

Abstract
Radiotherapy (RT) is an important anti-cancer treatment modality that activates innate and adaptive immune responses. When all-trans retinoic acid (RA) was administered with radiation, we observed superior antitumor responses compared to ionizing radiation (IR) alone or RA alone. The superior antitumor effects of combination treatment were accompanied by a dramatic increase of TNF-α– and inducible nitric oxide synthase (iNOS)–producing inflammatory macrophages in local and distal non-irradiated (distal) tumors. Inflammatory macrophages are essential for the therapeutic efficacy of combination treatment by inducing effector T cell infiltration and enhancing the effector T cell to regulatory T cell ratio in local and distal tumors. T cells and T cell-derived IFN-γ are crucial for increasing inflammatory macrophage levels in IR and RA treated tumors. Notably, whereas CD8+ T cells are required for the antitumor response to IR, CD4+ T cells are required for the effectiveness of the IR and RA combination. Combination treatment with RA enhanced the abscopal response when radiation and PD-L1 blockade were used together. The synergistic positive feedback loop of inflammatory macrophages and adaptive immunity is required for the antitumor efficacy of IR plus RA combination treatment. Our findings provide a translational and relatively nontoxic strategy for enhancing the local and systemic antitumor effects of IR.
One Sentence Summary:
All-trans retinoic acid induces inflammatory myeloid cells to enhance antitumor adaptive immunity following radiation therapy.
Introduction
Radiotherapy (RT) is employed in 50%-60% of patients with cancer. However, local and distal failure in patients with large localized tumors often occurs. The efficacy of IR depends in part on direct cytotoxic effects of DNA damage on the tumor and stroma, and in part on innate and adaptive immune responses (1-4). Radiation influences antitumor immunity through multiple mechanisms. For example, radiation-induced cellular stress and death generate damage-associated molecular pattern (DAMP) molecular signals, DNA, chemokines and cytokines which provide key links between innate and adaptive immunity (4-6). Emerging evidence also demonstrates that radiation increases the expression of immune suppressive molecules such as IL-10, PD-L1 and CTLA-4, in addition to elevating regulatory T cells (Treg) and myeloid-derived suppressor cell (MDSC) numbers within the tumor microenvironment (7-10). These observations have led to many clinical trials employing IO (Immuno-Oncology) agents in combination with radiotherapy. However, the optimization of IR and IO interactions needs further attention (11, 12). Exploring new strategies to combine radiotherapy with immunotherapy will provide an opportunity to improve the clinical outcomes for patients with cancer (4, 13).
All-trans retinoic acid (RA) is an active metabolite of vitamin A that is required for a wide range of physiological processes and plays a significant role in many pathologies (14-16). RA use in the treatment of acute promyelocytic leukemia (APL) is considered to be a paradigm shift, in which it works by targeting the oncoproteins PML-RARα and Pin1 to promote differentiation of malignant myeloid cells (17). However, there is a limited body of evidence on the clinical utility of RA to treat other leukemias and solid tumors (18). RA is essential for the development, differentiation, apoptosis and function of immune cells (19-21). Moreover, RA has been shown to regulate antitumor immunity through promoting survival of tumor specific CD8+ T cells (22) and increasing expression of MHC I on tumor cells (23, 24). Additionally, RA has been reported to eliminate MDSCs and promote their differentiation resulting in enhanced antitumor immunity (25).
A group of inflammatory TNF-α/iNOS producing myeloid cells have been shown to arise during inflammation and are critical mediators of innate and adaptive immune defenses. These cells, designated Tip-DCs for TNF-α/iNOS producing-DCs in some previous reports, are derived from bone marrow monocytes and are critical for activation of the T cell response (26, 27). Previous reports have shown that Tip-DCs are crucial to the antitumor effect of adoptive T cell therapy (ACT) (28). Furthermore, other reports have identified iNOS-expressing macrophages identified as responsible for the infiltration of T cells and enhanced antitumor immunity in ACT when combined with low dose radiation (29). A recent report has indicated that treatment with checkpoint inhibitors can also induce accumulation of iNOS-expressing macrophages (30). RA was reported to promote iNOS expression in dendritic cells in vitro (31) and LPS–stimulated iNOS expression in vivo (32).
Radiotherapy and RA alone are utilized in pre-clinical studies and clinical practice, yet the effect of combined IR and RA for the treatment of solid tumors has not been widely explored. In this report, we demonstrate that combining local ablative IR with RA enhances antitumor immunity and significantly inhibits the growth of local and distal tumors. We discovered that combination treatment leads to a dramatic increase of iNOS/TNFα-producing myeloid cells. These myeloid cells, whose generation relies on IFN-γ and adaptive immunity, also in turn promote T cell infiltration through a positive feedback loop. Combining two widely used therapies, our results hereby provide a valuable strategy for cancer therapy which has the potential to translate into clinical practice.
Results
Combining local ablative IR with RA enhances tumor radiosensitivity and antitumor immune memory response
To examine the effect of the combination of local ionizing radiation with RA on tumor growth, we treated established syngeneic mouse MC38 colon carcinoma tumors by oral gavage with oil (control), RA (400 μg/dose), local IR (15 Gy), or IR plus RA (IR + RA) (for treatment schedule see Fig. 1A). IR alone inhibited tumor growth, while RA had no significant effect on MC38 tumor growth. The combination of IR with RA synergistically inhibited tumor growth compared to either the untreated controls or any single treatment (Fig. 1B). Combination treatment resulted in complete tumor regression in 17 of 19 mice (Fig. 1C), whereas animals that were untreated or treated by IR or RA alone did not survive more than 50 days (Fig. 1D). The cured mice and naive mice were re-challenged with LLC cells or MC38 cells. We found that LLC grew at the same rate in naive mice and cured mice, whereas MC38 cells did not form tumors in the cured mice (Fig. 1E). These results suggest that the combined treatment led to tumor-specific immune memory. In the B16 melanoma tumor model, Renca (a renal cancer tumor model), and CT26 (a colon cancer model), IR and RA combination treatment also had a significant benefit compared with single treatments (Fig. 1F, 1G and fig. S1A respectively).
Fig. 1. Combining local ablative IR and RA inhibited tumor growth and induced antitumor memory.
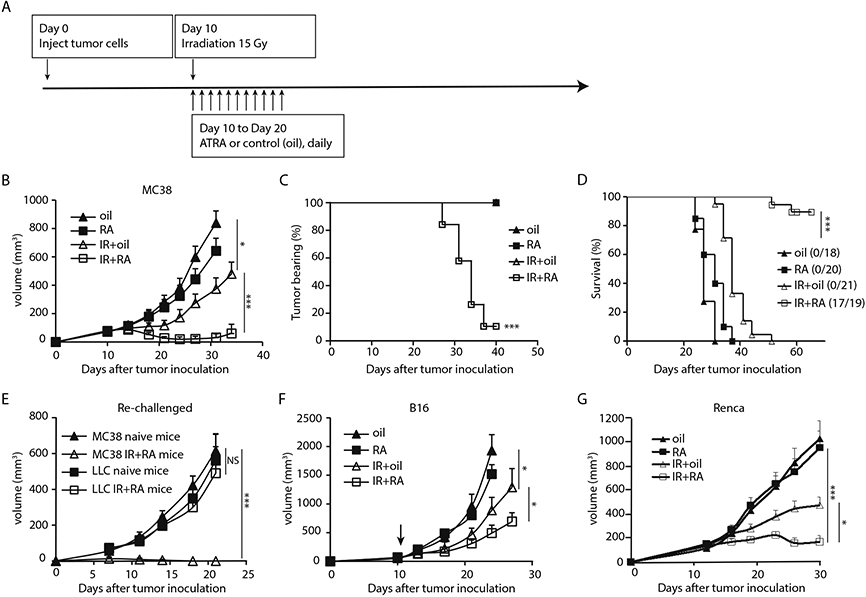
(A) Scheme of the treatment schedule. Daily administration of RA by gavage at 400 μg/dose.
(B) Growth curve of MC38 tumors treated with oil, RA, 15 Gy+oil or 15 Gy plus RA.
(C) Percentage of tumor bearing mice over time post indicated treatment. Animals were pooled from three separate experiments.
(D) Survival curve of mice in (C). Data from three independent experiments were pooled.
(E) Tumor growth curve in cured (MC38) and naive mice which were re-challenged with 5 X106 MC38 or 1X106 LLC cells respectively.
(F) Growth curve of B16 tumor in mice treated with oil, RA, 15Gy + oil or 15Gy and RA combined.
(G) Growth cure of kidney cancer Renca tumor treated with oil, RA, 15Gy + oil or 15Gy and RA combined.
Representative data are shown from two or three experiments conducted with 5-10 mice per group, except for C and D. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001. N.S., not significant.
