

Abstract
Background:
Renal ischemia-reperfusion injury (IRI) causes acute kidney injury as well as liver injury. Renal IRI depletes hepatic antioxidants, promotes hepatic inflammation and dysfunction through Tlr9 upregulation. There is no treatment available for liver injury during renal IRI. This study examines the hepatoprotective role of treprostinil, a prostacyclin analog, during renal IRI.
Methods:
Male Sprague-Dawley rats were divided into four groups: control, sham, IRI-placebo, or IRI-treprostinil and subjected to bilateral ischemia (45 minutes) followed by reperfusion (1–72 hours). Placebo or treprostinil (100 ng/kg/min) was administered subcutaneously via an osmotic minipump.
Results:
Treprostinil significantly reduced peak serum creatinine, BUN, alanine aminotransferase, and aspartate aminotransferase levels vs. IRI-placebo. Treprostinil also restored hepatic levels of superoxide dismutase, glutathione, catalase, and Gclc expression to baseline, while reducing lipid peroxidation vs. IRI-placebo. Additionally, treprostinil significantly reduced elevated hepatic Tlr9, Il-1β, Ccl2, Vcam1, and Serpine1 mRNA expression. Renal IRI increased hepatic apoptosis which was inhibited by treprostinil through reduced cytochrome c and cleaved caspase-3 protein expression. Treprostinil enhanced hepatic ATP concentrations and mitochondrial DNA copy number and improved mitochondrial dynamics by restoring Pgc-1α expression and significantly upregulating Mfn1, Mfn2, and Sirt3 levels, while reducing Drp-1 protein vs. IRI-placebo. Non-targeted semi-quantitative proteomics showed improved oxidative stress indices and ATP subunits in the IRI-treprostinil group.
Conclusions:
Treprostinil improved hepatic function and antioxidant levels, while suppressing the inflammatory response and alleviating Tlr9-mediated apoptotic injury during renal IRI. Our study provides evidence of treprostinil’s hepatoprotective effect, which supports the therapeutic potential of treprostinil in reducing hepatic injury during renal IRI.
Keywords: liver injury, prostacyclin, inflammation, oxidative stress, apoptosis
Graphical Abstract
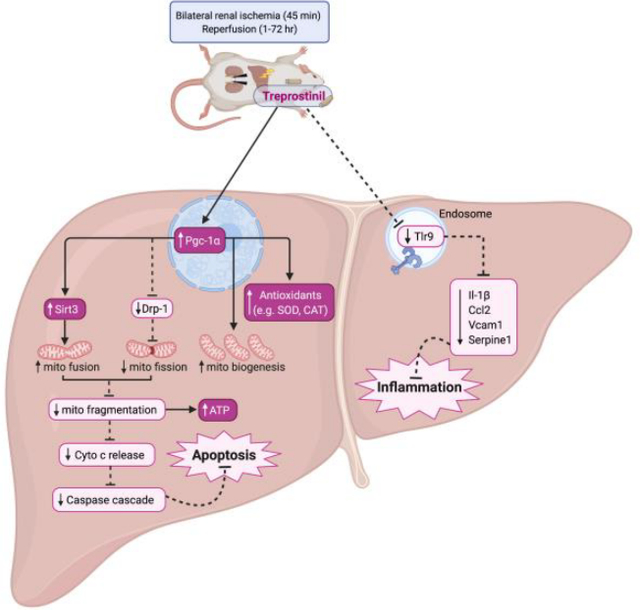
1. Introduction
Renal ischemia-reperfusion injury (IRI) is a major cause of morbidity and mortality in several clinical settings, and it is unavoidable during kidney transplantation [1]. Renal IRI leads to acute kidney injury (AKI) [1], often requiring subsequent retransplantation which further depletes the scarce donor pool. In addition to kidney injury, vital organs, particularly the liver [2], are also damaged as a complication of renal IRI. Consequences of renal IRI-induced hepatic injury include multi-organ injury and increased mortality due to complications of AKI and liver dysfunction [3].
The liver contains an abundance of antioxidants and renal IRI decreases hepatic antioxidant levels, particularly superoxide dismutase (SOD) [4], glutathione (GSH) [5], and catalase (CAT) [6]. Oxidative stress is one of the major mechanisms that contributes to renal IRI-induced hepatic injury [5]. In fact, a weakened antioxidant defense system causes mitochondrial dysfunction through uncontrolled reactive oxygen species (ROS) [7]. Mitochondrial dysfunction increases mitochondrial fission, depletes adenosine triphosphate (ATP), and increases the mitochondrial membrane permeability [7]. These events trigger the release of cytochrome c and subsequent activation of caspases, resulting in mitochondrial-mediated apoptosis [8]. During renal IRI, activation of pro-apoptotic proteins, e.g., caspase-3, contribute to liver cell death [9].
The network of mitochondrial proteins that regulate mitochondrial DNA (mtDNA) maintenance include fusion proteins, e.g., Mitofusin (Mfn)1, Mfn2, and Pgc-1α, along with the fission protein, e.g., dynamin-related protein-1 (Drp-1) [10]. Drp-1 promotes mitochondrial fragmentation, causing mtDNA leakage and cytosolic mtDNA stress [11]. During renal IRI, mitochondrial fusion proteins are downregulated, while Drp-1 is upregulated, resulting in disrupted mitochondrial dynamics [12].
Renal IRI-induced hepatic injury also leads to hepatic inflammation [13]. In particular, toll-like receptor (TLR)-9, which is located intracellularly in endosomes, recognizes pathogen- or damage-associated molecular patterns (DAMP), and triggers the inflammatory response [14]. During renal IRI, hepatic Tlr9 is upregulated by released mtDNA [15], a type of DAMP, which leads to the production of proinflammatory cytokines, including interleukins (IL), e.g. IL-1β [16]. The interaction between mtDNA and Tlr9 can activate neutrophil secretion and contribute to hepatic inflammation and subsequent hepatic injury [17].
A therapy to reduce renal IRI is needed to reduce hepatic oxidative stress and mitochondrial-mediated apoptosis, thereby, reduce the inflammatory response and liver cell death. Prostacyclin (PGI2) is an endogenous compound with vasodilatory, anti-coagulant, and anti-inflammatory effects [18]. Treprostinil (Remodulin®) is an FDA-approved PGI2 analog with potent vasodilatory and anti-platelet aggregation properties [19]. Importantly, treprostinil alleviates IRI during rat AKI [20] and orthotopic liver transplantation [21]. This study investigates the hepatoprotective effects of treprostinil during renal IRI.
2. Materials and methods
2.1. IRI animal model
Adult male Sprague Dawley rats weighing 200–250 gm (Charles River Laboratories, Wilmington, MA) were housed in a laminar-flow, specific pathogen-free atmosphere, and allowed a standard diet and water ad libitum (Rhode Island Hospital (RIH), Providence, RI). Animals were randomly divided into four groups: control, sham, IRI-placebo, and IRI-treprostinil. Briefly, animals were anesthetized with isoflurane and subjected to bilateral renal ischemia (45 minutes), followed by clamp removal and reperfusion (1–72 hours). Control animals were not subjected to any surgical manipulation; sham animals had incisions only to account for any influence from basic surgical handling or anesthesia and to serve as the baseline. All surgical procedures were performed by the same surgeon who was blinded to treatment. All procedures involving animals were performed with approval from RIH Institutional Animal Care and Use Committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
2.2. Animal treatment
Treprostinil (Remodulin®) or placebo (sodium chloride, metacresol, sodium citrate, water for injection) manufactured by United Therapeutics, Corp. (Durham, NC, USA) were administrated subcutaneously (100 ng/kg/min) via osmotic minipumps (Alzet Inc., Cupertino, CA) and implanted at 18–24 hours before renal IRI to ensure steady-state concentrations at the time of renal IRI. Post-reperfusion, animals were kept under a heating lamp for 2 hours and were given regular food and water ad libitum. The general condition of the rats was checked three times daily and animals were euthanized by inhalation of carbon dioxide followed by cervical dislocation if needed. Blood and liver tissue were collected at 1–72 hours post-reperfusion. Liver tissue was formalin-fixed or snap-frozen and stored at −80 °C for analysis.
