

Graphic abstract
Isoindigo, the structural isomer of the well-known dye indigo, has seen a major revival recently because of the increasing interest of its use as a potential drug core structure and for the development of organic photovoltaic materials. Highly beneficial for diverse applications are its facile synthesis, straightforward functionalisation and the broad absorption band in the visible range. Moreover, its intrinsic electron deficiency renders isoindigo a promising acceptor structure in bulk heterojunction architectures. Here we present new insights into the substituent effects of N-functionalised isoindigos, developing a reliable and fast in silico screening approach of a library of compounds. Using experimental UV–Vis and electrochemical data increased the accuracy of the TD-DFT method employed. This procedure allowed us to accurately predict the optical and electrochemical properties of N-functionalised isoindigos and the elucidation of the relationship between substituent effects and electronic properties.

Supplementary Information
The online version contains supplementary material available at 10.1007/s43630-021-00071-5.
Introduction
Isoindigo was first synthesised in 1842 [1] and initially identified with the name "indin" (iso-I, Fig. 1A) [2]. This structure belongs to the indigoid family of compounds, together with indigo itself, indirubin and the related thioindigo. Similarly to indirubin scaffolds [3], iso-I became attractive to the pharmaceutical industry due to the properties of some of its derivatives. N-substituted iso-Is have found use in leukaemia treatment under the commercial names Natura and Meisoindigo (Fig. 1B) [3, 4]. Other molecules based on the same motif showed promising activity against different tumour strains under in vitro conditions [3, 5]. Indigo- and thioindigo- compounds [6] are widely employed as dyes in the textile industry [7] and as photosensitisers [8, 9] and are studied regarding their photophysical and photochromic properties [10–14]. In contrast, the optical properties of iso-I were the subject of a more recent research effort [15–19] as it was realised that iso-I is highly suitable for organic photovoltaics (OPVs) [17] and optoelectronics [15–19]. The facile synthesis of iso-I and its derivatives opens numerous possibilities for various applications. Indeed, several 6,6′-substituted iso-I derivatives are reported as p-type donor in donor–acceptor–donor (DAD) materials and conjugated polymers due to its electron deficiency [4, 19]. On the other hand, installing substituents at the lactam nitrogen is largely unexplored [4]. A limited selection of N-alkylations is reported to overcome solubility issues in iso-I OPV research [4]. At the same time, aromatic substituents at the nitrogen are rare and only found in a few bio-active iso-Is [3, 5]. iso-I is very stable towards photobleaching [20], a property attributable to the ultrafast intramolecular singlet fission that the molecule undergoes after excitation [21]. This sub-ns event leads to efficient triplet pair separation in thiophene-functionalised derivatives, while in solution it affords radiationless deactivation of the excited state, recovering the starting material at the ground state. Isoindigo absorbs in the visible region of the UV–Vis absorption spectrum (3900 M−1 cm−1 in DMSO at the absorption maximum at 490 nm [15]) tailing in the near-infrared (NIR) [15, 22, 23]. Consequently, the molecule holds great potential for high conversion efficiency in photochemical processes using visible light.
Fig. 1.
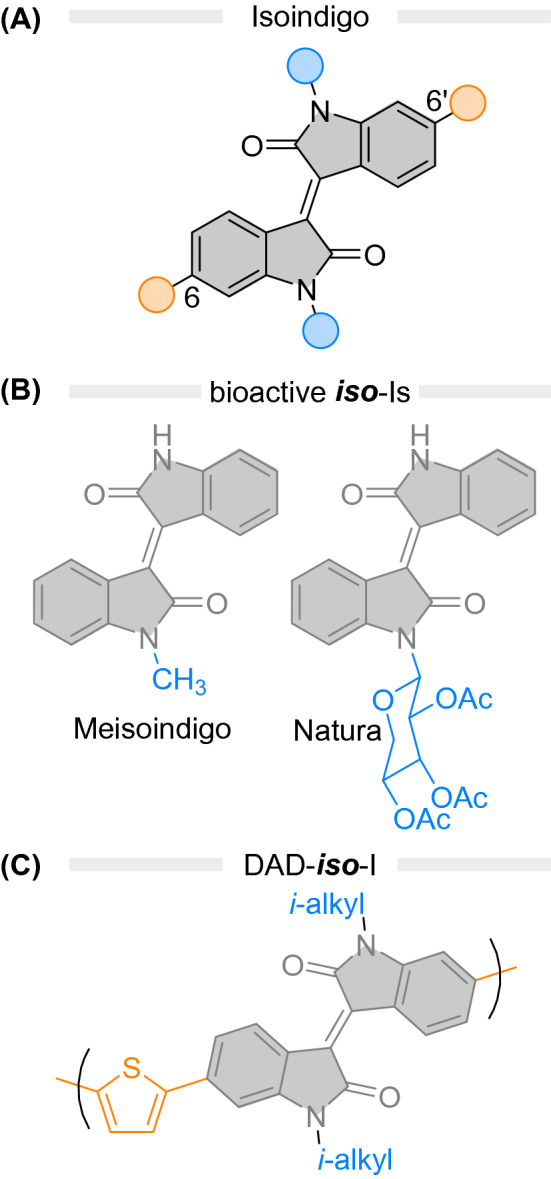
A Isoindigo with the 6,6′- and N,N′-substitution position highlighted. B Bioactive N-substituted iso-Is such as Meisoindigo (left) and Natura (right) are used in cancer treatment [4, 24]. C In recent OPV-research, the 6,6′-substitution pattern was used as a handle to obtain DAD-materials. The exemplary structure is one of the initial proposed OPV-polymers [17].
The introduction of substituents onto the iso-I core structure allows tuning its UV–Vis absorption profile [24, 25]. The π-system of iso-Is can be easily extended at the 6,6′-position (Fig. 1A, orange). Substituents at the lactam nitrogens (N,N′; see Fig. 1, blue), such as alkyl chains, prevent π–π stacking and lead to increased solubility of iso-I derivatives [19]. However, to the best of our knowledge, the influence of N,N′-substituents on the optical and electronic properties of iso-I were not systematically studied so far. Functionalisation on the lactam nitrogen is synthetically straightforward and would provide an additional handle for tuning the electronic properties of the core structure. A reliable prediction tool is crucial to efficiently judge the outcome of a synthetic modification and facilitate effective rational design for specific optoelectronic applications.