Significant yet suboptimal tumor inhibition effects are also observed when combining IR with RA administered by i.p. injection (fig. S1B), slow RA-releasing pellets implanted subcutaneously (fig. S1C) or when RA was given at a lower dose (200 μg/dose) by oral gavage (fig. S1D). Animals receiving higher dose RA (800 μg/dose) by gavage experienced weight loss 7-14 days post treatment, but the weight was regained by 3 weeks (fig. S1E). However, animals that received 10 days of 400 μg/dose RA post IR did not exhibit any weight loss (fig. S1F). If not otherwise indicated, we used a daily dose of 400 μg via gavage throughout the following experiments. Overall, our results demonstrate that the addition of RA improved the therapeutic efficacy of ablative IR, and led to a tumor-specific memory immune response.
Inflammatory macrophages are essential for the antitumor effect of local IR and RA treatment
To investigate whether RA has direct cytotoxic and/or radiosensitizing effects on tumor cells, we cultured MC38 cells with RA (0 to 2000 nM) after IR (0 to 15 Gy) for three days. RA had no direct effects on tumor cell death (fig. S2A) in vitro. Since both IR and RA regulate immune responses, we sought to examine how the combination treatment changes the tumor immune environment. Using a multiplex assay, we detected the level of chemokines and cytokines in tumors three days after treatment. Following IR + RA treatment we observed increased levels of CCL2, CCL3, CCL4 and CCL5 (by 2–3-fold compared to levels in any single treatment), which are involved in monocyte recruitment (fig. S2B). GM-CSF, the critical cytokine for DC generation, increased by more than 5-fold in whole tumor homogenates. Other cytokine levels, such as IFN-β, IL-6, IL-1 and IFN-γ, were also significantly elevated in IR + RA treated tumors compared to other treatments (fig. S2C), indicating that the combination treatment of IR and RA resulted in an inflamed tumor microenvironment.
We examined immune cells in tumors 4 days after the start of the treatment. Flow cytometry results showed the percentage of CD45+ and CD11b+ cells increased by 2–3-fold in tumors receiving IR + RA treatment (fig. S3A; Fig. 2A, 2B). In CD11b+ subsets, Ly6C+ cells, including CD11c+Ly6C+ cells, increased drastically by 4- fold in tumors receiving the combination treatment (Fig. 2C, fig. S3B-S3E). On day 4 post treatment, there was no significant change in the percentage of CD4+, and there was only a marginal change in CD8+ T cells (fig. S3F, S3G). Next, we examined the DC population by analyzing high-dimensional flow cytometry data using traditional DC markers. We detected MoDCs (Ly6C+CD11b+CD11c+ MHC-II+CXCR3+ CD64+ SIRPα+ CD206− cells), a low level of DC2s (Ly6C−CD11b+CD11c+MHC-II+ CXCR3+ CD64−SIRPα+CD206−) and a very low level of DC1s (Ly6C−CD11b−CD11c+MHC-II+ CXCR1+ CD103+CD64−) in IR and RA treated tumors (fig. S3H and I). While the DC1 and DC2 populations were not changed during IR and RA treatment, the level of MoDCs was significantly induced (fig. S3I).
Fig. 2. Combining local ablative IR with RA induces iNOS and TNF-α producing inflammatory macrophages.
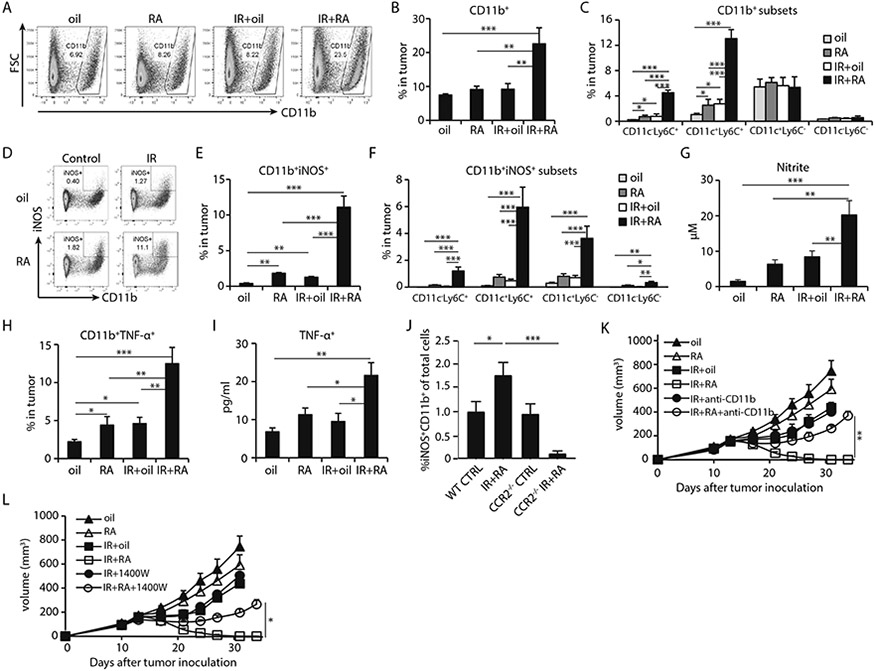
MC38 tumors were treated as indicated and harvested 4 days post start of the IR/RA treatments for analysis in A-J.
(A) The gating strategy of iNOS+ myeloid cells.
(B) Frequency of CD11b+ myeloid cells in total cells of tumors.
(C) Percentage of major subsets of CD11b+ cells in tumors received IR/RA treatment.
(D) Gating strategy of iNOS+ inflammatory macrophages (Inf-MACs).
(E) Percentage of iNOS+ inflammatory macrophages in total cells of tumors.
(F) Percentage of subsets of iNOS+ Inf-MACs in terms of CD11c and Ly6C marker in total cells.
(G) Concentration of iNOS product nitrite in tumor homogenate. Treated tumors were harvested on day 4 post treatment and tumor fragments were cultured in vitro. Nitrite level in culture media was measured after 2 hours.
(H) TNF-α+ CD11b+ population in tumor receiving indicated treatment.
(I) Concentration of TNF-α in tumor homogenate at 4 days after start of the indicated treatments.
(J) Percentage of Inf-MACs in tumors grown in WT or CCR2−/− mice, 4 days after start of IR and RA treatment.
(K) Tumor growth curve of MC38 tumors during treatments while CD11b+ cell recruitment was blocked.
(L) Tumor growth curve of MC38 tumors during treatments while iNOS inhibitor (1400W) was administered.
*, p<0.05; **, p<0.01; ***, p<0.001. Experiments were conducted 3 times with 3-5 mice in each group. Data in K, L are presented as mean ± SEM, the rest of the data are mean ± SD. Results from representative experiments are shown.
Flow cytometry results demonstrated that the combination of RA and IR treatment dramatically increased CD11b+iNOS+ cells in tumors compared to any single treatment alone (0.4%, 1.8%, 1.3% and 11.1% in tumors with oil, RA, IR or IR+RA treatment, respectively; Fig. 2D-2E). Overall, CD11c+Ly6C+ cells accounted for the most (80%) of increase in CD11b+iNOS+ cells (Fig. 2F) and approximately 70% of iNOS-producing myeloid cells are F4/80+ (fig. S4A). Since this group cells were traditionally named TNF-α/iNOS-producing DCs (Tip-DCs), we used panel of DC markers to further investigate this population. In our high-dimensional flow analysis, iNOS+CD11b+ cells do not belong to any categories of known DCs as most of them express CD64 and F4/80 (fig. S4A, gating strategy shown in fig. S4B). Co-expression of CD11c, Ly6C and F4/80 in myeloid cells is one of the characteristics of inflammatory macrophages. Therefore, we refer to these cells as “inflammatory macrophages” (Inf-MACs) in the rest of this report. A significant increase in nitrite levels was found in the culture media of IR and RA treated tumors when cultured ex vivo, suggesting an increase in nitric oxide (NO) levels in response to combination treatment (19.8 μM vs 9.5 μM, in IR+RA and IR treated tumors, respectively; Fig. 2G). Elevated expression of TNF-α was also observed in CD11b+ cells after combination treatment (Fig. 2H). Accordingly, the amount of TNF-α in tumor homogenates was significantly increased after IR and RA combination treatment (20.4 vs 11.5 pg/ml in IR+RA and RA treated tumors, respectively; Fig. 2I). Approximately 80% of iNOS+ myeloid cells are also TNF-α positive, while about 30% of TNF-α positive cells are double positive for iNOS (fig. S4C). We consider iNOS+ myeloid cells represent the majority of Inf-MAC cells. mRNA levels of iNOS and TNF-α were increased in CD11b+CD11c+ cells sorted from treated tumors (fig. S4D). On day 8 following the start of the treatment, more CD11b+ CD11c+ iNOS+ TNF-α+ cells were found in tumors treated with IR + RA compared to any other treatment group (fig. S5A and B). These findings suggest that the combination treatment of IR and RA can lead to enhanced recruitment/differentiation of Inf-MACs, which may play an important role for the antitumor response of IR and RA combination treatment. To verify that the source of Inf-MACs is monocytes, we employed CCR2 knockout mice in which CCR2-expressing monocytes fail to migrate out of bone marrow towards peripheral tissue, especially inflamed tissue (such as irradiated tumors). When treated with IR+RA, the number of iNOS-producing Inf-MACs was significantly lower in tumors grown in CCR2 KO hosts than tumors in WT mice (Fig. 2J). The result indicates that monocyte infiltration during the IR/RA treatment is required for Inf-MAC generation.
