

ABSTRACT
The functioning, health, and productivity of soil are intimately tied to a complex network of interactions, particularly in plant root-associated rhizosphere soil. We conducted a stable-isotope-informed, genome-resolved metagenomic study to trace carbon from Avena fatua grown in a 13CO2 atmosphere into soil. We collected paired rhizosphere and nonrhizosphere soil at 6 and 9 weeks of plant growth and extracted DNA that was then separated by density using ultracentrifugation. Thirty-two fractions from each of five samples were grouped by density, sequenced, assembled, and binned to generate 55 unique bacterial genomes that were ≥70% complete. We also identified complete 18S rRNA sequences of several 13C-enriched microeukaryotic bacterivores and fungi. We generated 10 circularized bacteriophage (phage) genomes, some of which were the most labeled entities in the rhizosphere, suggesting that phage may be important agents of turnover of plant-derived C in soil. CRISPR locus targeting connected one of these phage to a Burkholderiales host predicted to be a plant pathogen. Another highly labeled phage is predicted to replicate in a Catenulispora sp., a possible plant growth-promoting bacterium. We searched the genome bins for traits known to be used in interactions involving bacteria, microeukaryotes, and plant roots and found DNA from heavily 13C-labeled bacterial genes thought to be involved in modulating plant signaling hormones, plant pathogenicity, and defense against microeukaryote grazing. Stable-isotope-informed, genome-resolved metagenomics indicated that phage can be important agents of turnover of plant-derived carbon in soil.
IMPORTANCE Plants grow in intimate association with soil microbial communities; these microbes can facilitate the availability of essential resources to plants. Thus, plant productivity commonly depends on interactions with rhizosphere bacteria, viruses, and eukaryotes. Our work is significant because we identified the organisms that took up plant-derived organic C in rhizosphere soil and determined that many of the active bacteria are plant pathogens or can impact plant growth via hormone modulation. Further, by showing that bacteriophage accumulate CO2-derived carbon, we demonstrated their vital roles in redistribution of plant-derived C into the soil environment through bacterial cell lysis. The use of stable-isotope probing (SIP) to identify consumption (or lack thereof) of root-derived C by key microbial community members within highly complex microbial communities opens the way for assessing manipulations of bacteria and phage with potentially beneficial and detrimental traits, ultimately providing a path to improved plant health and soil carbon storage.
KEYWORDS: bacteriophages, metagenomics, plant-microbe interactions, rhizosphere, stable-isotope probing
INTRODUCTION
Plant-derived carbon provides the energetic basis for an intricate web of life in soil, among the world’s most complex microbial ecosystems. Many heterotrophic soil organisms are sustained primarily by the carbon that is fixed by plants and released into soil in the region surrounding growing roots, the rhizosphere. Root-derived carbon not only supports a bloom of microbial activity and biomass growth but also stimulates microbial interactions that play roles in plant immunity and nutrient acquisition and lead to organic matter associations with soil minerals (1).
The soil ecosystem is characterized by interactions occurring among organisms across trophic levels, which may direct the fate of plant-derived carbon in soil. These interactions can be difficult to investigate because of the tremendous physical and chemical heterogeneity of soil and resulting vast biological diversity. Much of the recent work on soil microbiology has been sequence based and focused on generating inventories of bacteria and archaea using 16S rRNA gene fragments (2, 3) or fungi using internal transcribed spacer region sequencing (4, 5). However, since DNA extracted from soil includes genes from virtually all organisms present, it is possible to use shotgun metagenome sequencing to profile complete soil communities, potentially with genomic resolution. This is important because genomes provide not only phylogenetic information but also a cache of functional predictions. However, only recently have studies achieved genomic resolution in soil, largely due to strain complexity and relatively even abundance levels (6–10). Bacterial genomes possess a huge variety of known and unknown genes, including those that comprise biosynthetic gene clusters (BGCs). Such biosynthetic pathways hold technological relevance but are also fundamental for soil ecology, as their products could mediate interorganismal interactions, including antagonistic interactions via antibiotics, mineral interactions via siderophores, and signaling compounds (11, 12). While there has been a surge of interest in viral diversity and ecology, there have been relatively few studies documenting bacterium-phage interactions in soil (13–15). Also present in soil are fungi, protists, and larger organisms that have been documented via 18S rRNA gene sequencing (16, 17) and more classical methods (18, 19). While some studies have documented bipartite interactions in soil, such as fungi and bacteria (20, 21), bacteria and phage (13, 22), and plant and fungi (23, 24), detailing complex cross-kingdom interactions in soil remains a huge challenge.
Stable-isotope probing (SIP) provides one means of tracing elemental flow among soil community members. SIP studies have been conducted in a variety of ecosystems, including hot springs and the animal gut, using a range of isotopes and labeled substrates (25–28). SIP techniques can use carbon-fixing organisms to generate biomass and complex mixes of compounds to investigate general processes such as decomposition of litter or C flow in the rhizosphere (29–33). Stable isotopes can also be monitored between trophic levels, allowing the study of microbial predation and phage activity (34, 35). A recently developed variant, quantitative SIP (qSIP), makes these measurements possible at the individual or population genome scale (36). This is facilitated by comparing the taxon-specific density for each sequenced entity in labeled and unlabeled DNA fractions (36, 37). From qSIP data, it is possible to estimate the gross growth rate of organisms assimilating labeled carbon substrates.
To better understand the movement of carbon introduced into soil as plant-derived rhizodeposits, we combined stable-isotope probing and genome-resolved metagenomics. We grew common wild oat grass, Avena fatua, for 6 and 9 weeks in a 13CO2-enriched atmosphere and tracked the isotopically enriched plant carbon as it was released into the surrounding soil community via exudation, decay of root biomass, and direct biotic transfers via root pathogen attack. We hypothesized that plant-derived carbon would be detected in multiple trophic levels and that genome analysis would provide clues as to the ecological means of this transfer. By separating extracted DNA based on density, we determined which populations consumed the isotopically heavy plant-derived carbon (or predated upon other organisms who were primary consumers) and incorporated it into their genomes during replication. Conversely, we also identified microorganisms present in substantial number that did not consume 13C-labeled root C. We used genome assembly and binning to analyze the genes at the organismal level. We chose to focus on binned bacterial genomes rather than studying all assembled scaffolds, as this allowed us to more accurately calculate population-level stable-isotope enrichment and interpret the functional significance of individual populations. In the metagenome-assembled genomes, we identified possible interaction signatures—such as genes for the production of plant hormones and modulation of hormone concentrations, secretion systems, and secondary metabolites. Assembled genomes also allowed us to predict whether an organism is a plant growth-promoting bacterium (PGPB) or a pathogen (38, 39).
RESULTS
13CO2 plant labeling and stable-isotope probing.
A. fatua plants were grown in a continuously regenerated 13CO2 plant growth chamber. After 6 weeks of growth, the plant shoots were highly 13C labeled (∼94 atom%) (40, 41). DNA was extracted from bulk soil samples collected at the beginning of the study, 6 and 9 weeks, and also from paired rhizosphere soil from plants grown for 6 and 9 weeks (see Fig. S1 in the supplemental material), and then all DNA was fractionated in a density gradient (37). We compared the density separation of rhizosphere DNA to bulk soil DNA to identify unenriched DNA (light), a mixture of enriched and unenriched DNA (middle), and highly 13C enriched (heavy); these fractions were then shotgun sequenced (see Fig. S2 and Table S1 in the supplemental material). After assembly of the individual metagenomes, the heavier fractions (middle and heavy) had overall larger assemblies with a greater percentage of reads aligning to the assembly than the light fraction assemblies (Table S1).
Experimental design of soil microcosms, stable-isotope probing density fractionation, and samples generated for sequencing from a 13CO2 labeling study of common wild oat, Avena fatua. Within the microcosms, hashed squares indicate root-excluding bulk soil bags. DNA fractionation plots show DNA density profiles from 0, 6, or 9 weeks of plant growth with unlabeled, partially labeled, and labeled community DNA indicated by gray, yellow, and peach shading, respectively. Brown lines and text indicate bulk soil samples, and green lines and text indicate rhizosphere samples. Download FIG S1, TIF file, 1.5 MB (1.5MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA density profiles from a 13CO2 plant labeling study illustrate stable-isotope separation in DNA 13C enrichment between labeled (rhizosphere) and unlabeled (bulk soil) treatments. The density (gram/milliliter) and concentration (nanogram/microliter) of DNA for each collected fraction from each sample is plotted to generate these SIP separation graphs. Fractions were subsequently combined into “light,” “middle,” and “heavy” bins prior to sequencing. Download FIG S2, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
SIP, sequencing, and metagenome assembly statistics. Download Table S1, PDF file, 0.03 MB (26.6KB, pdf) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Organisms identified from SIP metagenomes.
It is currently unrealistic to bin genomes for all organisms present in a soil sample, yet relatively extensive reconstruction of genome fragments is possible. We used an assembled marker gene approach to approximate microbial community composition, using the ribosomal protein S3 gene (rpS3), which is found in single copy on bacterial and archaeal genomes and has been used to profile microbial communities for phylogeny and abundance (42). The rpS3 gene tends to assemble well from metagenomes, and since it occurs as a single copy per genome, it provides a better abundance metric than 16S rRNA (42, 43). From each sample, we identified the rpS3 and dereplicated the sequences to a level of 99% nucleic acid identity (44). The resulting 314 distinct rpS3 sequences we identified represent a diverse array of soil bacteria (Fig. 1).
FIG 1.

Phylogenetic tree illustrating the bacterial rpS3 genes identified in this study in the context of published rpS3 sequences. Soil- and rhizosphere-derived metagenomic bacterial bins (55 bins; >70% complete) with an rpS3 gene and unbinned scaffolds are marked (314 total). Publicly available representative species to provide phylogenetic grounding are also included. Bacterial clades are highlighted in different colors.
