymerization and affect the tissue clearing process. Make sure all the air in the vial is replaced by nitrogen.
Perform hydrogel polymerization of the brain tissue/slices by using an X-clarity polymerization system or other similar polymerization instruments. Put a tube containing sample with hydrogel polymerization solution inside the chamber and run the instrument at −90 kPa vacuum and 37 °C for 3 hours. This will form a tissue-hydrogel polymer.
-
Shake the sample in a shaker for 1-2 minutes. Rinse the sample with PBS 3 times.
The sample can be stored in PBS at room temperature for 1-2 weeks.
Passive clarity clearing
The passive clarity clearing process is same for the slices and whole brains.
-
8.
Remove excess hydrogel by washing with PBS 3 times.
-
9.
Transfer the brain tissue into a vial containing a passive clarity clearing solution.
-
10.
Incubate the brain in passive clarity clearing solution at 37 °C until the brain clears completely. Change the solution every 2 days.
-
11.
Continue clearing until the tissue becomes nearly transparent.
Wash the cleared tissue with PBST 3 times. This will help to remove excessive SDS. The tissue can be stored in PBST at room temperature upto 6 months
Basic protocol title 2: Antibody labeling and refractive index matching
Introductory Paragraph:
This protocol discusses the steps to perform antibody labeling in FACT or passive clarity cleared whole brain tissue as well as region-specific slices. The cleared tissue will look slightly less transparent in antibody solution, but it will look clear after performing treatment with a 2,2′-Thiodiethanol or 2,2'-sulfanediyldi(ethan-1-ol) (TDE) solution after the antibody labeling. The first step of the protocol is to wash the tissue with PBS containing Triton X-100 (PBST). In the next step, permeabilization is achieved by treating the tissue with Tween-20 containing PBS solution (PBST-20). This helps penetration of the primary and secondary antibodies (see Table 1). Special attention should be given in selecting ‘astrocyte -specific’ antibodies as they can be cross-reactive. Table 2 provides a timing of the various wash steps.
Table 1:
Common astrocyte markers
| Protein/Marker | Description | Expression | Function | Antibody |
|---|---|---|---|---|
| Glial fibrillary acidic protein (GFAP) | An intermediate filament protein(Lee, Messing, Su, & Brenner, 2008) | Whole-brain astrocytes, overexpressed in reactive astrocytes | Important during brain development, Overexpresses during reactive astrocytosis | Anti-GFAP |
| EAAT1/GLAST | Astrocyte transporter (Miralles et al., 2001) | Whole-brain astrocytes | Transports L-Glutamate and Aspartate | Anti-GLAST, Anti-EAAT1 |
| EAAT2/GLT-1 | Astrocyte transporter (Miralles et al., 2001) | Whole-brain astrocytes but also expressed at lower levels on neurons(Petr et al., 2015) | Mediates uptake of L-Glutamate and Aspartate | Anti-EAAT2, Anti-GLT-1 |
| Glutamine synthetase | Astrocyte enzyme (Rose, Verkhratsky, & Parpura, 2013) | Whole-brain astrocytes | Catalyzes Glutamate to Glutamine conversion | Anti-Glutamine synthetase |
| S100 β | Calcium-binding protein (Z. Zhang et al., 2019) | Whole-brain astrocytes, *some Schwann cells and oligodendrocytes | Binds Zn2+ with high affinity and Ca2+ with medium affinity | Anti-S100 β |
| Alcohol dehydrogenase 1l1 (ALDH1L1) | Astrocyte enzyme (Cahoy et al., 2008) | Whole-brain astrocytes | Folate conversion | Anti-ALDH1L1 |
| Aquaporin-4 | Water channel protein (Mader & Brimberg, 2019) | End feet of astrocytes and some blood vessels | Water transportation and homeostatic control | Anti-Aquaporin-4 |
| N-myc downregulated gene 2 (NDRG2) | Tumor suppressor protein (Flügge et al., 2014) | Mature non-reactive astrocytes | Bind cytosol of protoplasmic and fibrous astrocytes | Anti-NDRG2 |
| Sulphorhodamine (SR101) ** | An astrocyte dye/Preferential astrocyte marker(Nimmerjahn, Kirchhoff, Kerr, & Helmchen, 2004; Rasmussen, Nedergaard, & Petersen, 2016) | Astrocytes and some oligodendrocytes | Spreads through the syncytium via astrocytic gap junctions(Rasmussen et al., 2016) | SR101 |
Counterstaining with another marker should be performed
SR101 is a rhodamine dye that has been commonly used for labeling astrocytes both in vitro and in vivo. Care must be taken as it is also shown to label oligodendrocytes in longer exposure (Hill & Grutzendler, 2014).
Table 2:
Reagents required for antibody labeling
| Process | Solution | Duration (for 1-2 mm slices) |
Duration (for hemibrain or whole brain) |
|---|---|---|---|
| Washing (PBST) | 0.1 % Triton X-100 in PBS | 1 hour (3 times) | 1 hour (3 times) |
| Permeabilization (PBST-20) | 0.2 % Tween-20 in PBS | 1 hour | 1 hour |
| Blocking | 0.2% Triton-X-100, 10% DMSO, 6% Normal Donkey Serum in PBS | 1 day | 1 day |
| Primary antibody | 0.2 % Triton X-100, 10% Normal Donkey Serum in PBS | 2-3 days | 1 week |
| Washing | 0.1 % Triton X-100 in PBS | 1 hour (3 times) | 1 hour (3 times) |
| Secondary antibody | 0.2 % Triton X-100, 10X Normal Donkey Serum in PBS | 2-3 days | 1 week |
| Washing | 0.1 % Triton X-100 in PBS | 1 hour (3 times) | 1 hour (3 times) |
| TDE clearing (TDE I) | 20% TDE for 1-2 mm slices, 30% TDE for hemibrain or whole brain | 1-2 hours for 1-2 mm slices, | 24 hours for hemibrain or whole-brain* |
| Refractive index matching (TDE II) | 47% TDE for 1-2 mm slices, 63% TDE for hemibrain or whole brain | 2 hours for 1-2 mm slices | 24-48 hours for hemibrain or whole-brain* |
hemibrain or whole brain should be imaged by using light-sheet microscopy. 1-2 mm slices can be imaged using confocal or multiphoton microscopy.