We further investigated the role of Inf-MACs and iNOS in the antitumor effect of combination treatment. We blocked CD11b+ cell infiltration into irradiated tumors by using an anti-CD11b antibody (33) starting on the day of treatment. The blockade was able to reduce CD11b levels after IR to the level of controls (fig. S5C). The Inf-MAC level was also reduced during CD11b blockade in IR+RA treated tumors (fig. S5D). Although CD11b blockade did not affect the efficacy of IR, the inhibition of CD11b+ cell recruitment into tumors significantly diminished the antitumor effect of IR and RA treatment (Fig. 2K). When production of NO was inhibited by the iNOS specific inhibitor 1400W in vivo, the antitumor efficacy of combined IR and RA treatment was significantly diminished (Fig. 2L). Our results indicate that Inf-MACs and iNOS induced by IR and RA treatment are important components for controlling solid tumor growth.
Combining IR and RA enhances tumor-specific adaptive immunity in a manner dependent on Inf-MACs
The adaptive immune response is critical for host antitumor immunity induced by radiation (34, 35). Eight days after treatment commencement, significantly greater CD8+ T cell infiltration was observed in tumors receiving IR + RA combination treatment (about 3-fold increase comparing IR+RA to IR treatment of tumors, p<0.05), resulting in increased percentages of CD8+IFN-γ+ and CD8+ granzyme B+ (GzmB) cells in tumors (Fig. 3A-3C) and draining lymph nodes (DLN) (fig. S6A, S6B, S6H) compared with those of tumors receiving single treatment. Similar increases in total CD4+ and CD4+IFN-γ+ T cells were also observed in tumors (Fig. 3D, 3E, S6H) and DLNs (fig. S6C). The percentage of Foxp3+ (Treg) cells in CD4+ T cells decreased (Fig. 3F), whereas the ratio of CD8+ IFN-γ+ cells to Treg increased significantly in tumors which received combination treatment (Fig. 3G) compared to those receiving any single treatment. By IFN-γ+ ELISPOT assay, we detected significantly enhanced tumor antigen-specific CD8+ T cell priming in draining lymph nodes (DLN) of mice receiving IR + RA combination treatment compared to control or single treatments alone (Fig. 3H). In addition, the percentage of naïve T cells (CD62L+CD44−) decreased and proliferating Ki67+ cells increased in DLN for both CD8+ and CD4+ cells after the combination treatment (fig. S6D-G). These results suggest that IR + RA treatment enhances the activation and priming of tumor-specific T cells in tumors, and augments T cell proliferation in the DLN.
Fig. 3. Combining ablative IR with RA enhances iNOS-dependent antitumor T cell responses.

MC38 tumors were harvested and analyzed at day 8 after start of treatments.
(A-C) Percentage of CD8+, CD8+ IFN-γ+ and CD8+ granzyme B+ (GzmB), respectively, in total cell of tumors received indicated treatment.
(D-E) Percentage of CD4+ and CD4+ IFN-γ+ in total cell of tumors received indicated treatment.
(F) Percentage of Foxp3+ cells in CD4+ cells in tumors received indicated treatment.
(G) Ratio of CD8+IFN-γ+ T cells to Foxp3+ T cells in tumors.
(H) IFN-γ ELISPOT assay for IFN-γ-secreting MC38 cell antigen-specific CD8+ T cells from tumor draining lymph nodes.
(I) qPCR analysis of mRNA levels of CXCL9, CXCL10 and CXCL11 in sorted CD11b+CD11c+ cells from tumors received indicated treatment
(J) CD4+ percentage in tumors received indicated treatment when iNOS activity was inhibited by inhibitor 1400W.
(K) CD8+ percentage in tumors received indicated treatment when iNOS activity was inhibited by using inhibitor 1400W.
(L) Percentages of CD4+, CD8+, IFN-γ+ CD4+ and IFN-γ+ CD8+ T cells in tumors grown in a CCR2−/− host which received a WT or iNOS−/− myeloid cell transfer. Cells were transferred one day prior to IR and tumors were harvested 8 days after control (oil) or combination treatment of IR and RA.
*, p<0.05; **, p<0.01; ***, p<0.001, ns, not significant. Experiments were conducted 3 times with 3-5 mice per group. Data in H and L are presented as mean ± SEM and the rest of the panels are mean ± SD. Results from representative experiments are shown.
We further investigated whether Inf-MACs play any role in shaping adaptive immunity. Transcription of CXCL9, CXCL10 and CXCL11, key chemokines which drive the trafficking of T cells, increased significantly in CD11c+ cells compared to controls (Fig. 3I). CXCL9 and CXCL10 levels in tumors were also elevated (fig. S2B). These results suggest that combining radiation and RA treatment can activate local adaptive immunity by enhancing T cell infiltration. Indeed, as shown in Fig. 3J and 3K, inhibiting iNOS activity abrogated increases in the levels of CD4+ and CD8+ cells induced by IR + RA. To further investigate the relationship between iNOS-producing Inf-MACs and T cell trafficking/function, bone marrow cells (mostly CD11b+ cells) from WT and iNOS KO mice were adoptively transferred into tumor-bearing CCR2 KO mice in which CCR2 null monocytes are not able to traffic to inflammatory sites (tumors receiving IR+RA treatment). This transfer allows iNOS deficient myeloid cells to populate the tumor during IR and RA treatment. Eight days after starting the IR+RA treatment, CD8+ and CD4+ T cells and their IFN-γ production were analyzed by flow cytometry. The results demonstrated that iNOS-producing Inf-MACs are required for increased CD4+ and CD8+ T cell infiltration as well as their increased IFN-γ production during IR+RA treatment (Fig. 3L). iNOS null myeloid cells promoted CD4+ T cell accumulation and their IFN-γ production in untreated tumors, compared to WT iNOS control (Fig. 3L), probably due to the defective T cell suppressive function of iNOS-null MDSCs in steady-state, compared to iNOS-WT MDSCs. These findings suggest that iNOS-producing Inf-MACs induced by IR + RA treatment in the tumor microenvironment are essential for launching antitumor adaptive immunity.