To study community dynamics across samples and fractions, we mapped the reads from each sample and fraction to scaffolds containing the rpS3 gene. We used the coverage of the scaffolds as a proxy for each organism’s relative abundance. Based on a principal coordinate analysis (PCoA), the fractions and samples show a clear community separation based on soil habitat (bulk versus rhizosphere) and SIP density fraction, but not time (Fig. 2). It is widely documented that density of DNA is affected both by its GC content (sequences with higher GC content are more dense than those with lower GC) and the enrichment level of the DNA (45). Because these two factors control DNA density, the bulk samples (even with no added 13C) separate into a lower GC light fraction cluster and a higher GC heavier fraction cluster. The bulk light fraction and rhizosphere light fraction samples group together. The rhizosphere middle fraction separates from both types of light fractions. The rhizosphere middle fraction also separates from the bulk soil heavier fraction, as it contains DNA from the high-GC organisms that did not incorporate the label and the community of lower-GC organisms that were labeled sufficiently to increase the density of their DNA. Finally, the rhizosphere heavy fractions separate from all other fractions—the bacteria present in this sample are almost entirely high-GC organisms that incorporated 13C into their DNA.
FIG 2.
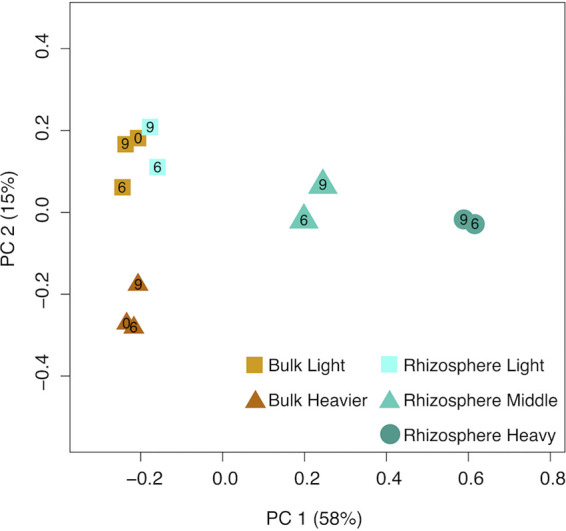
PCoA of bacterial rpS3 gene sequences from DNA from five soil samples fractionated into 12 SIP fractions collected from a 13CO2 plant labeling study. Symbols and colors represent the different samples and fractions. Numbers inside the symbols correspond to the week of sampling. See Fig. S1 in the supplemental material for additional explanation.
From our 12 metagenomes (Fig. S1), we reconstructed and binned 55 bacterial genomes that were ≥70% complete with ≤10% contamination, as measured by the inventory of 51 single copy genes from ggKbase (Fig. 3 and Fig. S3). In addition to bacteria, we also detected a number of eukaryotes and phage. We reconstructed 27 complete 18S rRNA gene sequences from soil eukaryotes (Fig. 3 and Fig. S4). The soil microeukaryotes fall into a variety of soil clades, including Amoebozoa, Fungi, Metazoa (nematodes and rotifers), Rhizaria, and Alveolata. We also identified phage-derived DNA in our samples and reconstructed 10 complete, circularized phage genomes (Fig. 3).
FIG 3.
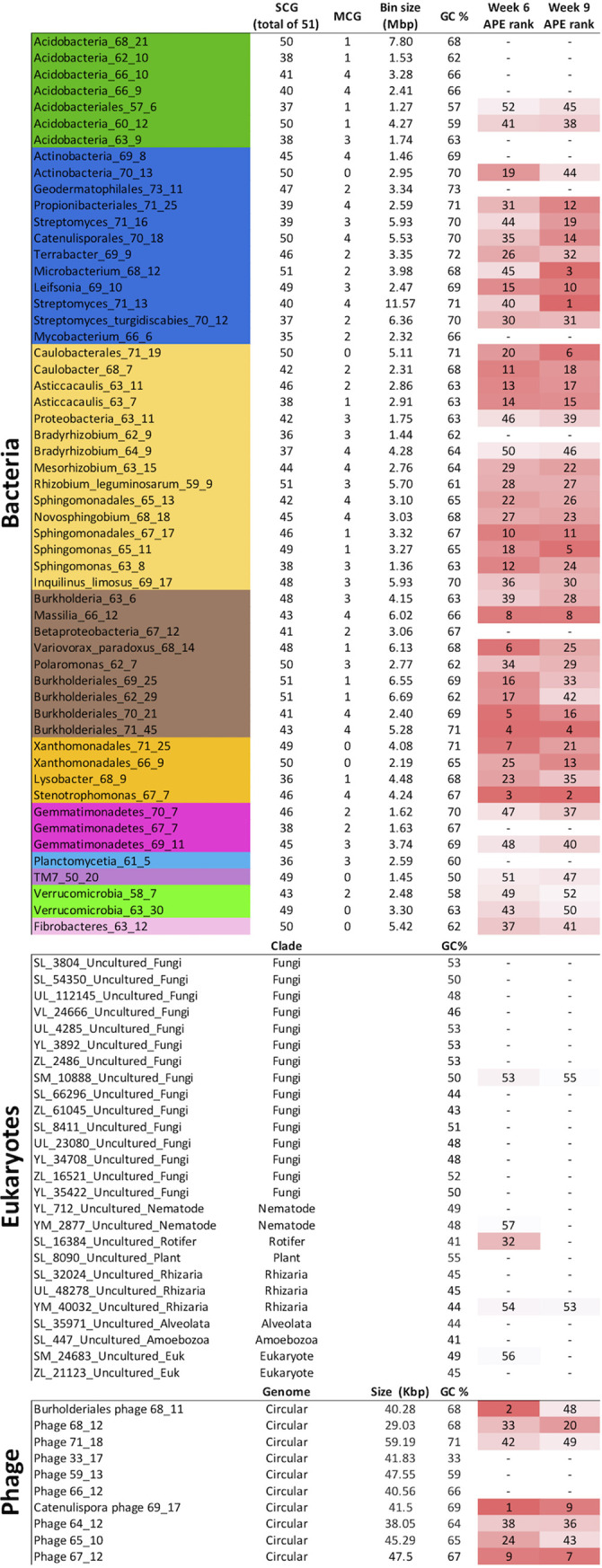
Genome and 13C isotope labeling statistics for metagenome-assembled bacterial genomes (colored by clade following the color scheme from Fig. 1), eukaryote scaffolds containing 18S rRNA genes, and complete phage genomes. Rank atom percent excess (APE) values (highlighted with red heat maps for each column) are derived from qSIP calculations. Bin completeness and contamination are presented as the number of 51 single copy genes (SCG) and number of multicopy genes (MCG).
Count of bacterial single copy genes in partial genome bins identified in this study. Color bar represents the clade of bin, colored according to the legend presented in Fig. 1. Download FIG S3, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
18S rRNA tree colored by clade. Black dots represent bootstrap values of ≥60. Long leaves were removed for ease of visualization, and 18S rRNA sequences identified in this study are shown in purple. Download FIG S4, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We used quantitative stable-isotope probing (qSIP) to estimate 13C atom percent excess (APE) for each taxon (36, 37). The qSIP method relies on tracking the shift in density, calculated by coverage in the different fractions, of a genome between unlabeled (bulk) and labeled (rhizosphere) samples, this metric is not influenced by the genome size. We mapped the reads of all samples against our dereplicated suite of 55 genome bins. We used the coverage of the scaffolds containing the 18S rRNA as a proxy for eukaryotic genome coverage because we were unable to bin eukaryotic genomes. Because of the lack of replicates and small number of fractions, we chose a conservative detection cutoff of 2.5% APE (see Materials and Methods); that is, any entities with an APE higher than this cutoff were interpreted as having detectably incorporated the 13C tracer (Fig. 4). We report the rank in order of enrichment (APE) for each sequence in Fig. 3. Many of the phage we identified appear to be highly labeled. Bacteria were also highly labeled, and a few of the eukaryote sequences were labeled, although to a much lesser degree than the bacterial genomes (Fig. 4). Of the 55 bacterial genomes we assembled, the majority (78%) were detectably 13C enriched.
FIG 4.

The rank of soil-derived phage genomes, bacterial genome bins, and scaffolds encoding eukaryotic 18S rRNA genes in week 6 and 9 in order of their atom percent excess (APE) based on the qSIP calculations. The gray region indicates unlabeled entities, and the pink region indicates predicted labeled DNA. The labeling cutoff is explained in Materials and Methods.
We calculated the gross growth rate for each taxon on labeled carbon. The gross growth rate was calculated by assuming linear growth and comparing the abundance of each taxon at week 0 to the other time points and incorporating the mass of the DNA extracted and the mass of DNA from the original soil sample (36). The gross growth rate is presented in the unit: mass (in nanograms) of DNA per day per gram of dry soil. These taxon-specific growth rate estimates can be thought of as a measure of which individual populations in the rhizosphere grew on 13C-labeled plant-derived carbon. The gross growth rates indicated bacterial and phage growth but very limited eukaryotic growth on plant-derived carbon. Indeed, only a few specific bacteria and phage had high gross growth rates on plant derived-carbon (Fig. S5).
Plots of atom percent excess (APR) versus gross growth rates of soil organisms (nanogram of DNA gram of soil−1 day−1) based on root-derived carbon. (Top) The log of gross growth per day on plant-derived carbon compared to APE, and (Bottom) violin plot of gross growth of different types of soil populations. Download FIG S5, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plant-soil community interactions.
Many of the bacterial genomes carrying plant interaction-mediating genes and pathways were also highly labeled, suggesting an intimate relationship between the plant roots and growing bacteria (Fig. 5). Bacteria that are closely associated with plants may degrade plant infection signaling hormones to avoid detection during plant colonization. Many of the bacterial genomes we binned carry genes that encode the ability to hydrolyze salicylic acid (Fig. 5), a common phenolic plant hormone used in pathogen defense signaling (46). However, this protein can also be used to degrade other phenolic compounds (47). Thus, we examined the nearby genomic regions for clues about the function of the gene. In one instance, the salicylate hydroxylase gene in the Microbacterium_68_12 genome is surrounded by a variety of glycosyl hydrolases and esterases that act on plant cell wall polymers, indicating that this region of the genome may be devoted to plant cell invasion and avoidance of detection. Additionally, many of the rhizosphere-dwelling populations that we detected carry genes that encode the ability to degrade nitric oxide gas (Fig. 5), another pathogen defense hormone (48).
FIG 5.

