

Abstract
Tau pathology in Alzheimer's disease (AD) preferentially afflicts the limbic and recently enlarged association cortices, causing a progression of mnemonic and cognitive deficits. Although genetic mouse models have helped reveal mechanisms underlying the rare, autosomal‐dominant forms of AD, the etiology of the more common, sporadic form of AD remains unknown, and is challenging to study in mice due to their limited association cortex and lifespan. It is also difficult to study in human brains, as early‐stage tau phosphorylation can degrade postmortem. In contrast, rhesus monkeys have extensive association cortices, are long‐lived, and can undergo perfusion fixation to capture early‐stage tau phosphorylation in situ. Most importantly, rhesus monkeys naturally develop amyloid plaques, neurofibrillary tangles comprised of hyperphosphorylated tau, synaptic loss, and cognitive deficits with advancing age, and thus can be used to identify the early molecular events that initiate and propel neuropathology in the aging association cortices. Studies to date suggest that the particular molecular signaling events needed for higher cognition—for example, high levels of calcium to maintain persistent neuronal firing‐ lead to tau phosphorylation and inflammation when dysregulated with advancing age. The expression of NMDAR‐NR2B (GluN2B)—the subunit that fluxes high levels of calcium—increases over the cortical hierarchy and with the expansion of association cortex in primate evolution, consistent with patterns of tau pathology. In the rhesus monkey dorsolateral prefrontal cortex, spines contain NMDAR‐NR2B and the molecular machinery to magnify internal calcium release near the synapse, as well as phosphodiesterases, mGluR3, and calbindin to regulate calcium signaling. Loss of regulation with inflammation and/or aging appears to be a key factor in initiating tau pathology. The vast expansion in the numbers of these synapses over primate evolution is consistent with the degree of tau pathology seen across species: marmoset < rhesus monkey < chimpanzee < human, culminating in the vast neurodegeneration seen in humans with AD.
Keywords: Alzheimer's disease, AT8, calcium, entorhinal cortex, p217Tau, prefrontal cortex
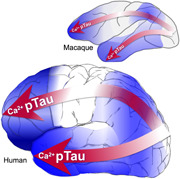
Magnified calcium signaling and tau pathology increase across the cortical hierarchy and across primate brain evolution.
Research Highlights
Studies of aging rhesus monkeys can help to reveal the molecular events that initiate Alzheimer's‐like tau pathology in the association cortex, including early phosphorylation events that are challenging to capture in postmortem human brains.
Evidence indicates that dysregulated calcium signaling is a key in initiating tau hyperphosphorylation, beginning in the entorhinal cortex and proceeding to the association cortices, with the same pattern and sequence as seen in humans.
Calcium signaling (e.g., NMDAR‐NR2B, calbindin, and calcium channels), sustains higher cognitive operations, and increases across the cortical hierarchy and across primate evolution, consistent with the expansion in tau pathology across primate species.
1. INTRODUCTION AND REVIEW OF ALZHEIMER'S DISEASE PATHOLOGY IN HUMANS
This review proposes the idea that Alzheimer's disease (AD) neuropathology has arisen in parallel with the evolution of the primate brain, and the great expansion of the association cortices underlying cognitive function (Figure 1). As nonhuman primates (NHPs) have extensive association cortices and are long‐lived, they offer a unique opportunity to learn about the causes of AD degenerative pathology that arise with advancing age. We have learned that neurons in the prefrontal association cortex (PFC) express the molecular machinery to magnify calcium signaling near the synapse, essential to sustain the persistent firing needed for higher cognitive operations. However, these molecular properties appear to confer vulnerability to degeneration when regulation of calcium is lost with advancing age.
Figure 1.
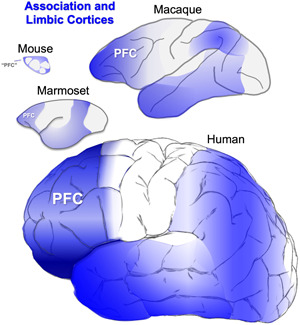
The association and limbic cortices (highlit in blue) expand across brain evolution
The hallmarks of AD neuropathology are extracellular amyloid Aβ plaques and intracellular neurofibrillary tangles (NFTs) comprised of hyperphosphorylated tau. These pathological events are inter‐related, as Aβ oligomers can drive tau phosphorylation (Um et al., 2013), and aggregations of phosphorylated tau may increase the production of Aβ (Arnsten et al., 2021; Paspalas et al., 2018), thus driving vicious cycles. Cognitive deficits correlate with NFTs, but not amyloid plaques (Nelson et al., 2012) suggesting that it is particularly important that we learn how tau phosphorylation arises in the aging association cortex. Tau normally serves to regulate microtubules, but with increasing phosphorylation, tau detaches from microtubules and aggregates, and with hyperphosphorylation, fibrillates within dendrites to form NFTs that then invade the soma. The cell eventually dies from autophagic degeneration, leaving a “ghost tangle” (reviewed in Arnsten et al. (2021)).
Tau pathology in AD shows a characteristic and intriguing progression across the cortical hierarchy. It preferentially afflicts glutamatergic neurons in the limbic and association cortices, beginning in the perirhinal and entorhinal limbic cortices, and proceeding to the neocortical association areas in later stages, but only afflicting neurons in primary sensory cortex at end‐stage disease (Braak & Braak, 1991; Hyman et al., 1984; Lewis et al., 1987; Pearson et al., 1985). The primate neuroanatomists who discovered this progression realized that they were seeing progression through an interconnected network (Hyman et al., 1984; Lewis et al., 1987; Pearson et al., 1985), consistent with more recent discoveries that phosphorylated tau can traffic between neurons to “infect” entire networks (Vogels et al., 2020). The selective vulnerability of these highly interconnected glutamatergic neurons is an important clue regarding the etiology of tau pathology.
There are also differences in tau pathology across species, where the degree of pathology corresponds with the extent of expansion of the association cortex: wild type rodents < marmosets < macaques < chimpanzees < humans (Braak & Braak, 1991; Carlyle et al., 2014; Edler et al., 2017; Leslie et al., 2020; Paspalas et al., 2018; Rodriguez‐Callejas et al., 2016). We propose that the evolutionary changes that expanded the association cortices and allowed the generation of sustained mental representations underlying abstract thought confer vulnerability for tau pathology when molecular regulation is lost with inflammation and advancing age. Specifically, we explore the ideas that increased vulnerability to tau pathology is associated with:
increased numbers of cortical‐cortical glutamatergic NMDAR‐NR2B synapses, where the glutamate synapse is the “engine” of pathology;
magnified calcium‐cAMP signaling near the glutamate‐NMDAR‐NR2B synapse, when regulation is lost with age, allowing toxic calcium actions;
increased oxidative stress from higher energy metabolism, driving calcium dysregulation in spines and dendrites; and
a long lifespan, giving time for pathology to build.
Studies of aging NHPs are particularly helpful for learning what causes tau pathology with advancing age in the common, sporadic form of AD (sAD), where pathology develops in the absence of autosomal dominant mutations. As rodents have little association cortex and very limited tau pathology, these animal models require genetic mutations to induce tau pathology, and thus are more limited in showing how pathology arises in the absence of mutations. This paper will review the contributions that research in aging NHPs can bring to the field, focusing on the age‐related changes in molecular signaling that lead to tau phosphorylation in the limbic and association cortices. We will describe how synapses in the PFC are uniquely regulated at the molecular level in a manner that confers susceptibility to tau pathology and degeneration when regulation is lost with advancing age, and how the vast expansion in numbers of glutamate synapses in the human evolutionary lineage can drive tau pathology to culminate in human brains as AD, with its massive neurodegeneration.
2. UNIQUE CONTRIBUTIONS OF RESEARCH IN NHPs
Research in NHPs provides the opportunity to capture early‐stage tau pathology that is not possible in rodent models or human brains. Cortical tau pathology in AD starts in layer pre‐α of the perirhinal cortex and the layer pre‐α (layer II) cell islands of the entorhinal cortex (ERC; Figure 2a; Braak & Braak, 1991; Braak et al., 1994; Hyman et al., 1984). It then arises in pyramidal cells in deeper layers of the ERC, in hippocampus, and in the association cortex, with pyramidal cells in primary sensory and motor cortex only affected at end‐stage disease (Figure 2b,c; Braak & Braak, 1991; Braak et al., 1994; Lewis et al., 1987). This differential vulnerability is challenging to address in rodents, where there is little association with cortex and genetic alterations that cause autosomal dominant AD and humanized tau that are needed to induce AD pathology (King, 2018). In contrast, rhesus monkeys naturally develop early‐stage cortical tau pathology with the same qualitative pattern and sequence as in humans, with striking pathology first arising in the ERC layer II cell islands (Figure 2d; Paspalas et al., 2018). As with humans, tau pathology later develops in pyramidal cells in the association cortices, while the primary visual cortex (V1) remains unaffected (see more detailed discussion below; Carlyle et al., 2014; Paspalas et al., 2018). Similar to tau pathology, there are age‐related reductions in spine density in the aged rhesus monkey dorsolateral prefrontal association cortex (dlPFC) but not in V1 (Young et al., 2014), consistent with dlPFC synapse loss in human AD (DeKosky & Scheff, 1990). Rhesus monkeys also naturally develop amyloid plaques with advancing age, of a similar size and shape as those seen in humans (Mufson et al., 1994; Paspalas et al., 2018; Uno & Walker, 1993). Importantly, rhesus monkeys of extreme age show classic NFTs in the ERC (Figure 2e; Paspalas et al., 2018), comprised of paired helical filaments identical to those in human (Figure 2f–i), and which are labeled by the AT8 antibody used to diagnose AD (Figure 2e–i). Thus, although rhesus monkeys have very few NFTs compared to chimpanzees and humans, the tau pathology is qualitatively similar to that in humans, and thus appropriate for the study of how tau pathology arises in aging association cortex. Early‐stage tau pathology with advancing age has also been seen in marmosets (Rodriguez‐Callejas et al., 2016, vervet monkeys (Latimer et al., 2019; and see Frye et al., 2021), and baboons (Schultz et al., 2000).