Computational chemistry provides valuable methods to predict the properties of a compound, supporting the development of new molecules, drugs and materials [26, 27]. To apply quantum mechanical calculations to a library of compounds, the availability of a fast and accurate method is a key requirement. In this context, density functional theory (DFT) is known as the "work-horse" of current theoretical studies in chemistry and physics [28, 29]. However, DFT methods often vary in their performances [30, 31], making the choice of the proper functional for a specific application of uttermost importance [29, 32]. Besides various attempts to categorise functionals by accuracy in a hierarchy as in the Perdew "Jacob's Ladder" [33], extensive benchmarks are needed to identify the functional best suited for a specific use.
Here, we present a library of 50 iso-I derivatives focusing on the N,N′-functionalisation pattern. The library was explored computationally, utilising an extensively benchmarked method to obtain insights into the substitution effects on the optical and electronic properties of iso-I. Additionally, we synthesised a limited subset of selected derivatives to experimentally validate and provide insights into the quality of the computational methods. We further utilised Koopmans’ theorem and parametrisation from experimental values to gain an accurate understanding of the dependencies of both optical and electrochemical properties of iso-I and its derivatives being part of the library. This methodology will serve as a predictive tool and consequently pave the way for the future rational design of iso-Is as chromophore in material sciences and organic photovoltaics.
Results and discussion
Design and synthesis
We explored a library of functionalised iso-I derivatives employing substituents with varying electronic and steric demand to rationalise their effect on the properties of the chromophore (Fig. 2). We focused on the less explored N,N′-functionalisation over the 6,6′-substitution of iso-I. We selected 4-methoxyphenyl as electron-donor and 4-nitrophenyl, 4-trifluoromethylphenyl, 4-fluorobenzyl and benzonitrile as electron-acceptor units, respectively, while the unsubstituted phenyl group serves as a reference. Additionally, t-butyl-acetyl and t-butyloxycarbonyl were selected for their different steric demand and electronic character. Permutation of the aforementioned N,N′-substituents resulted in a library of 45 derivatives (Fig. 2, blue, 1–45). N,N′-propyl and bis-propyl substituted derivatives were included, to serve as a reference for the commonly used alkylated iso-I derivatives in the OPV research (Fig. 2, blue, 46–47). For comparison, we included a nitrile group as electron-poor and a methoxy group as electron-rich substituents on the more frequently used 6,6′-position, providing three additional derivatives (Fig. 2, orange, 48–50). Compounds iso-Is 25, 27, 30, 31, 35, 36, 45, 46 and 47 (Fig. 2, grey boxes) were synthesised to reference our library. A detailed overview of all synthetic steps and procedures is available in the SI.
Fig. 2.

The calculation library consists of permutations of N,N′-substituents: hydrogen, 4-methoxyphenyl, 4-nitrophenyl, 4-trifluoromethylphenyl, 4-fluorophenyl, t-butyl-acetyl and t-butoxycarbonyl, as well as 6,6′-permutations of nitrile and methoxy substituents. The derivatives highlighted in the grey boxes have been synthesised in this work
Benchmark study
We benchmarked several functionals and basis sets to find the best-suited time-dependent (TD) DFT method to analyse the optical properties of a library of different iso-Is efficiently. The absorption spectra and the solvatochromism of unfunctionalised iso-I-45 served as reference for the benchmarking. The solvatochromism [34] of λmax in different solvents (toluene, methanol, chloroform, acetonitrile) relative to DMSO was computed at different levels of theory with the respective SMD solvent model [35] and 25 computed states and was compared to the experimental results. The average error of the solvatochromism (defined as “Avg.-Error” in Fig. 3), the average of the absolute deviations of λmax in the chosen solvents from experimental results (Abs.-Error), the computing wall time (CalcTime), the difference between the highest and lowest error of the solvatochromism (Error-range), and the lowest and highest error of the solvatochromism (Min.-Error and Max.-Error), were used to select the best suited functional. We aimed for the lowest errors (Avg.-Error, Abs.-Error, Min.-Error and Max.-Error), a uniform description in all solvents (small Error-range) and a computational efficient method (short CalcTime). A selection of the results is given in Fig. 3 and a detailed protocol is presented in the SI. Our study gave the TD-PBE0 [36]/cc-pVDZ [37] level of theory as the best-suited method for the simulations of the UV–Vis spectra of the library. The general applicability regarding excitation energies of TD-PBE0 in intramolecular excited state charge-transfer (CT), as the one observed in iso-I, was demonstrated earlier [23, 30, 38], even though in some cases TD-PBE0 has shown limitations in describing the CT character of the excitations [38]. In our case, visual inspection of the orbital contribution to the lower excited states confirmed a limited effect of the functional on the nature and order of the states. Therefore, we confidently chose PBE0 to proceed with our analysis. After selection of the functional and basis set, we extended the analysis to our library (see SI).
Fig. 3.