Possible interorganismal interactions encoded by 55 bacterial metagenome-assembled bacterial genomes identified in this study. Numbers indicate the number of individual genes or nearly complete pathways predicted to be used in interorganismal interaction. Asterisks signify genome bins with detectable 13C label, and pink highlighting indicates taxa that are among the top 15% of 13C-enriched populations.
Although plant-derived carbon is the main source of 13C used by the soil community, other organisms may be capable of fixing CO2. However, we found no evidence for carbon fixation pathways in the bacterial genomes, though some unbinned organisms could have the ability to fix carbon. The lack of a density shift in the bulk samples indicates that carbon fixation in the bulk soil was undetectable (Fig. S2).
Some bacteria, especially PGPB, promote plant growth through the production of hormones and other compounds. Two of the bacterial genomes we reconstructed encoded the pathway for indole-3-acetic acid production, a compound that increases plant growth and induces a variety of other physiological changes (Fig. 5). Eight of the genomes encode 1-aminocyclopropane-1-carboxylate (ACC) deaminase (Fig. 5), which prevents ACC from being converted to ethylene in the plant. However, ACC deaminase is also involved in the generation of propionate. In the Streptomyces_71_13 genome, the ACC deaminase gene is surrounded by plant carbon degradation genes such as pectin lyases and pectinesterases. We identified 18 genomes with the pathways for the production of acetoin and/or 2,3-butanediol from pyruvate (Fig. 5). These bacterially produced volatile organic compounds (VOCs) diffuse through soil and can act as growth-promoting factors and stimulate a plant systemic defense. Although these compounds increase resistance to plant pathogens, the pathway can also be involved in the anaerobic fermentation of glucose (49, 50).
Microbes can also promote plant growth through nutrient generation or mobilization. Microbially produced phytases release phosphorus from phytate, a phosphorus storage compound common in soil but inaccessible to mature plants (Fig. 5) (51). Several of the genomes encoded biosynthetic pathways to produce siderophores (Fig. S6). In addition to iron acquisition, siderophores can complex iron and other metals, thus promoting the release of phosphate from insoluble soil-associated minerals (52). We did not identify any N2-fixing pathways in the genomes or on the unbinned scaffolds.
Biosynthetic gene clusters encoded in bacterial genome bins identified in this paper. Signaling compounds includes homoserine lactone clusters, N-acyl amino acid clusters, and butyrolactone cluster. Download FIG S6, TIF file, 2.9 MB (2.9MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Interactions between bacteria and microeukaryotes.
Several of the bacterial genomes encoded secretion systems that are thought to be associated with bacterium-eukaryote interactions. Six genomes carry multiple genes from type III secretion systems, which are known to be important in symbiotic colonization and infection of eukaryotes (53). We do not know the intended targets of the type III secretion systems because of the diversity of possible hosts. However, we identified 58 type III effector proteins with sequence homology to known plant pathogen effector proteins (mostly from Ralstonia, Pseudomonas, and Xanthomonas) encoded on the Burkholderiales_62_29 genome (Fig. 5). We also identified 13 genomes with probable type IV secretion systems (Fig. 5), which are used in conjugation or the injection of protein-DNA complexes into eukaryotic cells (54).
Some of the assembled bacterial genomes encode systems that may influence interactions with soil fungi. Nine of the partially complete genomes encoded fusaric acid resistance proteins, which protect from the antibiotic produced by Fusarium species (Fig. 5). The Variovorax_paradoxus_68_14 genome appeared to encode three fusaric acid resistance modules; one was near an esterase and phospholipase C which hydrolyzes phosphatidylcholine, an important fungal phospholipid (55). In one Streptomyces_71_13 genome, the fusaric acid resistance module is near two glycoside hydrolases which may act on fungal cell walls. Included in this region is a ceramidase, which hydrolyzes glucosylceramides, a necessary metabolite for Fusarium pathogenesis and morphology (56). Another possible indication of bacterial-fungal relationships is bacterial genes for decomposition of fungal compounds. The Streptomyces_71_13 genome encodes 39 enzymes that target fungal biomass, 30 of which have an identifiable secretion signal, indicating they are exported extracellularly (Fig. 5).
We also identified some evidence for bacterially produced defenses against grazing. Bacteria often use extracellular polymeric substance (EPS) production, specific secondary metabolites, and active infection to deter grazing (57). EPS production is common in soil bacteria but difficult to infer from genome information. One indication of EPS formation is the production of proteinaceous adhesins (58). Many of the genomes we investigated encoded adhesins, with Verrucomicrobia_58_7, Verrucomicrobia_63_30, and Variovorax_paradoxus_68_14 genomes encoding the most (Fig. 5). Several genomes encode the pathway to produce hydrogen cyanide (HCN) (Fig. 5). Eight of the genomes encode a insecticidal toxin subunit TcC which is lethal to certain insects, and possibly nematodes (Fig. 5) (59). The Acidobacteria_68_21 genome encoded two complete Tc insecticidal toxin modules and an insecticidal crystal protein related to the bt toxin from Bacillus thuringiensis (Fig. 5).
Bacterium-bacterium interactions.
In addition to bacterial interaction, cooperation, and competition with eukaryotes, genomes can provide indications of interbacterial interactions in soil. We identified some of the best characterized mediators of interbacterial interactions, including signaling molecules such as acyl-homoserine lactones, autoinducing peptides, indoles, gamma-butyrolactones, and a variety of other compounds (Fig. S6). In addition, many genomes encoded one or more quorum-quenching genes, which may act either to degrade self-produced quorum molecules or as a means to disrupt other bacterial species communications.
We identified a large number of biosynthetic gene clusters (BGCs) in the bacterial genomes, especially polyketide and nonribosomal peptide biosynthetic gene clusters (Fig. S6). Several Streptomyces spp. encoded many BGCs, and several Burkholderiales and Acidobacteria genomes also encoded a high number of secondary metabolite clusters, as has been found in other soils (12, 60). Some of the BGCs were located near other genes of interest for instance, the Streptomyces_turgidiscabies_70_12 genome encoded three BGCs near 10 plant cell wall hydrolysis proteins. Evidence for competition between closely related strains comes in the form of bacteriocins, antibiotics that act on closely related bacteria. These were found in nearly 40% of the genomes. Eleven genomes encoded type VI secretion systems, which inject effectors into neighboring bacteria (Fig. 5). Many genomes had multiple VgrG proteins, the tip of the needle and effector transporter, near large proteins with Rhs repeat domains which may function as bacterial toxins (Fig. 5) (61).
Evidence of bacterium-phage interactions.
Many of the phage we identified appeared to be highly 13C labeled (Fig. 4), providing direct evidence of plant-derived C moving through trophic levels. Indeed, in the week 6 sample, the two most labeled entities were phage (Fig. 3). We focused on circularized phage genomes (as opposed to those integrated into the host genome as a prophage where they could become 13C labeled through host growth alone) as these are likely complete genomes and the product of active infection during our experiment.
We identified Burkholderiales_62_29 as the possible host for one of the most highly labeled phage, Burkholderiales phage 68_11, based on the match between a CRISPR-Cas spacer and the complete phage genome. The spacer hit the large terminase subunit with two mismatches (33 bp total). It appears the phage may be capable of a lysogenic life cycle because of the presence of a serine recombinase and a possible induction region consisting of a histone-like protein and lambda repressor-like gene.
Another phage-host connection we identified was based on a recent lateral gene transfer event. The Catenulispora phage 69_17 was highly 13C labeled and ranked as the 1st and 9th most labeled entity in the week 6 and 9 rhizosphere samples, respectively (Fig. 3). The probable host, Catenulisporales_70_18, encodes a glycoside hydrolase (GH25) which shares 77% amino acid identity with the phage protein, and a phylogenetic tree indicates that the phage and bacterial proteins are more related to one another than to other publicly available sequences (Fig. S7). This suggests the phage may have acquired the gene from this bacterial host population via lateral transfer. The acquired gene may be a lysis factor, as GH25 breaks down peptidoglycan. The phage has a lysogenic life cycle based on the presence of a gene annotated as a tyrosine recombinase and induction regulation genes.
Phylogenetic tree of GH25 gene. The GH25 genes from phages are highlighted in green, and the bacterial genome bin from this study is shown in blue. Download FIG S7, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The remaining eight complete phage genomes could not be linked to a specific host in our data set. Many carry DNA methylation genes that may protect the phage DNA from detection or destruction by host antiviral systems. Of the 55 draft bacterial genomes, only 2 contained identifiable CRISPR-Cas systems, the Burkholderiales described above and Acidobacteria_68_21, which encoded two Cas type III-B systems and a type I-C system (Fig. 5).
DISCUSSION
We combined stable-isotope probing with genome-resolved metagenomics to trace the flow of plant-derived carbon into bacterial primary consumers and then to phage and bacteriovores. Through the generation of genome bins, we were able to discover clues regarding the genomic potential and ecological roles of these organisms and to develop ideas about the nature of the movement of carbon through the system. The heavy fractions yielded larger assemblies than the lighter fractions, likely due to the reduced diversity of sequenced DNA. These larger assemblies and the reduced diversity likely improved binning by providing more complete sequencing of the present bacteria.
Our approach, which identified single copy rpS3 genes from different samples and their distribution across isotopically enriched fractions, indicates that the supply of labeled plant root carbon can help to identify assemblages of active rhizosphere organisms that are distinct from the background soil community. Based on the rpS3 analysis, a large portion of organisms in the rhizosphere were not detectably responsive to the influx of plant-derived carbon and their communities were indistinguishable from bulk samples. Successional shifts with time may have occurred, but the replication (and thus resolution) of our study was not sufficient to detect them. In a parallel highly replicated experiment, sequencing of 16S rRNA genes did identify effects of sampling time during plant growth on microbial composition (41). In addition to bacteria, we identified a number of soil eukaryotes, and though we recognize that the number we identified does not scratch the surface of soil eukaryotic diversity, our assembled metagenomes provide complete 18S rRNA sequences without the primer bias inherent to tag-based methods. We also identified 10 complete phage genomes which likely represent some of the most abundant phage in our system. This is a small number compared to the total diversity of phage likely present in the soil, yet these complete genomes allow us to predict lysogenic lifestyles and to identify possible hosts of some of the dominant phage.
