

Abstract
Chimeric antigen receptor (CAR) T cell therapy has been established in the treatment of hematological malignancies. However, in solid tumors its efficacy remains limited. The aim of this article is to give an overview of the field of cell therapy itself, to introduce the underlying concepts of CAR T cell-based treatment approaches and to address its limitations in advancing the treatment for solid malignancies.
Keywords: Adoptive T cell therapy, CAR T cells, Solid tumors, Immunotherapy, Tumor immunology
Background
Over the last decade, treatment of cancer has undergone a radical paradigm shift. Targeted therapies, either utilizing tyrosine kinase inhibitors (TKI) or therapeutic antibodies, have developed into integral elements of oncological treatment regimes. In contrast, cellular therapies are merely starting to enter clinical routine [1]. Of these, T cell-based methods, also known as adoptive T cell therapy (ACT), are the most advanced. ACT aims to combine the extraordinary specificity of the adaptive immune system and the natural antitumor response of T cells in the fight against cancer. To date, depending on the source of the T cells and the subsequent genetic or nongenetic manipulation, three main forms of T cell-based therapies can be distinguished:
Tumor-infiltrating lymphocytes (TIL),
T cell receptor (TCR)-engineered T cells and
Chimeric-antigen receptor (CAR) T cells [2].
TIL are T cells, found in the tumor tissue, which in most cases are equipped with endogenous TCR specific for tumor-associated antigens. In TIL-based ACT, these T cells are isolated from surgical tumor specimens, are expanded in vitro and re-infused into the patients [3]. However, one major limitation to this approach is the often low number of antigen-specific T cells found in tumor explants and the inability to retrieve and expand T cells from all patients. To overcome these limitations, in vitro engineering methods have been developed to create antigen-specific T cells without needing to isolate them from tumor tissues. As such, naïve, unspecific T cells are isolated from the peripheral blood of the patients via leukapheresis, are then genetically modified with a tumor-specific recognition construct (e.g., tumor-specific TCR, CAR), expanded and finally re-infused into the patient [4].
TCR-engineered T cells and CAR T cells
As described, TCR T cells are genetically modified to express an antigen-specific TCR. In the treatment of neoplastic diseases, the target is usually a tumor-specific antigen (TSA) or tumor-associated antigen (TAA). Ideally, these would be uniquely expressed in malignant cancer cells, but not in healthy cells. Peptides derived from TSA are generated through intracellular proteasome-mediated processing mechanisms and subsequently presented on the MHC‑I complex of the tumor cells [2]. TCR-engineered T cells are able to recognize the MHC-TSA-peptide complex, which leads to an activation of T cells and subsequent lysis of neoplastic cells. In contrast, CAR T cells are engineered through introduction of an artificial synthetic construct, which ultimately also leads to the activation of the T cells and tumor cell lysis [2]. Both strategies have certain advantages and disadvantages, which have been extensively reviewed elsewhere [2, 5].
The artificial CAR construct usually contains an antibody-derived single-chain variable fragment (scFv) as an extracellular domain, a hinge domain and a transmembrane domain, anchoring the receptor in the cell membrane. T cells expressing the construct are able to bind the respective TSA via the extracellular antibody-derived scFv domain of the CAR receptor. Activation of T cells is subsequently induced by an intracellularly located signaling domain, consisting of a CD3ζ chain and one or more co-stimulatory domains (e.g., CD28, 4‑1BB) [2]. The CD3ζ chain is physiologically part of the TCR-CD3 complex and is the major inducer of T cell activation following antigen recognition. Co-stimulatory domains were included in the second and third generations of CAR constructs, as augmented antitumor efficacy [6] and increased persistence of the transferred T cells [7] has been observed. Depending on the CAR receptor used, CAR T cells are classified in different generations, as depicted in Fig. 1.
Fig. 1.