Materials
Triton X-100 (Catalog number: 97063-864, VWR international)
Phosphate Buffer Saline (PBS, pH=7, BDH7447-1, VWR Chemicals)
Tween-20 (Catalog number: 97063-872, VWR international)
PBST solution (see recipe)
PBST-20 solution (see recipe)
Dimethyl Sulfoxide (DMSO, Catalog number: BDH1115-1LP, VWR international)
Normal Donkey Serum (Catalog number: 80054-446, VWR international)
Primary antibody (anti-GFAP, 1:100, Thermo-Fisher, PA1-10019, RRID: AB_1074611)
Secondary antibody (anti Rabbit Alexa 488, 1:200, NC0449336, RRID: AB_2313584)
The primary antibody incubation solution (see recipe)
The secondary antibody incubation solution (see recipe)
2,2′-Thiodiethanol or 2,2'-sulfanediyldi(ethan-1-ol) (TDE, 166782-100G, EC Number 203-874-3, Millipore Sigma)
TDE I solution (see recipe)
TDE II solution (see recipe)
Shaker with incubator (VWR, catalog number: 75874-516)
Ice
Protocol steps with step annotations:
Note: If you are using a transgenic/knock-in animal that expresses astrocytes labeled with a fluorescent protein, you do not need to perform antibody labeling steps. You can directly perform the refractive index matching steps with TDE. However, if you are trying to study the expression of a specific protein or want to use a second astrocyte marker, proceed with antibody labeling steps.
This protocol has two sections
Antibody labeling
- Prepare the necessary reagents before moving to the next step.
- Wash buffer
- Permeabilization buffer
- Blocking buffer
- Primary antibody solution
- Secondary antibody solution
- TDE I solution
TDE II solutionWash the whole brain or the slices with wash buffer3 times. Make sure the SDS is washed completely out. Complete removal of SDS will result in an absence of soapiness in the solution. Change the vial you have used for clearing and use a new vial for the rest of the steps.
Permeabilize the cleared whole brain or the tissues with the permeabilization buffer by incubating the brain tissue/slices at 37 °C for 1 hour.
Block the nonspecific binding sites in the cleared tissue using the blocking buffer. This should be performed by incubating the brain/slice tissue in the blocking buffer for 24 hours at 37 °C.
-
Incubate the tissue with 1:100 diluted primary antibody solution. The incubation should be performed at 37 °C for 48 to 72 hours for slices and 7 days for a hemibrain or a whole brain. Stain the slice with an astrocyte-specific primary antibody.
One important thing to consider while selecting a secondary antibody is to make sure the excitation and emission wavelength of the fluorophore conjugate in the secondary antibody matches the excitation and emission lasers available in the microscopy system you are planning to use.
Wash the tissue with washing buffer 3 times in an interval of 1 hour at room temperature.
Incubate the brain tissue with secondary antibody 1:100-1:500 concentration at 37 °C for 2-3 days for slices and 1 week for a hemibrain or a whole brain.
Wash the tissue with washing buffer 3 times in an interval of 1 hour at room temperature.
Refractive index matching
-
9.
Prepare 20 % and 47% of TDE solution for brain slices to image them in a confocal or multiphoton microscope.
-
10.
Prepare 30 % and 63 % of TDE solution for a whole-brain or a hemibrain to image them in a light-sheet microscope.
-
11.
Incubate the slices in 20 % TDE at 37 °C for 2 hours. Incubate the whole brain or hemibrain in 30 % TDE at 37° C for 24 hours.
-
12.
Incubate the slices in 47% TDE at 37 °C for 2 hours. Incubate the whole brain or the hemibrain in 63 % TDE at 37 °C for 24-48 hours.
The refractive index of TDE depends on its concentration which can be used to perform TDE clearing and refractive index matching (Aoyagi, Kawakami, Osanai, Hibi, & Nemoto, 2015).
Basic protocol 3: Fluorescence imaging and characterization of astrocyte morphology
Introductory paragraph:
Astrocyte morphology differs under different neuropathological conditions. Glial fibrillary acidic protein (GFAP) is one of the common markers of reactive astrocytosis. The sample should be in TDE II solution during fluorescence imaging. Imaris software can be used to perform a quantitative analysis of morphological parameters. Fluorescence imaging in a light sheet system or a confocal/multiphoton system should be performed by following specific steps which are provided by the respective instrument manufacturer and the microscopy core facility. We have, however, tried to provide general instructions/information which will be applicable for all the instruments.
Materials:
Cleared brain slices/ hemibrains/ whole brain
-
TDE II clearing solution (Refractive index matching solution)
Alternatively, RIMS solution from Logos biosystem or Focus clear can also be used.
# 1.5 Glass bottom dish (MatTek, Part No: P35G-1.5-14-C)
# 1.5 Glass bottom plate (Cellvis, Catalog: P24-1.5H-N)
Cyanoacrylate Glue (VWR, Catalog: 470024-626)
Tweezers (VWR, Catalog: 82027-442)
A circular PDMS holder or a 3D printed holder (with outer diameter 33 mm, thickness 2 mm, and inner diameter 19 mm.)
Heavy-Duty Staples (VWR, Catalog: 500025-679)
Staple (Item # 749601, Office Depot)
A light sheet microscope Z1 (Zeiss)
A confocal (Nikon A1R, RRID:SCR_020317 )/multiphoton (Zeiss 880, RRID:SCR_020925) microscope
Hard drive or a data storage device
Bitplane Imaris Viewer (Oxford instruments)
Bitplane Imaris (Oxford instruments, RRID:SCR_007370 )
ImageJ (NIH, RRID:SCR_001935)
Protocol steps with step annotations:
-
Mount the whole brain or slices in an appropriate dish or a holder (based on the holder requirement in the available microscopy system).
In general, #1.5 glass-bottom dishes can be used for mounting 1- or 2-mm thick slices. Put the slice holder (3-d printed) inside the glass bottom dish, make sure it is tight. Put the brain slice inside the holder and fill it with TDE II solution. Cover it with a # 1.5 glass coverslip for inverted multiphoton or confocal imaging. It can be sealed with nail polish for long-term imaging or shipping the sample from one location to another location. For an upright multiphoton or confocal microscope, leave the top open. The bottom of the brain can be glued to the glass-bottom dish. Optical wells can also be used for attaching brains.
-
Perform imaging of the cleared brain tissue with an available light sheet/confocal or multiphoton microscopy system.
For light sheet systems which require attaching the sample in a cylindrical holder, the brain can be glued to a staple and attached to a cylindrical holder which will be immersed in a refractive index matching solution and images will be recorded from the objective.
-
Choose an objective with an appropriate numerical aperture (NA) and magnification. Perform light-sheet imaging of the hemibrain or the whole brain OR perform confocal/multiphoton imaging of slices. Choose an appropriate wavelength in the microscopy system based on the antibody or the marker you used (check the excitation and emission wavelengths tables 3 and 4).