By assuming linear growth over the course of the experiment, we derived gross growth rate estimates for prokaryotes, microeukaryotes, and phage that rely on root-derived carbon. From our analysis, both phage and bacteria showed higher average growth rates than eukaryotes. This may be because the eukaryotes were not feeding on enriched rhizosphere bacteria, they were slow to replicate their genomes, or the process of sieving soil and constructing the microcosms decreased their population. While we acknowledge these estimated rates have associated uncertainty, as do estimates of the abundance of metagenome-assembled genomes in soil, this approach remains one of the only means to estimate population-specific growth rates in situ. The use of a larger number of density fractions does result in much reduced uncertainties in growth rate estimates (62). These quantitative metrics enable us to directly link previously undescribed populations of rhizosphere microbiota to plant-derived C fluxes in an intact plant-soil environment. Future development of this technique will allow us to better understand complex carbon utilization networks in soil.
Many of the most highly labeled organisms were those with probable plant interaction systems and may span the spectrum from mutualist to pathogen. By analyzing genomes with ≥70% completeness, we were able to identify genes and pathways involved in interaction but also investigate their genomic context in order to gain additional information to help predict the purpose of these genes (63, 64). For instance, in the Streptomyces_71_13 genome, the ACC deaminase gene is adjacent to plant cell wall hydrolysis genes and the biosynthetic pathway for producing ectoine, a compatible solute common in PGPB (40). This region of the genome may enable this Streptomyces sp. to limit plant stress responses by lowering ethylene levels and producing an osmoprotectant which would ensure greater plant growth (65, 66). The plant cell wall digestion enzymes may enable this PGPB to invade the plant to form a closer mutualistic association with the roots. Apart from that specific region of the genome, the Streptomyces_71_13 genome also encodes a number of other pathways and genes known to be important in PGPB, including production of indole-3-acetic acid and 2,3-butanediol. The possible close association with plant growth promotion may explain why Streptomyces_71_13 was the most labeled population in our samples collected at week 9.
In addition to possible PGPB we also identified probable plant pathogens in the group of 13C-enriched taxa. Based on the presence of a type III secretion system and plant effector proteins, Burkholderiales_62_29 may act as a plant pathogen. If so, this would have enabled the assimilation of large amounts of root-derived carbon and contributed to making this genome the 17th most labeled population in week 6. To further evaluate this hypothesis, we referred to the transcription of the effector proteins from a related study of the Avena rhizosphere metatranscriptome which used the Burkholderiales_62_29 genome as a reference; 8 of its 58 effectors were statistically upregulated in the rhizosphere compared to bulk soil transcription (67). In addition, we identified a labeled Fusarium sp. (SM_10888_Uncultured_Fungi) in the rhizosphere samples; several Fusarium spp. are known Avena pathogens and are able to obtain carbon directly through infection of plant roots (68). Bacteria with genomes that encode a large number of fungal cell wall hydrolysis genes may obtain carbon from fungal necromass or actively antagonize living fungi through digestion of their cell walls (69).
It appears that not all bacteria growing on root-derived C have identifiable genes that predict a close relationship with the plant. Burkholderiales_70_21, Leifsonia_69_10, and Sphingomonadales_67_17 were some of the most highly labeled bins, despite encoding few identifiable interaction systems. We identified at most 6 genes in these bins compared to the average number of 16 predicted interaction genes for all genome bins. It is possible some noninteracting organisms are well positioned to take advantage of the abundance of resources in the rhizosphere and grow quickly.
Several of the eukaryotes we identified were labeled with plant-derived 13C, including two nematodes, a rotifer and a rhizaria. Based on their phylogeny, these microeukaryotes may lead a bacterivorous lifestyle. Their 13C enrichment in our study indicates that they consumed rhizosphere bacteria that were actively consuming root-derived carbon. This represents a flow of root-derived carbon through two trophic levels from plant to primary consumers and into predators. Several of the labeled bacterial genomes encoded systems that may act as grazing deterrents, for instance the pathway to produce HCN which acts as a nematicidal agent (70).
In this study, we traced carbon movement though two trophic levels from plant root carbon into bacterial genomes and then into phage genomes. Stable-isotope probing enabled us to identify the most actively infectious phage in the rhizosphere. We infer that the complete phage genomes are derived from phage particles or phage in the process of replication because of their circularized genomes, rather than phage integrated into bacterial genomes. Interestingly, two of the most highly labeled phage were more highly isotopically labeled than their bacterial hosts. It is likely that recently synthesized nucleotide pools are more highly labeled that other cell structural components, and these nucleotides were shunted directly into the replicating phage genomes. The presence of highly labeled phage implies that phage predation may be a major source of bacterial death, and thus nutrient cycling, in the rhizosphere.
In the bacterial genomes we assembled, we also identified possible mediators of bacterium-bacterium communication and competition. In the labeled genomes, we identified many signaling compounds and quorum-quenching genes; although we cannot definitively verify their function, it appears communication systems could be critical for life in rhizosphere soil (71). The number and distribution of interbacterial killing systems in labeled genomes, including a possible facultative predatory bacteria Lysobacter (72), may indicate active competition for resources or space in the rhizosphere.
Finally, one of the more intriguing genomes we reconstructed is for an Acidobacterium. The Acidobacteria_68_21 genome encodes a flexible metabolism and many defensive capabilities, CRISPR-Cas systems, insecticidal proteins, and 40 BGCs. While this genome was not labeled, it was one of the most abundant bacteria in bulk soil, and these defense systems may serve to protect this dominant bacterium from grazing or parasitism.
By tracing the movement of the carbon from plant roots into the soil community, we can begin to understand rhizosphere ecology, which in turn informs us about the carbon cycle in soil. The possible interactions that we identify in rhizosphere soil have the ability to impact plant growth and shape the flow and stabilization of carbon in soil. Lysis of bacteria by phage or interbacterial killing systems may release easily metabolized compounds that could be respired and returned to the atmosphere. Also, bacteria may contribute to soil aggregate stability and carbon stabilization through the production of EPS and other types of organic matter (73). PGPB could enable the plant to fix more CO2, ultimately increasing the amount of carbon introduced into the soil (74). Only through a better understanding of the interdomain interactions occurring in soil can we begin to understand the functioning of soil.
Conclusions.
We used genome-resolved metagenomics applied in the context of a SIP study to generate insights into the active members of a rhizosphere soil community. We identified organisms and genetic sequences that suggest mechanisms of potential interaction; these provide fertile topics for further exploration and verification. In the long term, understanding of soil interaction networks may provide pathways to improve plant primary production and carbon compound sequestration in soil.
MATERIALS AND METHODS
Plant growth and 13CO2 labeling.
To generate the samples for this study, we grew common wild oat, Avena fatua, in greenhouse microcosms packed to field bulk density with grassland soil. Briefly, surface soil (0 to 10 cm) was collected from the University of California Hopland Research and Extension Center (Hopland, CA, USA), from a field dominated by Avena spp. The soil is a fine-loamy, mixed, active, mesic Typic Haploxeralf (75, 76). For this experiment, three separate microcosms were constructed (see Fig. S1 in the supplemental material) and maintained at approximately 15% soil moisture; additional details of the microcosm design and plant growth conditions have been documented previously (41). One microcosm was destructively harvested after 2 weeks of acclimation; before planting, this represents our time zero sample. Since this sample had no root influence, the soil is considered a “bulk” soil. In the other two microcosms, an A. fatua seedling was planted, and the microcosms were incubated in an isotope labeling chamber supplied with 99 atom% 13CO2 at 400 μl/liter CO2 during the day to achieve an overall atmospheric 13C enrichment of 90 atom%. After 6 and 9 weeks of growth, a single microcosm was destructively harvested. Rhizosphere samples were defined as soil attached to a root after gentle shaking. Paired bulk soil samples from weeks 6 and 9 came from a root exclusion mesh bag (1 μm) which was designed to exclude roots but allow the soil inside to otherwise experience identical microcosm conditions (moisture, temperature, etc.). Rhizosphere soil was washed off roots, and bulk soil was collected from the bulk soil bags; from these soils, 0.5 g was used for DNA extraction using a phenol-chloroform procedure (41). In sum, our analyses included five separate DNA samples, a time zero bulk sample, week 6 bulk and rhizosphere samples, and week 9 bulk and rhizosphere samples.
Density separation.
We used a CsCl density gradient centrifugation to separate each DNA sample based on density using previously described methods (77). Briefly, for each sample, 5.5 μg of DNA was added to a gradient buffer with a density of 1.735 g/ml. The solution was spun in ultracentrifuge tubes (Beckman Coulter Quick-Seal, 13 × 51 mm) in an Optima L-90K ultracentrifuge (Beckman Coulter, Brea, CA, USA) using a VTi65.2 rotor at 44,000 rpm (176,284 average relative centrifugal force [RCFavg]) at 20°C for 109 h with maximum acceleration and braking of the rotor to maintain the integrity of the density separations. The gradient was then separated into ∼32 fractions using a syringe pump delivering light mineral oil. Each fraction (∼144 μl) was measured for density using an AR200 digital refractometer (Reichert Inc., Depew, NY, USA), and DNA was precipitated and quantified as previously described (77). Fractions were then combined into three bins based on density and by comparison between the rhizosphere samples and the associated bulk soil (light = 1.692 to 1.737 g/ml; middle = 1.738 to 1.746 g/ml; heavy = 1.747 to 1.765 g/ml; see Fig. S2 and Table S1 in the supplemental material). The heavy bin was defined as any fraction with a density greater than the point at which the bulk sample DNA concentration reached 0 ng/μl DNA. The rhizosphere middle fractions (and bulk heavier fractions) were defined as every fraction between the point where the rhizosphere and bulk lines crossed and where the heavy fraction started (Fig. S2). For the rhizosphere samples, we sequenced all three bins; for the bulk samples, we sequenced only the light and heavier bins. Thus, from the five separate DNA samples described above, we generated 12 distinct fractionated DNA samples for sequencing.
Sequencing.
The 12 fractionated DNA samples were sequenced at the University of California (UC) Davis Genome Center on an Illumina HiSeq 3000 (Illumina Inc., Hayward, CA, USA) with paired-end libraries prepared with the Kapa Hyper protocol and a read length of 150 bp.