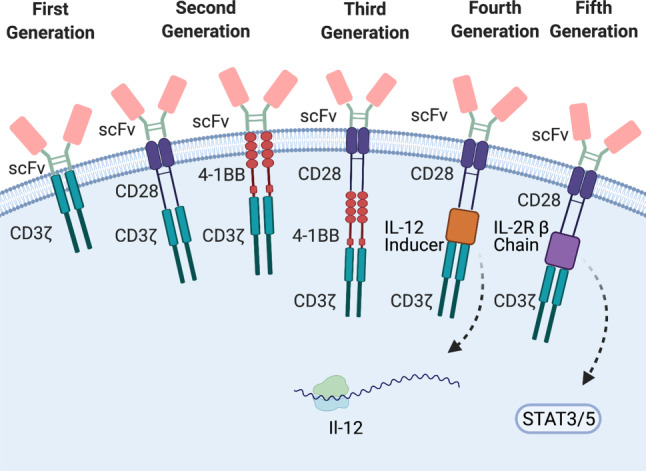
Structure and classification of CAR T cells. CAR T cells are grouped into different generations depending on the structure of the CAR. Recent advancements have added new CAR structures, which are extensively reviewed in [5, 8]. scFv single chain variable fragment, CD3ζ CD3 zeta chain, IL-2R β Chain IL‑2 receptor β chain
In recent years, further improvements of the CAR structures have been employed in order to improve efficacy of CAR T cells, especially in solid malignancies (see “CAR T cells in solid tumors” section). These innovative approaches have been extensively reviewed by us and other groups and interested readers are referred to the following literature for a more detailed overview [5, 8]. In short, fourth-generation CAR constructs incorporate a cassette into the intracellular domain to induce the secretion of pro-inflammatory cytokines. This strategy enables the innate immune system to contribute to the antitumor effect (Table 1; [9]). In contrast, fifth-generation CAR T cells followed a completely different approach and aimed to activate the Janus kinase-signal transducers and activators of transcription (JAK-STAT) signaling pathway to promote T cell proliferation. Fifth-generation CAR T cells were shown to have superior antitumor effect and persistence compared to second- and third-generations [10]. However, these developments are merely at the beginning and not part of the clinical routine. All U.S. Food & Drug Administration (FDA)- or European Medicines Agency (EMA)-approved ACT strategies are second-generation CAR-based approaches and only approved for the treatment of certain hematological malignancies.
Table 1.
Published clinical trials in solid tumors
| Trial number | Cancer entity | Published | CAR Target molecule |
Trial Phase |
Patients N |
Outcome | Reference |
|---|---|---|---|---|---|---|---|
| NCT01869166 | Biliary tract cancer | 2018 | EGFR | I | 19 |
1/17 complete remission 10/17 stable disease |
[36] |
| NCT02349724 | CRC | 2017 | CEA | I | 10 | 7/10 stable disease | [15] |
| NCT01212887 | GI tumors | 2017 | CEACAM5 | I | 14 |
No objective clinical response Terminated due to safety concerns |
[37] |
| NCT02541370 | GI tumors | 2018 | CD133 | I | 23 |
3/23 partial response 14/23 stable disease |
[38] |
| NCT00730613 | Glioblastoma | 2015 | IL13Rα2 | I | 3 | No objective clinical response | [16] |
| NCT02209376 | Glioblastoma | 2017 | EGFRvIII | I | 10 | Not available due to surgical intervention | [17] |
| NCT01109095 | Glioblastoma | 2017 | HER-2/neu | I | 17 |
1/17 partial response 7/17 stable disease |
[39] |
| NCT01454596 | Glioblastoma | 2019 | EGFRvIII | I | 18 | No objective clinical response | [21] |
| NCT02395250, NCT03146234 | HCC | 2020 | GPC3 | I | 13 |
2/13 partial response, 1/13 stable disease |
[40] |
| Park et al. | Neuroblastoma | 2007 | L1-CAM | I | 6 | 1/6 stable disease then partial response | [41] |
| Pule et al. | Neuroblastoma | 2008 | GD2 | I | 11 | 4/8 evidence of regression | [42] |
| NCT00085930 | Neuroblastoma | 2011 | GD2 | I | 19 | 3/19 complete remission | [43] |
| NCT01822652 | Neuroblastoma | 2017 | GD2 | I | 11 | 5/11 stable disease | [44] |
| NCT01869166 | NSCLC | 2016 | EGFR | I | 11 |
2/11 partial response 5/11 stable disease |