Benchmarks of the error of the change of λmax in different solvents (toluene, methanol, chloroform, acetonitrile) relative to DMSO (Avg.-Error) computed at the TD-HF level and at a selected number of TD-DFT functionals with the respective SMD solvent model and 25 computed states versus experimental results. The best fitting functional was chosen based on the minimisation of Avg.-Error, the absolute deviation of λmax in the chosen solvents from experimental results (Abs.-Error), the computing wall time (CalcTime), the difference between the highest and lowest relative error (Error-range) and the lowest and highest relative error (Min.-Error and Max.-Error), respectively
Optical properties
Our calculations revealed that all derivatives show the expected S0 → S1 π–π* vertical excitation (between 480 and 530 nm in methanol). This transition occurs with different probabilities determined by their oscillator strength (f). The second vertical transition to the S2 has, in most cases, a low oscillator strength, making this state a so-called dark state. This finding is in accordance with calculations on iso-I [24]. Introducing different substituents on the lactam nitrogen influences the position of λmax and the oscillator strength of the respective transition. For instance, an iso-I with two Boc-groups (iso-I-36) has an unusually high oscillator strength of 0.3019 to the S1. In contrast, the asymmetrically substituted iso-Is with only one Boc unit show a bright π–π* S0 → S2 transition that occurs in iso-I-38 and iso-I-27 with higher probability (f = 0.1343 and f = 0.1733 for iso-I-38 and iso-I-27 in toluene) than the excitation to the S1 (f = 0.1299 and f = 0.1051 for iso-I-38 and iso-I-27 in toluene). Though the π–π* transitions in most derivatives were not apparent to be CT from natural transition orbital analysis (see SI), the special case of iso-I-27 showed three consecutive bright and marked CT states. This could be of high interest for optoelectronics, as several excitations and thus a broader spectral range leads to productive charge separation. Moreover, a push–pull configuration like in iso-I-12 (4-methoxyphenyl and 4-nitrophenyl substituents), results in the most bathochromically shifted λmax of the library (530 vs. 481 nm of the unsubstituted iso-I-45 and vs. 495 nm of the bis-alkylated reference iso-I-47, in methanol). All derivatives containing an anisole substituent show an additional aromatic contribution to the frontier orbitals, whereas the frontier orbitals are primarily centred on both the oxindole halves of every other substitution patterns (see SI).
While a push–pull substitution pattern on the nitrogens induced a substantial bathochromic shift of λmax (vide supra), the calculations indicated less marked substituent effects at the 6,6′-position. Specifically, iso-I-48 (6,6′-bis-OMe) has the highest oscillator strength of the 6,6′-derivatives, but the lowest λmax with f = 0.3967 at 467 nm (toluene). By replacing the two electron-donating methoxy moieties by either one or two nitrile substituents, λmax increases with simultaneously decreasing f over iso-I-49 (f = 0.3421 at 478.51 nm in toluene) to iso-I-50 (f = 0.2404 at 484.01 nm in toluene). These findings emphasise the challenging balance of absorptivity and electronic transition maximum in the design of new isoindigo chromophores.
The synthesised iso-I derivatives were characterised regarding their UV–Vis absorption spectra. As an example, the experimental and predicted spectra of iso-I-27 and iso-I-36 are depicted in Fig. 4 (for all compounds we provided spectra and an excitation analysis in the SI; natural transition orbitals for all synthesised derivatives can be found in the SI). The experimental properties of all iso-I-derivatives are in good accordance with the calculated results (see Fig. 4) and are summarised in Table 1. In iso-I-27, the S0 → S2 excitation band is used as a reference as the first transition band does not correspond to a local maximum. The experimental spectra confirmed the discussed behaviour of Boc-N,N′-substitution based on the theoretical results (vide supra). iso-I-27 shows the increased transition probability to the S2 and iso-I-36 yields the highest attenuation coefficient for the π–π* transition, while the remaining derivatives show high and distinct attenuation coefficients at λmax with broad absorption bands ranging up to ca. 600 nm.
Fig. 4.

Experimental (solid lines) and calculated spectra (dashed lines) in the respective solvents are in good accordance. Both the vertical excitation energies and the overall shape is met with good accuracy. The S0 → S1 transition at ca. 475 nm in iso-I-27 does not correspond to a local maximum in both calculated and experimental spectra. The high attenuation of iso-I-36 was very accurately described by the TD-DFT model, along with the excellent prediction of the vertical excitation energy in toluene, tailing into the NIR
Table 1.
Comparison of experimental and theoretical optical and electrochemical properties
| iso-I | Solvent | λmax [nm] | Attenuation at λmax | E1/2 [V] (V vs Fc/Fc+) | Exciton binding energycalc. [eV] | ||
|---|---|---|---|---|---|---|---|
| Exp | Calc | fcalc | εexp [M−1 cm−1] | ||||
| 25 | Toluene | 508 | 508 | 0.1422 | 2882 | ||
| MeCN | 503 | 508 | 0.1366 | 2596 | − 1.12a | 4.30 | |
| MeOH | 512 | 526 | 0.0932 | 1936 | |||
| 27 | Tolueneb | 400 | 449 | 0.1733 | 12,006 | ||
| MeCNb | 396 | 451 | 0.1655 | 10,865 | − 1.01a | 4.18 | |
| MeOHb | 402 | 459 | 0.2239 | 13,313 | |||
| 30 | Toluene | 484 | 492 | 0.1748 | 4962 | ||
| MeCN | 485 | 497 | 0.1496 | 5277 | − 1.13 | 4.30 | |
| MeOH | 494 | 504 | 0.1650 | 4046 | |||
| 31 | Toluene | 485 | 490 | 0.1999 | 3985 | ||
| MeCN | 482 | 493 | 0.1712 | 3501 | − 1.11 | 4.69 | |
| MeOH | 483 | 497 | 0.174 | 2783 | |||
| 35 | Toluene | 487 | 486 | 0.1995 | 4689 | ||
| MeCN | 483 | 490 | 0.1783 | 4773 | − 1.15a | 4.64 | |
| MeOH | 482 | 497 | 0.1821 | 2780 | |||
| 36 | Toluene | 474 | 487 | 0.3019 | 9935 | ||
| MeCN | 472 | 487 | 0.2727 | 10,695 | − 0.90 | 4.82 | |
| 45 | Toluene | 484 | 482 | 0.3967 | 5105 | ||
| MeCN | 478 | 487 | 0.1884 | 4208 | − 1.16a | 4.74 | |
| MeOH | 481 | 496 | 0.1870 | 4473 | |||
| 46 | Toluene | 489 | 496 | 0.1492 | 5198 | ||
| MeCN | 483 | 501 | 0.1286 | 4440 | − 1.19 | 4.63 | |
| MeOH | 485 | 509 | 0.1256 | 4067 | |||
| 47 | Toluene | 493 | 501 | 0.1594 | 4154 | ||
| MeCN | 493 | 507 | 0.1361 | 3784 | − 1.20 | 4.56 | |
| MeOH | 495 | 515 | 0.1330 | 3671 | |||
The experimentally obtained λmax and the corresponding molar attenuation coefficient (εexp) are compared to the calculated S0 → S1 transition properties at the TD-PBE0/cc-pVDZ level of theory. The value of E1/2 obtained by cyclic voltammetry in acetonitrile are referenced versus Fc/Fc+. The exciton binding energy was obtained from the analysis of obtained data with the Multiwfn software. A complete list of computed optical and electrochemical data can be found in the SI
aDetermined from computational results, corrected by the parametrisation scheme described below
bλmax as S0–S2 transition band, since the S0–S1 transition does not build up to a local maximum, leading to a formal increased error
To understand the substituent effects on the nature of the electronic transition, we proceeded to analyse the library using transition density matrices (TDMs). This treatment allows accessing the electron and the hole distribution in the excited state and identifying their delocalisation [39]. In this way, one can visualise electronic excitation processes such as CT [39], which is of direct relevance in optoelectronic materials [24, 40, 41]. For this, we divided each compound into fragments to facilitate the interpretation of the results (Fig. 5A). The choice of the fragments allows us to ascribe the off-diagonal elements to CT and diagonal elements to local excitations. We used the Multiwfn software to analyse the TDMs and the exciton binding energies [42]. Inspecting the TDMs, we can assign four major transition behaviours to the first excited state. Symmetric and asymmetric TDMs, which can be further categorised by the presence of solely inductive or additional mesomeric effects. In symmetric TDMs, both the electron and the hole share a similar distribution around the central double bond, spreading on both oxindole halves of the molecule. On the contrary, asymmetric TDMs have a hole located on one half of the structure. The electron for all S0 → S1 TDMs is localised on fragments 3 and 4 (Fig. 5A).