Figure 2.
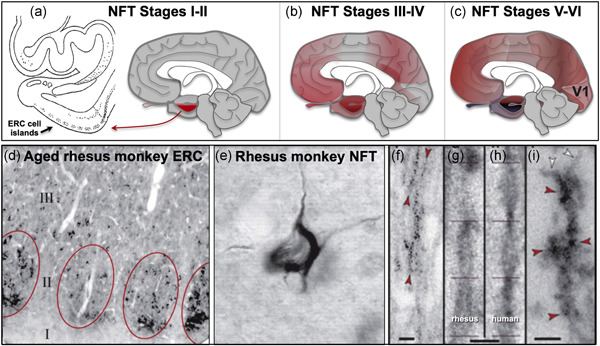
Tau pathology in humans and rhesus monkeys. (a) In human brain, cortical tau pathology begins in layer pre‐a in the perirhinal cortex and ERC (Braak stages I–II). The cell islands in layer II of the ERC are particularly prominent and indicated by a black arrow. (b) In Braak stages III–IV, tau pathology worsens in the ERC and medial temporal cortex, and builds in pyramidal cells of the association cortices. (c) Tau pathology only begins in the primary sensory cortices only at the latest stages, Braak stages V–VI. (d) Similar to humans, cortical tau pathology in aged rhesus monkeys begins in the layer II cell islands of the ERC. (e) Although not as frequent as in humans or chimpanzees, classic NFTs labeled by the AT8 antibody can be seen in the ERC of the oldest rhesus monkeys. (f–i) NFTs and pretangles in the aged rhesus monkey are comprised of paired helical filaments that label with the AT8 antibody used to diagnose AD (f), with the same helical frequency and size as human (g, h), ending abruptly as seen in human (i). ERC, entorhinal cortex; NFT, neurofibrillary tangle. (a–c) based on Arnsten et al. (2021) with permission; (d–i) from Paspalas et al. (2018) with permission
Another substantial advantage of NHP research is the opportunity to examine perfusion‐fixed brains with minimal or no PMI, which allows detection of early‐stage tau phosphorylation that is often lost in human brains due to dephosphorylation and membrane degradation postmortem (Wang et al., 2015). This preservation of tissue and phosphorylation state in fixed NHP brains allows high‐resolution immunoelectron microscopy (immunoEM) to observe the earliest stages of tau pathology in situ, and to examine what changes in aging association cortex initiate tau hyperphosphorylation.
3. KEY METHODOLOGICAL ISSUES THAT CAN CONFUSE THE FIELD
Early‐stage phosphorylated tau, before extensive fibrillation, can be lost due to a number of factors. Thus, issues such as PMI and tissue preparation can produce large effects on whether phosphorylated tau can be detected in tissue, and increases the chance of false‐negative findings. For example, we are learning that postmortem immersion fixation in formalin or paraffin‐embedding seems to destroy soluble, early‐stage tau pathology (Paspalas et al., 2018). As these are standard methods for processing human brain tissue, early‐stage tau pathology has been hard to assess in human brains. Furthermore, as brains from great apes, such as gorillas and chimpanzees, are usually collected in a similar manner, these factors may also obscure comparisons between great apes and perfusion‐fixed tissues from monkeys.
Another important issue is the interpretation of AT8 antibody labeling, and the confusion that can develop when comparing human postmortem tissue with monkey brains. The AT8 antibody is currently used to diagnose AD, as it labels NFTs. In human brains that are fixed postmortem in formalin, or frozen and embedded in paraffin, only highly fibrillated tau survives, and thus AT8 only labels fibrillated tau, for example, in neurites and NFTs. Thus, AT8 labeling in human neuropathology has become synonymous with NFTs. However, in perfusion‐fixed NHP tissue, we are able to see soluble (early) species that are usually absent in postmortem fixed human tissue, as well as later stage, fibrillated tau. Thus, AT8 labeling in rhesus monkeys can be seen along a continuum of nonfibrillar (diffuse early stage) through highly fibrillar, with morphological characteristics of classic NFTs (Paspalas et al., 2018). Similarly, in marmosets, Alz50 labeling is lighter and more diffuse in younger animals, and becomes more fibrillar in the oldest animals (Rodriguez‐Callejas et al., 2016). Our field will need to find a way to clearly distinguish these key details to better understand the evolution of tau pathology, and to accurately compare nonhuman and human primates. One possibility is to take advantage of the clarity provided by perfusion‐fixed primate tissue to examine AT8 labeling within the context of additional markers of cell pathology, for example, markers of mitochondrial dysfunction or autophagic degeneration, to provide a deeper assessment of cellular functional integrity and its relationship to tau phosphorylation state. In this review, we try to respect these details to begin to have a more refined analysis of how tau pathology arises in the aging association cortex.
An additional challenge in using NHPs for aging/AD research is trying to equate ages across species with different life spans, for example, how to compare an “aged” marmoset with an “aged” rhesus monkey. This can be especially difficult when trying to assess age span in animals in captivity, where living conditions and access to veterinary care can vary widely. The current review uses the following assumptions for ages across captive primate species: common marmosets: “early” aged is about 10 years, and extreme age limit is about 20 years (Nishijima et al., 2012; Rodriguez‐Callejas et al., 2016; Tardif et al., 2011); rhesus monkeys: “early” aged is about 20 years (18 years is often used as the cutoff for considering a rhesus monkey to be aged, as established decades ago by Bartus and Dean (1988)), and extreme age limit is about 40 years (Colman et al., 2009; Tigges et al., 1988); chimpanzees: “early” aged is about 30 years, and extreme age limit is almost 70 years (Havercamp et al., 2019). Luckily, there have been analyses of tau pathology in the association cortices from animals of extreme age in all of these species, allowing some comparisons of how far pathology can proceed with maximal life span.
4. ASSOCIATION CORTICES GENERATE PROLONGED REPRESENTATIONS NEEDED FOR COGNITION
Why does tau pathology preferentially afflict glutamatergic neurons in the association cortices? Several lines of evidence indicate that excitatory neurons in the association cortices have special properties that allow them to participate in higher cognitive operations, but make them vulnerable to degeneration (Arnsten et al., 2020). As discussed in this section, there appear to be molecular gradients across the cortical hierarchy from primary sensory to association to limbic cortices, with increasing reliance on magnified calcium mechanisms in the higher association and limbic cortices (Arnsten et al., 2020; Wang, 2020). This gradient is also reflected in increasing time scales in neuronal firing across the cortical hierarchy in rhesus monkeys (Monosov et al., 2020; Murray et al., 2014) and in transcriptomic expression patterns across the cortical hierarchy in humans (Burt et al., 2018). These differences in molecular and physiological properties are appropriate to the disparate functions of these cortical areas, where short time scales in primary sensory cortex are needed for accurate decoding of a sensory event, intermediate time scales are needed to integrate sensory information (e.g., in parietal association cortex), longer time periods to generate, maintain and manipulate internal representations in working memory without sensory stimulation (e.g., in the PFC), still longer to mediate mood (e.g., anterior cingulate cortex), and the longest of all to allow long‐term memory storage (e.g., ERC and hippocampus; Arnsten et al., 2020).
The dlPFC in rhesus monkeys has been the focus of intensive physiological, anatomical, and molecular research, and thus can be helpful in learning about the characteristics that allow cortical circuits to generate higher cognition. Neurons in the rhesus monkey dlPFC have the ability to represent a position in visual space across the delay period in a working memory task, maintaining neuronal firing without any “bottom‐up” sensory stimulation (Figure 3a). These so‐called “Delay cells” can maintain firing for upwards of 15 s (Fuster & Alexander, 1971), and are spatially tuned such that they fire only to their preferred spatial position (Funahashi et al., 1989). This persistent firing arises from extensive, recurrent excitatory circuits in deep layer III of the dlPFC with NMDAR synapses on dendritic spines, with lateral inhibition to refine the information held in working memory stores (Figure 3b; Goldman‐Rakic, 1995; González‐Burgos et al., 2000, 2005; Wang et al., 2013). As described below, these excitatory NMDAR connections increase across the cortical hierarchy and across primate evolution.
Figure 3.