2.3. Biochemical analysis
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity levels were determined using the Infinity ALT and AST assays (Thermo Fisher Scientific, Waltham, MA). Serum creatinine (SCr) and blood urea nitrogen (BUN) levels were measured by biochemical assays (Bioassay Systems, Hayward, CA), n = 3–10/group. The hepatic activity or concentration of SOD, GSH, and CAT were determined using biochemical assays (Cayman Chemical, Ann Arbor, MI). Tissue samples were homogenized, and protein quantification was calculated using bovine serum albumin as a standard. Hepatic lipid peroxidation was measured by the malondialdehyde (MDA) concentration in liver homogenates using a fluorometric thiobarbituric acid reactive substance (TBARS) assay (BioAssay Systems) per manufacturer’s instructions (n = 3–6/group). Fluorescence (λex/em 530/550 nm) was measured using a SpectraMax iD3 microplate reader (Molecular Devices, San Jose, CA).
2.4. Histological assessment of liver tissue
Liver tissue was harvested, fixed in 10% formalin, and embedded in paraffin. Sections were cut at 4 μm and stained with hematoxylin and eosin (H&E) then evaluated by a pathologist under light microscopy in a blinded fashion.
2.5. TUNEL assay
Paraffin-embedded liver tissue sections were used to perform the Click-iT™ Plus TUNEL Assay (Alexa Fluor™ 647 dye, Invitrogen, Carlsbad, CA). Nuclei were counterstained with Hoechst dye according to manufacturer’s instruction (Invitrogen). Liver sections were viewed under a Nikon Eclipse Ti2 inverted confocal microscope. Random fields (n = 3–5/section) with the same acquisition setting were imaged per slide (n = 3–4/group). Hepatic DNA fragmentation was semi-quantitated based on the intensity ratio of TUNEL:Hoechst staining using Image J (×200, scale bar = 100 μm).
2.6. Cytosolic and mitochondrial fractionations
Snap-frozen liver tissue was homogenized in 0.25 M sucrose buffer containing 0.1 mM EDTA, 10 mM HEPES, and protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific) using a Dounce homogenizer. The supernatant was centrifuged at 13,000 × g for 15 minutes and collected as the cytosolic fraction. The crude mitochondrial pellet was washed twice and resuspended in PBS containing protease and phosphatase inhibitor and 0.1% Triton X-100, then disrupted twice with a sonicator at 40% of maximum setting for 10 seconds. Following lysate centrifugation, the supernatant was collected as the mitochondrial fraction.
2.7. Liver microsome isolation
Liver microsomes were isolated from snap-frozen liver tissue using a differential centrifugation procedure as described by XenoTech (Sekisui XenoTech, Kansas City, KS). Briefly, tissue was homogenized in a buffer containing 0.25 M sucrose, 50 mM Tris-HCl (pH 7.4), 2 mM EDTA, and 150 mM potassium chloride, using a Bead Ruptor 24 (Omni International, Kennesaw, GA), then centrifuged at 10,000 × g for 20 minutes (4 °C). The supernatant was centrifuged at 100,000 × g for 65 minutes (4 °C), and the microsomal pellet was resuspended in a medium consisting of 0.25 M sucrose using a Dounce homogenizer.
2.8. Western blot analysis
Total, cytosolic, mitochondrial, and microsomal liver protein fractions were separated by electrophoresis using polyacrylamide gels and transferred to PVDF membranes. Membranes were incubated overnight (4 °C) with antibodies for caspase-3 (Cat# 14220, Cell Signaling Technology, Danvers, MA), Drp-1 (Cat# 8570), cytochrome c (Cat# sc-13156, Santa Cruz Biotechnology, Dallas, TX), Sirt3 (Cat# sc-365175), Tlr9 (Cat# sc-52966), β-actin (Cat# sc-47778), Gapdh (Cat# sc-32233) or Cox-IV (Cat# sc-517553), followed by IRDye® donkey anti-rabbit or goat anti-mouse IgG (H+L) 680 RD and goat anti-mouse IRDye® 800 CW (LI-COR Biosciences, Lincoln, NE) at room temperature for 1 hour. Blots were imaged using LI-COR Odyssey® Clx scanner. Protein band density was determined using Image J. Bands were normalized to β-actin (whole lysate), Gapdh (cytosol, microsomal), or Cox-IV (mitochondria) protein levels, and expressed as fold-change vs. sham (n = 3–5/group).
2.9. Hepatic ATP concentrations
Snap-frozen liver tissue was homogenized in ice-cold 2% trichloroacetic acid. The supernatants were collected and neutralized with Tris-acetate (100 mM) and EDTA (2 mM, pH, 7.8), n = 4–6/group. ATP concentrations were determined using the Enliten ATP Assay System Bioluminescence Detection Kit (Promega, Madison, WI).
2.10. Hepatic mtDNA copy number
DNA was extracted from snap-frozen liver tissue (n=4–5/group) using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). DNA concentration was determined using a Nanodrop OneC (Thermo Fisher Scientific). Mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) content were determined by mitochondrial encoded NADH-ubiquinone oxidoreductase chain 1 (mt-Nd1) and nuclear DNA (18S) using Taqman® probes (Applied Biosytems, Foster City, CA), respectively. Quantitative real-time PCR was performed using a ViiA 7 Real-Time PCR System (Life Technologies, Carlsbad, CA). The hepatic mtDNA copy number was calculated using the ratio between mitochondrial and nuclear DNA (mtDNA/nDNA) [22].
2.11. Quantitative real-time PCR analysis (qPCR)
RNA was extracted from snap-frozen liver sections using TRIzol™ (Invitrogen) according to manufacturer’s protocol. The purity and concentration of RNA were measured at 260/280 nm by Nanodrop (Thermo Fisher Scientific). Two micrograms of total RNA were used to generate cDNA using SuperScript™ IV First-Strand Synthesis System kit (Invitrogen). Hepatic mRNA levels of Gclc, Tlr9, Il-1β, Ccl2, Vcam1, Serpine1, Mfn1, Mfn2, Pgc-1α, and 18S were analyzed using Taqman® probes (Applied Biosystems). Relative mRNA expression was calculated using the ΔΔCt method and normalized to 18S expression (n = 4–5/group). Real-time qPCR was performed using a ViiA 7 Real-Time PCR System (Life Technologies).