Fig. 5.

A Fragments used to visualise and analyse the transition density matrices (TDMs). B An asymmetric TDM can be observed in the strongly mesomeric polarised iso-I-12 with its low exciton binding energy of 3.87 eV. C The symmetric inductive effect dominates iso-I-36, which shows a lower charge transfer with partial local excitation and a high exciton binding energy of 4.82 eV. D The orbital energies EHOMO (lower end of coloured bars) and ELUMO (upper end of coloured bars) mark the lower and upper bound of the band gap (coloured bars). The band gap of the N,N′-library correlate to the exciton binding energies (grey bars), while the 6,6′-substituted compounds (iso-I-48–50) does not follow this trend (EHOMO and ELUMO of derivative 50, out of range with − 0.558 and 0.928 eV).
The presence of electron-donating and -withdrawing groups interacting with the hole is the origin of the asymmetry in the TDMs. The donation of electron density to the hole via mesomeric effects by an electron donating moiety distorts the hole location. In this case, the TDM can be categorised as mesomerically influenced (Fig. 5B), rather than indirectly affected by only inductive effects (see SI). Vice versa, the inductively dominated TDM in Fig. 5C shows no localisation of the hole on one oxindole-half, without any significant TDM-elements on the substituent. The mesomeric contribution to the TDM is highly dependent on the polarity difference induced by the N,N′-substituents. The strong polarisation induced by the anisole and nitrophenyl substitution, and the mesomeric effects in iso-I-12 lead to a significant hole-density at the N,N′-substituents (Fig. 5B). The unpolarised, bis-anisole substituted iso-I-25 on the other hand does barely show any hole-density at the position of the substituent (see SI). The findings related to the enhanced resonance between the hole density and the anisole in iso-I-12 are in contrast to other examples where the rotation of the phenyl ring prevented an effective overlap, a trend encountered in 6,6′-substituted patterns [19, 24].
The known methodological limitation of the analysis of the excited state character and the excitation process [43, 44] can be observed from the electron and hole-densities in Fig. 5B, C. The CT is not always apparent from electron–hole or natural transition orbital analysis alone. This is due to the poorly defined localisation of the electron density around electron-donating and -withdrawing groups [38]. The TDM analysis, however, aided the identification of the CT and the substitution effects.
Next, we focused on the exciton binding energies, which represent the Coulomb attraction between the exciton quasiparticles (electron and hole). It is a measure for the separability of the exciton in free charges and is directly related to the generation of an effective current in optoelectronics [41]. The strong polarisation and the mesomeric effects in iso-I-12 (Fig. 5B) significantly lower the exciton binding energy to 3.87 eV, compared to 4.82 eV in the symmetric iso-I-36 (see Fig. 5B, C, along with Table 1). Analysing the HOMO and LUMO energies (EHOMO and ELUMO) provides further information on the effects of the N,N′-substitution on the electronic structure properties (see Fig. 5D). The variation of EHOMO and ELUMO in the N,N′-substituent library of 0.245 and 0.299 eV is more marginal than the one found in the 6,6′-substituted compounds (5.55 and 3.92 eV).
We further investigated the band gaps (EHOMO–ELUMO) of the N,N′-library varying by a range of 0.136 eV, with a maximum of 3.309 eV in the unsubstituted iso-I-45 and a minimum of 3.173 eV in iso-I-26. Investigation of the band gap and the exciton binding energy reveals evidence of a direct correlation between the two. The band gap increases with rising exciton binding energy (see Fig. 5D, all the compounds apart from iso-I-48–50, which belong to the 6,6′-substitution pattern). While some derivatives are outliers in this trend (e.g. iso-I-26, 30, 40), this finding indicates that the exciton binding energy correlates to the band gap of the N,N′-library, specifically. The 6,6′-substituted iso-I derivatives iso-I-48–50 are outliers in this series. iso-I-49 shows the second smallest band gap of the library (3.13 eV) while being positioned in the upper third of the library regarding the exciton binding energy. Additionally, iso-I-50 shows the lowest band gap in the series (0.371 eV), but is characterised by the third-highest exciton binding energy of 4.75 eV. These examples, combined with the above-mentioned variation in the EHOMO and ELUMO, emphasise further that 6,6′-substitution patterns cannot be categorised following the same trend observed for N,N′-substituted iso-I derivatives.
Electrochemical properties
According to Koopmans’ theorem [45], EHOMO and ELUMO can be approximated to the ionisation potential (IP) and electron affinity (EA), respectively (Fig. 6).
Fig. 6.