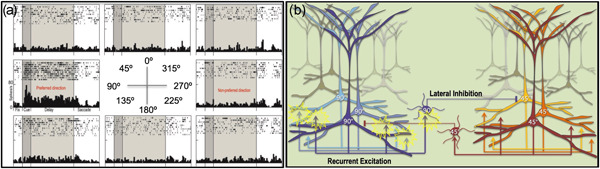
The cellular basis of working memory in the rhesus monkey dlPFC. (a) A “Delay cell” from the dlPFC of a rhesus monkey performing a visuospatial working memory task. Delay cells exhibit spatially tuned, persistent firing across the delay period when the monkey is remembering the spatial location that is the “preferred direction” for the neuron, but do not increase firing for nonpreferred directions. Thus, Delay cells are able to generate and sustain mental representations of visual space without sensory stimulation. (b) The recurrent excitatory microcircuits in deep layer III of rhesus monkey dlPFC are thought to underlie Delay cell firing. Pyramidal cells with a shared preferred direction excite each other through NMDAR synapses on spines to maintain firing across the delay period, while lateral inhibition from PV GABAergic interneurons helps to refine spatial tuning. dlPFC, dorsolateral prefrontal association cortex
The ability to generate and sustain mental representations requires different molecular mechanisms than those found in classic circuits such as those in V1 (Figures 4 and 5). For example, while V1 neurons rely heavily on AMPAR neurotransmission (Figures 4a and 5b), Delay cells in dlPFC depend on NMDAR neurotransmission (Figures 4b and 5a), including those with NR2B subunits (NMDAR2B), which flux the highest levels of calcium (Wang et al., 2013; Yang et al., 2018). ImmunoEM shows that NMDAR2B is expressed exclusively within the postsynaptic density (PSD) in layer III dlPFC spine synapses, and not at extrasynaptic locations (Wang et al., 2013). In classical neurons, stimulation of AMPAR depolarizes the postsynaptic membrane and ejects the magnesium block from the NMDAR, permitting NMDAR neurotransmission. However, dlPFC Delay cells do not rely on AMPAR for permissive excitation of NMDAR actions, but rather on acetylcholine (Yang et al., 2013). For example, nicotinic alpha7 receptors reside in the glutamate PSD and are needed for NMDAR excitation (Yang et al., 2013). As acetylcholine is only released during waking and not in deep sleep, Delay cell firing depends on arousal state, which may help to explain why we are unconscious during deep sleep (Yang et al., 2013).
Figure 4.
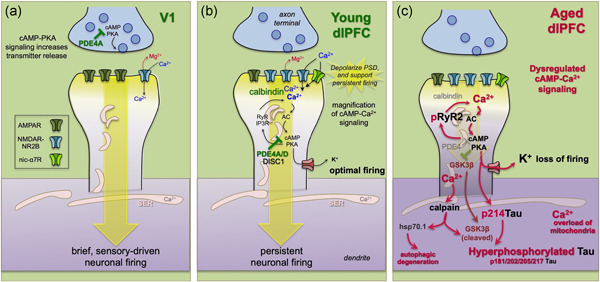
Neurotransmission and neuromodulation in rhesus monkey V1 versus dlPFC, and its dysregulation with advancing age. (a) V1 neurons show classic characteristics, with neurotransmission relying heavily on AMPAR. In layer III of V1, PDE4A regulation of cAMP signaling is concentrated in presynaptic glutamatergic terminals, where cAMP‐PKA signaling can enhance glutamate release. Consistent with the immunoEM, physiological studies show that increasing cAMP‐PKA signaling in V1 increases neuronal firing. (b) Neurotransmission and neuromodulation are very different in layer III of the dlPFC, where neurotransmission relies heavily on NMDAR, including those with slowly closing NR2B subunits that flux high levels of calcium. PDE4 is concentrated in layer III spines on the SER, positioned to regulate cAMP‐PKA drive on internal calcium release, which in turn increases cAMP production, creating a vicious cycle that must be tightly regulated by PDE4s. While moderate levels of calcium are necessary to sustain persistent firing, higher levels of cAMP‐calcium signaling open nearby potassium (K+) channels to weaken connectivity and reduce firing, for example, as occurs with uncontrollable stress. In young adult dlPFC, layer III dlPFC pyramidal cells express high levels of PDE4s to regulate cAMP drive on calcium release, and calbindin to regulate calcium levels when it is released into the cytosol. These regulatory factors are lost with advanced age. (c) The loss of PDE4s and calbindin in aged dlPFC pyramidal cells leads to excessive calcium‐cAMP‐PKA signaling and initiates a series of vicious cycles and toxic events including: (1) PKA phosphorylation of RyR2 to cause calcium leak into the cytosol, leading to more cAMP‐PKA signaling; (2) excessive opening of K+ channels that reduce neuronal firing; (3) calcium overload of mitochondria to induce inflammatory signaling (see Figure 6); (4) PKA phosphorylation of tau, which primes tau for hyperphosphorylation by GSK3β; (5) with sufficient cytosolic calcium, the activation of calpain, which disinhibits GSK3β to hyperphosphorylate tau at key sites that lead to tau fibrillation; and (6) calpain activation of heatshock protein 70.1 to drive autophagic degeneration. dlPFC, dorsolateral prefrontal association cortex
Figure 5.
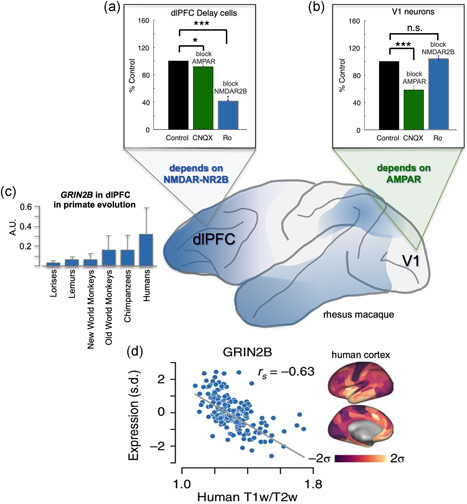
NMDAR‐NR2B neurotransmission increases across the cortical hierarchy and across primate evolution. (a) Delay cells in rhesus monkey dlPFC rely on NMDAR‐NR2B neurotransmission, with relatively little influence from AMPAR. Thus, blockade of AMPAR with CNQX has little effect, while blockade of NMDAR with NR2B subunits with Ro25‐6981 markedly reduces Delay cell firing as the monkey performs a working memory task. (b) In contrast, neurons in rhesus monkey V1 depend on AMPAR more than NMDAR. Neurons responding to a bar of light greatly reduce their firing when AMPAR is blocked with CNQX, but show little effect when NMDAR‐NR2B is blocked by Ro25‐6981. (c) Increased expression of GRIN2B, which encodes for the NMDAR‐NR2B, in the dlPFC across primate evolution. (d) GRIN2B expression increases across the human cortical hierarchy. dlPFC, dorsolateral prefrontal association cortex. Panels A and B from Yang et al. (2018) with permission. Panel C from Muntané et al. (2015) with permission. Panel D from Burt et al. (2018) with permission.
Synapses on dendritic spines in deep layer III dlPFC also show evidence of magnified internal calcium release (Figure 4b), where cAMP signaling increases calcium release from the smooth endoplasmic reticulum (SER, called the spine apparatus when it elaborates in the spine) into the cytosol (Carlyle et al., 2014). Calcium is released from the SER through IP3 receptors and ryanodine receptors (e.g., RyR2). Layer III dlPFC spines contain the molecular machinery for cAMP‐PKA signaling to increase the release of calcium from the SER, which in turn can increase cAMP production, creating feedforward signaling (reviewed in Arnsten et al. (2020)). For example, the phosphodiesterase PDE4A, which catabolizes cAMP, is localized presynaptically near glutamate‐like vesicles in area V1 (Figure 4a), but is concentrated on the SER spine apparatus in layer III dlPFC spines, positioned to regulate cAMP‐PKA drive on internal calcium release (Figure 4b). The magnification of internal calcium release near the synapse may help to maintain the PSD in a depolarized state needed for NMDAR‐dependent persistent firing. However, high levels of cAMP‐calcium firing have the opposite effects, opening nearby potassium channels to reduce firing (Arnsten et al., 2012; Galvin et al., 2020; Wang et al., 2007). This may provide important negative feedback in a recurrent excitatory circuit to prevent seizures, and also serves as a mechanism to dynamically regulate network inputs, and to take the PFC “offline” during uncontrollable stress when high levels of catecholamines greatly increase cAMP‐calcium signaling (Arnsten, 2015; Arnsten et al., 2012, 2019).