2.12. SWATH-MS proteomics analysis
The effects of liver injury during renal IRI ± treprostinil treatment on the hepatic proteome was assessed using sequential window acquisition of all theoretical mass spectra (SWATH-LC/MS/MS). Snap-frozen liver tissue were weighed on ice, placed in bead-mill tubes, and homogenized in a urea lysis buffer containing 8 M urea, 50 mM triethylammonium bicarbonate, 10 mM dithiothreitol, and protease inhibitor cocktail (Thermo Fisher Scientific) using a Bead Ruptor 24 (Omni International), then centrifuged at 12,000 × g (4 °C) and the supernatant was collected. Protein concentration was measured using a Pierce™ BCA Protein Assay Kit. The samples were diluted to 500 μg/100 μl before digestion. Proteins were digested with TPCK-treated trypsin (AB SCIEX, Framingham, MA) at trypsin/protein ratio of 1:20 under high pressure cycles in a Barocycler NEP2320 (Pressure BioSciences, South Easton, MA). The reaction was stopped by the addition of water:acetonitrile (50:50) with 5% formic acid. The supernatant containing peptides was collected for mass spectrometry analysis [23]. The samples were analyzed using a SCIEX 5600 TripleTOF mass spectrometer coupled to an Acquity UHPLC HClass System (Waters Corp., Milford, MA) [23]. Afterwards, proteins were identified, and relative quantification was performed through SWATH-MS acquisition, followed by data-dependent analysis using Spectronaut™ software (Biognosys AG, Schlieren, Switzerland).
2.13. Statistical analysis
All data are represented as the mean ± standard error of the mean (SEM). One- or two-way ANOVA followed by Tukey-multiple comparisons test was used to determine the differences between groups. Statistical significance was set at P<0.05. GraphPad Prism 8.3.0 (GraphPad Software, Inc., La Jolla, CA) was used for statistical analyses and graphing.
3. Results
3.1. Treprostinil improves renal and hepatic function
Renal IRI was demonstrated by measuring SCr and BUN levels, two classical markers of kidney injury. In IRI-placebo animals, SCr and BUN levels were significantly elevated and reached peak levels at 24-hr post-reperfusion vs. sham (1.8 ± 0.3 mg/dL vs. 0.6 ± 0.06 mg/dL and 151 ± 20.4 mg/dL vs. 32.5 ± 1.7 mg/dL, respectively, P<0.001). Treatment with treprostinil significantly reduced peak SCr (0.6 ± 0.06 mg/dL) and BUN (74 ± 12.8 mg/dL) vs. IRI-placebo (P<0.001), Figure 1A, 1B. Renal IRI-induced hepatic injury was evaluated by measuring serum ALT and AST levels, which are biomarkers for liver dysfunction. Serum ALT and AST activity were significantly elevated in IRI-placebo animals and reached peak levels at 6-hr post-reperfusion vs. sham (110 ± 6.9 U/L vs. 29 ± 1.6 U/L and 32 ± 3.6 U/L vs. 19 ± 0.6 U/L, respectively, P<0.001). In contrast, treatment with treprostinil significantly reduced ALT and AST activity at 6-hr post-reperfusion vs. placebo (33 ± 6.6 U/L and 20 ± 1.8 U/L, respectively, P<0.001), Figure 1C, 1D. These results demonstrate that treprostinil improves liver function during renal IRI.
Figure 1.

Treprostinil reduces kidney and liver dysfunction. (A) SCr, (B) BUN, (C) ALT, and (D) AST measured at pre-IRI (baseline) 1–72 hours post-reperfusion in control-, sham-, placebo-, and treprostinil-treated animals. Data are represented as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 vs. sham; ϕP<0.05, ϕϕP<0.01, ϕϕϕP<0.001 vs. IRI-placebo (n = 3–10/group). Two-way ANOVA, Tukey’s multiple comparisons test. (E) H&E (hematoxylin & eosin) staining of liver sections obtained from sham-operated, IRI-placebo or IRI-treprostinil rats at 6-hours post-reperfusion. Black circle and arrow indicate increased vacuolation in hepatocytes in IRI-placebo group. Data are representative of 4–5 sections per sample (n=4/group, ×400). SCr: Serum creatinine; BUN: Blood urea nitrogen; ALT: Alanine aminotransferase; AST: aspartate aminotransferase; CTRL: Control; PLB: IRI-placebo; TRE: IRI-treprostinil.
Livers from sham-operated, IRI-placebo and -treprostinil-treated animals were surgically removed, fixed in formalin, and embedded in paraffin. Sections stained with H&E were examined under light microscopy by a pathologist blinded to the treatment group from which each specimen was derived, Figure 1E. Light microscopic examination revealed focal coarse cytoplasmic vacuolation of hepatocytes, indicating acute cellular injury in livers from IRI-placebo animals. Injured hepatocytes were consistently localized specifically within peripheral areas of hepatic lobules, corresponding to hepatic acinus zone 1. In contrast, only minimal vacuolation was noted in hepatocytes from animals pretreated with treprostinil. No cytoplasmic vacuolation was observed in sham-operated animals.
3.2. Treprostinil alleviates hepatic oxidative stress and lipid peroxidation
We next studied the role of treprostinil in regulating hepatic oxidative stress during renal IRI. Hepatic SOD activity in IRI-placebo animals significantly decreased at 1-hr post-reperfusion vs. sham animals (29 ± 3.5 vs. 45 ± 3.8 U/mg protein, P<0.05), which treprostinil restored (44 ± 3.2 U/mg protein, P<0.05 vs. IRI-placebo), Figure 2A. Total hepatic GSH concentrations in IRI-placebo group significantly decreased at 6-hr post-reperfusion compared to sham (626 ± 123 vs. 1,057 ± 72 μM/mg protein, respectively, P<0.05). Treatment with treprostinil significantly improved total GSH content in the liver compared to placebo (1,130 ± 59 vs. 626 ± 123 μM/mg protein, P<0.01), Figure 2B. Similarly, hepatic CAT activity in IRI-placebo group significantly decreased compared to sham at 24-hr post-reperfusion (1,574 ± 126 vs. 2,195 ± 80 U/mg protein, P<0.01), which treprostinil also significantly improved (2,441 ± 116, P<0.001 vs. IRI-placebo), Figure 2C. Lipid peroxidation promotes the generation of toxic aldehydes, including MDA. The hepatic MDA concentration increased by 1.6-fold in the IRI-placebo group compared to control at 48-hr post-reperfusion (0.6 ± 0.07 vs. 0.36 ± 0.01 μM/mg protein, respectively, P=0.09). On the other hand, treprostinil significantly reduced hepatic MDA concentrations by 50% relative to IRI-placebo animals (0.3 ± 0.05 vs. 0.6 ± 0.07 μM/mg protein, P<0.05), which nearly restored levels to baseline, Figure 2D.
Figure 2.

Treprostinil alleviates hepatic oxidative stress levels. (A) SOD, (B) GSH, and (C) CAT activity levels at 1-, 6-, and 24-hours post-reperfusion; (D) Lipid peroxidation levels at 48-hours post-reperfusion; (E) Antioxidants at 24-hours post-reperfusion measured by SWATH-MS based proteomics normalized to Gapdh and expressed as a fold-change over control. (F) Gclc mRNA levels at 48-hours post-reperfusion normalized to 18S. Data represented are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 vs. control or sham; ϕP<0.05, ϕϕP<0.01, ϕϕϕP<0.001 vs. IRI-placebo (n = 3–6/group). One- or two-way ANOVA, Tukey’s multiple comparisons test. SOD: Superoxide dismutase; GSH: Glutathione; CAT: Catalase; SWATH-MS: Sequential windowed acquisition of all theoretical fragment ion mass spectra; MDA: Malondialdehyde; Gpx: Glutathione peroxidase; Gsta2: Glutathione S-transferase alpha 2; Gstm4: Glutathione S-transferase mu 4; Gclc: Glutamate-cysteine ligase catalytic subunit; CTRL: Control; PLB: IRI-placebo; TRE: IRI-treprostinil.