T cells mediate antitumor efficacy and induce Inf-MACs during IR and RA combination treatment
We next sought to determine whether T cell responses are crucial for the antitumor efficacy of IR + RA treatment. Tumors grown in Rag1−/− mice, which are deficient of T and B cells, did not respond to IR + RA combination treatment, in contrast to tumors in wild type mice (Fig. 4A). To evaluate the individual contribution of CD8+ and CD4+ T cells, we depleted them in the context of IR and IR + RA treatment altogether or separately (For depletion efficiency, see Fig. S7A and S7B). The effect of IR alone and IR + RA treatment on tumor growth was completely abrogated by depleting both CD4+ and CD8+ T cells (Fig. 4B). Anti-CD8 antibody completely abrogated the effect of IR alone, but only partially blocked the therapeutic effect IR + RA combination treatment (Fig. 4C). To determine roles of CD4+ T cells in RA induced antitumor effect, we depleted CD4+ T cells in tumors treated with higher dose of RA (800 μg/mouse/dose) for 7 days. Tumor growth was inhibited by the higher dose of RA and depletion of CD4+ T cells completely abolished the effect of RA (Fig. 4D). Anti-CD4 also diminished antitumor efficacy of IR + RA treatment to the effect of either single treatment (Fig. 4D). These results indicate that adaptive immunity, specifically the actions of CD4+ T cells together with CD8+ T cells, plays an essential role in the therapeutic effect of IR and RA combined treatment, during which CD4+ T cell function is required for RA antitumor activity. We further investigated whether newly infiltrated T cells are required for the efficacy by administering the sphingosine-1-phosphate receptor agonist FTY720. FTY720 effectively blocks T cells from exiting the thymus and secondary lymphoid organs toward sites of inflammation (36). The response to IR was not affected by FTY720 treatment (37)(Fig. 4E). However, without new lymphocyte tumor infiltration, the efficacy of IR + RA treatment was diminished to the level of IR alone (Fig. 4E). These results indicate that the superior tumor control of IR + RA treatment relies on newly infiltrated T cells, which are necessary for the antitumor activity of RA, but not radiation. We next studied the role of T cells on Inf-MAC induction. We found that T cell deficiency, in either Rag1−/− mice or mice depleted of T cells, abrogated the iNOS expression in myeloid cells in all treatments, suggesting that T cells play an essential role for the induction of Inf-MACs (Fig. 4F, fig. S7C-D). Specifically, CD8+ T cells are required for inducing/maintaining a high level of iNOS expression in tumors which received IR alone. CD4+ T cells, in addition to CD8+ T cells, are important for iNOS expression after RA + IR treatment (Fig. 4F). In addition, newly infiltrated T cells are required for a full induction of Inf-MACs in tumors treated with IR and RA as FTY720 administration abolishes increased iNOS production in Inf-MACs in tumors (Fig. 4G) and in circulating blood after IR+RA treatment (fig. S7E). Expression of TNF-α is also reduced when CD4+ and CD8+ T cells were depleted altogether or separately (fig. S7F). To determine whether NK cells contribute to Inf-MAC induction and the antitumor effect of IR and RA treatment, we analyzed Inf-MACs in tumors depleted of NK cells using anti-NK1.1 antibody four days after start of the treatment. Flow cytometry showed that NK depletion did not significantly diminish the induction capacity of IR+RA (fig. S7G). Tumor growth curves were monitored during NK depletion. The result showed that NK depletion did not alter antitumor efficacy of IR+RA treatment (Fig. 4H). Taken together, our results reveal that T cells are not only crucial to mediating the antitumor activity of IR and RA treatment, but are also essential for inducing iNOS expression in tumor-associated myeloid cells. Our results also indicate that by combining RA with IR, we can harness the antitumor power of both CD4+ and CD8+ T cells.
Fig. 4. T cells are essential for antitumor efficacy of IR and RA combination treatment and Inf-MAC induction.

(A) MC38 tumors established in WT mice and Rag1−/− mice were treated by oil, RA, 15Gy+oil or 15Gy and RA combined. Tumor growth curves are shown.
(B-D) MC38 tumor growth curve when T cells were depleted during indicated treatments. (B) WT mice bearing MC38 tumor were injected with both anti-CD4 and anti-CD8 antibodies; (C) CD8 depletion; and (D) CD4 depletion. In CD4 depletion, 800 μg/dose/day of RA was used.
(E) MC38 tumor growth curve when FTY720 was administered for 7 days from starting of the indicated treatment.
(F) iNOS expression in myeloid cells of tumors in mice received indicated treatment when CD4, CD8 or CD4+CD8 were depleted.
(G) Inf-MAC level in tumors treated with oil or IR and RA in FTY720 treated mice.
(H) Tumor growth curve of IR/RA during NK1.1 depletion. Anti-NK1.1 antibody was administered every 3 days for 3 doses from the start of the treatments.
*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; ns, not significant. Experiments were repeated 3 times with 3-5 mice per group. Data in F are presented as mean ± SD, the rest are presented as mean ± SEM. Results from representative experiments are shown.
IFN-γ synergistically promotes induction of Inf-MACs by RA
IFN-γ is a potent stimulus for iNOS expression in myeloid cells and is crucial for the generation of Tip-DCs in infection models (38). We observed that iNOS production increased significantly in bone marrow-derived DCs (BMDCs) cultured in the presence of RA, and that the addition of IFN-γ synergistically increased iNOS production in a dose dependent manner (Fig. 5A, 5B). We have shown above that IR + RA treatment induced more IFN-γ+ T cells, and that T cells are required for Inf-MAC generation in tumors (Fig. 4). In addition, IFN-γ levels increased significantly by more than 2-fold in tumors treated with IR compared to untreated tumors 2 days after treatment (Fig. 5C). When IFN-γ was neutralized in vivo using a neutralizing antibody, iNOS and TNF-α induction in myeloid cells after IR or IR plus RA treatments was significantly diminished in tumors (Fig. 5D-F) and in blood PBLs (fig. S7E). In addition, the antitumor efficacy of the combination treatment was completely abrogated by neutralizing IFN-γ (Fig. 5G). These results suggested that IFN-γ is a main mediator of Inf-MAC induction during treatment and a major determinant of the efficacy of IR + RA. To determine whether IFN-γ produced by T cells is required for Inf-MAC induction, we adoptively transferred T cells from WT or IFN-γ−/− mice into tumor-bearing Rag1−/− mice. Two days after transfer, tumors were treated by oil (vehicle) or IR plus RA. Inf-MAC induction by IR plus RA was significantly abrogated in tumors grown in IFN-γ−/− Rag1−/− hosts compared to tumors in WT/Rag1−/− mice (Fig. 5H). The result indicates that IFN-γ from T cells is required for Inf-MAC induction during IR and RA treatment. We also neutralized GM-CSF using neutralizing antibody as the level of GM-CSF was significantly elevated in tumors treated with IR combined with RA (fig. S2). Inf-MAC levels during the treatment were not significantly affected by GM-CSF depletion (fig. S7H). Taken together, our results indicate that T cell-produced IFN-γ promoted by radiation and RA converge to transform infiltrating monocytes into iNOS/TNF-α–producing inflammatory macrophages.
Fig. 5. T cells promote the generation of Inf-MACs through IFN-γ signaling in IR and RA combination treatment.
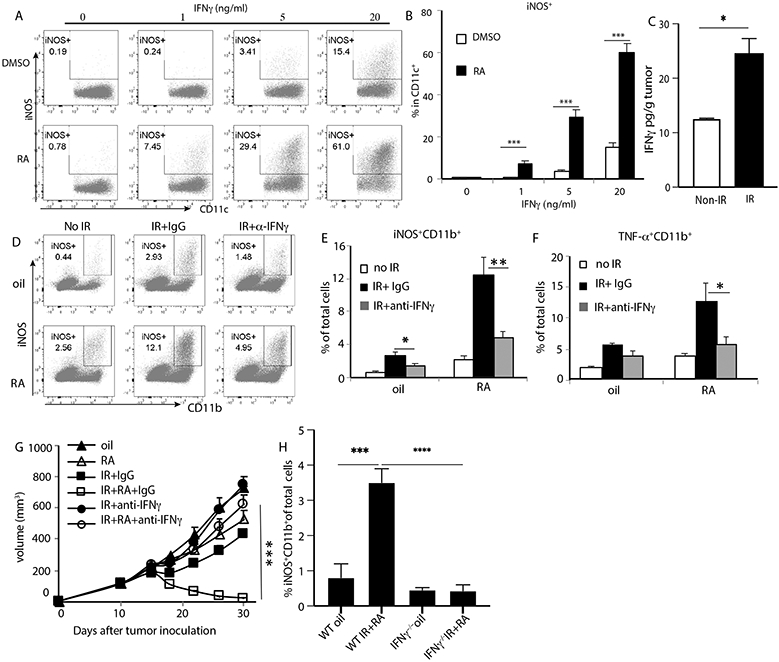
(A-B) iNOS induction in BMDC in the presence of RA and/or IFN-γ
(C) IFN-γ levels in tumors two days after receiving radiation.
(D-E) iNOS induction in myeloid cells of tumors treated with indicated treatments in the presence of anti-IFN-γ neutralizing antibody.
(F) TNF-α induction in in myeloid cells of tumors in the presence of IFN-γ neutralizing antibody.
(G) MC38 tumor growth curve when received indicated treatments during IFN-γ neutralization.
(H) iNOS-producing myeloid cell percentage in tumors 4 days after treatment with vehicle or IR plus RA in Rag1−/− mice that received an adoptive transfer of WT or IFN-γ−/− T cells.
*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; ns, not significant. Experiments were repeated 3 times with 3-5 mice per group. Data in C, G and H are presented as mean ± SEM, the rest are presented as mean ± SD. Results from representative experiments are shown.
IR and RA treatment leads to an abscopal response enhanced by PD-L1 blockade
To evaluate whether the combination of IR with RA could result in an abscopal effect on the non-irradiated tumors, we implanted MC38 tumor cells in both flanks of mice and only the tumors on the right flank (primary) were irradiated. We observed a slower growth rate of non-irradiated tumors in the group that received RA and IR treatment compared to any single treatment alone (Fig. 6A). This result indicates that RA not only improves the effect of IR on the primary tumor, but also enhances the systemic abscopal effect.
Fig. 6. PD-L1 blockade enhances abscopal effects of RA and IR combination treatment.