For a light-sheet microscope (5-20X), smaller objectives are used which captures a large field of view. NA refers to the ability of the objective to gather light and a larger NA objective allows to collect more light and hence increases the resolution. Magnification, on the other hand, refers to the size of the image in comparison to the size of the object. 60X Oil objective is a good objective for confocal or multiphoton imaging of astrocytes. The wavelength should be selected based on the absorbance of the secondary antibody.
-
Start with lower power and gain and increase them slowly to obtain the best possible image with no saturation. For the light sheet, choose different values for left and right light-sheet offset and find the optimum value where the best possible image is obtained.
Try to minimize high power as it can cause photobleaching and tissue damage. Try to increase the gain for increasing signal intensity. Excessive increase in gain may cause lower signal/noise so optimization is necessary. Pixel dwell time is another way of increasing signal but it can also cause photobleaching and increase the time of imaging.
-
Define upper and lower limits for Z-stacks and save the file in the appropriate folder or your hard drive.
Make sure to collect z stacks so that 3-dimensional information will be recorded. Choose the thickness of z stack less than 3 micrometers so that astrocyte morphology can be easily These kinds of files have typically large sizes so a hard drive is required to save them.
-
Collect tiles for visualizing all the parts of the slices or the whole brain. Make sure each tile has exactly the same number of z stacks.
Tiles can later be stitched using microscopy data analysis software. Make sure to keep a note of the number of rows, columns, overlap percentage, and the number of z-stacks per tiles. This information is important for image analysis
-
Open the z stacks in Imaris viewer and convert the original file format into “ims” file format.
In the case of tiles, stitch them using appropriate software before analyzing them in imaris. Imaris viewer can be used for viewing the z stacks. For morphological analysis, convert the file into ims using Imaris viewer.
Plug the hard drive into a computer that has Imaris preinstalled on it. Open the files converted in “ims” format in the software.
Go to 3 D view.
Select surfaces.
-
Create an artificial surface by adjusting parameters.
These parameters will depend on sample fluorescence and need to be optimized based on your sample.
-
Choose threshold which allows to select full astrocytic feature (3 dimensional) and construct artificial surface. Testen et al 2020 have detailed a protocol to image individual astrocytes by using a confocal microscope(Testen, Kim, & Reissner, 2020).
A sphericity filter can be used to remove if some blood vasculatures are also labeled in the image. Using a 0.3-1 sphericity filter seems to work well for astrocytes.
Measure area, volume, and other morphological parameters.
-
Save the statistical data and make plots.
For studying GFAP expression by fluorescence imaging, special attention should be given. DAPI can be used for normalization. A region of interest can be selected from the tissue sample and both GFAP and DAPI fluorescence should be measured. The GFAP fluorescence should be normalized against DAPI fluorescence to compare across different samples.
Table 3:
Excitation and emission wavelengths of common fluorophores
| Fluorophore | Absorption wavelength (nm) |
Emission wavelength (nm) |
Visible color |
|---|---|---|---|
| Hydroxycoumarin | 325 | 386 | Blue |
| Methoxy coumarin | 360 | 410 | Blue |
| Alexa fluor | 345 | 442 | Blue |
| DAPI | 345 | 455 | Blue |
| Hoechst 33258 | 345 | 478 | Blue |
| Aminocoumarin | 350 | 455 | Blue |
| SYTOX blue | 431 | 480 | Blue |
| Hoechst 33342 | 343 | 483 | Blue |
| YOYO-1 | 489 | 509 | Dark green |
| Cynin 2 | 490 | 510 | Dark green |
| FAM | 495 | 516 | Dark green |
| Alexa fluor 488 | 494 | 517 | Light green |
| Fluorescein FITC | 495 | 518 | Light green |
| SYTOX green | 504 | 533 | Light green |
| TOTO 1, TO-PRO-1 | 509 | 533 | Light green |
| Alexa fluor 430 | 430 | 545 | Light green |
| Alexa fluor 532 | 530 | 555 | Light green |
| HEX | 535 | 556 | Light green |
| Cynin 3 | 550 | 570 | Yellow |
| SYTOX Orange | 547 | 570 | Yellow |
| TRITC | 547 | 572 | Yellow |
| Alexa fluor 546 | 556 | 573 | Yellow |
| Alexa fluor 555 | 555 | 565 | |
| Chromomycin A3 | 445 | 575 | Yellow |
| Mithramycin | 445 | 575 | Yellow |
| R-phycoerythrin (PE) | 480,565 | 578 | Yellow |
| Rhodamine Red-X | 560 | 580 | Orange/Red |
| Tamara | 565 | 580 | Orange/Red |
| Cynin 3.5 | 581 | 596 | Red |
| Rox | 575 | 602 | Red |
| Alexa fluor 568 | 578 | 603 | Red |
| Red 613 | 480,565 | 613 | Red |
| Texas Red | 589 | 615 | Red |
| Alexa fluor 594 | 590 | 617 | Red |
| Alexa fluor 633 | 621 | 639 | Red |
| Cynin 5 | 650 | 670 | Red |
| Alexa fluor 660 | 663 | 690 | Red |
| Cynin 5.5 | 675 | 694 | Red |
| Propidium iodide | 536 | 617 | Red |
| Ethidium bromide | 493 | 620 | Red |
| Alexa fluor 680 | 679 | 702 | Red |
| Cynin 7 | 743 | 770 | Red |
Table 4:
Excitation and emission wavelengths of common fluorescent proteins
| Fluorescent Protein |
Absorption Wavelength (nm) |
Emission Wavelength (nm) |
Visible color | Reference |
|---|---|---|---|---|