To further investigate the role of treprostinil in restoring hepatic antioxidants during renal IRI, SWATH-MS-based proteomic analysis was performed. Consistent with our biochemical data, hepatic antioxidants, e.g., Gpx1, Gpx3, Gsta2, Gstm4, Sod2, Cat, and Gclc, were significantly reduced in IRI-placebo at 24-hr post-reperfusion vs. control (P<0.05), all of which treprostinil upregulated, Figure 2E. In addition, hepatic Gclc mRNA expression decreased in the IRI-placebo group by 30% relative to sham at 48-hr post-reperfusion, whereas treatment with treprostinil upregulated Gclc expression by 2.0 ± 0.6-fold, relative to sham, Figure 2F. Taken together, these results demonstrate that renal IRI reduced hepatic antioxidant levels and that treprostinil alleviates hepatic oxidative stress induced by renal IRI.
3.3. Treprostinil reduces hepatic TLR9 and inflammation
Hepatic TLR9 is upregulated during severe renal IRI and TLR9 activation by mtDNA released from damaged mitochondria contributes to inflammation [15]. In this study, hepatic Tlr9 mRNA and protein expression increased in IRI-placebo animals by 30% and 37% (P<0.05) vs. sham, respectively, Figure 3A, 3B. A striking contrast was observed in the IRI-treprostinil group, where Tlr9 mRNA and protein expression were significantly reduced by 61% (P<0.05) and 70% (P<0.001) vs. IRI-placebo, respectively. These findings indicate that treprostinil inhibits Tlr9-mediated hepatic injury during renal IRI.
Figure 3.

Treprostinil reduces hepatic Tlr9 mRNA and protein expression. (A) Tlr9 at 48-hours post-reperfusion was measured by qPCR, normalized to 18S, and expressed as fold-change over sham. (B) Tlr9 at 48-hours post-reperfusion was measured by western blot normalized to Gapdh, and quantification was performed using Image J software; Data are represented as mean ± SEM. *P<0.05 vs. sham; ϕϕP<0.01, ϕϕϕP<0.001 vs. IRI-placebo (n = 3–5/group). One-way ANOVA, Tukey’s multiple comparisons test. Tlr9: Toll-like receptor 9; Gapdh: Glyceraldehyde 3-phosphate dehydrogenase; PLB: IRI-placebo; TRE: IRI-treprostinil.
Proinflammatory cytokines and adhesion molecules play an important role in the pathophysiology of liver injury during renal IRI [24]. In this study, Il-1β mRNA expression significantly increased in the IRI-placebo group at 6-hr post-reperfusion by 1.6 ± 0.5-fold relative to sham (P<0.05), Figure 4A. Pre-treatment with treprostinil decreased Il-1β expression by 22% compared to placebo. Ccl2 expression significantly increased in the IRI-placebo group 24-hr post-reperfusion by 3.4 ± 1.5-fold relative to sham (P<0.05), Figure 4B. Subsequently, treprostinil significantly reduced Ccl2 expression by 76% compared to placebo (P<0.05). Vcam1 and Serpin E1 recruit neutrophils to injured tissue to further promote inflammation [25, 26]. Hepatic mRNA Vcam1 levels increased in the IRI-placebo group by 1.4 ± 0.2-fold relative to sham (P=0.06) at 24-hr post-reperfusion, which pretreatment with treprostinil significantly reduced by 30% vs. IRI-placebo (P<0.05) which restored levels to baseline, Figure 4C. In IRI-placebo, Serpine1 mRNA expression was significantly increased by 2.9 ± 1.4-fold relative to sham (P<0.05), which treprostinil reduced by 54% vs. placebo (P<0.05), Figure 4D. These results support the anti-inflammatory role of treprostinil during renal IRI and the reduction in hepatic Tlr9 expression suggests that hepatic injury decreases by suppressing the inflammatory response.
Figure 4.

Treprostinil reduces hepatic inflammation. (A) Il-1β at 6-hours post-reperfusion, (B) Ccl2 at 6-hours post-reperfusion, (C) Vcam1 at 24-hours post-reperfusion, and (D) Serpine1 at 24-hours post-reperfusion were measured by qPCR, normalized to 18S, and expressed as a fold-increase over sham. Data are represented as mean ± SEM. *P<0.05 vs. sham; ϕP<0.05 vs. IRI-placebo (n = 4–5/group). One-way ANOVA, Tukey’s multiple comparisons test. Il-1β: Interleukin-1 beta; Ccl2: Chemokine ligand 2: Vcam1: Vascular cell adhesion molecule 1; Serpine1: Serine protease inhibitor, family E, member 1; PLB: IRI-placebo; TRE: IRI-treprostinil.
3.4. Treprostinil inhibits hepatic apoptosis
Liver injury due to renal IRI causes mitochondrial dysfunction which leads to hepatic apoptosis [27]. Hepatocyte apoptosis was evaluated by TUNEL staining, an indicator of DNA fragmentation. In the IRI-placebo group, TUNEL-positive cells were significantly increased by 40% vs. sham (P<0.05) at 6-hr post-reperfusion. In contrast, treprostinil significantly reduced DNA fragmentation to nearly that of sham, reflecting a 30% reduction in IRI-induced apoptosis vs. IRI-placebo (P<0.05), Figure 5A.
Figure 5.

Treprostinil inhibits hepatic apoptosis. (A) Hepatic apoptosis in situ, measured by TUNEL staining (red fluorescence) in rat liver sections at 6-hours post-reperfusion (×200, scale bar = 100 μm). Nuclei were counterstained with Hoechst (blue fluorescence). (B) Hepatic cytochrome c protein levels at 3-hours post-reperfusion measured by western blot, normalized to Gapdh; (C) Cleaved (activated) caspase-3 protein levels at 48-hours post-reperfusion measured by western blot, normalized to β-actin, and quantified using Image J software. Data are represented as mean ± SEM. *P<0.05, **P<0.01 vs. sham; ϕP<0.05, ϕϕP<0.01 vs. IRI-placebo (n = 3–5/group). One-way ANOVA, Tukey’s multiple comparisons test. TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling; Cyto c: cytochrome c; Gapdh: Glyceraldehyde 3-phosphate dehydrogenase; PLB: IRI-placebo; TRE: IRI-treprostinil.
Mitochondria fragmentation releases cytochrome c into the cytosol, which initiates caspase activation [8]. We found that the cytosolic protein expression of cytochrome c in the liver increased in the IRI-placebo group by 1.7 ± 0.2-fold vs. sham at 3-hr post-reperfusion, which treprostinil decreased by 24% compared to IRI-placebo, Figure 5B. Subsequent activation of caspase-3 by cleavage from procaspase-3 triggers the apoptotic cascade [8]. Here, cleaved caspase-3 protein expression was significantly increased in the IRI-placebo group by 12.4 ± 4.5-fold at 48-hr post-reperfusion vs. sham (P<0.01), Figure 5C. On the other hand, treprostinil reduced the cleavage of caspase-3 by 80% compared to placebo (P<0.01). These results suggest that treprostinil inhibits hepatic apoptosis by suppressing the release of mitochondrial cytochrome c and caspase-3 activation.
3.5. Treprostinil restores hepatic ATP and ATP synthases
ATP is required to maintain mitochondrial homeostasis and disrupted ATP production contributes to dysfunctional mitochondria [28]. In this study, hepatic ATP levels were reduced in the IRI-placebo group by more than 50% of sham levels at 6-hr post-reperfusion, Figure 6A. Similarly, hepatic proteomic analyses revealed downregulated ATP synthase subunits, specifically 5f1c, 5f1d, 5i (P<0.001), 5j2 (P<0.01), 5l (P<0.001), and 5o (P<0.05) in IRI-placebo, vs. control at 24-hr post-reperfusion, Figure 6B. In contrast, treprostinil attenuated the loss of hepatic ATP levels which corresponded with upregulated hepatic ATP synthase subunits. Together, these results demonstrate treprostinil restores ATP production, which is necessary to maintain mitochondrial function.