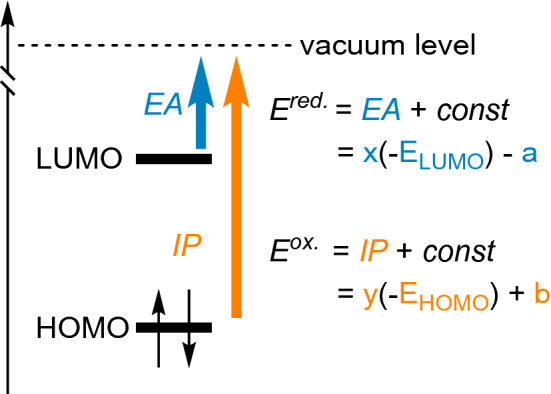
EHOMO and ELUMO can be approximated to electron affinities (EA) and ionisation potential (IP). The reduction and oxidation potentials can be obtained from the EA and IP
In our study, we decided to utilise the linear dependency of the EA and the reduction potential combined with Koopmans’ theorem to apply a parameterisation method on the computed values. We derived the ELUMOs from experimental reduction potentials () of a small sub-library of compounds that we use to parametrise the complete dataset of ELUMOs obtained computationally (). This approach is extensively described in the literature [24, 46–50]. We derived s from the reduction half-wave potential (E1/2) versus Fc/Fc+, obtained by cyclic voltammetry in acetonitrile, by applying Eq. (1), using the value of 5.1 eV vs. vacuum for ferrocene [47]. The experimental values were plotted against the theoretical ones and the parameters of the relation between and were obtained by linear regression (Fig. 7). Applying Eq. (1), we inferred the parametrisation coefficients (cf. x and a in Fig. 6) to correct the calculated results via Eq. (2) to better analyse the reduction potentials in the library.
| 1 |
| 2 |
Fig. 7.
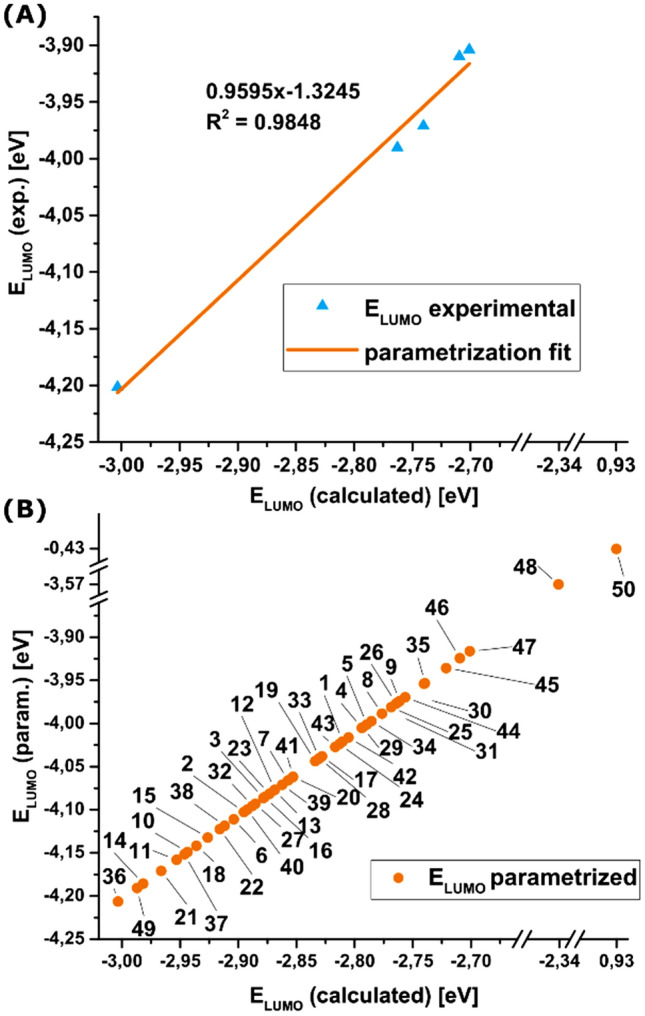
A Parametrisation of the calculated ELUMOs with an experimental subset using a previously described method. B Application of the parametrisation to the library. While the N,N′-library spans a very local energy range, the 6,6′-library shows more pronounced changes in the ELUMO and thus its reduction potentials. The 6,6′-substitution pattern does not follow the same trend as the N,N′-substitution pattern regarding the ELUMO (see discussion in the text). Thus, we did not further analyse the 6,6′-substitution patterns with the method mentioned above, but we included the parametrised values only for a general overview.
A correlation between N,N′-substitution pattern and the ELUMO is observed. The N,N′-substituted iso-I derivatives show more stable LUMOs (lower energy relative to the vacuum level) with electron-withdrawing groups and more destabilised ones by introducing electron-donating groups. Contrary to the considerations taken concerning the TDMs and the exciton binding energy, we do not observe a significant difference from inductive or mesomeric effects. Applying Koopmans’ theorem, these findings were used to obtain an accurate prediction of the reduction potentials. Electron-withdrawing groups increase (iso-I-36 E1/2(param./exp.) = − 0.89 V/− 0.90 V), while weakly electron-donating groups decrease the reductions potential (iso-I-47 E1/2(param./exp.) = − 1.20 V/− 1.18 V). While the electron-donating bis-anisole substituted iso-I-25 shows a higher reduction potential (E1/2(param.) = − 1.12 V) and thus seems to oppose the trend, the push–pull configurations support the findings, as their reduction potentials range is in between the pull–pull and push–push configurations (e.g. E1/2(param.) = − 1.02 V, − 1.06 V and − 1.06 V for iso-I-12, iso-I-19 and iso-I-28).
As the reduction potential is directly linked to the open-circuit voltage of photovoltaics [41], our method could aid the design of enhanced photovoltaic materials.
Conclusion
In this work, we conducted a detailed investigation into the substitution effects on the N,N′-site of iso-I, providing an alternative for optical and electronic tuning to the already well-studied 6,6′-site. To achieve a comprehensive study of a broad range of substituents, a combination of computational and experimental methods on a library of molecules was applied and a subset of these molecules was synthesised to allow for parametrisation.