There are a number of mechanisms that regulate these powerful, feedforward signaling events within spines in the young dlPFC (Figure 4b): for example, alpha‐2AR and mGluR3 inhibit cAMP production, PDE4s catabolize cAMP once it is formed, and calbindin binds calcium once it is released into the cytosol, or enters through NMDAR2B (Carlyle et al., 2014; Jin et al., 2018; Wang et al., 2007). However, many of these regulatory mechanisms are lost with age (Figure 4c; Datta et al., 2021) and/or inflammation (Arnsten et al., 2020), leading to dysregulation of cAMP‐calcium signaling in the aging association cortex (Carlyle et al., 2014; Datta et al., 2021; Hof & Morrison, 1991; Lally et al., 1997). This includes calcium leak from PKA‐phosphorylated RyR2 (pRyR2) from the SER into the cytosol (Lacampagne et al., 2017; Paspalas et al., 2018; Datta et al., 2021) and reduced calcium binding in the cytosol as the calcium‐binding protein, calbindin, is lost with age from pyramidal cells in aged rhesus monkey dlPFC (Datta et al., 2021). It is these calbindin‐containing pyramidal cells in young cortex that are preferentially lost in AD (Hof & Morrison, 1991; Lally et al., 1997). Dysregulated cAMP‐calcium signaling can induce a cascade of detrimental actions (Datta et al., 2021) as summarized in Figure 4c, including: (1) increased potassium channel opening and a reduction in dlPFC Delay cell firing (Wang et al., 2011), (2) mitochondrial dysfunction and inflammatory complement signaling which coordinates the removal of spines (Datta, Leslie, et al., 2020; Duan et al., 2003; Dumitriu et al., 2010; Morozov et al., 2017), 3) cognitive deficits (Morrison & Baxter, 2012), and, as reviewed in the next section, (4) increased phosphorylation of tau by kinases that are activated by dysregulated PKA and high levels of cytosolic calcium (Datta et al., 2021). Although much of this study has focused on the aging dlPFC, early signs of calcium dysregulation also can be seen in ERC neurons even in middle age (Gant et al., 2018; Paspalas et al., 2018), consistent with the very early expression of tau pathology in these neurons in humans.
It is likely that a number of factors contribute to the reduced regulation of calcium signaling with advancing age. Acute, very high levels of cytosolic calcium, such as occurs with excitotoxicity during stroke, induce apoptosis via mitochondrial death signaling (Hoye et al., 2008). However, there is little evidence of apoptosis in AD; rather, neurons die from autophagic degeneration, which likely involves more subtle yet prolonged calcium dysregulation (Correia et al., 2015). One possible contribution to calcium dysregulation may arise from oxidative stress in mitochondria (Figure 6), where excessive reactive oxygen species (ROS) can initiate p38‐MK2 signaling (Hoye et al., 2008). MK2 can inhibit and unanchor the PDE4s that normally serve to regulate cAMP drive on calcium release from the SER (Houslay et al., 2017; MacKenzie et al., 2011). This may set up a vicious cycle, as the increased calcium release from SER into mitochondria may cause further inflammatory signaling (López‐Armada et al., 2013). Animal models show that mitochondrial oxidative stress is associated with increased phosphorylation of tau (Melov et al., 2007). Consistent with these animal data, there is extensive evidence of oxidative stress in AD brains, including early in the disease process, supporting this hypothesis (Yin et al., 2016; Zhu et al., 2003; Zhu et al., 2007), as well as data showing that p38 is co‐expressed with tau pathology in AD brains (Zhu et al., 2000). In addition to oxidative stress, calcium dysregulation may also arise from inflammatory increases in GCPII signaling (Zhang et al., 2016), which would lessen mGluR3 regulation of cAMP‐calcium signaling (Jin et al., 2018). Finally, there is some evidence that emotional stress can decrease calbindin expression (Li et al., 2017), which would elevate free cytosolic calcium levels, for example, to activate calpain. Thus, both physiological and emotional stressors may contribute to dysregulated calcium signaling, initiating a cascade of cellular events that increase the risk of AD.
Figure 6.
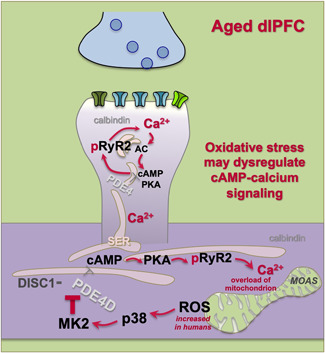
Schematic illustration of the hypothesis that elevated ROS (oxidative stress) in dysmorphic mitochondria can induce p38‐MK2 signaling, which can dysregulate calcium signaling by unanchoring and inhibiting PDE4s, which in turn can create calcium overload of mitochondria, perpetuating a vicious cycle. Calcium dysregulation and p38 signaling can both contribute to the phosphorylation of tau. Higher rates of energy metabolism in human brain may increase these factors in humans, propelling AD pathology. MOAS, mitochondria‐on‐a‐string, an abnormal phenotype seen in AD and aged monkey association cortex; AD, Alzheimer's disease; ROS, reactive oxygen species
5. EARLY STAGE TAU PATHOLOGY IN ASSOCIATION CORTICES—A KEY ROLE FOR CALCIUM
There is a longstanding hypothesis that dysregulated calcium signaling drives neuropathology in AD, in both sAD and the early onset, autosomal dominant forms of AD caused by presenilin mutations that cause calcium leak from the SER (Area‐Gomez & Schon, 2017; Gibson & Thakkar, 2017; Khachaturian, 1991; Mattson, 2007; Stutzmann, 2007). For example, high levels of cytosolic calcium increase the activity of BACE, the critical enzyme for cleavage of APP to Aβ (Hayley et al., 2009). High levels of cAMP‐calcium signaling can also drive the phosphorylation of tau, including early events that can be captured in aging rhesus monkey cortex, where postmortem interval (PMI) can be minimized or abolished by perfusion fixation (Carlyle et al., 2014; Paspalas et al., 2018). A likely sequence of tau phosphorylation based on both in vitro findings and data from aging rhesus monkeys is illustrated in Figure 4c. An important early event involves PKA phosphorylation of tau at S214, causing tau to detach from microtubules and aggregate in dendrites (Carlyle et al., 2014; Jicha et al., 1999; Paspalas et al., 2018), and priming tau for hyperphosphorylation by GSK3β (Liu et al., 2006; Liu et al., 2004). As shown in Figure 7, pS214tau is first observed in the layer II cell islands in ERC in middle age (Figure 7a; Paspalas et al., 2018), and then in layer III pyramidal cells in aged dlPFC (Figure 7b; Carlyle et al., 2014), but not in aged V1 (Figure 7c; Carlyle et al., 2014), consistent with the progression of general tau pathology seen in humans. Ultrastructural analysis with immunoEM revealed that pS214Tau is prominent on the SER near glutamate‐like synapses in spines and dendrites of glutamatergic neurons, consistent with an early role of calcium (Carlyle et al., 2014; Datta et al., 2021; Paspalas et al., 2018). pS214tau is also found within the glutamate PSD (Carlyle et al., 2014; Paspalas et al., 2018), suggesting that these synapses serve as an early “engine” of early tau pathology.
Figure 7.
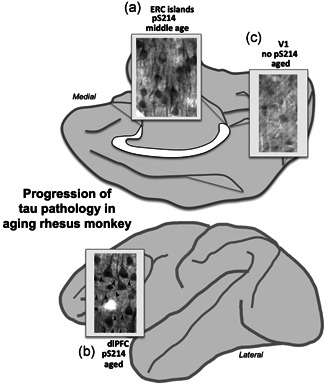
Early stage tau pathology (PKA phosphorylation of tau at pS214Tau) across the aging rhesus monkey cortex shows the same pattern and progression as in human, starting in the ERC layer II cell islands in middle age (a), and then with advanced age in the association cortices such as the dlPFC (b), but not in V1 even in very old animals (c). dlPFC, dorsolateral prefrontal association cortex. Image of tau pathology in A from Paspalas et al. (2018) with permission; images of tau pathology in B and C from Carlyle et al. (2014) with permission
Glutamate synapses in the association cortex also appear to provide sites for “seeding” tau pathology within a neuronal network. Studies of aging rhesus monkeys have documented pS214tau trafficking between neurons near or within glutamate synapses, where it may “infect” a network of higher cortical glutamatergic neurons. This phenomenon has been documented in layer II ERC in middle‐aged rhesus monkeys (Figure 8a; Paspalas et al., 2018), and in dlPFC in older age (Figure 8b; Carlyle et al., 2014). Tau “seeding” between neurons has also been found in studies of human AD brain tissue, where the ERC was the most effective region for “infecting” host tissues (Kaufman et al., 2018). Evidence of tau trafficking has also been seen in mouse models (Gibbons et al., 2019). The results from rhesus monkeys actually visualize this process, and indicate a mechanism whereby tau pathology can spread through association cortical networks very early in the disease process.
Figure 8.