Sequence preparation and analysis.
Reads were trimmed using Sickle (https://github.com/najoshi/sickle; version 1.33) with default parameters; BBtools (https://sourceforge.net/projects/bbmap/; version 35) was used with default parameters to remove Illumina adaptors and phiX sequences. Each sample was assembled individually using IDBA-UD (-step 20, -maxk 140, -mink 40) (78). Only scaffolds larger than 1,000 bp were included in further analyses. Genes were predicted using Prodigal (79). The open reading frames (ORFs) were annotated using a combined approach. Sequence similarity searches were performed using USEARCH (version 7.0.959) (80) against UniRef100 (July 2014) (81), UniProt (June 2014) (82), and the KEGG (June 2015) (83) databases. Additional gene annotations were done using hidden Markov models (HMMs) that were constructed based on KEGG Orthologies (KO) as outlined in reference 84. Briefly, all proteins assigned to a KO were clustered using MCL (85) with inflation parameter (-I) of 1.1, based on global percent identity. Individual trusted thresholds were calculated by running HMM search of all the proteins with assigned KOs against the HMM database. Clusters were aligned using MAFFT v7 (86), and HMMs were constructed using the HMMER suite (87). Protein domain-level analysis was conducted using InterProScan (88). Carbohydrate active enzymes were identified using dbCAN2 (89, 90). Secondary metabolite clusters were found using antiSMASH 4.0 (91, 92). tRNAs were predicted using tRNAScan-SE (93). The 16S and 18S rRNA sequences were found and aligned using ssu_tree.py (https://github.com/christophertbrown/bioscripts27; version 1.0). Eukaryotic 18S rRNA genes were dereplicated and clustered at 98% nucleic acid identity representing a possible species-level designation (94, 95), aligned using SSU-ALIGN (96), and trees were generated using RAxML on CIPRES (97, 98). Genomes were binned using a combined approach. We used abawaca (https://github.com/CK7/abawaca; version 1.00), MaxBin2 (99), and MetaBAT (100), the most complete bins with the least amount of contamination (as calculated by the number of 51 single copy genes [Fig. S3]) were chosen using DAS Tool (101). Further genome curation was conducted in ggKbase (101, 102), such as the removal of taxonomically divergent scaffolds which were binned incorrectly from algorithms which do not incorporate phylogenetic information (https://ggkbase.berkeley.edu/). Bins were dereplicated to the species level based on rpS3.
The rpS3 genes were identified and dereplicated to the species level (99% nucleotide identity), and the longest scaffold was chosen using rpS3_trckr (https://github.com/AJProbst/rpS3_trckr; version 1.0). Each sample was mapped to each scaffold using Bowtie2 (--sensitive and --rfg 200,300), the reads were filtered for two mismatches, and the coverage was calculated using calculate_breadth.py (https://github.com/banfieldlab/mattolm-public-scripts/blob/master/calculate_breadth.py; version 1.0), which calculates the coverage of supplied scaffolds based on mapping files produced with Bowtie2 (103). The coverage values for the rpS3 scaffolds were normalized for total read depth from the corresponding sample. Principal coordinate analysis was conducted in the R programming environment with the vegan package (104, 105). The R script is publicly available (106). rpS3 amino acid sequences were aligned using MAFFT v7.402 (86) with the E-INS-i options on Cipres (98). Trees were generated on Cipres using RaxML with the Jones-Taylor-Thornton (JTT) protein substitution model, and figures were generated using iTOL (97, 98, 107).
Phage genomes were identified using VirSorter and manually on ggKbase (108). Phage genome completeness was checked by mapping reads, as described above, and visualizing on Geneious R9.1 (109). Complete, circularized phage genomes will have uniform read coverage across the genome and reads paired across the entire span of the scaffold without repetitive elements on the end of the scaffold which could cause long paired reads instead of a circular sequence. CRISPR spacers were found using CRISPRDetect (110).
Enrichment, sip, and growth rate.
The coverage and relative abundance of individual soil populations were calculated based on mapping reads using Bowtie2 (--sensitive and --rfg 200,300) (103). For bacterial bins, the reads were mapped to all scaffolds in the bin. For eukaryotes, we were only able to identify individual eukaryote scaffolds with the 18S rRNA gene, but we were not able to bin genomes from the eukaryotes. For eukaryotes, reads were mapped to the whole scaffold containing the 18S rRNA gene. For phage, the reads were mapped to the complete phage genomes. Reads were filtered for two mismatches, and the coverage was calculated using calculate_breadth.py. The coverage values were normalized for total read depth from the corresponding sample.
We estimated the atom percent excess (APE) 13C enrichment for each taxon following the procedures detailed in Hungate et al. (37), with the following adjustments for metagenome-assembled genomes instead of 16S rRNA genes. The density of an organism’s DNA was compared between labeled (rhizosphere) and unlabeled (bulk) samples, and a model of isotope substitution in DNA was used to convert the observed change in density to isotope enrichment (36, 37). In previous qSIP experiments, amplicon sequencing of the 16S rRNA gene has been used to estimate the relative abundances of bacterial and archaeal taxa. Those relative abundances were then converted to estimates of absolute abundance by multiplying by the total number of 16S rRNA gene copies using the universal 16S rRNA primer for qPCR for each density fraction in each replicate gradient. Here, we used the relative coverage (i.e., coverage normalized for read depth) of metagenome-assembled genomes as a proxy for relative abundance and total DNA concentration in place of total 16S copies in order to calculate a metric of abundance (y) for each taxon (i) in each density fraction (k), for each replicate (j) as
where p is relative coverage and f is the total DNA concentration. We acknowledge that this metric of taxon-specific abundance (y) is incomplete because the total rhizosphere DNA presumably represents many more taxa beyond the metagenome-assembled genomes and eukaryotic scaffolds that we identified. The qSIP analyses were conducted separately for the week 6 and week 9 data. For each time point, we used the bulk soil data from the corresponding week as the unlabeled treatment. Because our experiment was not replicated, we were unable to calculate confidence intervals for APE 13C based on means and standard errors, an approach that has been used previously to estimate minimum detectable differences in isotope incorporation (37). However, in our data, the relationship between population rank and APE 13C value exhibited a spline function pattern (Fig. S8), suggesting there is a threshold above which 13C-label incorporation in the bacterial genome bins, phage genomes, and eukaryotic scaffolds was detectable above background variation in the qSIP-derived estimates of 13C uptake. We used breakpoint analysis to test this spline function and identify a threshold value above which 13C incorporation could be confidently inferred. We used the “segmented” package in R, and the segmented function to identify the breakpoint, with the Davies test for significance. We focused this analysis on the 90 populations exhibiting the lowest values of qSIP-estimated 13C atom percent excess (i.e., from ranks 1 to 90), thereby avoiding more enriched regions where the slope of the rank-APE relationship declined (Fig. S8B). We found that including these high-rank APE values skewed the estimate of the breakpoint to near 0 APE, whereas focusing on the data surrounding the visually obvious breakpoint of interest resulted in a more conservative estimate of the detection threshold. We identified the breakpoint threshold at rank 71.1, with a standard error of 0.6. We used this breakpoint plus three times the standard error (the upper 99.7% confidence limit of the breakpoint estimate) to identify the threshold above which 13C uptake could be reasonably inferred. The 13C APE value associated with the threshold was 2.5%. Thus, we interpreted values above that APE value as having detectably incorporated the 13C tracer. We note that this is somewhat lower than threshold values estimated using confidence intervals calculated from replicated measurements in a 16S rRNA gene qSIP study (5.6% for 13C in Hungate et al. [37]).
Breakpoint analysis was used to determine the minimum detectable difference of isotope incorporation for the bacterial genome bins, phage genomes, and eukaryotic scaffolds analyzed in this study. (A) Atom fraction excess (AFE) compared to the rank of bacterial bins, phage genomes, and eukaryotic scaffolds based on AFE. (B) In the inset region—focused around the predicted breakpoint region—we calculated the breakpoint in the spline curve as an indication of the lowest enrichment where 13C incorporation could be confidently inferred. Download FIG S8, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
For each taxon, we also estimated the gross growth rate based on plant-derived 13C-enriched substrates. Because some taxa may have used nonlabeled substrates for growth, this metric does not capture all growth that may have occurred during the incubation. Accordingly, these taxon-specific estimates can be thought of as measuring the degree to which individual microbial populations in the rhizosphere grew on plant-derived carbon. We used the approach outlined by Koch et al. (36), but for 13C instead of 18O (37), and we did not estimate taxon-specific mortality rates. We used a linear growth model:
where NTOTALit and NTOTALi0 represent the total (labeled plus unlabeled) abundance of taxon i at time t and time zero, respectively, and ri is the net growth rate of taxon i with units of nanogram of DNA gram of soil−1 day−1. Abundances were estimated as described above (for the atom percent excess calculations). We further assumed that all carbon atoms in newly created DNA could originate from the labeled C substrates in the rhizosphere, thus we set U = 1.0 (36).
Data availability.
All sequence data have been made public and can be found on NCBI using the following accession numbers: BioProject, PRJNA419965; SRA, SRX6701119 to SRX6701130; complete phage genomes, MN304815.1 to MN304824.1; eukaryotic 18S rRNA, MT533858 to MT533883, and the metagenome-assembled genome bins, JAEKKE000000000 to JAEKMG000000000. Additional data, including raw trees and genome annotations, are available on Figshare (https://doi.org/10.6084/m9.figshare.c.5405805).
ACKNOWLEDGMENTS
This work was made possible by samples and expertise provided by personnel at the Hopland Research and Extension Center (Hopland, CA, USA). Plant growth and labeling were conducted at the UC Berkeley Oxford Facility. Labeling, sampling efforts, and technical expertise were provided by Katerina Estera-Molina and Donald J. Herman. Sequencing was done at the UC Davis Genome Center, and technical advice was provided by Lutz Froenicke. The HMM pipeline was developed by David Burstein.