| EBFP2 | 383 | 448 | Blue | (Ai, Shaner, Cheng, Tsien, & Campbell, 2007) |
| mTagBFP2 | 399 | 454 | Blue | (Subach, Cranfill, Davidson, & Verkhusha, 2011) |
| mTurquoise2 | 434 | 474 | Cyan | (Goedhart et al., 2012) |
| Cerulean3 | 433 | 475 | Cyan | (Markwardt et al., 2011) |
| TagGFP | 476 | 496 | Green | (Xia et al., 2002) |
| TurboGFP | 482 | 502 | Green | (Shagin et al., 2004) |
| Azami Green | 492 | 505 | Green | (Karasawa, Araki, Yamamoto-Hino, & Miyawaki, 2003) |
| TagGFP2 | 482 | 506 | Green | (Subach et al., 2008; Xia et al., 2002) |
| Emerald | 487 | 509 | Green | (Cubitt, Woollenweber, & Heim, 1998) |
| Superfolder GFP | 488 | 510 | Green | (Pédelacq, Cabantous, Tran, Terwilliger, & Waldo, 2006) |
| Clover | 505 | 515 | Green | (Lam et al., 2012) |
| mNeonGreen | 506 | 517 | Green | (Shaner et al., 2013) |
| mYFP | 514 | 527 | Yellow | (Kremers, Goedhart, van Munster, & Gadella, 2006) |
| mVenus | 515 | 528 | Yellow | (Nagai et al., 2002) |
| mCitrine | 516 | 529 | Yellow | (Griesbeck, Baird, Campbell, Zacharias, & Tsien, 2001) |
| TurboYFP | 525 | 538 | Yellow | (Shagin et al., 2004) |
| mPapaya1 | 530 | 541 | Yellow | (Hoi et al., 2013) |
| Kusabira Orange2 | 551 | 565 | Orange | (Karasawa, Araki, Nagai, Mizuno, & Miyawaki, 2004) |
| mOrange 2 | 549 | 565 | Orange | (Shaner et al., 2008) |
| TagRFP | 555 | 584 | Orange | (Merzlyak et al., 2007) |
| TagRFP-T | 555 | 584 | Orange | (Shaner et al., 2008) |
| TurboRFP | 553 | 574 | Red | Merzlyak et al., 2007) |
| tdTomato | 554 | 581 | Red | (Shaner et al., 2004) |
| mRuby2 | 559 | 600 | Red | (Lam et al., 2012) |
| FusionRed | 580 | 610 | Red | (Shemiakina et al., 2012) |
| mCherry | 587 | 610 | Red | (Shaner et al., 2004) |
| mKate2 | 588 | 630 | Red | (Shcherbo et al., 2009) |
| E2-Crimson | 605 | 646 | Far-Red | (Strack et al., 2009) |
| eqFP650 | 592 | 650 | Far-Red | (Shcherbo et al., 2010) |
| mNeptune2 | 599 | 651 | Far-Red | (Chu et al., 2014) |
| mCardinal | 604 | 659 | Far-Red | (Chu et al., 2014) |
REAGENTS AND SOLUTIONS:
4 % (w/v) PFA solution (pH 7.4)
4% Paraformaldehyde
96 % PBS
NaOH
HCl
Dissolve 20 g of Paraformaldehyde in 400 ml of PBS. Use a magnetic bar and stirrer to dissolve by maintaining temperature around 60° C (do not heat higher than 60°). If it does not dissolve add 2-3 pellets of NaOH. Adjust the volume to 500 ml and pH to 7-7.5. The solution can be stored at 4°C for few months.
Fatal plus injection solution 10% (v/v):
10 % Fatal plus
90 % PBS
Dissolve 1 ml of fatal plus in 9 ml of PBS. It can be stored at room temperature for 6 months.
FACTS clearing solution:
8% (w/v) Sodium dodecyl sulfate
92% PBS
Dissolve 8 mg of SDS in 100 ml PBS. FACTS solution can be stored at room temperature for 6 months.
Hydrogel fixation solution
4% Acrylamide
0.05% Bisacrylamide
0.25% VA-044 Initiator
0.2 M boric acid
4% sodium dodecyl sulfate (SDS)
PBS
4 % PFA
Prepare 400 ml of hydrogel fixation solution by using 40 ml of 40% acrylamide which will results in 4% final concentration of acrylamide, 10 ml of 2% Bisacrylamide which will result in 0.05% of Bisacrylamide, 16 g of paraformaldehyde powder, 40 ml of 10X PBS, 210 ml of distilled water and 1 g of VA-044 thermal initiator which results in 0.25% (w/v) final concentration. All the reagents should be kept on ice. The ratio of acrylamide to bisacrylamide should be maintained at approximately 80:1. A lower total percentage of acrylamide and bisacrylamide can be used by maintaining the same ratio ( acryalammide to bisacrylamide of approximately 80:1) but lower concentration of both in the whole solution. A lower ratio results in larger pore sizes and increases the speed of clearing and more efficient antibody penetration. A higher ratio, on the other hand, helps to preserve the DNA and RNA content of the tissue. The reagents should be stored at −20°C.
Passive clarity clearing solution stock (pH =8.5):
1 M boric acid
40% sodium dodecyl sulfate (SDS)
dH2O
NaOH
A stock solution with 40 % SDS in distilled water and 1 M boric acid buffer can be prepared for long-term storage. The pH of the solution should be adjusted to 8.5 by adding NaOH pellets. This stock solution can be stored at room temperature and can be diluted 5 times in distilled water for a fresh application.
The final concentration of diluted clearing solution is:
0.2 M boric acid
8% sodium dodecyl sulfate (SDS)
dH2O
NaOH
Wash buffer
0.1 % (v/v) Triton X-100
PBS
Dissolve 100 μl of Triton X-100 in PBS to make 100 ml total volume.
Permeabilization buffer
0.2 % (v/v) Tween-20
PBS
Dissolve 40 μl of Tween-20 in 20 ml of PBS.
Blocking buffer
0.2% (v/v) Triton-X-100
10% (v/v) DMSO
6% (v/v) Normal Donkey Serum
PBS
Mix 20 μl of Triton X-100 with 1 ml DMSO and 600 μl of Donkey serum to make total 10 ml of solution.
Primary antibody solution
0.2 % Triton X-100
(rabbit, anti-GFAP, 1:100, Thermo-Fisher, PA1-10019, RRID: AB_1074611)
Secondary antibody solution
0.2 % Triton X-100
(anti Rabbit Alexa 488, 1:200, NC0449336, RRID: AB_2313584)
TDE I solution (for slices)
20 % TDE
80 % water
TDE I solution (for a hemibran or a wholebrain)
30 % TDE
70 % water
TDE II solution (for slices)
47 % TDE
53 % water
TDE II solution (for a hemibrain or a wholebrain)
63 % TDE
37 % water
Prepare 20%, 30%, 47% and 63% TDE solutions. For thick slices 1-2 mm prepare 20% and 47 % (w/v) TDE solutions. For whole brain prepare 30% and 63% of TDE solutions.