Figure 6.

Treprostinil restores hepatic ATP and ATP synthase levels. (A) Hepatic ATP levels at 6-hours post-reperfusion measured by bioluminescent assay; (B) ATP synthase subunits at 24-hours post-reperfusion measured by SWATH-MS based proteomics, normalized to Gapdh, and expressed as fold-change over control. Data represented are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 vs. sham; ϕP<0.05 vs. IRI-placebo (n = 4–6/group). One- or two-way ANOVA, Tukey’s multiple comparisons test. ATP: Adenosine triphosphate; SWATH-MS: Sequential windowed acquisition of all theoretical fragment ion mass spectra; Atp5f1c: ATP synthase F1 subunit gamma; Atp5f1d: ATP synthase F1 subunit delta; Atp5i: ATP synthase subunit e; Atp5j2: ATP synthase subunit f; Atp5l: ATP synthase subunit g; Atp5o: ATP synthase subunit o; CTRL: Control; PLB: IRI-placebo; TRE: IRI-treprostinil.
3.6. Treprostinil preserves mitochondrial biogenesis and mtDNA content
Mitochondrial biogenesis is necessary for liver recovery and generation after acute injury [29]. The peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) is considered one of the master regulators of mitochondrial biogenesis [29]. In this study, hepatic Pgc-1α mRNA expression was significantly decreased in IRI-placebo group to nearly 50% of sham levels at 24-hr post-reperfusion (P<0.05). Conversely, treprostinil upregulated Pgc-1α mRNA expression by 1.9-fold (P<0.01 vs. IRI-placebo), which restored mRNA expression to baseline, Figure 7A.
Figure 7.

Treprostinil preserves mitochondrial biogenesis and mtDNA. (A) Hepatic mRNA expression of Pgc-1α at 24-hours post-reperfusion was measured by qPCR, normalized to 18S and is expressed as a fold-change over sham. (B) Hepatic mtDNA copy number at 6-hours post-reperfusion, measured by real-time qPCR; Data are represented as mean ± SEM. *P < 0.05 vs. sham; ϕϕP < 0.01 vs. IRI-placebo (n = 4–5/group). One-way ANOVA, Tukey’s multiple comparisons test. Pgc-1α: Peroxisome proliferator-activated receptor gamma coactivator 1 alpha; mtDNA: mitochondrial DNA; PLB: IRI-placebo; TRE: IRI-treprostinil.
Pgc-1α also regulates the mitochondrial DNA copy number [30], which is released into the circulation following renal IRI and activates hepatic TLR9 [15]. Here, we demonstrated that the hepatic mtDNA copy number in the IRI-placebo group significantly decreased to 70% of sham (P<0.05) at 6-hr post-reperfusion which was nearly restored by treprostinil to sham levels, Figure 7B. These findings suggest that a greater amount of hepatic mtDNA content is released into the circulation during renal IRI, which in turn activates Tlr9. In contrast, treprostinil preserves hepatic mtDNA, which is likely mediated through the upregulation of hepatic Pgc-1α.
3.7. Treprostinil improves hepatic mitochondrial dynamics
To further investigate the role of treprostinil in alleviating hepatic mitochondrial injury during renal IRI, we studied hepatic mitochondrial dynamics. We found that the facilitators of outer mitochondrial membrane fusion, Mfn1 and Mfn2 [29], were compromised in the liver following renal IRI. Specifically, hepatic mRNA expression of Mfn1 and Mfn2 decreased in IRI-placebo group by 30% (P=0.08) and 18% relative to sham, respectively, at 24-hr post-reperfusion, Figure 8A, 8B. In contrast, treprostinil upregulated Mfn1 and Mfn2 expression by 30% (P<0.01) and 40% (P=0.08), respectively, in comparison to sham.
Figure 8.

Treprostinil improves hepatic mitochondrial dynamics after renal IRI. Hepatic mRNA expressions of (A) Mfn1 and (B) Mfn2 at 24-hours post-reperfusion measured by qPCR, normalized to 18S and expressed as fold-change over sham. Hepatic protein expression of (C) Drp-1 and (D) Sirt3 at 3- and 6-hours post-reperfusion, respectively measured by western blot, normalized to Cox-IV and quantified using Image J software. Data represented are mean ± SEM. *P<0.05, **P<0.01 vs. sham; ϕϕP<0.01 vs. IRI-placebo (n = 4–5/group). One-way ANOVA, Tukey’s multiple comparisons test. Mfn1: Mitofusin-1; Mfn2: Mitofusin-2; Drp-1: Dynamin-related protein 1; Sirt3: NAD-dependent deacetylase sirtuin-3; Cox-IV: Cytochrome c oxidase subunit 4; PLB: IRI-placebo; TRE: IRI-treprostinil.
Following stimulus, such as cellular stress due to renal IRI, the mitochondrial fission protein Drp-1 translocates to the outer mitochondrial membrane where it causes mitochondrial fragmentation [31]. Early post-reperfusion, mitochondrial protein levels of hepatic Drp-1 in IRI-placebo animals significantly increased by 1.8 ± 0.2-fold vs. sham animals (P<0.01), which treprostinil significantly decreased (P<0.01) and returned to baseline, Figure 8C. Notably, NAD-dependent deacetylase sirtuin-3 (Sirt3), the master regulator of mitochondrial fusion, significantly decreased in IRI-placebo animals by 44% vs. sham (P<0.05) at 6-hr post-reperfusion, Figure 8D, which treprostinil restored to sham levels (P<0.01 vs. IRI-placebo). Together, these data demonstrate that treprostinil improves mitochondrial dynamics by mitigating Drp-1-mediated mitochondrial fission while favorably contributing to Sirt3-mediated mitochondrial fusion.
4. Discussion
Renal IRI occurs during several clinical situations, including kidney transplantation, and results in kidney injury as well as liver injury [13, 32]. In fact, a correlation between the severity of renal IRI and the severity of liver injury and cell death has been found [13], which further supports the evidence of hepatic damage during renal IRI. Left untreated, hepatic injury during renal IRI can lead to a comorbidity which elevates the risk of mortality [3]. Prostacyclin analogs, including treprostinil (Remodulin®), have been studied for their efficacy in reducing IRI, particularly during liver transplantation [33, 34] and, more recently, during acute kidney injury [20]. Therefore, we hypothesized that treprostinil would protect the liver against injury during renal IRI. This study demonstrates a novel mechanism by which treprostinil improves hepatic function, reduces hepatic oxidative stress, inflammation, mitochondrial damage, and apoptosis during renal IRI.
The liver is highly susceptible to renal damage as a result of IRI during AKI [13]. Animal studies have shown elevated serum ALT and AST levels during renal IRI [2, 6, 9, 35], supporting secondary liver dysfunction. Prostacyclin analogs are effective in reducing liver dysfunction during hepatic IRI, including mice preconditioned with beraprost [36] when subjected to hepatic IRI or pretreated with treprostinil before rat [33] and clinical orthotopic liver transplantation [34]. Treprostinil is a stable PGI2 analog at room temperature with potent vasodilatory and anti-platelet properties [19] and a half-life of 3–4 hours [37], whereas beraprost is not FDA-approved.