This research was supported by the U.S. Department of Energy Office of Science, Office of Biological and Environmental Research Genomic Science program under awards DE-SC0010570 and DE-SC0016247 to M.K.F., DE-SC10010566 to J.F.B., DE-SC0020172 to B.A.H., and SCW1589 and SCW1632 to J.P.-R. A.J.P. was supported by the German Science Foundation under DFG1603-1/1. Work conducted at Lawrence Livermore National Laboratory was done under the auspices of the U.S. Department of Energy under contract DE-AC52-07NA27344. E.P.S. was supported by a grant from the National Science Foundation Graduate Research Fellowships Program and by the National Science Foundation CZO EAR-1331940 grant for the Eel River Critical Zone Observatory.
S.S., J.P.-R., and M.K.F. designed the labeling experiment. S.S. carried out the labeling. E.P.S. and S.J.B. conducted the density centrifugation. B.J.K. and B.A.H. conducted the qSIP and growth rate analyses and interpretation. E.P.S., M.K.F., and J.F.B. analyzed the data, with input from A.J.P. and J.P.-R. E.P.S., M.K.F., and J.F.B. wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Mary K. Firestone, Email: mkfstone@berkeley.edu.
Jillian F. Banfield, Email: jbanfield@berkeley.edu.
Vincent B. Young, University of Michigan—Ann Arbor
REFERENCES
- 1.Pett-Ridge J, Shi S, Estera-Molina K, Nuccio E, Yuan M, Rijkers R, Swenson T, Zhalnina K, Northen T, Zhou J, Firestone MK. 2021. Rhizosphere carbon turnover from cradle to grave: the role of microbe–plant interactions, p 51–73. In Gupta VVSR, Sharma AK (ed), Rhizosphere biology: interactions between microbes and plants. Springer, Singapore, Singapore. [Google Scholar]
- 2.Bahram M, Hildebrand F, Forslund SK, Anderson JL, Soudzilovskaia NA, Bodegom PM, Bengtsson-Palme J, Anslan S, Coelho LP, Harend H, Huerta-Cepas J, Medema MH, Maltz MR, Mundra S, Olsson PA, Pent M, Põlme S, Sunagawa S, Ryberg M, Tedersoo L, Bork P. 2018. Structure and function of the global topsoil microbiome. Nature 560:233–237. doi: 10.1038/s41586-018-0386-6. [DOI] [PubMed] [Google Scholar]
- 3.Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK, Fierer N. 2018. A global atlas of the dominant bacteria found in soil. Science 359:320–325. doi: 10.1126/science.aap9516. [DOI] [PubMed] [Google Scholar]
- 4.Gao C, Montoya L, Xu L, Madera M, Hollingsworth J, Purdom E, Hutmacher RB, Dahlberg JA, Coleman-Derr D, Lemaux PG, Taylor JW. 2019. Strong succession in arbuscular mycorrhizal fungal communities. ISME J 13:214–226. doi: 10.1038/s41396-018-0264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor DL, Walters WA, Lennon NJ, Bochicchio J, Krohn A, Caporaso JG, Pennanen T. 2016. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing. Appl Environ Microbiol 82:7217–7226. doi: 10.1128/AEM.02576-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butterfield CN, Li Z, Andeer PF, Spaulding S, Thomas BC, Singh A, Hettich RL, Suttle KB, Probst AJ, Tringe SG, Northen T, Pan C, Banfield JF. 2016. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 4:e2687. doi: 10.7717/peerj.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diamond S, Andeer PF, Li Z, Crits-Christoph A, Burstein D, Anantharaman K, Lane KR, Thomas BC, Pan C, Northen TR, Banfield JF. 2019. Mediterranean grassland soil C–N compound turnover is dependent on rainfall and depth, and is mediated by genomically divergent microorganisms. Nat Microbiol 4:1356−1367. doi: 10.1038/s41564-019-0449-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White RA, III, Bottos EM, Roy Chowdhury T, Zucker JD, Brislawn CJ, Nicora CD, Fansler SJ, Glaesemann KR, Glass K, Jansson JK. 2016. Moleculo long-read sequencing facilitates assembly and genomic binning from complex soil metagenomes. mSystems 1:e00045-16. doi: 10.1128/mSystems.00045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kroeger ME, Delmont TO, Eren AM, Meyer KM, Guo J, Khan K, Rodrigues JLM, Bohannan BJM, Tringe SG, Borges CD, Tiedje JM, Tsai SM, Nüsslein K. 2018. New biological insights into how deforestation in Amazonia affects soil microbial communities using metagenomics and metagenome-assembled genomes. Front Microbiol 9:1635. doi: 10.3389/fmicb.2018.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue Y, Jonassen I, Øvreås L, Taş N. 2019. Bacterial and archaeal metagenome-assembled genome sequences from Svalbard permafrost. Microbiol Resour Announc 8:e00516-19. doi: 10.1128/MRA.00516-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyc O, Song C, Dickschat JS, Vos M, Garbeva P. 2017. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol 25:280–292. doi: 10.1016/j.tim.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Crits-Christoph A, Diamond S, Butterfield CN, Thomas BC, Banfield JF. 2018. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558:440–444. doi: 10.1038/s41586-018-0207-y. [DOI] [PubMed] [Google Scholar]
- 13.Emerson JB, Roux S, Brum JR, Bolduc B, Woodcroft BJ, Bin Jang H, Singleton CM, Solden LM, Naas AE, Boyd JA, Hodgkins SB, Wilson RM, Trubl G, Li C, Frolking S, Pope PB, Wrighton KC, Crill PM, Chanton JP, Saleska SR, Tyson GW, Rich VI, Sullivan MB. 2018. Host-linked soil viral ecology along a permafrost thaw gradient. Nat Microbiol 3:870–880. doi: 10.1038/s41564-018-0190-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starr EP, Nuccio EE, Pett-Ridge J, Banfield JF, Firestone MK. 2019. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc Natl Acad Sci USA 116:25900–25908. doi: 10.1073/pnas.1908291116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trubl G, Bin Jang H, Roux S, Emerson JB, Solonenko N, Vik DR, Solden L, Ellenbogen J, Runyon AT, Bolduc B, Woodcroft BJ, Saleska SR, Tyson GW, Wrighton KC, Sullivan MB, Rich VI. 2018. Soil viruses are underexplored players in ecosystem carbon processing. mSystems 3:e00076-18. doi: 10.1128/mSystems.00076-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L, Xu J, Feng Y, Wang J, Yu Y, Brookes PC. 2015. Responses of soil microeukaryotic communities to short-term fumigation-incubation revealed by MiSeq amplicon sequencing. Front Microbiol 6:1149. doi: 10.3389/fmicb.2015.01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lara E, Berney C, Harms H, Chatzinotas A. 2007. Cultivation-independent analysis reveals a shift in ciliate 18S rRNA gene diversity in a polycyclic aromatic hydrocarbon-polluted soil. FEMS Microbiol Ecol 62:365–373. doi: 10.1111/j.1574-6941.2007.00387.x. [DOI] [PubMed] [Google Scholar]
- 18.Crossley DA, Blair JM. 1991. A high-efficiency, “low-technology” Tullgren-type extractor for soil microarthropods. Agric Ecosyst Environ 34:187–192. doi: 10.1016/0167-8809(91)90104-6. [DOI] [Google Scholar]
- 19.Macfadyen A. 1953. Notes on methods for the extraction of small soil arthropods. J Anim Ecol 22:65. doi: 10.2307/1691. [DOI] [Google Scholar]
- 20.Warmink JA, van Elsas JD. 2009. Migratory response of soil bacteria to Lyophyllum sp. strain Karsten in soil microcosms. Appl Environ Microbiol 75:2820–2830. doi: 10.1128/AEM.02110-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deveau A, Antony-Babu S, Le Tacon F, Robin C, Frey-Klett P, Uroz S. 2016. Temporal changes of bacterial communities in the Tuber melanosporum ectomycorrhizosphere during ascocarp development. Mycorrhiza 26:389–399. doi: 10.1007/s00572-015-0679-7. [DOI] [PubMed] [Google Scholar]
- 22.Ashelford KE, Day MJ, Fry JC. 2003. Elevated abundance of bacteriophage infecting bacteria in soil. Appl Environ Microbiol 69:285–289. doi: 10.1128/AEM.69.1.285-289.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang A, Liu N, Tian Q, Bai W, Williams M, Wang Q, Li L, Zhang WH. 2015. Rhizosphere bacterial communities of dominant steppe plants shift in response to a gradient of simulated nitrogen deposition. Front Microbiol 6:789. doi: 10.3389/fmicb.2015.00789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonfante P, Genre A. 2010. Mechanisms underlying beneficial plant–fungus interactions in mycorrhizal symbiosis. Nat Commun 1:48. doi: 10.1038/ncomms1046. [DOI] [PubMed] [Google Scholar]
- 25.Schubotz F, Hays LE, Meyer-Dombard DR, Gillespie A, Shock EL, Summons RE. 2015. Stable isotope labeling confirms mixotrophic nature of streamer biofilm communities at alkaline hot springs. Front Microbiol 6:42. doi: 10.3389/fmicb.2015.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godwin S, Kang A, Gulino L-M, Manefield M, Gutierrez-Zamora M-L, Kienzle M, Ouwerkerk D, Dawson K, Klieve AV. 2014. Investigation of the microbial metabolism of carbon dioxide and hydrogen in the kangaroo foregut by stable isotope probing. ISME J 8:1855–1865. doi: 10.1038/ismej.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herrmann E, Young W, Rosendale D, Reichert-Grimm V, Riedel CU, Conrad R, Egert M. 2017. RNA-based stable isotope probing suggests Allobaculum spp. as particularly active glucose assimilators in a complex murine microbiota cultured in vitro. Biomed Res Int 2017:1829685. doi: 10.1155/2017/1829685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egert M, De Graaf AA, Maathuis A, De Waard P, Plugge CM, Smidt H, Deutz NEP, Dijkema C, De Vos WM, Venema K. 2007. Identification of glucose-fermenting bacteria present in an in vitro model of the human intestine by RNA-stable isotope probing. FEMS Microbiol Ecol 60:126–135. doi: 10.1111/j.1574-6941.2007.00281.x. [DOI] [PubMed] [Google Scholar]
- 29.Murase J, Shibata M, Lee CG, Watanabe T, Asakawa S, Kimura M. 2012. Incorporation of plant residue-derived carbon into the microeukaryotic community in a rice field soil revealed by DNA stable-isotope probing. FEMS Microbiol Ecol 79:371–379. doi: 10.1111/j.1574-6941.2011.01224.x. [DOI] [PubMed] [Google Scholar]
- 30.Wilhelm RC, Singh R, Eltis LD, Mohn WW. 2019. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J 13:413–429. doi: 10.1038/s41396-018-0279-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haichar FEZ, Roncato M-A, Achouak W. 2012. Stable isotope probing of bacterial community structure and gene expression in the rhizosphere of Arabidopsis thaliana. FEMS Microbiol Ecol 81:291–302. doi: 10.1111/j.1574-6941.2012.01345.x. [DOI] [PubMed] [Google Scholar]