TD-PBE0/cc-pVDZ emerged as the best-suited method to simulate the experimental properties of isoindigo, reaching a good agreement between the simulated and calculated ones. Based on the computational data, we discovered trends relating the optical properties to the substitution patterns. Notably, derivatives iso-I-12 and 36 showed the highest λmax and ε(λmax), respectively. The otherwise typical dark S2 state could be turned into a bright one by asymmetric substitution with one Boc and one other substituent tested.
The analysis of the excitation behaviour by TDMs elucidated the difference between symmetric and asymmetric hole distributions and inductive and resonance contributions to the hole-density. In mesomeric push–pull TDMs we observed a significant contribution of anisole moieties to the hole-density, in contrast to related 6,6′-iso-Is.
The analysis of the exciton binding energy showed that strongly polarised derivatives have lower exciton binding energies, with iso-I-12 having the lowest exciton Coulomb interaction of 3.87 eV, and the unpolarised iso-I-36 showing the highest (4.82 eV). We discovered a relation in the N,N′-library between the band gap and the exciton binding energy. The lower the band gap, the lower the Coulomb attraction between the electron and the hole. However, this trend does not hold for the 6,6′-substitution patterns.
Finally, a parametrization method for ELUMO was employed to obtain accurate predictions for the reduction potentials of the whole library. We can conclude from the observed trend in ELUMO that electron-withdrawing (Boc) groups give rise to low ELUMOs, while the propyl substituted iso-Is have the highest ELUMOs of the N,N′-library.
The method here reported allows to obtain the reduction potentials for a library of 47 derivatives, utilising a limited subset of experimental data in combination with computational available data. We could enhance the prediction by discovering trends [51] towards the accurate calculation of electrochemical properties. Furthermore, our data did elucidate a relationship between electron-donating and -withdrawing groups to decrease and increase the reduction potential in the N,N′-substitution patterns. This approach shows potential to be useful for rational design and library screening of similar compounds.
In conclusion, we successfully elucidated several N,N′-substitution effects in isoindigo, which shows potential for more extensive use in material sciences. Taking iso-I-27 as an example, the three consecutive CT states, in combination with one of the lowest exciton binding energies of 4.18 eV, and a low band gap could enable broad spectral operating OPV-materials. We expect that this study will foster more detailed research on the electronic structure of substituted isoindigos. Additionally, we see major potential in the use of N,N′-substitution for optical and electronic tuning in cases where the 6,6′-site is already occupied [52].
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1Supporting Information containing synthetic procedures, characterisation of novel compounds, a full set of spectroscopic and electrochemical studies, details regarding DFT calculations is available free of charge (DOCX 44221 KB)
Acknowledgements
We thank Mr. Emmanuel P. R. Vrionakis (University of Groningen) for his assistance regarding the synthesis of compounds 46 and 47. We gratefully acknowledge the generous support from the Horizon 2020 Framework Program (ERC Advanced Investigator Grant no. 694345 to BLF), the Marie Skłodowska-Curie Actions (Individual Fellowship 838280 to SC), the Alexander-von-Humboldt Foundation (Feodor Lynen Fellowship to NAS) and the Ministry of Education, Culture and Science of the Netherlands (Gravitation Program No. 024.001.035 to BLF). We would like to thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high-performance computing cluster.
Declarations
Conflict of interest
There are no conflicts of interest to declare.
Contributor Information
Ben L. Feringa, Email: b.l.feringa@rug.nl
Stefano Crespi, Email: s.crespi@rug.nl.
References
- 1.Laurent Ueber einige neue Verbindungen aus der Reihe des Indigo. Journal für Praktische Chemie. 1842;26(1):123–125. doi: 10.1002/prac.18420260125. [DOI] [Google Scholar]
- 2.Sander L. Über “Indin”, Isoindigo, “Hydrindin” und das innere Anhydrid der α, β-Bis-[o- amino-phenyl]-äpfelsäure. Berichte der deutschen chemischen Gesellschaft (A and B Series) 1925;58(5):820–824. doi: 10.1002/cber.19250580506. [DOI] [Google Scholar]
- 3.Wee XK, Yeo WK, Zhang B, Tan VBC, Lim KM, Tay TE, Go ML. Synthesis and evaluation of functionalized isoindigos as antiproliferative agents. Bioorganic and Medicinal Chemistry. 2009;17(21):7562–7571. doi: 10.1016/j.bmc.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Bogdanov AV, Musin LI, Mironov VF. Advances in the synthesis and application of isoindigo derivatives. ARKIVOC. 2015;2015(6):362–392. doi: 10.3998/ark.5550190.p009.090. [DOI] [Google Scholar]
- 5.Zhao P, Li Y, Gao G, Wang S, Yan Y, Zhan X, Liu Z, Mao Z, Chen S, Wang L. Design, synthesis and biological evaluation of N-alkyl or aryl substituted isoindigo derivatives as potential dual cyclin-dependent kinase 2 (CDK2)/glycogen synthase kinase 3β (GSK-3β) phosphorylation inhibitors. European Journal of Medicinal Chemistry. 2014;86:165–174. doi: 10.1016/j.ejmech.2014.08.049. [DOI] [PubMed] [Google Scholar]
- 6.Friedländer P. Ueber schwefelhaltige Analoga der Indigogruppe. Berichte der deutschen chemischen Gesellschaft. 1906;39(1):1060–1066. doi: 10.1002/cber.190603901167. [DOI] [Google Scholar]