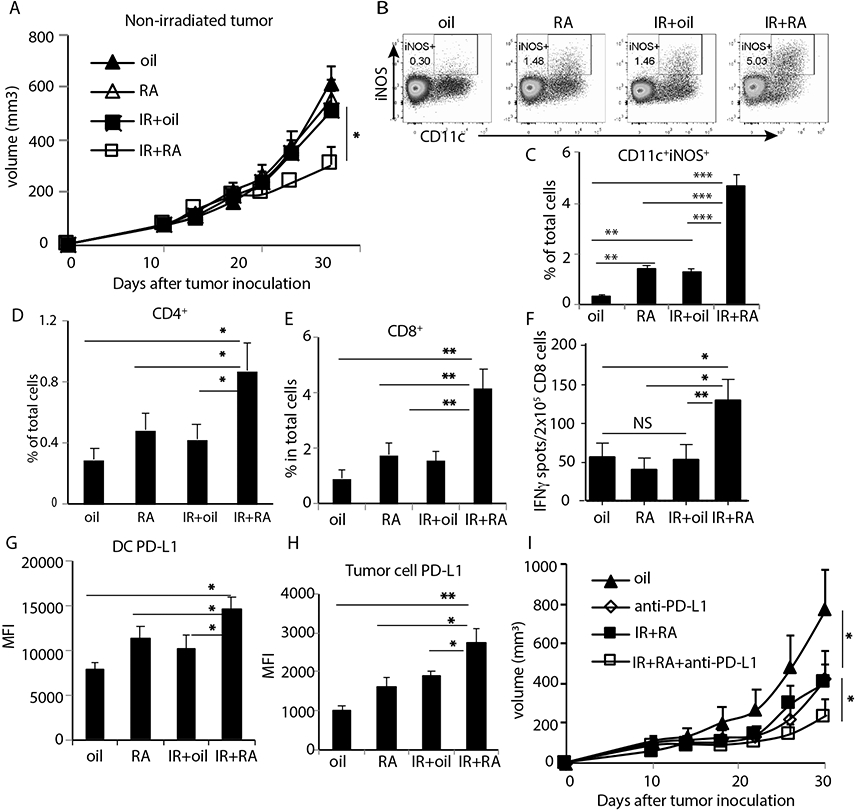
(A) Growth curve of non-irradiated MC38 tumors in two tumor model with primary tumors treated with oil, RA, 15 Gy+oil and 15 Gy plus RA.
(B-C) Inf-MAC level in non-irradiated tumors 8 days post IR.
(D-E) Percentage changes in CD4 and CD8 levels during treatments in non-irradiated tumors.
(F) IFN-γ producing spots of tumor antigen-specific CD8 T cells isolated from DLN of non-irradiated tumor.
(G-H) PD-L1 induction in DCs and tumor cells in non-irradiated tumors 3 days post starting of indicated treatments.
(I) Growth curve of non-irradiated tumors in MC38 two-tumor model when primary tumors received indicated treatment. *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant. Experiments were repeated 3 times with 3-5 mice per group. Data in I are presented as mean ± SEM, and the rest are presented as mean ± SD. Results from representative experiments are shown.
In the non-irradiated tumors 8 days after combination treatment, we observed a significantly increased proportion of CD11b+ cells (fig. S8A) and CD11b+CD11c+Ly6C+ cells (fig. S8B). CD11c+iNOS+ cells also increased in the non-irradiated tumor, but not in other distal organs of the mice which received combination treatment (Fig. 6B, 6C; fig. S8C). We also observed more CD8+ T and CD4+ T cells (Fig. 6D, 6E) in the non-irradiated tumors when primary tumors received IR + RA treatment. Whereas the proportion of CD8+IFN-γ+, CD8+GzmB+ and CD4+IFN-γ+ cells increased within the tumor, there was a marked decrease of Foxp3+CD4+ T cells (fig. S8D-G). IFN-γ production of antigen-specific T cells isolated from the DLN of non-irradiated tumors revealed that priming of CD8+ T cells was also augmented in the non-irradiated tumor when the primary tumor received combination treatment (Fig. 6F). These results indicate that combination treatment of IR and RA shifted the tumor microenvironment of non-irradiated tumors towards augmented states of antitumor innate and adaptive immunity.
Consistent with previously reports (39, 40), we observed increased PD-L1 expression on DCs and tumor cells in non-irradiated tumors 3 days post IR + RA treatment (Fig. 6G, 6H and fig. S8H, S8I). We hypothesized that employing adaptive immune checkpoint blockade by neutralizing PD-L1 could further enhance the abscopal effect of IR and RA treatment. As expected, the combination of anti-PD-L1 with IR and RA further suppressed the growth of non-irradiated tumors compared to PD-L1 blockade or IR and RA treatment alone (Fig. 6I). These results suggest that the combination of RA and anti-PD-L1 treatment can overcome tumor radioresistance and achieve superior control of distal tumors.
The significant correlation of iNOS expression with cancer patient prognosis is attested to by analyzing the survival of kidney and colon cancer patients in the TCGA database. Patients who had higher iNOS expression in their tumors exhibited a higher survival rate (Fig. 7A, 7B). Further analysis revealed that iNOS expression in a colon cancer patient cohort is significantly correlated with expression of genes related to adaptive immunity including CD3E, CD8A, IFNG and PRF1 (Fig. 7C). In summary, combining local ablative radiation and RA to treat solid tumors revealed a positive feedback loop between iNOS/TNF-α-producing inflammatory macrophages and adaptive immunity that led to local and systemic antitumor responses (Fig. 7D). We propose the loop starts at IFN-γ produced by tumor pre-existing T cells (37) immediately following radiation. IFN-γ, together with administered RA, drives newly infiltrated monocytes into inflammatory macrophages and further inflames the TME. The inflamed TME calls in more T cells which in turn induces more Inf-MACs. The positive feedback loop generates inflammation and high level of T cell priming to achieve tumor local and distal control.
Fig. 7. High iNOS expression in tumor tissue correlates with favorable survival and T cell signature in colon and kidney cancer patients.

Overall survival analysis of cancer patients with (A) kidney renal clear cell carcinoma (KIRC) or (B) colon adenocarcinoma (COAD) and either high or low mRNA expression for NOS2 (iNOS). Patients from each dataset were split into two groups by median expression level of NOS2. Normalized gene expression (RNAseq) and corresponding clinical data on patients were obtained from The Cancer Genome Atlas (TCGA). Survival curves were compared by log rank (Mantel-Cox) test.
(C) Correlation between expression of NOS2 and CD3E, CD8A, IFNG and PRF1 in Colorectal Adenocarcinoma patients (n=592) from TCGA PanCancer data. Spearman r and P values are shown on the individual plots. Gene expression levels are presented in the log2 form.
(D) Schematic summary of mechanism. We propose that during combined treatment with radiation and exogenous RA, the infiltrating monocytes which are induced by radiation are transformed into iNOS/TNF-α–producing inflammatory macrophages. Through iNOS production the inflammatory macrophages enhance CD4+ and CD8+ T cell infiltration and the newly arrived T cells produce more IFN-γ to induce even higher levels of Inf-MACs. This positive feedback loop between Inf-MACs and T cells amplifies antitumor innate and adaptive immunity and leads to superior tumor control compared to either treatment alone.
Discussion
In this study, we report that the combination of radiation and RA reprograms the immune contexture of the tumor microenvironment by synergistically inducing an iNOS/TNF-α–producing subset of CD11b+ inflammatory myeloid cells. This increase in iNOS expression mediated a dramatic increase of T cell infiltration and priming in IR + RA treated tumors. iNOS has been demonstrated to be immunosuppressive in certain contexts. Specifically, NO production is a major mechanism by which MDSCs inhibit T cells. Conversely, iNOS expression is also the hallmark marker of the M1 macrophage, which can activate T cell responses. Moreover, NO is important for the differentiation of T cells (41, 42). In the instance of RA and IR combination treatment, inhibiting iNOS activity, especially in myeloid cells, diminished the T cell infiltration, IFN-γ production and antitumor efficacy effect.