[18] |
| Kershaw et al. | Ovarian Carcinoma | 2006 | FRα | I | 14 | No objective clinical response | [45] |
| NCT01897415 | PDAC | 2018 | MSLN | I | 6 | 2/6 stable disease | [46] |
| NCT01869166 | PDAC | 2020 | EGFR | I | 14 |
4/14 partial response 8/14 stable disease |
[14] |
| Junghans et al. | Prostate cancer | 2016 | PSMA | I | 5 (6) | 2/5 partial response | [47] |
| Lamers et al. | RCC | 2016 | CAIX | I | 12 | No objective clinical response | [48] |
| NCT00902044 | Sarcomas | 2015 | HER-2/neu | I | 17 (19) | 4/17 stable disease | [49] |
| NCT02159716 | Solid tumors | 2019 | MSLN | I | 15 | 11/15 stable disease | [50] |
CRC Colorectal Carcinoma, GI tumor, Gastrointestinal Tumor, HCC Hepatocellular Carcinoma, NSCLC Non-Small Cell Lung Cancer, PDAC Pancreatic Ductal Adenocarcinoma, RCC Renal Cell Carcinoma, IL13Ra2 Interleukin-13 receptor subunit alpha 2, L1-CAM L1 Cell Adhesion Molecule, HER2/neu Human epidermal growth factor receptor 2, EGFR(vIII) Epidermal Growth Factor Receptor (variant III), CEACAM5 Carcinoembryonic antigen-related cell adhesion molecule 5, MSLN Mesothelin, CEA Carcino-Embryonic Antigen, GPC3 Glypican‑3, CAIX Carboxyanhydrase-IX, FRα α-folate Receptor, PSMA Prostate-specific membrane antigen
CAR T cells in hematological malignancies
Axicabtagene ciloleucel and Tisagenlecleucel, both approved in 2017 by the FDA and in 2018 by the EMA, are CAR T cells engineered to target the B cell lineage antigen CD19. CD19 is exclusively expressed on both healthy and malignant B cells. Consequently, these CAR T cells can be used to treat B cell malignancies such as diffuse large B cell lymphomas (DLBCL) and B cell acute lymphoblastic leukemia (B-ALL, only Tisagenlecleucel). Approval was granted after astonishing initial response rates of up to 93% in ALL and 54% in DLBCL were observed [5]. Importantly, these response rates were reached in extensively pretreated patients with chemotherapy-refractory or relapsed malignant disease and many were durable [2]. A third T cell product was just recently approved by both the FDA and the EMA for the treatment of mantle cell lymphoma (MCL; Tecartus, brexucabtagene autoleucel) (NCT02601313) [11]. Recent long-term follow-up studies revealed sustained response rates in patients. However, disease relapse is seen in up to 41% of patients suffering from ALL [12]. In contrast, response rates in DLBCL seems to be more durable as the majority of responding patients do not experience relapse during the 12-month follow-up period [5, 13]. In summary, CAR T cell therapy has emerged as an important therapeutic option for hematological malignancies. However, in nonhematological malignancies CAR T cell therapy has so far failed to demonstrate comparable treatment responses.
CAR T cells in solid tumors
Encouraged by the striking results seen in DLBCL and ALL, new CAR T cells targeting different epithelial antigens were developed and clinically tested. As such, CAR-based ACT was evaluated in different gastrointestinal malignancies (pancreatic cancer, NCT01869166; colorectal cancer, NCT02349724) [14, 15], glioblastoma (NCT00730613; NCT02209376) [16, 17] and non-small cell lung cancer (NSCLC, NCT01869166) [18]. Table 1 gives a summary of already conducted clinical trials in solid malignancies.