COMMENTARY:
Background Information:
Optical imaging of thick tissue, particularly from intact organs, is challenging with traditional microscopy approaches. To enhance the imaging depth in biological tissues, researchers have optimized chemical solutions with biological samples to develop reagents for tissue clearing methods (Chance, Liu, Kitai, & Zhang, 1995). There are three primary approaches to conduct tissue clearing: using hydrophobic reagents, hydrophilic reagents, and hydrogel-based methods (Ueda et al., 2020). The first step in each of these clearing processes is to harvest the brain tissue, via cardiac perfusion to remove blood from the brain which can otherwise interfere with the imaging process (Gage, Kipke, & Shain, 2012). Thus, a good perfusion is the key to success for any tissue clearing method. Subsequently, fixation and post-fixation is performed to preserve morphological and anatomical features at the cellular and subcellular level in the brain by terminating ongoing biochemical reactions (McFadden et al., 2019). The fixed brain is then subjected to tissue clearing. Two of the major steps in the clearing process are lipid removal and refractive index matching. Lipid removal can be achieved in a variety of ways (Ueda et al., 2020b). In the hydrogel-based technique, a polymeric hydrogel is formed which preserves subcellular components of the tissue such as DNA and RNA. In hydrophobic methods, water is removed from the tissue by using water-miscible solvents which is then followed by dilipidation. Hydrophilic methods, on the other hand, use mild detergents to remove lipids from the lipid bilayer (Ueda et al., 2020b). During the dilipidation process the size of the brain tissue expands. Incubation in the mounting solution is used to shrink the brain back to the normal size (Richardson & Lichtman, 2017).
In this protocol SDS, an ionic detergent is used to remove lipids from the lipid bilayer in order to increase the transparency of the tissue. This also improves the penetration of antibodies providing access to the inner parts of the tissue. TDE solution is used as a refractive index matching solution. TDE is dissolved in water and its refractive index depends on its concentration (Figure 3) so it can be used for mounting the brain or the slices (Costantini et al., 2015). The tissue clearing process removes opaque molecules such as lipids from the tissue without significantly influencing the cellular and subcellular contents of the tissue. The highest efficiency in the clearing is achieved at a temperature of approximately 37 °C.
FIGURE 3:
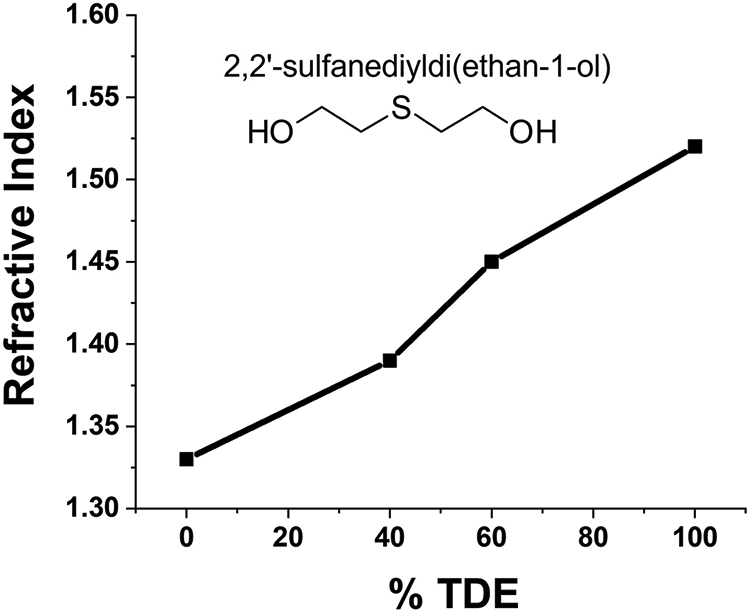
Variation of the refractive index of TDE solution at different concentrations
One major advantage of this technique is it allows one to preserve astrocyte morphology in the cleared tissue. It is a rapid process in comparison to other clearing techniques, and there is no forceful removal of lipid using electrophoresis. It also does not require hydrogel and is faster than other passive clearing methods such as passive Clarity and Scales. For studying DNA and RNA via fluorescence markers, we have included a protocol for passive clarity which is better than the hydrophilic method. This method has already been used to study microglia morphology and blood vasculature. We believe this technique can be used to study other cell types in the brain as well as other biological tissues in the body.
Critical Parameters and Troubleshooting:
As discussed above, perfusion to clear the brain of blood is essential to both the tissue clearing process and to reducing background fluorescence that will hinder visualization. A thorough understanding of cardiac ananotomy is essential. If the right atrium is used in error the fluid will be pushed to the lungs which results in their inflation. The perfusion liquid is then pushed out through the nose or mouth. This can be avoided by carefully clamping the needle in the heart after insertion into the correct chamber to avoid inadvertently piercing the other chambers (Gage et al., 2012). Also, insertion of the needle to far into the heart can result in the aorta being punctured which will also result in leakage of blood and fluid and insufficient clearling of blood from the brain. Perfusion should be continued until the liver turns tan-colored and the solution is clear, a flow rate of approximately 5-10 ml/min can be used in mice. 40-50 ml of PBS is typically sufficient to clear blood from a mouse for tissue clearing. Subsequently, perfusion with 20-30 ml of 4% PFA (in PBS) should be performed for fixation. During post-extraction fixation, a the volume of fixative should be at least 10-15 times the volume of tissue.
Special attention should be given to the temperature and speed of agitation during the clearing process, as lower temperatures result in a significantly longer time to clear the tissue. Agitation is performed to allow the clearing solution to mix thoroughly throughout the sample and to enter the brain tissue. In the case of samples containing a fluorescent protein label, aluminum foil or other light blocking material can be used to minimize the samples exposure to light which can otherwise cause loss of fluorescence. The clearing solution should be changed every 2 days which helps to remove SDS that contains a higher concentration of lipids that were extracted from the tissue and replenishes the solvent which speeds up the clearing process. The volume of the clearing solution should be sufficient to immerse the tissue during agitation.
Antibody selection and penetration of antibodies deep into the brain tissue is another key factor for imaging of astrocytes. Antibody selection should be evaluated carefully as antibody cross-reactivity may result in fluorescence from unintended parts of the sample. Another factor is allowing sufficient time for antibodies to label the interior of the brain. A relatively high concentration (approximately 10-100 fold higher than used for labeling typical slices (~20-40 μm thick) of antibodies should be used for whole-brain imaging. Penetration of antibodies throughout the tissue typically requires 7 days of incubation. We have discussed several potential problems and troubleshooting steps in the subsequent section. See Table 5 for Troubleshooting.