RA has been used as a breakthrough treatment in acute promyelocytic leukemia (APL). In addition to promoting differentiation of myeloid cells by targeting PML-RARα and Pin1(17, 43, 44), RA is also essential for the development, differentiation, apoptosis and function of other immune cells (19-21, 44), including promoting the survival of tumor-specific CD8+ T cells (22) and eliminating myeloid-derived suppressor cells (23, 24). As indicated in our in vitro (RA+IFN-γ) and in vivo (use of CCR2−/− mice) studies, Ly6C+ CCR2+ monocytes are likely the main target cells of RA treatment when radiation is administered. We propose that tumor growth and local tumor irradiation create a highly inflammatory microenvironment that is crucial for recruitment and differentiation of inflammatory macrophages. Although these cells also circulate in blood and enter distal tumors, the local effects at the site of irradiation are much stronger. We cannot exclude the possibility that RA may also affect intratumoral Treg development as Treg levels also decrease in IR and RA-treated tumors. RA is reported to modulate T helper cell maturation and differentiation (45), especially by inducing Foxp3 expression. In this study, we demonstrated that when RA is combined with IR, the Treg cell population did not increase and the ratio of IFN-γ+ CD8+ T cells to Treg cells significantly increased. In CD4/CD8 depletion experiments, we observed that the depletion of CD4+ cells diminished the antitumor effect of RA treatment and reversed iNOS induction, similar to the response to IR observed when CD8+ T cells were depleted. A recent report showed that CD4+ T cells are essential for immunotherapy by recognizing MHC-II-restricted neoantigens and priming CD8+ T cells (46). In our study, we hypothesize that therapy (RA) induced newly infiltrated CD4+ T cells which act as helper cells to prime CD8+ T cells. The exact role of CD4+ T cells in antitumor immunity induced by combination treatment of IR and RA warrants further study.
Our results demonstrate that T cells are essential to generate iNOS-expressing inflammatory myeloid cells in IR and RA treated tumors, due to production of IFN-γ, whereas inflammatory myeloid cells are critical for further T cell infiltration and priming. The innate and adaptive mechanisms orchestrate a positive feedback cycle to inhibit tumor growth following IR and RA treatment. We hypothesize that the varieties of immune cell recruitment as well as timing of activation are important for combined radio- and immunotherapy. Kidney and colon cancer patients who had higher iNOS expression exhibited prolonged survival and iNOS expression correlates with T cell adaptive immunity markers in colon cancer patients, suggesting that iNOS expression in the tumor microenvironment might provide a prognostic marker for immunotherapy. It is particularly noteworthy that RA, which is relatively nontoxic compared to most chemotherapies, can act as an immune modulator in reprogramming myeloid cell-rich environment induced by IR. Our report has potential clinical significance in using RA as a non-specific T cell targeting reagent to improve the local and systemic effects of radiotherapy, thereby converting IR from a purely local modality into a systemic treatment.
Materials and Methods
Study design
The objective of this study was to determine whether and how combining RA with IR enhanced antitumor immunity and efficacy. The effect of IR and RA treatment and the relationship between Inf-MACs and IFN-γ+ T cells were assessed in MC38-tumor–bearing mice (WT, iNOS KO, IFN-γ KO, CCR2 KO, RAG1 KO), treated with anti-CD4, anti-CD8, anti-NK, anti-IFN-γ, anti-PD-L1 or anti-GM-CSF neutralizing/blocking antibodies or the FTY720 and 1400W inhibitors. Sample sizes for flow analysis, protein and gene expression analysis (n = 3 to 5, repeated two to five times), tumor monitoring (n = 4 to 7, repeated three times), and the choices of time points for analysis were based on previous experiments (10, 39, 47) and consultation with the Biostatistics core at the University of Chicago to ensure sufficient statistical power to compare the differences. Investigators were blinded when performing and analyzing the data. For tumor growth monitoring, age-matched mice were assigned to experimental cohorts based on randomized matching tumor volumes, and data presented include all outliers. Investigators were not blinded when monitoring tumor burden. Biological replicates are indicated in the figure captions by n. Three or more independent trials were performed. The endpoint of all experiments followed guidelines set by the Institutional Animal Care and Use Committee of the University of Chicago.
Mice, Cell lines and Reagents
Six- to eight-week-old C57BL/6J mice were purchased from Envigo. Rag1 knockout mice, iNOS knockout, IFN-γ knockout and CCR2 knockout mice were from The Jackson Laboratory. Mice were maintained under specific pathogen-free conditions and used in accordance to the animal experimental guidelines set by the Institutional Animal Care and Use Committee of the University of Chicago and Xuzhou Medical University. MC38, B16/F1, Renca, and LLC cell lines were acquired from ATCC, tested free of murine pathogen and authenticated. Cells were cultured in DMEM medium with 10% FCS and 1% of Pen/Strep antibiotics. RA (Cat# PHR1187) was purchased from Sigma-Aldrich. 1400W was purchased from Cayman Chemical Company (Cat# 214358-33-5) and prepared in PBS before injection. Depleting or blocking antibodies against PD-L1 (BE0101), CD8α (BE0004-1), CD4 (BP0003-1), NK1.1 (BE0036), GM-CSF (BE0259), CD11b (BE0007) and IFN-γ (BE0055) were purchased from BioXCell. Anti-mouse Pacific Blue–anti-CD45 (103126), fluorescein isothiocyanate (FITC)–anti-CD45 (103108), PE–anti-CD4 (116005), APC/CY7–anti-CD8α (100714), APC-anti-IFN-γ ( 505810), Alexa Fluor 700-anti-CD44(103025), Brilliant Violet 605-antiCD62L (104437), Zombie UV (77474), Brilliant Violet 605-anti-CX3CR1(149027), Brilliant Violet 650-anti-XCR1 (148220), Brilliant Violet-510-anti-CD24(101831), Brilliant Violet 711-anti-CD64 (139311), Brilliant Violet 785-anti-F4/80 (123141), Pacific Blue-anti-CD11b (101224), PerCP-Cy5.5-anti-I-A/I-E (107626), PE-Cy7-anti-CD11c (117318), FITC-anti-Ly6C (128006), PE-Dazzle 594-anti-CD103 (121429), Alexa Fluor 700-anti-CD206 (141734), APC-Cy7-anti-SIRPα (144017), Alexa Fluor 488-anti-Ki-67 were purchased from BioLegend. Alexa Fluor 532-anti-CD45 (58-0451-82), PerCP-eFluor710-anti-Flt3 (46-1351-82), PE-anti-iNOS (12-5920-82), and APC-anti-TNF (17-7321-81) are from ThermoFisher Scientific. PE-anti-iNOS (D6B6S, 14792) and Griess Reagent Nitrite Measurement Kit (13547) were from Cell Signaling Technology. CD11c isolation kit (18780), and CD8 T cell selection kit (18953) were purchased from STEMCELL Technologies. Nitric oxide detection kit (ADI-917-020), and FTY720 (BML-SL233-0025) were purchased from Enzo. LEGENDplex Mouse chemokine assay kit (740451) and mouse inflammation panel (740446) were purchased from BioLegend. Mouse IFN-γ CBA Flex Set (BDB558296) and Mouse IFN-γ ELISPOT Set (551083) were from Fisher Scientific.
Tumor Growth and Treatments
1X106 MC38, B16/F1, Renca or CT26 cells were subcutaneously implanted into flanks of mice. Tumors were irradiated using a Phillips IR250 orthovoltage X-ray generator operating at 250 kV 15 mA. Subcutaneous local tumors were irradiated by shielding bodies with a cylindrical lead cover with only flank tumors exposed for radiation. ATRA (400 μg/mice/day) was administered by oral gavage for 10 days. For the two tumor model, 1 X 106 and 0.7 X 106 MC38 cells were subcutaneously implanted into the right and left flank of mice, respectively. For the tumor re-challenge assay, both the cured mice and the naive mice were re-challenged with 5 X106 MC38 or 1X106 LLC cells, respectively. Tumors were measured along two diameters (length and width) and depth twice weekly and volumes were calculated as length x width x depth/2. For CD8+ and CD4+ T cell depletion experiments, 200 μg of antibodies were injected i.p. on day 0, 2, 5 and 8 after radiation. Anti-CD11b antibody was injected i.p. every two days starting 1 day before radiation. Anti-PD-L1 antibody was administered i.p. on day 0, 3, and 7 after radiation at 200 μg/mouse. Anti-IFN-γ was administered i.p. at 500 μg/mouse every 7 days. Anti-NK1.1 antibody was administered via i.p. at 25 μg/mouse every 3 days for 3 doses. Anti-GM-CSF antibody was given by i.p. injection at 250 μg/mouse every 2 days. FTY720 was delivered via gavage at 20 μg per dose every day for 7 days.
Flow Cytometric Analysis
Single cell suspensions were obtained as previously described (40). Cells were blocked with anti-Fc receptor (2.4G2, BioXCell) and stained for live/dead with Zombie UV, followed staining using antibodies against CD45, CD4, CD8, NK1.1, CD11b, CD11c, Ly6C, Ly6G, F4/80,CD44, CD62L.