Treatment and outcomes of patients suffering from carcinomas, per definition derived from epithelial tissues, however, differs from the treatment of hematological malignancies. To at least some extent, target antigens are usually co-expressed on healthy tissues [5]. Application of anti-CD19 CAR T cells for example can lead to sustained B cell aplasia. In the clinical setting however, this is not a life-threatening side effect and can be managed with regular substitution of immunoglobulins [19]. In comparison, in solid malignancies the target antigen can be expressed at low levels on other epithelial cells (e.g., lung, heart) and thus potentially lead to serious adverse effects, as observed by Morgan et al. [20]. Furthermore, high-dose treatment with CAR T cells against an epithelial cell antigen (EGFRvIII) has also been reported to lead to the congestion of pulmonary vasculature and lethal respiratory failure (NCT01454596) [21].
As a consequence, several newly initiated clinical trials have primarily focused on the safety of the newly developed CAR T cells, directed against various target antigens of solid tumors (e.g., EGFRvIII, MUC‑1, MAGE, CEA, GD2, CA125, MSLN; Table 1; [22]). These studies were most often conducted in malignancies with poor overall survival such as glioblastoma and pancreatic ductal adenocarcinoma; however, as depicted in Table 1, therapies are assessed in a wide range of different solid malignancies. While most treatments were shown to be safe, the overall response rates observed in these trials, especially compared to the impressive clinical benefit obtained in ALL and DLBCL, were rather disappointing. Overall mortality remained approximately the same and the patients usually only benefited from the treatment temporarily [8].
Hurdles of CAR T cell therapy in solid tumors
As described above the responsiveness of solid malignancies to CAR T cell therapy is bleak at best.
Over the last years, researchers have identified several underlying mechanisms responsible for the lack of treatment efficacy in solid tumors and have identified three major hurdles: (1) trafficking of T cells as the first key limiting step, (2) the choice of target antigen and antigen loss (tumor cell recognition) and (3) the hostile tumor microenvironment [8].
Trafficking of the transferred T cells into solid tumors is a limiting factor dampening therapeutic efficacy. As such, different strategies have been applied to improve T cell trafficking into the tumors. Direct application (intratumoral injection) of T cells has been employed to directly deliver the CAR T cells to the tumor site (NCT00730613) [16]. The need for invasive interventional procedures as well as the often inaccessible tumors however limit these approaches. Alternative strategies make use of physiological processes of immune cell trafficking: Immune cell recruitment to the site of inflammation is mediated by the chemokine–chemokine receptor axis. High levels of chemokine ligands secreted at the site of inflammation lead to the recruitment of immune cells expressing matching receptors. As solid tumors tend to show enhanced levels of chemokine ligands, co-transduction of chemokine receptors commonly not present on T cells, and CAR receptors into T‑cells, has been employed by us and another groups [23, 24]. This has been shown to enhance both T cell infiltration and therapeutic efficacy in preclinical models. This concept is currently under investigation in clinical trials (NCT03602157).
Loss of the target antigen on tumor cells is a problem common in treatment of both hematological and nonhematological malignancies. Relapse with CD19-negative disease, for example, is frequently observed after treatment with CD19 CAR T cells. In solid tumors, down-regulation of the target antigen following CAR T cell therapy has also been reported in different clinical trials [17]. Targeting of multiple antigens (e.g. CD19 plus CD20; CD19 plus CD22) or alternatively sequential targeting strategies have shown benefit in different clinical trials [25–27].
Finally, solid tumors exhibit a complex, often hostile tumor microenvironment (TME). Besides cancer cells, the TME of solid tumors comprises infiltrating and resident immune cells, stromal cells as well as many pro- and anti-inflammatory mediators [28]. The interactions between the different components of the TME are complex and cannot be described in detail here. For further information please see the following literature [28–30]. In general, the components of the TME suppress an appropriate immune response against cancer cells, thus, creating a conducive environment for the tumor cells to proliferate.