- 32.Hünninghaus M, Dibbern D, Kramer S, Koller R, Pausch J, Schloter-Hai B, Urich T, Kandeler E, Bonkowski M, Lueders T. 2019. Disentangling carbon flow across microbial kingdoms in the rhizosphere of maize. Soil Biol Biochem 134:122–130. doi: 10.1016/j.soilbio.2019.03.007. [DOI] [Google Scholar]
- 33.Pett-Ridge J, Firestone MK. 2017. Using stable isotopes to explore root-microbe-mineral interactions in soil. Rhizosphere 3:244–253. doi: 10.1016/j.rhisph.2017.04.016. [DOI] [Google Scholar]
- 34.Haig S-J, Schirmer M, D'Amore R, Gibbs J, Davies RL, Collins G, Quince C. 2015. Stable-isotope probing and metagenomics reveal predation by protozoa drives E. coli removal in slow sand filters. ISME J 9:797–808. doi: 10.1038/ismej.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee CG, Watanabe T, Fujita Y, Asakawa S, Kimura M. 2012. Heterotrophic growth of cyanobacteria and phage-mediated microbial loop in soil: examination by stable isotope probing (SIP) method. Soil Sci Plant Nutr 58:161–168. doi: 10.1080/00380768.2012.658739. [DOI] [Google Scholar]
- 36.Koch BJ, McHugh TA, Hayer M, Schwartz E, Blazewicz SJ, Dijkstra P, Gestel N, Marks JC, Mau RL, Morrissey EM, Pett‐Ridge J, Hungate BA. 2018. Estimating taxon‐specific population dynamics in diverse microbial communities. Ecosphere 9:e2090. doi: 10.1002/ecs2.2090. [DOI] [Google Scholar]
- 37.Hungate BA, Mau RL, Schwartz E, Caporaso JG, Dijkstra P, van Gestel N, Koch BJ, Liu CM, McHugh TA, Marks JC, Morrissey EM, Price LB. 2015. Quantitative microbial ecology through stable isotope probing. Appl Environ Microbiol 81:7570–7581. doi: 10.1128/AEM.02280-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsurumaru H, Okubo T, Okazaki K, Hashimoto M, Kakizaki K, Hanzawa E, Takahashi H, Asanome N, Tanaka F, Sekiyama Y, Ikeda S, Minamisawa K. 2015. Metagenomic analysis of the bacterial community associated with the taproot of sugar beet. Microbes Environ 30:63–69. doi: 10.1264/jsme2.ME14109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wylezich C, Papa A, Beer M, Höper D. 2018. A versatile sample processing workflow for metagenomic pathogen detection. Sci Rep 8:13108–13112. doi: 10.1038/s41598-018-31496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Starr EP, Shi S, Blazewicz SJ, Probst AJ, Herman DJ, Firestone MK, Banfield JF. 2018. Stable isotope informed genome-resolved metagenomics reveals that Saccharibacteria utilize microbially-processed plant-derived carbon. Microbiome 6:122. doi: 10.1186/s40168-018-0499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi S, Nuccio E, Herman DJ, Rijkers R, Estera K, Li J, da Rocha UN, He Z, Pett-Ridge J, Brodie EL, Zhou J, Firestone M. 2015. Successional trajectories of rhizosphere bacterial communities over consecutive seasons. mBio 6:e00746. doi: 10.1128/mBio.00746-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharon I, Kertesz M, Hug LA, Pushkarev D, Blauwkamp TA, Castelle CJ, Amirebrahimi M, Thomas BC, Burstein D, Tringe SG, Williams KH, Banfield JF. 2015. Accurate, multi-kb reads resolve complex populations and detect rare microorganisms. Genome Res 25:534–543. doi: 10.1101/gr.183012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Probst AJ, Ladd B, Jarett JK, Geller-McGrath DE, Sieber CMK, Emerson JB, Anantharaman K, Thomas BC, Malmstrom RR, Stieglmeier M, Klingl A, Woyke T, Ryan MC, Banfield JF. 2018. Differential depth distribution of microbial function and putative symbionts through sediment-hosted aquifers in the deep terrestrial subsurface. Nat Microbiol 3:328–336. doi: 10.1038/s41564-017-0098-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. 2013. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23:111–120. doi: 10.1101/gr.142315.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lueders T, Manefield M, Friedrich MW. 2004. Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ Microbiol 6:73–78. doi: 10.1046/j.1462-2920.2003.00536.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang D, Pajerowska-Mukhtar K, Culler AH, Dong X. 2007. Salicylic acid inhibits pathogen growth in plants through repression of the auxin signaling pathway. Curr Biol 17:1784–1790. doi: 10.1016/j.cub.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 47.Balashova NV, Stolz A, Knackmuss HJ, Kosheleva IA, Naumov AV, Boronin AM. 2001. Purification and characterization of a salicylate hydroxylase involved in 1-hydroxy-2-naphthoic acid hydroxylation from the naphthalene and phenanthrene-degrading bacterial strain Pseudomonas putida BS202-P1. Biodegradation 12:179–188. doi: 10.1023/a:1013126723719. [DOI] [PubMed] [Google Scholar]
- 48.Mur LAJ, Carver TLW, Prats E. 2006. NO way to live; the various roles of nitric oxide in plant–pathogen interactions. J Exp Bot 57:489–505. doi: 10.1093/jxb/erj052. [DOI] [PubMed] [Google Scholar]
- 49.Farag MA, Ryu C-M, Sumner LW, Paré PW. 2006. GC–MS SPME profiling of rhizobacterial volatiles reveals prospective inducers of growth promotion and induced systemic resistance in plants. Phytochemistry 67:2262–2268. doi: 10.1016/j.phytochem.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 50.Ryu C-M, Farag MA, Hu C-H, Reddy MS, Wei H-X, Pare PW, Kloepper JW. 2003. Bacterial volatiles promote growth in Arabidopsis. Proc Natl Acad Sci USA 100:4927–4932. doi: 10.1073/pnas.0730845100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alori ET, Glick BR, Babalola OO. 2017. Microbial phosphorus solubilization and its potential for use in sustainable agriculture. Front Microbiol 8:971. doi: 10.3389/fmicb.2017.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cervini-Silva J, Kearns J, Banfield J. 2012. Steady-state dissolution kinetics of mineral ferric phosphate in the presence of desferrioxamine-B and oxalate ligands at pH=4–6 and T=24±0.6°C. Chem Geol 320-321:1–8. doi: 10.1016/j.chemgeo.2012.05.022. [DOI] [Google Scholar]
- 53.Nazir R, Mazurier S, Yang P, Lemanceau P, van Elsas JD. 2017. The ecological role of type three secretion systems in the interaction of bacteria with fungi in soil and related habitats is diverse and context-dependent. Front Microbiol 8:38. doi: 10.3389/fmicb.2017.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Green ER, Mecsas J. 2016. Bacterial secretion systems: an overview. Microbiol Spectr 4. doi: 10.1128/microbiolspec.VMBF-0012-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y, Li B, Cen K, Lu Y, Zhang S, Wang C. 2018. Diverse effect of phosphatidylcholine biosynthetic genes on phospholipid homeostasis, cell autophagy and fungal developments in Metarhizium robertsii. Environ Microbiol 20:293–304. doi: 10.1111/1462-2920.13998. [DOI] [PubMed] [Google Scholar]
- 56.Rittenour WR, Chen M, Cahoon EB, Harris SD. 2011. Control of glucosylceramide production and morphogenesis by the Bar1 ceramide synthase in Fusarium graminearum. PLoS One 6:e19385. doi: 10.1371/journal.pone.0019385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matz C, Kjelleberg S. 2005. Off the hook – how bacteria survive protozoan grazing. Trends Microbiol 13:302–307. doi: 10.1016/j.tim.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Berne C, Ducret A, Hardy GG, Brun YV. 2015. Adhesins involved in attachment to abiotic surfaces by Gram-negative bacteria. Microbiol Spectr 3. doi: 10.1128/microbiolspec.MB-0018-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen W-J, Hsieh F-C, Hsu F-C, Tasy Y-F, Liu J-R, Shih M-C. 2014. Characterization of an insecticidal toxin and pathogenicity of Pseudomonas taiwanensis against insects. PLoS Pathog 10:e1004288. doi: 10.1371/journal.ppat.1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Esmaeel Q, Pupin M, Kieu NP, Chataigné G, Béchet M, Deravel J, Krier F, Höfte M, Jacques P, Leclère V. 2016. Burkholderia genome mining for nonribosomal peptide synthetases reveals a great potential for novel siderophores and lipopeptides synthesis. Microbiologyopen 5:512–526. doi: 10.1002/mbo3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ho BT, Dong TG, Mekalanos JJ. 2014. A view to a kill: the bacterial type VI secretion system. Cell Host Microbe 15:9–21. doi: 10.1016/j.chom.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sieradzki ET, Koch BJ, Greenlon A, Sachdeva R, Malmstrom RR, Mau RL, Blazewicz SJ, Firestone MK, Hofmockel KS, Schwartz E, Hungate BA, Pett-Ridge J. 2020. Measurement error and resolution in quantitative stable isotope probing: implications for experimental design. mSystems 5:e00151-20. doi: 10.1128/mSystems.00151-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huynen M, Snel B, Lathe W, Bork P. 2000. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res 10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galperin MY, Koonin V. 2000. Who’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol 18:609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- 65.Singh RP, Shelke GM, Kumar A, Jha PN. 2015. Biochemistry and genetics of ACC deaminase: a weapon to “stress ethylene” produced in plants. Front Microbiol 6:937. doi: 10.3389/fmicb.2015.00937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bouskill NJ, Wood TE, Baran R, Ye Z, Bowen BP, Lim H, Zhou J, Van Nostrand JD, Nico P, Northen TR, Silver WL, Brodie EL. 2016. Belowground response to drought in a tropical forest soil. I. Changes in microbial functional potential and metabolism. Front Microbiol 7:525. doi: 10.3389/fmicb.2016.00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nuccio EE, Starr E, Karaoz U, Brodie EL, Zhou J, Tringe SG, Malmstrom RR, Woyke T, Banfield JF, Firestone MK, Pett-Ridge J. 2020. Niche differentiation is spatially and temporally regulated in the rhizosphere. ISME J 14:999–1014. doi: 10.1038/s41396-019-0582-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Warzecha T, Zieliński A, Skrzypek E, Wójtowicz T, Moś M. 2012. Effect of mechanical damage on vigor, physiological parameters, and susceptibility of oat (Avena sativa) to Fusarium culmorum infection. Phytoparasitica 40:29–36. doi: 10.1007/s12600-011-0196-y. [DOI] [Google Scholar]