- 7.Steingruber E. Ullmann’s encyclopedia of industrial chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; 2000. Indigo and Indigo colorants. [Google Scholar]
- 8.Hosseinnezhad M, Moradian S, Gharanjig K. Novel organic dyes based on thioindigo for dye-sensitized solar cells. Dyes and Pigments. 2015;123:147–153. doi: 10.1016/j.dyepig.2015.07.016. [DOI] [Google Scholar]
- 9.Rajan AK, Cindrella L. Studies on new natural dye sensitizers from Indigofera tinctoria in dye-sensitized solar cells. Optical Materials. 2019;88:39–47. doi: 10.1016/j.optmat.2018.11.016. [DOI] [Google Scholar]
- 10.Petermayer C, Thumser S, Kink F, Mayer P, Dube H. Hemiindigo: Highly bistable photoswitching at the biooptical window. Journal of the American Chemical Society. 2017;139(42):15060–15067. doi: 10.1021/jacs.7b07531. [DOI] [PubMed] [Google Scholar]
- 11.Gerwien A, Mayer P, Dube H. Green light powered molecular state motor enabling eight-shaped unidirectional rotation. Nature Communications. 2019;10(1):1–7. doi: 10.1038/s41467-019-12463-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerwien A, Reinhardt T, Mayer P, Dube H. Synthesis of double-bond-substituted hemithioindigo photoswitches. Organic Letters. 2018;20(1):232–235. doi: 10.1021/acs.orglett.7b03574. [DOI] [PubMed] [Google Scholar]
- 13.Sailer A, Ermer F, Kraus Y, Lutter FH, Donau C, Bremerich M, Ahlfeld J, Thorn-Seshold O. Hemithioindigos for cellular photopharmacology: Desymmetrised molecular switch scaffolds enabling design control over the isomer-dependency of potent antimitotic bioactivity. ChemBioChem. 2019;20(10):1305–1314. doi: 10.1002/cbic.201800752. [DOI] [PubMed] [Google Scholar]
- 14.Wiedbrauk S, Dube H. Hemithioindigo—An emerging photoswitch. Tetrahedron Letters. 2015;56(29):4266–4274. doi: 10.1016/j.tetlet.2015.05.022. [DOI] [Google Scholar]
- 15.Luňák S, Horáková P, Lyčka A. Absorption and fluorescence of arylmethylidenoxindoles and isoindigo. Dyes and Pigments. 2010;85(3):171–176. doi: 10.1016/j.dyepig.2009.11.001. [DOI] [Google Scholar]
- 16.Huang J, Chen Z, Mao Z, Gao D, Wei C, Lin Z, Li H, Wang L, Zhang W, Yu G. Tuning frontier orbital energetics of azaisoindigo-based polymeric semiconductors to enhance the charge-transport properties. Advanced Electronic Materials. 2017;3(11):1700078. doi: 10.1002/aelm.201700078. [DOI] [Google Scholar]
- 17.Stalder R, Mei J, Reynolds JR. Isoindigo-based donor–acceptor conjugated polymers. Macromolecules. 2010;43(20):8348–8352. doi: 10.1021/ma1018445. [DOI] [Google Scholar]
- 18.Zhang G, Fu Y, Xie Z, Zhang Q. Synthesis and photovoltaic properties of new low bandgap isoindigo-based conjugated polymers. Macromolecules. 2011;44(6):1414–1420. doi: 10.1021/ma102357b. [DOI] [Google Scholar]
- 19.Randell NM, Kelly TL. Recent advances in isoindigo-inspired organic semiconductors. The Chemical Record. 2019;19(6):973–988. doi: 10.1002/tcr.201800135. [DOI] [PubMed] [Google Scholar]
- 20.Munshi MU, Martens J, Berden G, Oomens J. Protoisomerization of indigo and isoindigo dyes confirmed by gas-phase infrared ion spectroscopy. Journal of Physical Chemistry A. 2019;123(38):8226–8233. doi: 10.1021/acs.jpca.9b06858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Bai S, Wu Y, Liu Y, Yao J, Fu H. Revealing the nature of singlet fission under the veil of internal conversion. Angewandte Chemie International Edition. 2020;59(5):2003–2007. doi: 10.1002/anie.201912202. [DOI] [PubMed] [Google Scholar]
- 22.Wang D, Ying W, Zhang X, Hu Y, Wu W, Hua J. Near-infrared absorbing isoindigo sensitizers: Synthesis and performance for dye-sensitized solar cells. Dyes and Pigments. 2015;112:327–334. doi: 10.1016/j.dyepig.2014.07.017. [DOI] [Google Scholar]
- 23.Perpète EA, Preat J, André JM, Jacquemin D. An ab initio study of the absorption spectra of indirubin, isoindigo, and related derivatives. Journal of Physical Chemistry A. 2006;110(17):5629–5635. doi: 10.1021/jp060069e. [DOI] [PubMed] [Google Scholar]
- 24.Estrada LA, Stalder R, Abboud KA, Risko C, Brédas JL, Reynolds JR. Understanding the electronic structure of isoindigo in conjugated systems: A combined theoretical and experimental approach. Macromolecules. 2013;46(22):8832–8844. doi: 10.1021/ma4013829. [DOI] [Google Scholar]
- 25.Rout Y, Chauhan V, Misra R. Synthesis and characterization of isoindigo-based push–pull chromophores. Journal of Organic Chemistry. 2020;85(7):4611–4618. doi: 10.1021/acs.joc.9b03267. [DOI] [PubMed] [Google Scholar]
- 26.Cavasotto CN, Aucar MG, Adler NS. Computational chemistry in drug lead discovery and design. International Journal of Quantum Chemistry. 2019;119(2):e25678. doi: 10.1002/qua.25678. [DOI] [Google Scholar]
- 27.Chen L-Q, Chen L-D, Kalinin SV, Klimeck G, Kumar SK, Neugebauer J, Terasaki I. Design and discovery of materials guided by theory and computation. NPJ Computational Materials. 2015;1(1):15007. doi: 10.1038/npjcompumats.2015.7. [DOI] [Google Scholar]
- 28.Geerlings P, De Proft F. Conceptual DFT: The chemical relevance of higher response functions. Physical Chemistry Chemical Physics. 2008;10(21):3028. doi: 10.1039/b717671f. [DOI] [PubMed] [Google Scholar]
- 29.Mai S, Atkins AJ, Plasser F, González L. The influence of the electronic structure method on intersystem crossing dynamics. The case of thioformaldehyde. Journal of Chemical Theory and Computation. 2019;15(6):3470–3480. doi: 10.1021/acs.jctc.9b00282. [DOI] [PubMed] [Google Scholar]