Antioxidants are responsible for protecting the liver from oxidative damage and their depletion serves as a key indicator of the generation of oxygen radicals during renal IRI [38]. Consistent with previous animal studies [5, 6, 13], our results demonstrate that hepatic oxidative injury occurs during renal IRI in vivo. We confirmed hepatic oxidative injury through biochemical analyses, particularly the reduced activity and concentrations of SOD, GSH, and CAT, in addition to increased MDA levels. Reduced Gclc mRNA expressions patterns also confirmed oxidative injury. Furthermore, proteomic analyses showed that treprostinil restored the depleted antioxidants through increased expression patterns of Gpx1, Gpx3, Gsta2, Gstm4, Sod2, Cat, and Gclc. These data provide further support that PGI2 restores the oxidant status during hepatic IRI.
Toll-like receptors play a major role in regulating the innate immune system’s response to pathogen- or DAMP [14]. Specifically, activation of TLR9 by mtDNA released from apoptotic cells [39] initiates a signaling cascade involving nuclear factor kappa B (NF-κB) which upregulates proinflammatory cytokines, including Il-1β [16]. Tlr9 mediates hepatic inflammation by increasing proinflammatory cytokines e.g., Il-6, Ccl2, and Tnf-α [40]. Additionally, Bakker et al. [15] showed that hepatic Tlr9 was selectively upregulated at 24-hr post-reperfusion during renal IRI, further supporting our hypothesis that Tlr9 mediates hepatic injury during renal IRI. Our results show that treprostinil suppresses the mRNA expression of Il-1β and Ccl2 early post-reperfusion. Overexpression of hepatic adhesions molecules, e.g., Vcam1, Icam1, E- and P-selectin, have also been found to increase following renal [24, 35] and hepatic IRI [41], which promote neutrophil-mediated organ injury and contribute to inflammation [42]. Volpes et al. [26] demonstrated that VCAM1 is highly expressed in the inflamed liver and Serpine1 is involved in inflammation [25]. In agreement with these findings, our study shows remarkable elevations of hepatic Vcam1 and Serpine1 mRNA expressions in IRI-placebo animals, which treprostinil reduces to alleviate hepatic inflammation. In addition, our findings also show that treprostinil suppresses the elevated hepatic Tlr9 expression, supporting our hypothesis of Tlr9-mediated hepatic inflammation during renal IRI.
Some PGI2 analogs are ligands for the peroxisome proliferator-activated receptor (PPAR)-alpha [43], a nuclear transcription factor that is predominantly expressed in the liver. In a mouse model of drug-induced liver injury, Fan et al. [44] demonstrated that hepatic Tlr9 protein expression is increased while Ppar-α expression is downregulated. Interestingly, treprostinil activates PPAR-β [45], which downregulates NF-κB-induced expression of IL-1β [46]. Crosstalk between some PPAR isoforms and TLRs have been studied since they both regulate the inflammatory NF-κB signaling cascade [47], but, to date, the relationship between PPARβ and TLR9 has not yet been examined.
Mitochondrial homeostasis is maintained through continuous fission and fusion events, and an imbalance of these events leads to apoptosis and cell death [48]. Importantly, injury to the liver during renal IRI disrupts the balance of mitochondrial dynamics [27]. During mitochondrial injury, cytochrome c is released from the inner mitochondrial membrane into the cytosol, thereby initiating the intrinsic apoptotic pathway and caspase activation [8]. Cytochrome c, a marker for circulating mitochondrial DNA, was decreased in Tlr9 knock-out mice subjected to severe renal ischemia [15]. Peerapanyasut et al. [27] showed that Drp-1, a regulator for mitochondrial fission (fragmentation), increases in hepatic tissue after renal IRI. Additionally, mitochondrial fragmentation depletes ATP due to an overproduction of ROS [28]. Consistent with these findings, our data demonstrate that hepatic ATP levels decreased in the IRI-placebo group, which is restored by treprostinil. Proteomic analyses confirmed the hepatic mitochondrial injury by the protein expression patterns of several ATP synthase subunits in IRI-placebo animals, all of which were upregulated by treprostinil. Furthermore, our data show that Drp-1 and cytochrome c protein expression are upregulated in the mitochondria and cytosol, respectively. Moreover, a decrease in procaspase-3 (inactive) and increase in the cleaved (active) forms indicate apoptotic injury [8]. Cleaved-caspase-3 promotes apoptosis by further triggering the apoptotic cascade in the intrinsic apoptotic pathway [8]. In agreement with Lai et al. [9], our study also demonstrates that renal IRI significantly increased hepatic cleaved caspase-3 protein expression, which treprostinil significantly reduced. Altogether, our data indicate that treprostinil restores hepatic ATP and reduces mitochondrial-mediated apoptosis during renal IRI.
Downregulation of PGC-1α, a major regulator of mitochondrial biogenesis, decreases mitochondrial content which exacerbates mitochondrial-mediated apoptosis [30]. In this study, treprostinil counteracted renal IRI-induced hepatic mitochondrial injury and restored hepatic Pgc-1α expression and mtDNA copy number. PGC-1α also regulates mitochondrial antioxidant activity and Sirt3 activity [49]. Deficiencies in Sirt3 promote further oxidative stress by inducing lipid peroxidation and decreasing SOD activity [50]. Sirt3 also downregulates murine Mfn1 and Mfn2 protein in the brain [51], illustrating the importance of maintaining hepatic Pgc-1α and Sirt3 levels. Here, we demonstrate that hepatic Sirt3 protein expression is reduced in the IRI-placebo group as well as Mfn1 and Mfn2. Collectively, our data demonstrate that treprostinil alleviates hepatic mitochondrial injury by improving mitochondrial dynamics during renal IRI.
Although this study demonstrates that treprostinil inhibits hepatic inflammation, the mechanisms of treprostinil in the TLR9-mediated pathway of renal IRI remain unknown. Indeed, having demonstrated the antioxidative and anti-apoptotic effects of treprostinil in the liver, the mechanisms of treprostinil that regulate hepatic antioxidation and mitochondrial apoptosis during renal IRI warrant further investigation.
5. Conclusion
In conclusion, there is currently no treatment for liver injury during kidney IRI. As such, patients who develop liver injury following AKI are at a higher risk for multi-organ injury and subsequent mortality. This study demonstrates the hepatoprotective effects of treprostinil during renal IRI in vivo. Specifically, the improved hepatic function, restored antioxidant levels, reduced inflammatory response, reduced mitochondrial-mediated apoptosis, and improved mitochondrial dynamics strongly indicate that treprostinil protects the liver during renal IRI by reducing the downstream effects of Tlr9-mediated liver injury. The hepatic proteome data provide further insight to the protein network and pathways regulated by treprostinil in reducing mitochondrial injury proteins. Combined with its advantageous properties, the findings in this study support the efficacy of treprostinil in reducing hepatic dysfunction during renal IRI and, further, support treprostinil as potential therapy to alleviate hepatic injury during clinical AKI.
Highlights.
What is already known about this subject:
Hepatic injury occurs during renal IRI and increases in-hospital mortality, no treatment available.
What this study adds:
Treprostinil inhibits renal IRI-induced hepatic inflammation and mitochondrial-mediated apoptosis in vivo.
Clinical significance:
Treprostinil is a novel and viable treatment to ameliorate renal IRI-induced hepatic injury.
Acknowledgements
We thank Meiwen Ding and Dr. Benjamin J. Barlock for technical support. The graphical abstract was created with BioRender.com.