- 69.Olanrewaju OS, Glick BR, Babalola OO. 2017. Mechanisms of action of plant growth promoting bacteria. World J Microbiol Biotechnol 33:197. doi: 10.1007/s11274-017-2364-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paiva G, Proença DN, Francisco R, Verissimo P, Santos SS, Fonseca L, Abrantes IMO, Morais PV. 2013. Nematicidal bacteria associated to pinewood nematode produce extracellular proteases. PLoS One 8:e79705. doi: 10.1371/journal.pone.0079705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deangelis KM, Lindow SE, Firestone MK. 2008. Bacterial quorum sensing and nitrogen cycling in rhizosphere soil. FEMS Microbiol Ecol 66:197–207. doi: 10.1111/j.1574-6941.2008.00550.x. [DOI] [PubMed] [Google Scholar]
- 72.Seccareccia I, Kost C, Nett M. 2015. Quantitative analysis of Lysobacter predation. Appl Environ Microbiol 81:7098–7105. doi: 10.1128/AEM.01781-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sher Y, Baker NR, Herman D, Fossum C, Hale L, Zhang X, Nuccio E, Saha M, Zhou J, Pett-Ridge J, Firestone M. 2020. Microbial extracellular polysaccharide production and aggregate stability controlled by switchgrass (Panicum virgatum) root biomass and soil water potential. Soil Biol Biochem 143:107742. doi: 10.1016/j.soilbio.2020.107742. [DOI] [Google Scholar]
- 74.Nie M, Bell C, Wallenstein MD, Pendall E. 2015. Increased plant productivity and decreased microbial respiratory C loss by plant growth-promoting rhizobacteria under elevated CO2. Sci Rep 5:9212–9216. doi: 10.1038/srep09212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whitman T, Neurath R, Perera A, Chu‐Jacoby I, Ning D, Zhou J, Nico P, Pett‐Ridge J, Firestone M. 2018. Microbial community assembly differs across minerals in a rhizosphere microcosm. Environ Microbiol 20:4444–4460. doi: 10.1111/1462-2920.14366. [DOI] [PubMed] [Google Scholar]
- 76.Placella SA, Brodie EL, Firestone MK. 2012. Rainfall-induced carbon dioxide pulses result from sequential resuscitation of phylogenetically clustered microbial groups. Proc Natl Acad Sci USA 109:10931–10936. doi: 10.1073/pnas.1204306109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blazewicz SJ, Schwartz E, Firestone MK. 2014. Growth and death of bacteria and fungi underlie rainfall-induced carbon dioxide pulses from seasonally dried soil. Ecology 95:1162–1172. doi: 10.1890/13-1031.1. [DOI] [PubMed] [Google Scholar]
- 78.Peng Y, Leung HCM, Yiu SM, Chin FYL. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 79.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 81.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. 2007. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 82.Magrane M, UniProt Consortium. 2011. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. 1999. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Probst AJ, Castelle CJ, Singh A, Brown CT, Anantharaman K, Sharon I, Hug LA, Burstein D, Emerson JB, Thomas BC, Banfield JF. 2017. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO 2 concentrations. Environ Microbiol 19:459–474. doi: 10.1111/1462-2920.13362. [DOI] [PubMed] [Google Scholar]
- 85.Van Dongen S. 2008. Graph clustering via a discrete uncoupling process. SIAM J Matrix Anal Appl 30:121–141. doi: 10.1137/040608635. [DOI] [Google Scholar]
- 86.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zdobnov EM, Apweiler R. 2001. InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 89.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. 2012. DbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40:W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. 2011. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de los Santos ELC, Kim HU, Nave M, Dickschat JS, Mitchell DA, Shelest E, Breitling R, Takano E, Lee SY, Weber T, Medema MH. 2017. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res 45:W36–W41. doi: 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hadziavdic K, Lekang K, Lanzen A, Jonassen I, Thompson EM, Troedsson C. 2014. Characterization of the 18s rRNA gene for designing universal eukaryote specific primers. PLoS One 9:e87624. doi: 10.1371/journal.pone.0087624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu S, Xiong J, Yu YY. 2015. Taxonomic resolutions based on 18S rRNA genes: a case study of subclass Copepoda. PLoS One 10:e0131498. doi: 10.1371/journal.pone.0131498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nawrocki EP. 2009. Structural RNA homology search and alignment using covariance models. PhD thesis. Washington University in St. Louis, St. Louis, MO. [Google Scholar]
- 97.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees, p 1–8. In 2010 Gateway Computing Environments Workshop (GCE). Institute of Electrical and Electronics Engineers, New York, NY. [Google Scholar]
- 99.Wu YW, Simmons BA, Singer SW. 2016. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 100.Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF. 2018. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Raveh-Sadka T, Thomas BC, Singh A, Firek B, Brooks B, Castelle CJ, Sharon I, Baker R, Good M, Morowitz MJ, Banfield JF. 2015. Gut bacteria are rarely shared by co-hospitalized premature infants, regardless of necrotizing enterocolitis development. Elife 4:e05477. doi: 10.7554/eLife.05477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Szoecs E, Maintainer HW. 2019. Package “vegan” Title Community Ecology Package.
- 105.R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 106.Weinmaier T, Probst AJ, La Duc MT, Ciobanu D, Cheng J-F, Ivanova N, Rattei T, Vaishampayan P. 2015. A viability-linked metagenomic analysis of cleanroom environments: eukarya, prokaryotes, and viruses. Microbiome 3:62. doi: 10.1186/s40168-015-0129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roux S, Enault F, Hurwitz BL, Sullivan MB. 2015. VirSorter: mining viral signal from microbial genomic data. PeerJ 3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Biswas A, Staals RHJ, Morales SE, Fineran PC, Brown CM. 2016. CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics 17:356. doi: 10.1186/s12864-016-2627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental design of soil microcosms, stable-isotope probing density fractionation, and samples generated for sequencing from a 13CO2 labeling study of common wild oat, Avena fatua. Within the microcosms, hashed squares indicate root-excluding bulk soil bags. DNA fractionation plots show DNA density profiles from 0, 6, or 9 weeks of plant growth with unlabeled, partially labeled, and labeled community DNA indicated by gray, yellow, and peach shading, respectively. Brown lines and text indicate bulk soil samples, and green lines and text indicate rhizosphere samples. Download FIG S1, TIF file, 1.5 MB (1.5MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DNA density profiles from a 13CO2 plant labeling study illustrate stable-isotope separation in DNA 13C enrichment between labeled (rhizosphere) and unlabeled (bulk soil) treatments. The density (gram/milliliter) and concentration (nanogram/microliter) of DNA for each collected fraction from each sample is plotted to generate these SIP separation graphs. Fractions were subsequently combined into “light,” “middle,” and “heavy” bins prior to sequencing. Download FIG S2, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
SIP, sequencing, and metagenome assembly statistics. Download Table S1, PDF file, 0.03 MB (26.6KB, pdf) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Count of bacterial single copy genes in partial genome bins identified in this study. Color bar represents the clade of bin, colored according to the legend presented in Fig. 1. Download FIG S3, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
18S rRNA tree colored by clade. Black dots represent bootstrap values of ≥60. Long leaves were removed for ease of visualization, and 18S rRNA sequences identified in this study are shown in purple. Download FIG S4, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plots of atom percent excess (APR) versus gross growth rates of soil organisms (nanogram of DNA gram of soil−1 day−1) based on root-derived carbon. (Top) The log of gross growth per day on plant-derived carbon compared to APE, and (Bottom) violin plot of gross growth of different types of soil populations. Download FIG S5, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Biosynthetic gene clusters encoded in bacterial genome bins identified in this paper. Signaling compounds includes homoserine lactone clusters, N-acyl amino acid clusters, and butyrolactone cluster. Download FIG S6, TIF file, 2.9 MB (2.9MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic tree of GH25 gene. The GH25 genes from phages are highlighted in green, and the bacterial genome bin from this study is shown in blue. Download FIG S7, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Breakpoint analysis was used to determine the minimum detectable difference of isotope incorporation for the bacterial genome bins, phage genomes, and eukaryotic scaffolds analyzed in this study. (A) Atom fraction excess (AFE) compared to the rank of bacterial bins, phage genomes, and eukaryotic scaffolds based on AFE. (B) In the inset region—focused around the predicted breakpoint region—we calculated the breakpoint in the spline curve as an indication of the lowest enrichment where 13C incorporation could be confidently inferred. Download FIG S8, TIF file, 2.8 MB (2.8MB, tif) .
Copyright © 2021 Starr et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
All sequence data have been made public and can be found on NCBI using the following accession numbers: BioProject, PRJNA419965; SRA, SRX6701119 to SRX6701130; complete phage genomes, MN304815.1 to MN304824.1; eukaryotic 18S rRNA, MT533858 to MT533883, and the metagenome-assembled genome bins, JAEKKE000000000 to JAEKMG000000000. Additional data, including raw trees and genome annotations, are available on Figshare (https://doi.org/10.6084/m9.figshare.c.5405805).