Table 5:
Troubleshooting
| Problem | Possible Cause | Solution |
|---|---|---|
| Inflated lungs during perfusion, liquid coming from mouth or nose | The needle might have inserted into the right atrium or right ventricle instead of the left ventricle | Remove the needle and insert it into the left ventricle |
| Fixation tremor is not observed during paraformaldehyde perfusion/fixation | The needle might have inserted into the wrong place | Re-insert the needle into the left ventricle |
| Blood vessels can be seen in the perfused brain | Perfusion was unsuccessful | Perform perfusion by inserting a syringe needle in the left ventricle and make a cut in the right atrium Increase the volume of solutions used or the timing of the perfusion process |
| The brain is too soft in clearing solution | The brain was not fixed properly | Perform post-extraction fixation for a longer time 48-72 hours in 4 % paraformaldehyde (in PBS) |
| Brain tissue becomes unclear/opaque after storing in PBS for a long time | The tissue might have absorbed salt | Perform TDE clearing for 24-48 hours |
| Fluorescence is too low | Antibody concentration might not have been enough, Exposure duration was not enough |
Use a higher concentration of antibody and incubate for a longer period |
| Vasculature/blood vessels are observed during astrocyte imaging | Perfusion was unsuccessful | Perform perfusion by inserting a syringe needle in the left ventricle and make a cut in the right atrium Perform perfusion with a higher volume of PBS |
| The fluorescence signal is not obtained from the inner part of the tissue | Antibody might have difficulty in reaching inner sections of the brain | Perform longer incubation and use 1:50-1:100 concentration of antibodies Use polyclonal booster antibodies (such as anti-GFP alexa 488 which will enhance GFP fluorescence at the same fluorescence channel) If possible, perform brain sectioning (hemibrains) or slicing so that antibodies can reach to inner parts easily |
Understanding Results:
Figure 4 shows regions in the mouse brain. Figure 5 illustrates cleared tissue mounting in different holders for confocal, multiphoton or light sheet microscopy. Figure 6 demonstrates the results from this tissue clearing technique. Tissue clearing will result in clear brain tissue or slices. Figure 6A shows a cleared brain slice of 2 mm thickness. There will be some expansion in the volume but it will return to normal after putting the brain in refractive index matching solution (TDE solutions). The final cleared tissue will be transparent. Confocal microscopy (Nikon A1R) was used for visualizing astrocyte morphology and Imaris (Bitplane) was used for image processing and data analysis. We have included images of astrocytes and morphological data from cleared brain tissue (Figure 6). These studies used anti-gfap Alexa 488 atibody and tdTomato fluorescent protein attached to aldh1l1, which are astrocyte-specific markers. 2 mm thick slices were prepared from the mouse brain and cleared with FACT reagents. Figure 6B shows astrocytes in aldh1l1-tdTomato mice and figure 6C shows astrocytes from a cleared tissue treated with anti-gfap Alexa 488 antibody. Surface area and volume plots were generated from imaris as shown in Figures 6E and 6F.
FIGURE 4:

Illustration of a mouse brain and various brain regions in the slices
FIGURE 5:

Illustration of sample holders for fluorescence microscopy of mouse brain tissue. (A) Mouse brain slices placed in a glass bottom dish with mounting solution and sealed with a coverslip. (B) A hemibrain mounted on a staple pin and (C) placed inside a light sheet microscope chamber. (D) A light sheet microscope imaging chamber which has (E) a magnet in an upper chamber and (F) a drawer to put the sample and refractive index matching solution in a chamber
FIGURE 6:

Example results and data for the study of astrocytes by the FACT tissue clearing method. (A) A cleared tissue of 2 mm thickness. (B)-(C) 3-dimensional image of astrocytes labeled with aldh1l1-tdTomat (knock-in) and anti-GFAP Alexa 488. (D) A 2D image of astrocytes labeled with anti-GFAP Alexa 488 showing astrocytic processes. (E)-(F) Example data for morphology characterization by Imaris.
Sometimes blood vessels might also appear while imaging astrocytes which might be due to the brain tissue not being perfused properly or because of resident autofluorescence in the tissue. Blood vessels are much longer and have cylindrical morphology with a lot of branching so they can be easily identified while imaging astrocytes. Blood vessels can be filtered during the data analysis process as these have different morphology and size range. The sphericity filter helps to remove nonspherical structures during data analysis in imaris.
Time Considerations:
The perfusion and fixation take 30-40 minutes, while the post-extraction fixation is performed for 1-2 days. The FACT clearing process takes a week for slices and 3 weeks for a whole-brain/hemibrain. Antibody staining and imaging also depend on the type of brain tissue used (2-3 weeks). On average perfusion, clearing, imaging, and data analysis take 2 months.
ACKNOWLEDGEMENTS:
The authors would like to acknowledge the UKY Light microscopy core and Imaging center for the use of their facilities. C. I. R. acknowledges support from the National Institutes of Health (DA038817). Schematic images were created with the help of Biorender (https://biorender.com/). Chemical structures were drawn with the help of Chemsketch.
Abbreviation
- GFAP
Glial fibrillary acidic protein
- FACT
fast free-of-acrylamide clearing tissue
- PFA
Paraformaldehyde
- PBS
Phosphate buffer saline
- PBST
Phoshpate buffer saline with 0.1% Triton X-100 (v/v)
- PBST-20
Phosphate buffer saline with 0.2% Tween-20 (v/v)
- TDE
2,2′-ThiodiethanolSDS: Sodium dodecyl sulfate
Footnotes
CONFLICT OF INTEREST STATEMENT:
The authors declare no conflict of interest.
KEY REFERENCES:
https://www.jove.com/t/3564/whole-animal-perfusion-fixation-for-rodents
This is a protocol article which shows how to perform perfusion in rats
https://www-nature-com.ezproxy.uky.edu/articles/s41583-019-0250-1.pdf
This is a review paper on different clearing techniques
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0160391
This article shows how to perform analysis of astrocyte morphology in cleared tissue.
INTERNET RESOURCES:
https://einsteinmed.org/research/facilities/fluorescent/fluorescent-proteins/common-proteins.aspx
https://www.abcam.com/ps/pdf/protocols/Fluorophore%20table.pdf
https://www.abcam.com/secondary-antibodies/fluorochrome-chart-a-complete-guide
https://mouse.brain-map.org/static/atlas
This is Allen brain atlas which helps to locate specific regions of the mouse brain.
https://www.setabiomedicals.com/2-photon-microscopy.html
This provides 2-photon wavelengths of some fluorophores.
https://imaris.oxinst.com/tutorials
This provides tutorials for imaris.
This provides a manual for Nikon A1R confocal microscope.
https://www.biotech.cornell.edu/sites/default/files/2020-06/LSM880%20Operating%20Manual.pdf
This is a manual for Zeiss 880 multiphoton microscope.
https://imaging.as.uky.edu/sites/default/files/LIGHTSHEET%20BASIC%20Z-STACK%20%20TIME-SERIES.pdf
This provides a manual for Zeiss light-sheet Z1 microscope.