The following antibodies were used for high-dimensional flow analysis: BV605-CX3CR, BV650-XCR1, PB-CD11b, BV510-CD24, BV711-CD64, BV785-F4/80, AF532-CD45, PerCP-Cy5.5-I-A/I-E, PerCP-eFluor710-Flt3, PE-Cy7-CD11c, FITC-Ly6C, PE-iNOS, PE-Dazzle 594-CD103, APC-TNF, Alexa Fluor 700-CD206, and APC-Cy7-SIRPα. For IFN-γ and GzmB (clone GB11, Biolegend) staining, cells were cultured with PMA/ionomycin and brefeldin A (423303, Biolegend) for 6 hours followed by intracellular staining using BD Cytofix/Cytoperm intracellular staining kit (554714, BD Biosciences). For TNF-α staining, cells were stimulated by PMA/ionomycin and brefeldin A for 4 hours followed by intracellular staining. Foxp3 (clone FJK-16s, ThermoFisher), Ki67 (clone 16-A8, Biolegend) and iNOS staining were performed following Foxp3 staining protocol (00-5523-00, ThermoFisher). Cells were analyzed on a Fortessa flow cytometer (BD Biosciences) or on an Aurora spectral cytometer (Cytek). Data was analyzed with FlowJo software (Tree Star).
ELISPOT Assay for IFN-γ-secreting CD8+ T cells
MC38 cells were irradiated (40 Gy) and cultured overnight. CD8+ T cells were isolated with EasySep mouse CD8+ positive selection kit (18953, STEMCELL) from draining lymph nodes of tumor-bearing mice on day 8 after treatment. 2x105 isolated CD8+T cells and 2X104 irradiated MC38 cells were plated onto 96-well PVDF-backed microplates coated with monoclonal antibody specific for mouse IFN-γ for 72 hours. Spots were developed by following the manufacturer’s protocol (BD Biosciences).
Generation of bone marrow-derived dendritic cells (BMDC) and Tip DC induction in vitro
Femurs and tibias from 8- to 10-week-old mice were harvested. BMDCs were derived as described (40). The cultured BMDC were collected on day 6 for further experiments. For Inf-MAC induction in vitro, bone marrow cells were cocultured with the presence of 200 μM RA. On day 6, indicated amount of IFNγ was added directly to the culture. Cells were stained for iNOS and TNF-α after 24-36 hours and analyzed using flow cytometry.
Cytokine/chemokine and nitrite assay
Whole tumors were excised at the indicated time point post treatment. Tumors were weighed and homogenized in PBS with the presence of protease inhibitor cocktails at a ratio of 1 ml per gram of tissue. 25 μl of supernatants were subjected to bead-based cytokine and chemokine assay (LEGENDplex mouse inflammation panel and chemokine panel). The assays were analyzed using a LSRII flow cytometer and LEGENDplex software. IFN-γ was measured using IFN-γ flex CBA set (BD Biosciences). TNF-α concentration was measured using ELISA kit from R&D (MTA00B). For nitrite assay, equal weight of tumors were cut into pieces and cultured in DMEM medium for 2 hours. The production of NO in the supernatants was measured indirectly by measuring the concentration of nitrite by using the Griess reagent nitrite measurement kit (Cell Signaling Technology, #13547).
Immune cell adoptive transfer
For T cell transfer, total T cells were isolated from lymph nodes and spleens using a T cell isolation kit (19851, STEMCELL Technologies). 2x106 T cells were adoptively transferred into each Rag1−/− mouse via retroorbital i.v. injection 2 days prior to start of the treatments. For myeloid cell transfer, bone marrow cells were harvested and washed twice with PBS. 2x106 cells were transferred via retroorbital i.v. injection 1 day prior to the start of the treatments.
RNA extraction and Quantitative Real-Time PCR
Total RNA from tumor and CD11c+ cells isolated from tumors with Easysep™ Mouse CD11c positive selection kit II was extracted with Quick-RNA™ Miniprep Plus kit (R1058, Zymo Research). cDNA was synthesized with High Capacity cDNA Reverse Transcription kit (4368814, Applied Biosystems). Real-time PCR was performed with SYBR Green PCR Master Mix (4309155, Applied Biosystems) and results were normalized to β-actin. The following primers were used:
iNOS For- CACCAACAATGGCAACATCAG Rev- GTCGATGCACAACTGGGTG
TNFa For- GCCTATGTCTCAGCCTCTTCT Rev-TCTGGGCCATAGAACTGATGA
CXCL9 For- CGCTGTTCTTTTCCTCTTGG Rev- AGTCCGGATCTAGGCAGGTT
CXCL10 For- CCAAGTGCTGCCGTCATTTTC Rev- GGCTCGCAGGGATGATTTCAA
CXCL11. For- AGTAACGGCTGCGACAAAGT Rev- GTCAGACGTTCCCAGGATGT
β-actin For- ACAGCTTCTTTGCAGCTCCT Rev- ATACAGCCCGGGGAGCA
Relative gene expression was calculated using the 2−ΔΔCT approach.
Statistical analysis and patient analysis
The RSEM (RNA-seq by expectation-maximization) normalized gene expression data from TCGA PanCancer were downloaded from the cBioPortal (February 2018 version) for colorectal adenocarcinoma. For correlation analysis, a log2 transformation was applied to the gene expression values, and then the Spearman r and P values for the correlation of all genes with NOS2 expression were computed with GraphPad Prism.
Data from animal experiments were analyzed by one-way analysis of variance (ANOVA) or two-way ANOVA with multiple comparison test or Student’s t test. Kolmogorov-Smirnov test was used to test for normality. Data are presented as means ± SEM. We indicated the level of statistical significance using these symbols: * P < 0.05, ** P < 0.01, and *** P < 0.001; **** P<0.0001; NS indicates no significant difference.
Supplementary Material
Data file S1. Raw data file (Excel spreadsheet)
Fig. S1. Related to Fig. 1. Mouse colon tumor response to RA treatment by various delivery methods.
Fig. S2. Related to Fig. 2. Local MC38 tumor microenvironment changes in IR and RA treated tumors 3 days after start of the treatments.
Fig. S3. Related to Fig. 2. Immune cell profile in MC38 tumors treated with indicated treatments 4 days after IR and start of RA treatment.
Fig. S4. Related to Fig. 2. iNOS and TNF-α production in Inf-MACs of MC38 tumors treated with IR and RA.
Fig. S5. Related to Fig. 2. iNOS-producing cells in MC38 tumors 8 days after start of the treatments, and efficacy of CD11b blockade of myeloid cell.
Fig. S6. Related to Fig. 3. Immune profile in MC38 tumor draining lymph nodes 8 days post starting of the treatments.
Fig. S7. Related to Fig. 4 and Fig. 5. Frequencies of Immune cells in MC38 tumors after T and NK cell depletion and IFN-ɣ and GM-CSF neutralization.
Fig. S8. Related to Fig. 6. Immune profile changes in distal MC38 tumor and other distal tissues.
Acknowledgments:
We thank Mr. Rolando Torres for technical assistance. We thank everyone in the Weichselbaum lab for helping hands and stimulating discussions.
Funding:
This work was supported by Ludwig Cancer Research, and in part by Foglia Family Foundation from Mr. and Mrs. Vincent Foglia and NIH grant NCI-R21 CA195075 to R.R.W. This work was also supported by the Research Foundation of Xuzhou Medical University (Grant No. D2019026), the social development projects of Xuzhou (KC19144), the Jiangsu Distinguished Professorship Program, the Jiangsu distinguished medical experts program and the Xuzhou Jinlonghu Distinguished Talents Program. This work was in part supported by National Natural Science Foundation of China (No. 82073176 to Y.Z.H). Flow cytometry was performed at flow core facility of University of Chicago (RRID: SCR_017760) and the cost was covered in part by University of Chicago Cancer Center Support Grant (P30CA014599). J.B. was supported by a Clinical Therapeutics Training Grant (T32GM007019).