Funding
The material presented herein is supported by Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health, which funds Brown University Advance-CTR (U54GM115677), and University of Rhode Island Core Lab (P20GM103430). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- IRI
Ischemia-reperfusion injury
- AKI
Acute kidney injury
- SOD
Superoxide dismutase
- GSH
Glutathione
- CAT
Catalase
- ROS
Reactive oxygen species
- ATP
Adenosine triphosphate
- IL
Interleukin
- TLR
Toll-like receptor
- mtDNA
Mitochondrial DNA
- Mfn
Mitofusin
- Pgc-1α
Peroxisome proliferator-activated receptor gamma coactivator 1 alpha
- Drp-1
Dynamin-related protein 1
- PGI2
Prostacyclin
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- SCr
Serum creatinine
- BUN
Blood urea nitrogen
- MDA
Malondialdehyde
- H&E
Hematoxylin & eosin
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- Sirt3
NAD-dependent deacetylase sirtuin-3
- Gapdh
Glyceraldehyde 3-phosphate dehydrogenase
- Cox-IV
Cytochrome c oxidase subunit 4
- mt-Nd1
mitochondrial encoded NADH-ubiquinone oxidoreductase chain 1
- Vcam1
Vascular cell adhesion molecule 1
- Serpine1
Serine protease inhibitor, family E, member 1
- Gclc
Glutamate-cysteine ligase catalytic subunit
- Ccl2
Chemokine ligand 2
- 18S
18S ribosomal RNA
- SWATH-MS
Sequential windowed acquisition of all theoretical fragment ion mass spectra
- Gpx
Glutathione peroxidase
- Gsta2
Glutathione S-transferase alpha 2
- Gstm4
Glutathione S-transferase mu 4
Footnotes
CRediT authorship contribution statement
Joyce Hou: Investigation, Formal analysis, Writing - Original Draft, Visualization; Evelyn Tolbert: Investigation; Mark Birkenbach: Formal analysis, Visualization, Writing - Original Draft; Nisanne Ghonem: Conceptualization, Resources, Methodology, Writing - Original Draft, Visualization, Supervision, Project administration, Funding acquisition. All authors approved the final version of the manuscript.
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Smith SF, Hosgood SA, Nicholson ML, Ischemia-reperfusion injury in renal transplantation: 3 key signaling pathways in tubular epithelial cells, Kidney Int 95(1) (2019) 50–56. [DOI] [PubMed] [Google Scholar]
- [2].Elshazly S, Soliman E, PPAR gamma agonist, pioglitazone, rescues liver damage induced by renal ischemia/reperfusion injury, Toxicol Appl Pharmacol 362 (2019) 86–94. [DOI] [PubMed] [Google Scholar]
- [3].Lee SA, Cozzi M, Bush EL, Rabb H, Distant Organ Dysfunction in Acute Kidney Injury: A Review, Am J Kidney Dis 72(6) (2018) 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Awad AS, Kamel R, Sherief MA, Effect of thymoquinone on hepatorenal dysfunction and alteration of CYP3A1 and spermidine/spermine N-1-acetyl-transferase gene expression induced by renal ischaemia-reperfusion in rats, J Pharm Pharmacol 63(8) (2011) 1037–42. [DOI] [PubMed] [Google Scholar]
- [5].Farag MM, Khalifa AA, Elhadidy WF, Rashad RM, Hepatorenal protection in renal ischemia/reperfusion by celecoxib and pentoxifylline, J Surg Res 204(1) (2016) 183–91. [DOI] [PubMed] [Google Scholar]
- [6].Gholampour F, Sadidi Z, Hepatorenal protection during renal ischemia by quercetin and remote ischemic perconditioning, J Surg Res 231 (2018) 224–233. [DOI] [PubMed] [Google Scholar]
- [7].Kim SH, Kim H, Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction-A Mini-Review, Nutrients 10(9) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ow YP, Green DR, Hao Z, Mak TW, Cytochrome c: functions beyond respiration, Nat Rev Mol Cell Biol 9(7) (2008) 532–42. [DOI] [PubMed] [Google Scholar]
- [9].Lai Y, Deng J, Wang M, Wang M, Zhou L, Meng G, Zhou Z, Wang Y, Guo F, Yin M, Zhou X, Jiang H, Vagus nerve stimulation protects against acute liver injury induced by renal ischemia reperfusion via antioxidant stress and anti-inflammation, Biomed Pharmacother 117 (2019) 109062. [DOI] [PubMed] [Google Scholar]
- [10].El-Hattab AW, Craigen WJ, Scaglia F, Mitochondrial DNA maintenance defects, Biochim Biophys Acta Mol Basis Dis 1863(6) (2017) 1539–1555. [DOI] [PubMed] [Google Scholar]
- [11].Yu R, Lendahl U, Nister M, Zhao J, Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges, Front Endocrinol (Lausanne) 11 (2020) 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dusabimana T, Kim SR, Kim HJ, Park SW, Kim H, Nobiletin ameliorates hepatic ischemia and reperfusion injury through the activation of SIRT-1/FOXO3a-mediated autophagy and mitochondrial biogenesis, Exp Mol Med 51(4) (2019) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Golab F, Kadkhodaee M, Zahmatkesh M, Hedayati M, Arab H, Schuster R, Zahedi K, Lentsch AB, Soleimani M, Ischemic and non-ischemic acute kidney injury cause hepatic damage, Kidney Int 75(8) (2009) 783–92. [DOI] [PubMed] [Google Scholar]
- [14].Anders HJ, Toll-like receptors and danger signaling in kidney injury, J Am Soc Nephrol 21(8) (2010) 1270–4. [DOI] [PubMed] [Google Scholar]
- [15].Bakker PJ, Scantlebery AM, Butter LM, Claessen N, Teske GJ, van der Poll T, Florquin S, Leemans JC, TLR9 Mediates Remote Liver Injury following Severe Renal Ischemia Reperfusion, PLoS One 10(9) (2015) e0137511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gao B, Innate immunity and steatohepatitis: a critical role of another toll (TLR-9), Gastroenterology 139(1) (2010) 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ, Circulating mitochondrial DAMPs cause inflammatory responses to injury, Nature 464(7285) (2010) 104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dorris SL, Peebles RS Jr., PGI2 as a regulator of inflammatory diseases, Mediators Inflamm 2012 (2012) 926968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lindegaard Pedersen M, Kruger M, Grimm D, Infanger M, Wehland M, The prostacyclin analogue treprostinil in the treatment of pulmonary arterial hypertension, Basic Clin Pharmacol Toxicol (2019). [DOI] [PubMed] [Google Scholar]
- [20].T.E. Ding M, Birkenbach M, Gohh R, Akhlaghi F, and Ghonem NS., Treprostinil, a prostacyclin analog, ameliorates renal ischemia-reperfusion injury: Preclinical studies in a rat model of acute kidney injury, Nephrol Dial Transp. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ghonem N, Yoshida J, Murase N, Strom SC, Venkataramanan R, Treprostinil Improves Hepatic Cytochrome P450 Activity during Rat Liver Transplantation, J Clin Exp Hepatol 2(4) (2012) 323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Quiros PM, Goyal A, Jha P, Auwerx J, Analysis of mtDNA/nDNA Ratio in Mice, Curr Protoc Mouse Biol 7(1) (2017) 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jamwal R, Barlock BJ, Adusumalli S, Ogasawara K, Simons BL, Akhlaghi F, Multiplex and Label-Free Relative Quantification Approach for Studying Protein Abundance of Drug Metabolizing Enzymes in Human Liver Microsomes Using SWATH-MS, J Proteome Res 16(11) (2017) 4134–4143. [DOI] [PubMed] [Google Scholar]