DATA AVAILABILITY STATEMENT:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
LITERATURE CITED:
- Ai H.-w., Shaner NC, Cheng Z, Tsien RY, & Campbell REJB (2007). Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. 46(20), 5904–5910. [DOI] [PubMed] [Google Scholar]
- Allen NJ, & Barres BA (2009). Glia—more than just brain glue. Nature, 457(7230), 675–677. [DOI] [PubMed] [Google Scholar]
- Aoyagi Y, Kawakami R, Osanai H, Hibi T, & Nemoto T (2015). A rapid optical clearing protocol using 2, 2′-thiodiethanol for microscopic observation of fixed mouse brain. PLoS One, 10(1), e0116280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal S, Fu X, Masud A, Neupane K, & Richards C (2021). Single-Molecule Studies of Membrane Receptors from Brain Region Specific Nanovesicles. Bio-protocol, 11(10), e4018. doi: 10.21769/BioProtoc.4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal SP, Fu X, Sandin JN, Neupane KR, Lakes JE, Grady ME, & Richards CI (2021). Nicotine induces morphological and functional changes in astrocytes via nicotinic receptor activity. Glia. doi: 10.1002/glia.24011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badia-Soteras A, Octeau JC, Verheijen MH, & Khakh BS (2020). Assessing Neuron–Astrocyte Spatial Interactions Using the Neuron–Astrocyte Proximity Assay. Current protocols in neuroscience, 91(1), e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K, Jährling N, Saghafi S, Weiler R, & Dodt H-U (2012). Chemical clearing and dehydration of GFP expressing mouse brains. PLoS One, 7(3), e33916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, … Krupenko S. A. J. J. o. N. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. 28(1), 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Liu H, Kitai T, & Zhang Y. J. A. b. (1995). Effects of solutes on optical properties of biological materials: models, cells, and tissues. 227(2), 351–362. [DOI] [PubMed] [Google Scholar]
- Chu J, Haynes RD, Corbel SY, Li P, González-González E, Burg JS, … Baird M. A. J. N. m. (2014). Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. 11(5), 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini I, Ghobril J-P, Di Giovanna AP, Mascaro ALA, Silvestri L, Müllenbroich MC, … Sacconi L. J. S. r. (2015). A versatile clearing agent for multi-modal brain imaging. 5(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SW, Carter RE, Aronson JD, Kodandaramaiah SB, Ebner TJ, & Chen CC (2021). Through the looking glass: a review of cranial window technology for optical access to the brain. Journal of neuroscience methods, 109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubitt AB, Woollenweber LA, & Heim R. J. M. i. c. b. (1998). Understanding structure—function relationships in the Aequorea victoria green fluorescent protein. 58, 19–30. [DOI] [PubMed] [Google Scholar]
- Flügge G, Araya-Callis C, Garea-Rodriguez E, Stadelmann-Nessler C, Fuchs EJC, & research, t. (2014). NDRG2 as a marker protein for brain astrocytes. 357(1), 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage GJ, Kipke DR, & Shain W (2012). Whole animal perfusion fixation for rodents. JoVE (Journal of Visualized Experiments)(65), e3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J, Von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, … Royant A. J. N. c. (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. 3(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, & Tsien R. Y. J. J. o. b. c. (2001). Reducing the environmental sensitivity of yellow fluorescent protein: mechanism and applications. 276(31), 29188–29194. [DOI] [PubMed] [Google Scholar]
- Hill RA, & Grutzendler J. J. N. m. (2014). In vivo imaging of oligodendrocytes with sulforhodamine 101. 11(11), 1081–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GE, Murphy KJ, & Sita L. V. J. C. p. i. n. (2016). The importance of titrating antibodies for immunocytochemical methods. 76(1), 2.12. 11–12.12. 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoi H, Howe ES, Ding Y, Zhang W, Baird MA, Sell BR, … biology. (2013). An engineered monomeric Zoanthus sp. yellow fluorescent protein. 20(10), 1296–1304. [DOI] [PubMed] [Google Scholar]
- Karasawa S, Araki T, Nagai T, Mizuno H, & Miyawaki AJBJ (2004). Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. 381(1), 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa S, Araki T, Yamamoto-Hino M, & Miyawaki A. J. J. o. B. C. (2003). A green-emitting fluorescent protein from Galaxeidae coral and its monomeric version for use in fluorescent labeling. 278(36), 34167–34171. [DOI] [PubMed] [Google Scholar]
- Khakh BS, & Deneen B. J. A. r. o. n. (2019). The emerging nature of astrocyte diversity. 42, 187–207. [DOI] [PubMed] [Google Scholar]
- Khoradmehr A, Mazaheri F, Anvari M, & Tamadon A (2019). A simple technique for three-dimensional imaging and segmentation of brain vasculature using fast free-of-acrylamide clearing tissue in murine. Cell Journal (Yakhteh), 21(1), 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers G-J, Goedhart J, van Munster EB, & Gadella TWJB (2006). Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. 45(21), 6570–6580. [DOI] [PubMed] [Google Scholar]
- Lam AJ, St-Pierre F, Gong Y, Marshall JD, Cranfill PJ, Baird MA, … Schnitzer M. J. J. N. m. (2012). Improving FRET dynamic range with bright green and red fluorescent proteins. 9(10), 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, & Brenner MJG (2008). GFAP promoter elements required for region-specific and astrocyte-specific expression. 56(5), 481–493. [DOI] [PubMed] [Google Scholar]
- Lia A, Henriques VJ, Zonta M, Chiavegato A, Carmignoto G, Gómez-Gonzalo M, & Losi G (2021). Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Frontiers in cellular neuroscience, 15, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader S, & Brimberg LJC (2019). Aquaporin-4 water channel in the brain and its implication for health and disease. 8(2), 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt ML, Kremers G-J, Kraft CA, Ray K, Cranfill PJ, Wilson KA, … Rizzo M. A. J. P. o. (2011). An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. 6(3), e17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden WC, Walsh H, Richter F, Soudant C, Bryce CH, Hof PR, … McKenzie A. T. J. A. n. c. (2019). Perfusion fixation in brain banking: a systematic review. 7(1), 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, … Gadella T. W. J. N. m. (2007). Bright monomeric red fluorescent protein with an extended fluorescence lifetime. 4(7), 555–557. [DOI] [PubMed] [Google Scholar]
- Miralles VJ, Martínez-López I, Zaragozá R, Borrás E, García C, Pallardó FV, & Viña J. R. J. B. r. (2001). Na+ dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) in primary astrocyte cultures: effect of oxidative stress. 922(1), 21–29. [DOI] [PubMed] [Google Scholar]