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Goldstein M, Kastan MB, The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med 66, 129–143 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Demaria S, Formenti SC, Radiotherapy effects on anti-tumor immunity: implications for cancer treatment. Frontiers in oncology 3, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weichselbaum RR, Liang H, Deng L, Fu YX, Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol 14, 365–379 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Herrera FG, Bourhis J, Coukos G, Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin 67, 65–85 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P, Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 12, 860–875 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu YX, Weichselbaum RR, STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmed MM, Hodge JW, Guha C, Bernhard EJ, Vikram B, Coleman CN, Harnessing the potential of radiation-induced immune modulation for cancer therapy. Cancer Immunol Res 1, 280–284 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Zitvogel L, Kroemer G, Subversion of anticancer immunosurveillance by radiotherapy. Nat Immunol 16, 1005–1007 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Vatner RE, Formenti SC, Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol 25, 18–27 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, Zheng W, Mauceri H, Mack M, Xu M, Fu YX, Weichselbaum RR, Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun 8, 1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JJ, Tannock IF, Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer 5, 516–525 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Diehn M, Clarke MF, Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst 98, 1755–1757 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Sharma RA, Plummer R, Stock JK, Greenhalgh TA, Ataman O, Kelly S, Clay R, Adams RA, Baird RD, Billingham L, Brown SR, Buckland S, Bulbeck H, Chalmers AJ, Clack G, Cranston AN, Damstrup L, Ferraldeschi R, Forster MD, Golec J, Hagan RM, Hall E, Hanauske AR, Harrington KJ, Haswell T, Hawkins MA, Illidge T, Jones H, Kennedy AS, McDonald F, Melcher T, O'Connor JP, Pollard JR, Saunders MP, Sebag-Montefiore D, Smitt M, Staffurth J, Stratford IJ, Wedge SR, Clinical development of new drug-radiotherapy combinations. Nat Rev Clin Oncol 13, 627–642 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Altucci L, Gronemeyer H, The promise of retinoids to fight against cancer. Nature Reviews Cancer 1, 181–193 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Tang XH, Gudas LJ, Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol 6, 345–364 (2011). [DOI] [PubMed] [Google Scholar]
- 16.di Masi A, Leboffe L, De Marinis E, Pagano F, Cicconi L, Rochette-Egly C, Lo-Coco F, Ascenzi P, Nervi C, Retinoic acid receptors: from molecular mechanisms to cancer therapy. Mol Aspects Med 41, 1–115 (2015). [DOI] [PubMed] [Google Scholar]
- 17.de The H, Pandolfi PP, Chen Z, Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 32, 552–560 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Schenk T, Stengel S, Zelent A, Unlocking the potential of retinoic acid in anticancer therapy. Brit J Cancer 111, 2039–2045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larange A, Cheroutre H, Retinoic Acid and Retinoic Acid Receptors as Pleiotropic Modulators of the Immune System. Annu Rev Immunol 34, 369–394 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Erkelens MN, Mebius RE, Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends Immunol 38, 168–180 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Mora JR, Iwata M, von Andrian UH, Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 8, 685–698 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo YX, Pino-Lagos K, Ahonen CA, Bennett KA, Wang JS, Napoli JL, Blomhoff R, Sockanathan S, Chandraratna RA, Dmitrovsky E, Turk MJ, Noelle RJ, A Retinoic Acid-Rich Tumor Microenvironment Provides Clonal Survival Cues for Tumor-Specific CD8(+) T Cells. Cancer Res 72, 5230–5239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE, Krois CR, Kenkel JA, Pham TD, Carmi Y, Tolentino L, Choi O, Hulett R, Wang J, Winer DA, Napoli JL, Engleman EG, Normalizing Microbiota-Induced Retinoic Acid Deficiency Stimulates Protective CD8(+) T Cell-Mediated Immunity in Colorectal Cancer. Immunity 45, 641–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin W, Song Y, Liu Q, Wu Y, He R, Topical treatment of all-trans retinoic acid inhibits murine melanoma partly by promoting CD8(+) T-cell immunity. Immunology 152, 287–297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI, All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res 66, 9299–9307 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong SZ, Wong KL, Lin G, Yang CM, Wong SC, Angeli V, Macary PA, Kemeny DM, Human CD8(+) T cells drive Th1 responses through the differentiation of TNF/iNOS-producing dendritic cells. Eur J Immunol 41, 1639–1651 (2011). [DOI] [PubMed] [Google Scholar]
- 27.De Trez C, Magez S, Akira S, Ryffel B, Carlier Y, Muraille E, iNOS-producing inflammatory dendritic cells constitute the major infected cell type during the chronic Leishmania major infection phase of C57BL/6 resistant mice. PLoS Pathog 5, e1000494 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marigo I, Zilio S, Desantis G, Mlecnik B, Agnellini AH, Ugel S, Sasso MS, Qualls JE, Kratochvill F, Zanovello P, Molon B, Ries CH, Runza V, Hoves S, Bilocq AM, Bindea G, Mazza EM, Bicciato S, Galon J, Murray PJ, Bronte V, T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 30, 651 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, Klapproth K, Schakel K, Garbi N, Jager D, Weitz J, Schmitz-Winnenthal H, Hammerling GJ, Beckhove P, Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 24, 589–602 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Xiong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, Nickles D, Cubas R, Anti-PD-L1 Treatment Results in Functional Remodeling of the Macrophage Compartment. Cancer Res 79, 1493–1506 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Bhatt S, Qin J, Bennett C, Qian S, Fung JJ, Hamilton TA, Lu L, All-trans retinoic acid induces arginase-1 and inducible nitric oxide synthase-producing dendritic cells with T cell inhibitory function. J Immunol 192, 5098–5108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devaux Y, Grosjean S, Seguin C, David C, Dousset B, Zannad F, Meistelman C, De Talance N, Mertes PM, Ungureanu-Longrois D, Retinoic acid and host-pathogen interactions: effects on inducible nitric oxide synthase in vivo. Am J Physiol Endocrinol Metab 279, E1045–1053 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM, Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A 107, 8363–8368 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, Weichselbaum RR, Fu YX, Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 114, 589–595 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang H, Deng L, Chmura S, Burnette B, Liadis N, Darga T, Beckett MA, Lingen MW, Witt M, Weichselbaum RR, Fu YX, Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J Immunol 190, 5874–5881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halin C, Scimone ML, Bonasio R, Gauguet JM, Mempel TR, Quackenbush E, Proia RL, Mandala S, von Andrian UH, The S1P-analog FTY720 differentially modulates T-cell homing via HEV: T-cell-expressed S1P1 amplifies integrin activation in peripheral lymph nodes but not in Peyer patches. Blood 106, 1314–1322 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arina A, Beckett M, Fernandez C, Zheng W, Pitroda S, Chmura SJ, Luke JJ, Forde M, Hou Y, Burnette B, Mauceri H, Lowy I, Sims T, Khodarev N, Fu YX, Weichselbaum RR, Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat Commun 10, 3959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bosschaerts T, Guilliams M, Stijlemans B, Morias Y, Engel D, Tacke F, Herin M, De Baetselier P, Beschin A, Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog 6, e1001045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng L, Liang H, Burnette B, Weicheslbaum RR, Fu YX, Radiation and anti-PD-L1 antibody combinatorial therapy induces T cell-mediated depletion of myeloid-derived suppressor cells and tumor regression. Oncoimmunology 3, e28499 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, Zhang Y, Yu X, Xu M, Mauceri H, Arina A, Weichselbaum RR, Fu YX, Non-canonical NF-kappaB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 49, 490–503 e494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bogdan C, Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 36, 161–178 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Xue Q, Yan Y, Zhang R, Xiong H, Regulation of iNOS on Immune Cells and Its Role in Diseases. Int J Mol Sci 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei S, Kozono S, Kats L, Nechama M, Li W, Guarnerio J, Luo M, You MH, Yao Y, Kondo A, Hu H, Bozkurt G, Moerke NJ, Cao S, Reschke M, Chen CH, Rego EM, Lo-Coco F, Cantley LC, Lee TH, Wu H, Zhang Y, Pandolfi PP, Zhou XZ, Lu KP, Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat Med 21, 457–466 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang ZY, Chen Z, Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 111, 2505–2515 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, Grigg ME, Kastenmayer R, Schwartzberg PL, Belkaid Y, Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34, 435–447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, Runci D, Ward JP, Gubin MM, Medrano RFV, Arthur CD, White JM, Sheehan KCF, Chen A, Wucherpfennig KW, Jacks T, Unanue ER, Artyomov MN, Schreiber RD, MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574, 696–701 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng LF, Liang H, Xu M, Yang XM, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, Huang XN, Gajewski TF, Chen ZJJ, Fu YX, Weichselbaum RR, STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Raw data file (Excel spreadsheet)
Fig. S1. Related to Fig. 1. Mouse colon tumor response to RA treatment by various delivery methods.
Fig. S2. Related to Fig. 2. Local MC38 tumor microenvironment changes in IR and RA treated tumors 3 days after start of the treatments.
Fig. S3. Related to Fig. 2. Immune cell profile in MC38 tumors treated with indicated treatments 4 days after IR and start of RA treatment.
Fig. S4. Related to Fig. 2. iNOS and TNF-α production in Inf-MACs of MC38 tumors treated with IR and RA.
Fig. S5. Related to Fig. 2. iNOS-producing cells in MC38 tumors 8 days after start of the treatments, and efficacy of CD11b blockade of myeloid cell.
Fig. S6. Related to Fig. 3. Immune profile in MC38 tumor draining lymph nodes 8 days post starting of the treatments.
Fig. S7. Related to Fig. 4 and Fig. 5. Frequencies of Immune cells in MC38 tumors after T and NK cell depletion and IFN-ɣ and GM-CSF neutralization.
Fig. S8. Related to Fig. 6. Immune profile changes in distal MC38 tumor and other distal tissues.