- [24].Kim M, Park SW, Kim M, D’Agati VD, Lee HT, Isoflurane activates intestinal sphingosine kinase to protect against renal ischemia-reperfusion-induced liver and intestine injury, Anesthesiology 114(2) (2011) 363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zou W, Roth RA, Younis HS, Malle E, Ganey PE, Neutrophil-cytokine interactions in a rat model of sulindac-induced idiosyncratic liver injury, Toxicology 290(2–3) (2011) 278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Volpes R, Van Den Oord JJ, Desmet VJ, Vascular adhesion molecules in acute and chronic liver inflammation, Hepatology 15(2) (1992) 269–75. [DOI] [PubMed] [Google Scholar]
- [27].Peerapanyasut W, Kobroob A, Palee S, Chattipakorn N, Wongmekiat O, Activation of Sirtuin 3 and Maintenance of Mitochondrial Integrity by N-Acetylcysteine Protects Against Bisphenol A-Induced Kidney and Liver Toxicity in Rats, Int J Mol Sci 20(2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hasnat M, Yuan Z, Ullah A, Naveed M, Raza F, Baig M, Khan A, Xu D, Su Y, Sun L, Zhang L, Jiang Z, Mitochondria-dependent apoptosis in triptolide-induced hepatotoxicity is associated with the Drp1 activation, Toxicol Mech Methods 30(2) (2020) 124–133. [DOI] [PubMed] [Google Scholar]
- [29].Ramachandran A, Umbaugh DS, Jaeschke H, Mitochondrial Dynamics in Drug-Induced Liver Injury, Livers 1(3) (2021) 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bi J, Zhang J, Ren Y, Du Z, Li Q, Wang Y, Wei S, Yang L, Zhang J, Liu C, Lv Y, Wu R, Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress, Redox Biol 20 (2019) 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li N, Wang H, Jiang C, Zhang M, Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway, Exp Cell Res 369(1) (2018) 27–33. [DOI] [PubMed] [Google Scholar]
- [32].Park SW, Chen SW, Kim M, Brown KM, Kolls JK, D’Agati VD, Lee HT, Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy, Lab Invest 91(1) (2011) 63–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ghonem N, Yoshida J, Stolz DB, Humar A, Starzl TE, Murase N, Venkataramanan R, Treprostinil, a prostacyclin analog, ameliorates ischemia-reperfusion injury in rat orthotopic liver transplantation, Am J Transplant 11(11) (2011) 2508–16. [DOI] [PubMed] [Google Scholar]
- [34].Almazroo OA, Miah MK, Pillai VC, Shaik IH, Xu R, Dharmayan S, Johnson HJ, Ganesh S, Planinsic RM, Demetris AJ, Al-Khafaji A, Lopez R, Molinari M, Tevar AD, Hughes C, Humar A, Venkataramanan R, An evaluation of the safety and preliminary efficacy of peri- and post-operative treprostinil in preventing ischemia and reperfusion injury in adult orthotopic liver transplant recipients, Clin Transplant 35(6) (2021) e14298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mohammadi M, Najafi H, Mohamadi Yarijani Z, Vaezi G, Hojati V, Piperine pretreatment attenuates renal ischemia-reperfusion induced liver injury, Heliyon 5(8) (2019) e02180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Deng J, Feng J, Liu T, Lu X, Wang W, Liu N, Lv Y, Liu Q, Guo C, Zhou Y, Beraprost sodium preconditioning prevents inflammation, apoptosis, and autophagy during hepatic ischemia-reperfusion injury in mice via the P38 and JNK pathways, Drug Des Devel Ther 12 (2018) 4067–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].O’Connell C, Amar D, Boucly A, Savale L, Jais X, Chaumais MC, Montani D, Humbert M, Simonneau G, Sitbon O, Comparative Safety and Tolerability of Prostacyclins in Pulmonary Hypertension, Drug Saf 39(4) (2016) 287–94. [DOI] [PubMed] [Google Scholar]
- [38].Serteser M, Koken T, Kahraman A, Yilmaz K, Akbulut G, Dilek ON, Changes in hepatic TNF-alpha levels, antioxidant status, and oxidation products after renal ischemia/reperfusion injury in mice, J Surg Res 107(2) (2002) 234–40. [DOI] [PubMed] [Google Scholar]
- [39].Zhu M, Barbas AS, Lin L, Scheuermann U, Bishawi M, Brennan TV, Mitochondria Released by Apoptotic Cell Death Initiate Innate Immune Responses, Immunohorizons 2(11) (2018) 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP, Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury, Hepatology 51(2) (2010) 621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Taki-Eldin A, Zhou L, Xie HY, Chen KJ, Yu D, He Y, Zheng SS, Triiodothyronine attenuates hepatic ischemia/reperfusion injury in a partial hepatectomy model through inhibition of proinflammatory cytokines, transcription factors, and adhesion molecules, J Surg Res 178(2) (2012) 646–56. [DOI] [PubMed] [Google Scholar]
- [42].Lassailly G, Bou Saleh M, Leleu-Chavain N, Ningarhari M, Gantier E, Carpentier R, Artru F, Gnemmi V, Bertin B, Maboudou P, Betbeder D, Gheeraert C, Maggiotto F, Dharancy S, Mathurin P, Louvet A, Dubuquoy L, Nucleotide-binding oligomerization domain 1 (NOD1) modulates liver ischemia reperfusion through the expression adhesion molecules, J Hepatol 70(6) (2019) 1159–1169. [DOI] [PubMed] [Google Scholar]
- [43].Chen HH, Chen TW, Lin H, Prostacyclin-induced peroxisome proliferator-activated receptor-alpha translocation attenuates NF-kappaB and TNF-alpha activation after renal ischemia-reperfusion injury, Am J Physiol Renal Physiol 297(4) (2009) F1109–18. [DOI] [PubMed] [Google Scholar]
- [44].Fan S, Huang X, Wang S, Li C, Zhang Z, Xie M, Nie S, Combinatorial usage of fungal polysaccharides from Cordyceps sinensis and Ganoderma atrum ameliorate drug-induced liver injury in mice, Food Chem Toxicol 119 (2018) 66–72. [DOI] [PubMed] [Google Scholar]
- [45].Ali FY, Davidson SJ, Moraes LA, Traves SL, Paul-Clark M, Bishop-Bailey D, Warner TD, Mitchell JA, Role of nuclear receptor signaling in platelets: antithrombotic effects of PPARbeta, FASEB J 20(2) (2006) 326–8. [DOI] [PubMed] [Google Scholar]
- [46].Wang Y, Nakajima T, Gonzalez FJ, Tanaka N, PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice, Int J Mol Sci 21(6) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dana N, Vaseghi G, Haghjooy Javanmard S, Crosstalk between Peroxisome Proliferator-Activated Receptors and Toll-Like Receptors: A Systematic Review, Adv Pharm Bull 9(1) (2019) 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Carelli V, Maresca A, Caporali L, Trifunov S, Zanna C, Rugolo M, Mitochondria: Biogenesis and mitophagy balance in segregation and clonal expansion of mitochondrial DNA mutations, Int J Biochem Cell Biol 63 (2015) 21–4. [DOI] [PubMed] [Google Scholar]
- [49].Rius-Perez S, Torres-Cuevas I, Millan I, Ortega AL, Perez S, PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism, Oxid Med Cell Longev 2020 (2020) 1452696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Li X, Song S, Xu M, Hua Y, Ding Y, Shan X, Meng G, Wang Y, Sirtuin3 deficiency exacerbates carbon tetrachloride-induced hepatic injury in mice, J Biochem Mol Toxicol 33(2) (2019) e22249. [DOI] [PubMed] [Google Scholar]
- [51].Tyagi A, Nguyen CU, Chong T, Michel CR, Fritz KS, Reisdorph N, Knaub L, Reusch JEB, Pugazhenthi S, SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain, Sci Rep 8(1) (2018) 17547. [DOI] [PMC free article] [PubMed] [Google Scholar]