- Mohammad Rezazadeh F, Saedi S, Rahmanifar F, Namavar MR, Dianatpour M, Tanideh N, … Tsutsui K (2018). Fast free of acrylamide clearing tissue (FACT) for clearing, immunolabelling and three-dimensional imaging of partridge tissues. Microscopy research and technique, 81(12), 1374–1382. [DOI] [PubMed] [Google Scholar]
- Moore MM, & Alejandro EUJCP (2021). Aortic Cross-Clamping to Provide Differential Fixation by Perfusion. 1(3), e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi F, Stankiewicz AJ, & Zhdanova I. V. J. B.-p. (2019). Looking through brains with fast passive CLARITY: Zebrafish, rodents, non-human primates and humans. 9(15), e3321–e3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, & Miyawaki A. J. N. b. (2002). A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. 20(1), 87–90. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, & Helmchen F. J. N. m. (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. 1(1), 31–37. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Goldman SA, & Nedergaard MJA (2012). Heterogeneity of astrocytic form and function. 23–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, & Khakh BS (2018). An optical neuron-astrocyte proximity assay at synaptic distance scales. Neuron, 98(1), 49–66. e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC, & Waldo G. S. J. N. b. (2006). Engineering and characterization of a superfolder green fluorescent protein. 24(1), 79–88. [DOI] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, … Armsen W. J. J. o. N. (2015). Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. 35(13), 5187–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen R, Nedergaard M, & Petersen N. C. J. S. r. (2016). Sulforhodamine 101, a widely used astrocyte marker, can induce cortical seizure-like activity at concentrations commonly used. 6(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DS, & Lichtman JW (2015). Clarifying tissue clearing. Cell, 162(2), 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DS, & Lichtman JWJC (2017). SnapShot: tissue clearing. 171(2), 496–496. e491. [DOI] [PubMed] [Google Scholar]
- Rose CF, Verkhratsky A, & Parpura VJBST (2013). Astrocyte glutamine synthetase: pivotal in health and disease. 41(6), 1518–1524. [DOI] [PubMed] [Google Scholar]
- Rusnakova V, Honsa P, Dzamba D, Ståhlberg A, Kubista M, & Anderova MJPO (2013). Heterogeneity of astrocytes: from development to injury–single cell gene expression. 8(8), e69734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shagin DA, Barsova EV, Yanushevich YG, Fradkov AF, Lukyanov KA, Labas YA, … evolution. (2004). GFP-like proteins as ubiquitous metazoan superfamily: evolution of functional features and structural complexity. 21(5), 841–850. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, & Tsien R. Y. J. N. b. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. 22(12), 1567–1572. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, … Israelsson M. J. N. m. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. 10(5), 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, & Tsien R. Y. J. N. m. (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. 5(6), 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbo D, Murphy CS, Ermakova GV, Solovieva EA, Chepurnykh TV, Shcheglov AS, … Roche P. M. J. B. j. (2009). Far-red fluorescent tags for protein imaging in living tissues. 418(3), 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbo D, Shemiakina II, Ryabova AV, Luker KE, Schmidt BT, Souslova EA, … Britanova O. V. J. N. m. (2010). Near-infrared fluorescent proteins. 7(10), 827–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemiakina I, Ermakova G, Cranfill P, Baird M, Evans R, Souslova E, … Gorodnicheva T. J. N. c. (2012). A monomeric red fluorescent protein with low cytotoxicity. 3(1), 1–7. [DOI] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Wegrzynowicz M, Lee E, Bowman AB, & Aschner M (2011). Role of astrocytes in brain function and disease. Toxicologic pathology, 39(1), 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack RL, Hein B, Bhattacharyya D, Hell SW, Keenan RJ, & Glick BSJB (2009). A rapidly maturing far-red derivative of DsRed-Express2 for whole-cell labeling. 48(35), 8279–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Cranfill PJ, Davidson MW, & Verkhusha V. V. J. P. o. (2011). An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. 6(12), e28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, Grüenwald D, … biology. (2008). Conversion of red fluorescent protein into a bright blue probe. 15(10), 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testen A, Kim R, & Reissner K. J. J. C. p. i. n. (2020). High-Resolution Three-Dimensional Imaging of Individual Astrocytes Using Confocal Microscopy. 91(1), e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer R, Ye L, Hsueh B, & Deisseroth K. J. N. p. (2014). Advanced CLARITY for rapid and high-resolution imaging of intact tissues. 9(7), 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda HR, Ertürk A, Chung K, Gradinaru V, Chédotal A, Tomancak P, & Keller PJ (2020a). Tissue clearing and its applications in neuroscience. Nature Reviews Neuroscience, 21(2), 61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda HR, Ertürk A, Chung K, Gradinaru V, Chédotal A, Tomancak P, & Keller PJJNRN (2020b). Tissue clearing and its applications in neuroscience. 21(2), 61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Khoradmehr A, & Tamadon A (2019). FACT or PACT: A comparison between free-acrylamide and acrylamide-based passive sodium dodecyl sulfate tissue clearing for whole tissue imaging. Cell Journal (Yakhteh), 21(2), 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia N-S, Luo W-X, Zhang J, Xie X-Y, Yang H-J, Li S-W, … Ng M.-H. J. M. b. (2002). Bioluminescence of Aequorea macrodactyla, a common jellyfish species in the East China Sea. 4(2), 155–162. [DOI] [PubMed] [Google Scholar]
- Xu N, Tamadon A, Liu Y, Ma T, Leak RK, Chen J, … Feng Y (2017). Fast free-of-acrylamide clearing tissue (FACT)—an optimized new protocol for rapid, high-resolution imaging of three-dimensional brain tissue. Scientific reports, 7(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Treweek JB, Kulkarni RP, Deverman BE, Chen C-K, Lubeck E, … Gradinaru V (2014). Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell, 158(4), 945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C-X, Habegger KM, Chowen JA, Stern J, & Tschöp MH (2011). A role for astrocytes in the central control of metabolism. Neuroendocrinology, 93(3), 143–149. [DOI] [PubMed] [Google Scholar]
- Zhang X, Alnafisah RS, Hamoud A-RA, Shukla R, Wen Z, McCullumsmith RE, & O’Donovan SM (2021). Role of astrocytes in major neuropsychiatric disorders. Neurochemical research, 1–16. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Ma Z, Zou W, Guo H, Liu M, Ma Y, & Zhang L. J. B. r. i. (2019). The appropriate marker for astrocytes: comparing the distribution and expression of three astrocytic markers in different mouse cerebral regions. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.