- 30.Laurent AD, Jacquemin D. TD-DFT benchmarks: A review. International Journal of Quantum Chemistry. 2013;113(17):2019–2039. doi: 10.1002/qua.24438. [DOI] [Google Scholar]
- 31.Leang SS, Zahariev F, Gordon MS. Benchmarking the performance of time-dependent density functional methods. Journal of Chemical Physics. 2012;136(10):104101. doi: 10.1063/1.3689445. [DOI] [PubMed] [Google Scholar]
- 32.Ohto T, Dodia M, Xu J, Imoto S, Tang F, Zysk F, Kühne TD, Shigeta Y, Bonn M, Wu X, Nagata Y. Accessing the accuracy of density functional theory through structure and dynamics of the water-air interface. Journal of Physical Chemistry Letters. 2019;10(17):4914–4919. doi: 10.1021/acs.jpclett.9b01983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perdew, J. P. (2003). Jacob’s ladder of density functional approximations for the exchange-correlation energy. In AIP Conference Proceedings (vol. 577, pp. 1–20). AIP Publishing. 10.1063/1.1390175
- 34.Marini A, Muñoz-Losa A, Biancardi A, Mennucci B. What is solvatochromism? The Journal of Physical Chemistry B. 2010;114(51):17128–17135. doi: 10.1021/jp1097487. [DOI] [PubMed] [Google Scholar]
- 35.Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. The Journal of Physical Chemistry B. 2009;113(18):6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 36.Adamo C, Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. Journal of Chemical Physics. 1999;110(13):6158–6170. doi: 10.1063/1.478522. [DOI] [Google Scholar]
- 37.Dunning TH. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. The Journal of Chemical Physics. 1989;90(2):1007–1023. doi: 10.1063/1.456153. [DOI] [Google Scholar]
- 38.Demoulin B, El-Tahawy MMT, Nenov A, Garavelli M, Le Bahers T. Intramolecular photo-induced charge transfer in visual retinal chromophore mimics: Electron density-based indices at the TD-DFT and post-HF levels. Theoretical Chemistry Accounts. 2016;135(4):1–10. doi: 10.1007/s00214-016-1815-y. [DOI] [Google Scholar]
- 39.Li Y, Ullrich CA. Time-dependent transition density matrix. Chemical Physics. 2011;391(1):157–163. doi: 10.1016/j.chemphys.2011.02.001. [DOI] [Google Scholar]
- 40.Sun H, Liu D, Wang T, Li P, Bridgmohan CN, Li W, Lu T, Hu W, Wang L, Zhou X. Charge-separated sensitizers with enhanced intramolecular charge transfer for dye-sensitized solar cells: Insight from structure-performance relationship. Organic Electronics. 2018;61:35–45. doi: 10.1016/j.orgel.2018.06.045. [DOI] [Google Scholar]
- 41.Li H-W, Guan Z, Cheng Y, Lui T, Yang Q, Lee C-S, Chen S, Tsang S-W. On the study of exciton binding energy with direct charge generation in photovoltaic polymers. Advanced Electronic Materials. 2016;2(11):1600200. doi: 10.1002/aelm.201600200. [DOI] [Google Scholar]
- 42.Lu T, Chen F. Multiwfn: A multifunctional wavefunction analyzer. Journal of Computational Chemistry. 2012;33(5):580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 43.Mu X, Wang J, Sun M. Visualization of photoinduced charge transfer and electron-hole coherence in two-photon absorption. Journal of Physical Chemistry C. 2019;123(23):14132–14143. doi: 10.1021/acs.jpcc.9b00700. [DOI] [Google Scholar]
- 44.Dawei Y, Xiaojuan Z, Zhiming W, Bing Y, Yuguang M, Yuyu P. Theoretical investigation of the effects of various substituents on the large energy gap between triplet excited-states of anthracene. RSC Advances. 2018;8(49):27979–27987. doi: 10.1039/c8ra04264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koopmans T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica. 1934;1(1–6):104–113. doi: 10.1016/S0031-8914(34)90011-2. [DOI] [Google Scholar]
- 46.Zhan CG, Nichols JA, Dixon DA. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: Molecular properties from density functional theory orbital energies. Journal of Physical Chemistry A. 2003;107(20):4184–4195. doi: 10.1021/jp0225774. [DOI] [Google Scholar]
- 47.Cardona CM, Li W, Kaifer AE, Stockdale D, Bazan GC. Electrochemical considerations for determining absolute frontier orbital energy levels of conjugated polymers for solar cell applications. Advanced Materials. 2011;23(20):2367–2371. doi: 10.1002/adma.201004554. [DOI] [PubMed] [Google Scholar]
- 48.Pavlishchuk VV, Addison AW. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorganica Chimica Acta. 2000;298(1):97–102. doi: 10.1016/S0020-1693(99)00407-7. [DOI] [Google Scholar]
- 49.Albuquerque LS, Arias JJR, Santos BP, Marques MDFV, Monteiro SN. Synthesis and characterization of novel conjugated copolymers for application in third generation photovoltaic solar cells. Journal of Materials Research and Technology. 2020;9(4):7975–7988. doi: 10.1016/j.jmrt.2020.05.009. [DOI] [Google Scholar]
- 50.Leonat L, Sbârcea G, Brânzoi IV. Cyclic voltammetry for energy levels estimation of organic materials. UPB Scientific Bulletin Series B. 2013;75:112–118. [Google Scholar]
- 51.Karakawa M, Nagai T, Adachi K, Ie Y, Aso Y. Precise control over reduction potential of fulleropyrrolidines for organic photovoltaic materials. RSC Advances. 2017;7(12):7122–7129. doi: 10.1039/c6ra27661j. [DOI] [Google Scholar]
- 52.Liesfeld P, Garmshausen Y, Budzak S, Becker J, Dallmann A, Jacquemin D, Hecht S. Highly cooperative photoswitching in dihydropyrene dimers. Angewandte Chemie International Edition. 2020;59(43):19352–19358. doi: 10.1002/anie.202008523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1Supporting Information containing synthetic procedures, characterisation of novel compounds, a full set of spectroscopic and electrochemical studies, details regarding DFT calculations is available free of charge (DOCX 44221 KB)