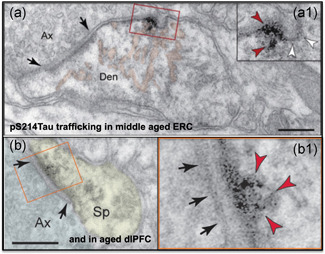
pS214Tau trafficking between neurons near a glutamatergic synapse in layer II of the middle‐aged ERC (a), and within a glutamatergic synapse in the aged dlPFC (b). Enlarged to show details. dlPAC, dorsolateral prefrontal association cortex; ERC, entorhinal cortex. (a) from Paspalas et al. (2018) with permission; (b) from Carlyle et al. (2014) with permission
Later stage hyperphosphosphorylation and fibrillation of tau are performed by multiple kinases, with phosphorylation by GSK3β playing an especially important role. Interestingly, PKA can inhibit GSK3β activity (Fang et al., 2000), which would hold subsequent hyperphosphorylation in check (Figure 4c). A key event in the induction of later stage tau pathology appears to be sufficient calcium build up in the cytosol to activate calpain and release GSK3β from inhibition by PKA (Figure 4c; Goñi‐Oliver et al., 2007; Jin et al., 2015): calpain cleaves off the regulatory site on GSK3β such that it can no longer be inhibited, and is free to hyperphosphorylate tau at key sites, including S202 and T205 recognized by the AT8 antibody currently used to diagnose AD, and T217, a potential early blood biomarker heralding subsequent AD pathology (Palmqvist et al., 2020; Janelidze et al., 2021). In addition to driving tau hyperphosphorylation, calpain can also cleave heatshock protein Hsp70.1 to drive autophagic neurodegeneration (Figure 4c; Yamashima, 2013). Thus, calpain may activate the events most linked to neurodegeneration. Importantly, activated calpain is highly expressed in AD cortex in correspondence with the rise in tau pathology (Kurbatskaya et al., 2016; Saito et al., 1993; Taniguchi et al., 2001), supporting findings from basic research.
An important link between calcium and tau pathology can also be seen in anatomical gradients across cortex, where there is a striking correspondence between the calcium‐binding protein, calbindin, and the amount of tau pathology across the cortical hierarchy. Calbindin is typically expressed in GABAergic cells such as interneurons, but it is also expressed in a subset of glutamatergic neurons, including pyramidal cells in the association cortex and stellate cells in entorhinal cortex (reviewed in Arnsten et al. (2020)). As described above, tau pathology and subsequent neurodegeneration in AD specifically afflicts glutamatergic neurons in the association cortex, starting in the perirhinal and entorhinal cortices (layer II), then expanding more generally in pyramidal cells of the association cortices, with primary visual and auditory cortices only afflicted at end‐stage disease (Braak & Braak, 1991; Hyman et al., 1984; Lewis et al., 1987; Pearson et al., 1985). The expression of calbindin in pyramidal cells shows a similar relationship, with the most expression in cortical areas at the top of the hierarchy, and the least in primary sensory cortices (Kondo et al., 1999; Suzuki & Porteros, 2002). These data are consistent with the idea that glutamatergic neurons engaged in higher cognitive operations utilize higher levels of calcium, and that the loss of calbindin with advancing age renders these neurons particularly vulnerable to calcium's toxic effects on tau phosphorylation and neurodegeneration. Indeed, detailed analyses show that these are the neurons that are preferentially lost in AD (Hof & Morrison, 1991; Ichimiya et al., 1988). Overall, these data suggest that neurons that require high levels of internal calcium signaling for their normative functioning, must express correspondingly high levels of calbindin in their cytosol for adequate protection, and that loss of calbindin with stress and/or age confers tremendous vulnerability to AD pathology (Arnsten et al., 2021).
Overall, the degree of AD‐like neuropathology over a lifespan likely depends upon the balance of detrimental (e.g., inflammation, mutations that increase Aβ production, head injury, emotional distress, and insulin resistance) versus protective (e.g., levels of calbindin, phosphatases, and PDE4) actions, and whether detrimental actions are sufficiently amplified by large numbers of synapses over a long life span to engage the vicious cycles that drive disease progression.
6. RELATIONSHIP TO EVOLUTION OF THE ASSOCIATION CORTICES
There is a vast difference in the extent of association cortex between rodents and primates, and major differences within primate species, humans having much more than NHPs (Figures 1 and 9; Donahue et al., 2018; Glasser et al., 2013; Passingham & Smaers, 2014; Wei et al., 2019). The expansion of the association cortices corresponds with an increase in AD neuropathology that can be seen across species (Table 1). There is little tau pathology in wild‐type rodents, with aged rats, but not mice, producing pS214, but not more hyperphosphorylated sites (Carlyle et al., 2014; Leslie et al., 2020). Very old marmosets express hyperphosphorylated tau labeled by the Alz50 and AT100 antibodies (Rodriguez‐Callejas et al., 2016), but this labeling appears to become fibrillar only in the very oldest animals (Rodriguez‐Callejas et al., 2016). The very oldest rhesus macaques express classic NFTs in the ERC, but they are rare, with fibrillar AT8 mostly found in neurons with pretangle characteristics (Paspalas et al., 2018). Finally, NFTs are more common in very old chimpanzees than they are in rhesus macaques (Edler et al., 2017), but not as numerous as those in humans (Braak & Braak, 1991). Thus, there is an increase in tau pathology across species that is related to both longer lifespan and larger and more elaborated association cortices.
Figure 9.
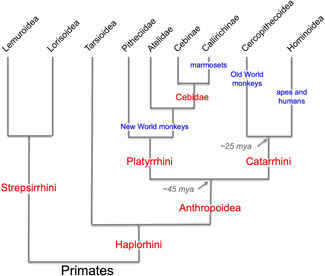
Evolutionary relationships among primates. Humans and apes comprised a higher‐order group, Hominoidea, which is related to another higher‐order group, Cercopithecoidea, which includes macaque monkeys and other Old World monkeys. Collectively, these are the Old World anthropoid primates, or the Catarrhini. The New World anthropoids, or Platyrrhini, include primates such as marmosets, squirrel monkeys, and capuchins. The species discussed in this review are highlighted in blue. Figure modified from Preuss (2019) with permission
Table 1.
Highest levels of tau pathology across species
| Mouse | Rat | Marmoset | Rhesus macaque | Chimpanzee | Human |
|---|---|---|---|---|---|
| pT231 | pS214 | Soluble AT8 | Fibrillar AT8 | Multiple NFTs | Vast number NFTs |
| (Carlyle et al., 2014) | (Leslie et al., 2020) | (Rodriguez‐Callejas et al., 2016) | A few NFTs | (Edler et al., 2017) | (Braak & Braak, 1991) |
| (Paspalas et al., 2018) |
Abbreviation: NFT, neurofibrillary tangle.
Tau pathology can be seen in both the aged marmoset (12–15 years) and aged rhesus monkey (31 years) in medial temporal lobe and dlPFC neurons (Figures 10, 11, 12). These preliminary comparisons are limited to only two aged marmosets, but the findings are similar to published results with a larger cohort examined with antibodies that detect hyperphosphorylated tau (Rodriguez‐Callejas et al., 2016). An important caveat is that it is challenging to know if these are equivalent ages across species (see above), but a comparison is still of interest. Figures 10 and 11 show labeling with the AT8 antibody currently used to diagnose AD postmortem, comparing the ERC, hippocampal CA3 and CA1 subfields, and layer III dlPFC in both species. In the aged rhesus monkey (31 years), there is dense AT8 labeling, including aggregations of likely fibrillated tau in the soma and dendrites (pretangles), reflected in “twisting” dendrites in layer V ERC (Figure 10). Aged marmosets also express AT8 labeling (Figure 11), although with lighter expression and a more diffuse pattern than seen in rhesus monkeys (Figure 11). Both aged marmosets and rhesus monkeys exhibit dense labeling with an antibody that recognizes tau phosphorylated at pT217 (Figure 12), the focus of a new potential CSF and blood diagnostic for AD (Janelidze et al., 2021, 2020; Palmqvist et al., 2020). Studies of pT217 in brain are just beginning, but its early appearance in CSF in AD suggests it may be an intermediate tau species, arising after pS214Tau and before AT8 labeled tau becomes dominant. This would be consistent with the extensive pT217 labeling in both species (Figure 12). However, the T217 labeling in the rhesus monkey dlPFC appears more advanced, with fibrillar aggregations in twisting dendrites similar to those seen in humans (Figure 12d). This may be related to the much more elaborated dendrites of the rhesus monkey versus marmoset in dlPFC layer III pyramidal cells, as described in the following paragraph.
Figure 10.
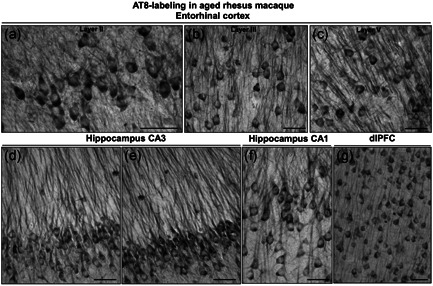
Tau fibrillation (AT8 labeling) in aged rhesus macaque (31 years). Dense tau fibrillation is observed aggregating in entorhinal cortex layer II cell islands (a), extending into the deeper layers, including layer III (b) and layer V (c). The fibrillated tau‐labeling pattern is especially prominent in the perisomatic compartment and along apical and basilar dendrites (pretangles) of excitatory neurons, including parallel bundles of “twisting” apical dendrites in entorhinal cortex layer III and layer V, indicative of possible neurodegeneration. Prominent aggregated AT8 labeling is visualized in AD‐related vulnerable brain regions, including hippocampus pyramidal cell CA3 (d, e) and CA1 (f) subfields, and recurrent excitatory circuits in dlPFC layer III (g). Scale bars, 25 µm (a–c) and 30 µm (d–g). AD, Alzheimer's disease; dlPFC, dorsolateral prefrontal association cortex
Figure 11.