The nowadays commonly used checkpoint inhibitors (e.g., pembrolizumab, nivolumab, ipilimumab) boost the activation and function of T cells through blockade of inhibitory receptors on T cells (e.g., PD‑1, CTLA-4). As such, combining CAR T cells with immune checkpoint inhibitors or other drugs influencing the immunosuppressive nature of the TME are currently being investigated [8]. In addition, genetic engineering can be employed to lift immune-suppressive effects on the transferred T cells. Our group has developed a fusion receptor, switching the inhibitory signal of PD‑1 into a T cell activating signal [31]. Alternatively, CRISPR-Cas9-mediated disruption of the PD‑1 locus in CAR T cells has been shown to increase therapeutic efficacy of CAR T cells in vitro and in vivo. Several clinical trials are currently investigating these strategies (NCT03081715, NCT02867332, NCT02867345, NCT02793856, NCT03044743) [32].
Lastly, preclinical research has demonstrated the feasibility of redirecting CAR T cells not against tumor cells, but at immunosuppressive cells in the tumor microenvironment. Thus, CAR T cells targeting cancer-associated fibroblasts or tumor-associated macrophages have been shown to delay cancer progression in preclinical mouse models [33, 34]. However, to date no clinical data on these strategies are available, so the value remains uncertain.
Conclusion
The clinical transition of CAR T cell therapy has started a new era in oncology. Although these approaches have already given hope to incurable cancer patients suffering from hematological malignancies, it still remains to be proven in the comprehensive field of solid malignancies. As one might infer from our short overview, glioblastoma was often targeted in clinical studies. Due to limitations arising from its anatomical location and quick progression rate, glioblastoma remains clinically challenging to this date. Even a tumor as aggressive as glioblastoma was reported to be fully regressed in a case collection by Brown et al. [35]. The overall results of clinical studies might seem disappointing, but such case reports highlight the potential of CAR T cell therapy in solid cancers and maybe give a glimpse into what can be achieved in the future. Consequent advancement of promising preclinical strategies into clinical testing is now crucial to broaden the scope of cellular therapies and to increase the efficacy in solid tumors, with the hope that these therapies will not only be effective in single patients, but present a real clinical alternative for so many incurable cancer patients in daily oncological routine.
Take home message
Adoptive T cell therapy has emerged has an important treatment option in relapsed and chemotherapy-refractory hematological malignancies.
Clinical trials in solid tumors have primarily focused on establishing the safety of CAR T cell therapy; however, secondary endpoint analyses have so far only revealed modest efficacy.
Preclinical research has been able to identify major caveats of CAR T cell therapy in solid tumors. Clinical trials will now have to determine whether this can be translated into clinically relevant improvements in patient outcome.
Acknowledgments
Acknowledgements
All figures were created at Biorender.com under a paid academic subscription.