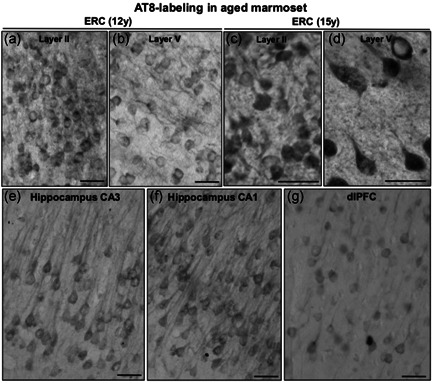
Tau fibrillation (AT8 labeling) in aged marmoset (12–15 years). In early aged marmoset (12 years), mild AT8 labeling is observed in stellate cell layer II islands (a) and deeper layer V (b) in entorhinal cortex. The labeling pattern is diffuse and visualized in the cell soma and the proximal segment along apical dendrites. With more advanced age in marmosets (15 years), more fibrillated AT8 labeling is observed aggregating in entorhinal cortex layer II (c) and layer V (d). Mild tau fibrillation is visualized in pyramidal neurons in hippocampus CA3 (e) and CA1 (f) subfields, and dlPFC layer III (g) in aged marmoset. Scale bars, 25 µm
Figure 12.
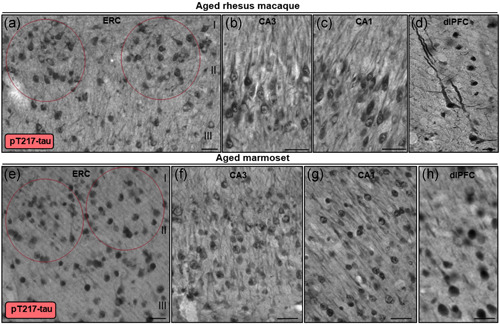
Multiregional pT217‐tau labeling in aged rhesus macaque and marmoset. In aged rhesus macaque (30–31 years), stellate cell layer II islands (red ovals) in entorhinal cortex (a), hippocampus CA3 (b), hippocampus CA1 (c), and dlPFC layer III (d) are reactive against pT217‐tau. Immunoreactivity for pT217‐tau is visualized in the cytoplasm of the perisomatic compartment, along apical and basilar dendrites, and within the nucleoplasm. Note, in aged rhesus macaque dlPFC (30 years), pT217‐tau labeling reveals a highly fibrillated pattern, reflected in “twisting” apical dendrites, similar to AT8. In aged marmoset (12 years), stellate cell layer II islands (red ovals) in entorhinal cortex (e), hippocampus CA3 (f), hippocampus CA1 (g), and dlPFC layer III (h) are reactive against pT217‐tau. Immunoreactivity for pT217‐tau in aged marmosets is more diffuse compared to rhesus macaques, but observed prominently in the neuronal soma, and proximal and distal segments of apical dendrites. Scale bars, 25 µm (a–c, e–g), 10 µm (d), 20 µm (h)
The association cortices expand not only in the increased area, but in the dendritic elaboration of their pyramidal cells to support the increased number of neural connections. The work of Elston (2000, 2006) and Elston et al. (2001, 2011) has shown that the association cortices have greater numbers of spines than the primary visual cortex (Figure 13a), and spine numbers in the association cortices (but not in V1) expand greatly across primates, for example, where total numbers of spines on pyramidal cells in the association cortices increase from marmoset to rhesus macaque to human (Figure 13b). A similar pattern is seen when comparing chimpanzee to human, where pyramidal cells in human association cortex are more complex than their counterparts in chimpanzee (Figure 14a; Bianchi et al., 2013), including greater levels of neuropil (Spocter et al., 2012), and white matter measures of connectivity are greater in human association cortices relative to chimp (Figure 14b; Ardesch et al., 2019). Of particular relevance to AD, these comparisons show greater connectivity of the human ERC as well, which may reflect the funneling of association cortical connections into layer II of the ERC (Ardesch et al., 2019).
Figure 13.
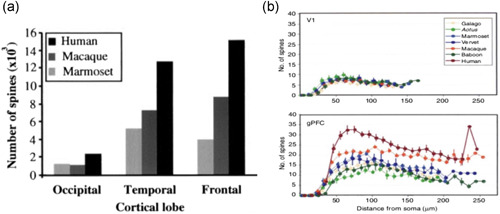
(a) Bar graph of the numbers of spines in V1 (occipital cortex), and the temporal and prefrontal association cortices from marmoset, macaque, and human, showing an evolutionary expansion across the cortical hierarchy and across primate evolution. From Elston et al. (2001) with permission. (b) The numbers of spines in V1 versus dlPFC across primate species, with no differences in V1, and a great expansion across species in dlPFC. dlPFC, dorsolateral prefrontal association cortex. From Elston (2006) with permission
Figure 14.
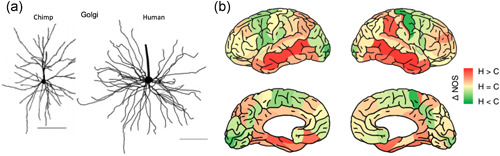
(a) dlPFC pyramidal cell basal dendrites in the chimpanzee are less complex than those in human. From Bianchi et al. (2013) with permission. (b) Measures of connectivity determined from MRI assays of white matter in chimpanzees versus humans. Cortical regions with greater connectivity in humans are shown in red, those with greater connectivity in chimpanzees are shown in green. dlPFC, dorsolateral prefrontal association cortex; MRI, magnetic resonance imaging. From Ardesch et al. (2019) with permission
The expanded numbers of spines across the evolution of the primate association cortex correspond with an expansion in the expression of NMDAR synapses with NR2B subunits, which flux high levels of calcium (Erreger et al., 2005). As described above, electrophysiological recordings and immunoEM analyses show that layer III dlPFC Delay cells in rhesus monkeys depend on NMDAR2B neurotransmission (Figure 5a; Wang et al., 2013) while neurons in V1 have much less NMDAR dependence (Figure 5b; Yang et al., 2018). A similar gradient can be seen in transcriptomic assays of human cortex, where GRIN2B (which encodes for the NMDAR2B subtype) is most enriched in higher cortical areas and least in primary sensory cortices (Figure 5d; Burt et al., 2018). Similarly, the expression of GRIN2B in the PFC increases in evolution with increasing closeness to humans, with low levels in New World monkeys, greater levels in Old World monkeys and chimpanzees, and the most in humans (Figure 5c; Muntané et al., 2015). Thus, the extent of tau pathology across cortical regions in AD, as well as the degree of tau pathology reached across species, corresponds with the expression of the NMDAR2B with the greatest calcium penetrance. In this regard, it is of note that the treatment for late‐stage AD, memantine, blocks NMDAR2B (Song et al., 2018; Winblad et al., 2007). It is possible that this compound is not helpful in earlier stages of AD because its therapeutic effects (reducing calcium influx into neurons) would also impair cognitive abilities in patients who still have functionally effective dlPFC circuits, negating its helpful actions.
In addition to gradients in calbindin and NMDAR2B across the cortical hierarchy, there appear to be gradients in cAMP signaling as well. For example, proteomic analysis shows a greater expression of PDE4D and mGluR3 in the human dlPFC than in V1 (B.C. Carlyle et al., 2017). Research in rhesus monkeys shows that PDE4D, PDE4A, and mGluR3 are concentrated on or near the SER where they regulate cAMP drive on calcium release (B.C. Carlyle et al., 2014; Datta, Enwright, et al., 2020; Datta, Leslie, et al., 2020; Jin et al., 2018). Importantly, the expression of these regulatory proteins is reduced in the aged PFC in both animals and humans (Carlyle et al., 2014; Datta, Enwright, et al., 2020; Datta, Leslie, et al., 2020; García‐Bea et al., 2016; Hernandez et al., 2018; Leslie et al., 2020; Lu et al., 2004), rendering the association cortices vulnerable to dysregulated cAMP‐calcium signaling.
Transcriptomic analyses suggest a number of changes in the human association cortex that may amplify AD neurodegeneration in human brains compared to NHPs. Transcriptomic comparisons of humans, chimpanzees, and macaques using next‐generation sequencing (Berto et al., 2019; Konopka et al., 2012; Mendizabal et al., 2019) have augmented earlier results from microarray studies that identified differences in levels of gene expression in the PFC of humans. The latter studies have also identified a number of human‐specific transcriptional “modules” (or more formally, “weighted gene co‐expression networks”) in PFC. Particularly notable is the gene ELAVL2, which is differentially expressed in humans compared to chimpanzees and macaques, and the human‐specific transcription module with which it is associated (Konopka et al., 2012). The module enriched with genes having ELAVL2 binding sites includes genes related to calcium channels (e.g., RYR2 and the voltage‐dependent L‐type calcium channels CACNB4 and CACNG2) and AD‐associated genes, such as BACE1 and GSK3B, and the NMDAR encoded by GRIN2A (Konopka et al., 2012). In addition, there is evidence from comparative genomics and comparative molecular biological studies strongly suggesting that humans underwent selective changes that increased levels of cerebral energy metabolism (Duka et al., 2014; Grossman et al., 2004; Sterner et al., 2013; Uddin et al., 2008). It is reasonable to assume that increased oxidative energy metabolism is associated with increased oxidative stress (ROS) in mitochondria. As described above (Figure 6), elevated ROS can initiate signaling events that dysregulate calcium signaling. Thus, the increased metabolic activity in the human brain may exacerbate calcium dysregulation and AD pathology, contributing to the extensive degenerative cascade in human versus nonhuman brains.
In summary, there is an increase in calcium‐related signaling across the cortical hierarchy needed for higher cognitive operations, but loss of calbindin and other regulatory proteins with advancing age may render these association neurons particularly vulnerable to toxic calcium actions, including tau phosphorylation. As this pathology builds slowly in the absence of genetic mutations, a long lifespan is needed to reach the extent of pathology seen in AD. As humans have the largest number of cortical‐cortical glutamatergic NMDAR2B synapses in their association cortices, as well as genetic differences that would magnify calcium dysregulation over the longest life span, these factors may contribute to the greater expression of AD pathology in humans compared to nonhuman species.
7. OUTSTANDING QUESTIONS AND FUTURE DIRECTIONS
There are many outstanding questions in this field. For example, why are the glutamatergic neurons in layer pre‐α (layer II) in the primate perirhinal and entorhinal cortices especially vulnerable to tau pathology? Is this due to unique molecular characteristics and/or to their receipt of such extensive cortical‐cortical glutamatergic inputs? ImmunoEM of layer II of the rhesus monkey ERC shows expanded SER under glutamate synapses on dendrites that are labeled very early by S214tau (Paspalas et al., 2018), but is this a general characteristic of layer II ERC neurons that is an initiating site for cortical tau pathology? Does tau pathology arise independently in other association cortices, and/or how much does it require seeding from the ERC? What are the temporal sequences of molecular changes in the different brain regions? There is a great need to expand analyses beyond the dlPFC and V1 to learn about the molecular, physiological and anatomical characteristics of cortical neurons that are vulnerable versus resilient to AD pathology.
The creation of transgenic NHPs will also be game‐changing, allowing us to see how autosomal dominant mutations, or mutations that increase the risk of AD, alter neuronal cell biology and physiology within the cellular context of elaborated association cortical circuits that express native AD‐like pathology. Understanding the mechanisms that initiate sAD pathology will be essential for finding strategies to slow or prevent this devastating disease.
AUTHOR CONTRIBUTIONS
Amy F. T. Arnsten: Conceptualization (lead); data curation (equal); formal analysis (equal); project administration (lead); visualization (lead); writing original draft (lead); writing review & editing (equal). Dibyadeep Datta: Data curation (equal); investigation (lead); methodology (lead); writing review & editing (equal). Todd M. Preuss: Data curation (equal); validation (lead); writing review & editing (equal).
ETHICS STATEMENT
Yale and AFTA receive royalties from Shire/Takeda from the United States sales of Intuniv. They do not receive royalties from non‐US sales nor sales of generic Intuniv or guanfacine.
ACKNOWLEDGMENTS
The authors thank Guoping Feng, Qiangge Zhang, Jitendra Sharma, and the members of the marmoset team at MIT for their support in providing marmoset brain tissues. This study was supported by NIH grants Pioneer Award DP1AG047744‐01 and R01AG061190 and NSF 2015276 (AFTA); Alzheimer's Association Research Fellowship AARF‐17‐533294 (DD), American Federation for Aging Research/Diamond Postdoctoral Fellowship (DD); and NIH ORIP/OD P51 OD011132 (TMP).
Arnsten, A. F. T. , Datta, D. , & Preuss, T. M. (2021). Studies of aging nonhuman primates illuminate the etiology of early‐stage Alzheimer's‐like neuropathology: An evolutionary perspective. Am J Primatol, 83, e23254. 10.1002/ajp.23254
REFERENCES
- Ardesch, D. J. , Scholtens, L. H. , Li, L. , Preuss, T. M. , Rilling, J. K. , & van den Heuvel, M. P. (2019). Evolutionary expansion of connectivity between multimodal association areas in the human brain compared with chimpanzees. Proceedings of the National Academy of Sciences of the United States of America, 116, 7101–7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area‐Gomez, E. , & Schon, E. A. (2017). On the pathogenesis of Alzheimer's disease: The MAM Hypothesis. FASEB Journal, 31, 864–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten, A. F. (2015). Stress weakens prefrontal networks: Molecular insults to higher cognition. Nature Neuroscience, 18, 1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten, A. F. T. , Datta, D. , Del Tredici, K. , & Braak, H. (2021). Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimer's & Dementia, 17, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten, A. F. T. , Datta, D. , & Wang, M. (2020). The genie in the bottle‐magnified calcium signaling in dorsolateral prefrontal cortex. Molecular Psychiatry. 10.1038/s41380-020-00973-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten, A. F. T. , Jin, L. E. , Gamo, N. J. , Ramos, B. , Paspalas, C. D. , Morozov, Y. M. , Kata, A. , Bamford, N. S. , Yeckel, M. F. , Kaczmarek, L. K. , & El‐Hassar, L. (2019). Role of KCNQ potassium channels in stress‐induced deficit of working memory. Neurobio Stress, 11, 100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten, A. F. T. , Wang, M. , & Paspalas, C. D. (2012). Neuromodulation of thought: Flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron, 76(1), 223–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus, R. T. , & Dean, R. L. (1988). Tetrahydroaminoacridine, 3,4 diaminopyridine and physostigmine: Direct comparisons of effects on memory in aged primates. Neurobiology of Aging, 9, 351–356. [DOI] [PubMed] [Google Scholar]
- Berto, S. , Mendizabal, I. , Usui, N. , Toriumi, K. , Chatterjee, P. , Douglas, C. , Tamminga, C. A. , Preuss, T. M. , Yi, S. V. , & Konopka, G. (2019). Accelerated evolution of oligodendrocytes in the human brain. Proceedings of the National Academy of Sciences of the United States of America, 116, 24334–24342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, S. , Stimpson, C. D. , Bauernfeind, A. L. , Schapiro, S. J. , Baze, W. B. , McArthur, M. J. , Bronson, E. , Hopkins, W. D. , Semendeferi, K. , Jacobs, B. , Hof, P. R. , & Sherwood, C. C. (2013). Dendritic morphology of pyramidal neurons in the chimpanzee neocortex: Regional specializations and comparison to humans. Cerebral Cortex, 23, 2429–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, E. , Braak, H. , & Mandelkow, E. M. (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathologica, 87, 554–567. [DOI] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1991). Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathologica, 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Burt, J. B. , Demirtaş, M. , Eckner, W. J. , Navejar, N. M. , Ji, J. L. , Martin, W. J. , Bernacchia, A. , Anticevic, A. , & Murray, J. D. (2018). Hierarchy of transcriptomic specialization across human cortex captured by structural neuroimaging topography. Nature Neuroscience, 21, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyle, B. C. , Kitchen, R. R. , Kanyo, J. E. , Voss, E. Z. , Pletikos, M. , Sousa, A. M. M. , Lam, T. T. , Gerstein, M. B. , Sestan, N. , & Nairn, A. C. (2017). A multiregional proteomic survey of the postnatal human brain. Nature Neuroscience, 20, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyle, B. C. , Nairn, A. C. , Wang, M. , Yang, Y. , Jin, L. E. , Simen, A. A. , Ramos, B. P. , Bordner, K. A. , Craft, G. E. , Davies, P. , Pletikos, M. , Šestan, N. , Arnsten, A. F. T. , & Paspalas, C. D. (2014). cAMP‐PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proceedings of the National Academy of Sciences of the United States of America, 111, 5036–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman, R. J. , Anderson, R. M. , Johnson, S. C. , Kastman, E. K. , Kosmatka, K. J. , Beasley, T. M. , Allison, D. B. , Cruzen, C. , Simmons, H. A. , Kemnitz, J. W. , & Weindruch, R. (2009). Caloric restriction delays disease onset and mortality in rhesus monkeys. Science, 325, 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia, S. C. , Resende, R. , Moreira, P. I. , & Pereira, C. M. (2015). Alzheimer's disease‐related misfolded proteins and dysfunctional organelles on autophagy menu. DNA and Cell Biology, 34, 261–273. [DOI] [PubMed] [Google Scholar]