Funding
This study was supported by the Marie-Sklodowska-Curie Program Training Network for the Immunotherapy of Cancer funded by the H2020 Program of the European Union (Grant 641549, to S.E. and S.K.), the Marie-Sklodowska-Curie Program Training Network for Optimizing Adoptive T Cell Therapy of Cancer funded by the H2020 Program of the European Union (Grant 955575, to S.K.), the Hector Foundation, the International Doctoral Program i‑Target: Immunotargeting of Cancer funded by the Elite Network of Bavaria (S.K. and S.E.); Melanoma Research Alliance Grants 409510 (to S.K.); the Else Kröner-Fresenius-Stiftung (S.K.); the German Cancer Aid (S.K.); the Ernst-Jung-Stiftung (S.K.); LMU Munich’s Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative (S.E. and S.K.); the Bundesministerium für Bildung und Forschung Project Oncoattract (S.E. and S.K.); by the European Research Council Grant 756017, ARMOR‑T (to S.K.), by the German Research Foundation (DFG to S.K.), the Fritz-Bender Foundation (to S.K.) and the José-Carreras Foundation (to S.K.).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
Ö. Umut and A. Gottschlich declare that they have no competing interests. S. Kobold has received honoraria from Novartis and GSK. S. Kobold and S. Endres are inventors of several patents in the field of immuno-oncology. S. Kobold and S. Endres received research support from TCR2 Inc. and Arcus Bioscience for work unrelated to this manuscript.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the European society for blood and marrow transplantation (EBMT) and the joint accreditation committee of ISCT and EBMT (JACIE) Haematologica. 2020;105(2):297–316. doi: 10.3324/haematol.2019.229781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houot R, Schultz LM, Marabelle A, Kohrt H. T-cell-based immunotherapy: adoptive cell transfer and checkpoint inhibition. Cancer Immunol Res. 2015;3(10):1115–1122. doi: 10.1158/2326-6066.CIR-15-0190. [DOI] [PubMed] [Google Scholar]
- 5.Lesch S, Benmebarek MR, Cadilha BL, Stoiber S, Subklewe M, Endres S, et al. Determinants of response and resistance to CAR T cell therapy. Semin Cancer Biol. 2020;65:80–90. doi: 10.1016/j.semcancer.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. 2019;120(1):26–37. doi: 10.1038/s41416-018-0325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 10.Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–359. doi: 10.1038/nm.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Guo Y, Wu Z, Feng K, Tong C, Wang Y, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: a phase I clinical trial. Cytotherapy. 2020;22(10):573–580. doi: 10.1016/j.jcyt.2020.04.088. [DOI] [PubMed] [Google Scholar]
- 15.Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA(+) metastatic colorectal cancers. Mol Ther. 2017;25(5):1248–1258. doi: 10.1016/j.ymthe.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. 2016;59(5):468–479. doi: 10.1007/s11427-016-5023-8. [DOI] [PubMed] [Google Scholar]
- 19.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126–135. doi: 10.1097/CJI.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov. 2016;6(2):133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rapp M, Grassmann S, Chaloupka M, Layritz P, Kruger S, Ormanns S, et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. OncoImmunology. 2016;5(3):e1105428. doi: 10.1080/2162402X.2015.1105428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood. 2020;136(14):1632–1644. doi: 10.1182/blood.2020005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang N, Hu X, Cao W, Li C, Xiao Y, Cao Y, et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood. 2020;135(1):17–27. doi: 10.1182/blood.2019000017. [DOI] [PubMed] [Google Scholar]
- 27.Pan J, Zuo S, Deng B, Xu X, Li C, Zheng Q, et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood. 2020;135(5):387–391. doi: 10.1182/blood.2019003293. [DOI] [PubMed] [Google Scholar]
- 28.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobold S, Grassmann S, Chaloupka M, Lampert C, Wenk S, Kraus F, et al. Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J Natl Cancer Inst. 2015;107(8):djv146. doi: 10.1093/jnci/djv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255–2266. doi: 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2(2):154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. 2021;12(1):877. doi: 10.1038/s41467-021-20893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q, et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res. 2018;24(6):1277–1286. doi: 10.1158/1078-0432.CCR-17-0432. [DOI] [PubMed] [Google Scholar]
- 37.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66(11):1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. OncoImmunology. 2018;7(7):e1440169. doi: 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2 specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clin Cancer Res. 2020;26(15):3979–3989. doi: 10.1158/1078-0432.CCR-19-3259. [DOI] [PubMed] [Google Scholar]
- 41.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 42.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25(9):2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155(1):29–32. doi: 10.1053/j.gastro.2018.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. 2016;76(14):1257–1270. doi: 10.1002/pros.23214. [DOI] [PubMed] [Google Scholar]
- 48.Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans. 2016;44(3):951–959. doi: 10.1042/BST20160037. [DOI] [PubMed] [Google Scholar]
- 49.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)—specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33(15):1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther. 2019;27(11):1919–1929. doi: 10.1016/j.ymthe.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]