- Datta, D. , Enwright, J. F. , Arion, D. , Paspalas, C. D. , Morozov, Y. M. , Lewis, D. A. , & Arnsten, A. F. T. (2020). Mapping phosphodiesterase 4D (PDE4D) in macaque dorsolateral prefrontal cortex: Postsynaptic compartmentalization in higher‐order layer III pyramidal cell circuits. Frontiers in Neuroanatomy, 14, 578483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, D. , Leslie, S. , Wang, M. , Yang, S. T. , Morozov, Y. , Mentone, S. , & Arnsten, A. F. T. (2021). Age‐related calcium dysregulation linked with tau pathology and impaired cognition in non‐human primates. Alzheimer's & Dementia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, D. , Leslie, S. N. , Morozov, Y. M. , Duque, A. , Rakic, P. , van Dyck, C. H. , Nairn, A. C. , & Arnsten, A. F. T. (2020). Classical complement cascade initiating C1q protein within neurons in the aged rhesus macaque dorsolateral prefrontal cortex. Journal of Neuroinflammation, 17, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky, S. T. , & Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer's disease: Correlation with cognitive severity. Annals of Neurology, 27, 457–464. [DOI] [PubMed] [Google Scholar]
- Donahue, C. J. , Glasser, M. F. , Preuss, T. M. , Rilling, J. K. , & Essen, D. C. V. (2018). Quantitative assessment of prefrontal cortex in humans relative to nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America, 115, E5183–E5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, H. , Wearne, S. L. , Rocher, A. B. , Macedo, A. , Morrison, J. H. , & Hof, P. R. (2003). Age‐related dendritic and spine changes in corticocortically projecting neurons in macaque monkeys. Cerebral Cortex, 13, 950–961. [DOI] [PubMed] [Google Scholar]
- Duka, T. , Anderson, S. M. , Collins, Z. , Raghanti, M. A. , Ely, J. J. , Hof, P. R. , Wildman, D. E. , Goodman, M. , Grossman, L. I. , & Sherwood, C. C. (2014). Synaptosomal lactate dehydrogenase isoenzyme composition is shifted toward aerobic forms in primate brain evolution. Brain Behavior and Evolution, 83, 216–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu, D. , Hao, J. , Hara, Y. , Kaufmann, J. , Janssen, W. G. M. , Lou, W. , Rapp, P. R. , & Morrison, J. H. (2010). Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging‐related cognitive impairment. Journal of Neuroscience, 30, 7507–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edler, M. K. , Sherwood, C. C. , Meindl, R. S. , Hopkins, W. D. , Ely, J. J. , Erwin, J. M. , Mufson, E. J. , Hof, P. R. , & Raghanti, M. A. (2017). Aged chimpanzees exhibit pathologic hallmarks of Alzheimer's disease. Neurobiology of Aging, 59, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston, G. N. (2000). Pyramidal cells of the frontal lobe: All the more spinous to think with. Journal of Neuroscience, 20, RC95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston, G. N. (2006). Specialization of the neocortical pyramidal cell during primate evolution. In Kaas J. H., Striedter G. F., Bullock T. H., Preuss T. M., Rubenstein J., & Krubutzer L. A. (Eds.), Evolution of nervous systems (pp. 191–242). Academic Press. [Google Scholar]
- Elston, G. N. , Benavides‐Piccione, R. , & DeFelipe, J. (2001). The pyramidal cell in cognition: A comparative study in human and monkey. Journal of Neuroscience, 21, RC163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston, G. N. , Benavides‐Piccione, R. , Elston, A. , Manger, P. R. , & Defelipe, J. (2011). Pyramidal cells in prefrontal cortex of primates: Marked differences in neuronal structure among species. Frontiers in Neuroanatomy, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger, K. , Dravid, S. M. , Banke, T. G. , Wyllie, D. J. , & Traynelis, S. F. (2005). Subunit‐specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. Journal of Physiology, 563(Pt 2), 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, X. , Yu, S. X. , Lu, Y. , Bast, R. C. J. , Woodgett, J. R. , & Mills, G. B. (2000). Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proceedings of the National Academy of Sciences of the United States of America, 97, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye, B. M. , Craft, S. , Latimer, C. S. , Keene, C. D. , Montine, T. J. , Register, T. C. , Orr, M. E. , Kavanagh, K. , Macauley, S. L. , & Shively, C. A. (2021). Aging‐related Alzheimer's disease‐like neuropathology and functional decline in captive vervet monkeys (Chlorocebus aethiops sabaeus). American Journal of Primatology, e23260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi, S. , Bruce, C. J. , & Goldman‐Rakic, P. S. (1989). Mnemonic coding of visual space in the monkey's dorsolateral prefrontal cortex. Journal of Neurophysiology, 61, 331–349. [DOI] [PubMed] [Google Scholar]
- Fuster, J. M. , & Alexander, G. E. (1971). Neuron activity related to short‐term memory. Science, 173, 652–654. [DOI] [PubMed] [Google Scholar]
- Galvin, V. C. , Yang, S.‐T. , Paspalas, C. D. , Yang, Y. , Jin, L. E. , Datta, D. , Morozov, Y. M. , Lightbourne, T. C. , Lowet, A. S. , Rakic, P. , Arnsten, A. F. T. , & Wang, M. (2020). Muscarinic M1 receptors modulate working memory performance and activity via KCNQ potassium channels in primate prefrontal cortex. Neuron, 106, 649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant, J. C. , Kadish, I. , Chen, K. C. , Thibault, O. , Blalock, E. M. , Porter, N. M. , & Landfield, P. W. (2018). Aging‐related calcium dysregulation in rat entorhinal neurons homologous with the human entorhinal neurons in which Alzheimer's disease neurofibrillary tangles first appear. Journal of Alzheimer's Disease, 66, 1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Bea, A. , Walker, M. A. , Hyde, T. M. , Kleinman, J. E. , Harrison, P. J. , & Lane, T. A. (2016). Metabotropic glutamate receptor 3 (mGlu3; mGluR3; GRM3) in schizophrenia: Antibody characterisation and a semi‐quantitative western blot study. Schizophrenia Research, 177, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons, G. S. , Lee, V. M. Y. , & Trojanowski, J. Q. (2019). Mechanisms of cell‐to‐cell transmission of pathological tau: A review. JAMA Neurology, 76, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, G. E. , & Thakkar, A. (2017). Interactions of mitochondria/metabolism and calcium regulation in Alzheimer's disease: A Calvinist point of view. Neurochemical Research, 42, 1636–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser, M. F. , Goyal, M. S. , Preuss, T. M. , Raichle, M. E. , & Essen, D. C. V. (2013). Trends and properties of human cerebral cortex: Correlations with cortical myelin content. NeuroImage, 93(Pt 2), 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman‐Rakic, P. S. (1995). Cellular basis of working memory. Neuron, 14(3), 477–485. [DOI] [PubMed] [Google Scholar]
- Goñi‐Oliver, P. , Lucas, J. J. , Avila, J. , & Hernández, F. (2007). N‐terminal cleavage of GSK‐3 by calpain—A new form of GSK‐3 regulation. Journal of Biological Chemistry, 282, 22406–22413. [DOI] [PubMed] [Google Scholar]
- González‐Burgos, G. , Barrionuevo, G. , & Lewis, D. A. (2000). Horizontal synaptic connections in monkey prefrontal cortex: An in vitro electrophysiological study. Cerebral Cortex, 10, 82–92. [DOI] [PubMed] [Google Scholar]
- González‐Burgos, G. , Krimer, L. S. , Povysheva, N. V. , Barrionuevo, G. , & Lewis, D. A. (2005). Functional properties of fast spiking interneurons and their synaptic connections with pyramidal cells in primate dorsolateral prefrontal cortex. Journal of Neurophysiology, 93, 942–953. [DOI] [PubMed] [Google Scholar]
- Grossman, L. I. , Wildman, D. E. , Schmidt, T. R. , & Goodman, M. (2004). Accelerated evolution of the electron transport chain in anthropoid primates. Trends in Genetics, 20, 578–585. [DOI] [PubMed] [Google Scholar]
- Havercamp, K. , Watanuki, K. , Tomonaga, M. , Matsuzawa, T. , & Hirata, S. (2019). Longevity and mortality of captive chimpanzees in Japan from 1921 to 2018. Primates, 60, 525–535. [DOI] [PubMed] [Google Scholar]
- Hayley, M. , Perspicace, S. , Schulthess, T. , & Seelig, J. (2009). Calcium enhances the proteolytic activity of BACE1: An in vitro biophysical and biochemical characterization of the BACE1‐calcium interaction. Biochimica et Biophysica Acta/General Subjects, 1788, 1933–1938. [DOI] [PubMed] [Google Scholar]
- Hernandez, C. M. , McQuail, J. A. , Schwabe, M. R. , Burke, S. N. , Setlow, B. , & Bizon, J. L. (2018). Age‐related declines in prefrontal cortical expression of metabotropic glutamate receptors that support working memory. eNeuro, 5(3):ENEURO.0164‐18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof, P. R. , & Morrison, J. H. (1991). Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Experimental Neurology, 111, 293–301. [DOI] [PubMed] [Google Scholar]
- Houslay, K. F. , Christian, F. , MacLeod, R. , Adams, D. R. , Houslay, M. D. , & Baillie, G. S. (2017). Identification of a multifunctional docking site on the catalytic unit of phosphodiesterase‐4 (PDE4) that is utilised by multiple interaction partners. Biochemical Journal, 474, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoye, A. T. , Davoren, J. E. , Wipf, P. , Fink, M. P. , & Kagan, V. E. (2008). Targeting mitochondria. Accounts of Chemical Research, 41, 87–97. [DOI] [PubMed] [Google Scholar]
- Hyman, B. T. , Van Hoesen, G. W. , Damasio, A. R. , & Barnes, C. L. (1984). Alzheimer's disease: Cell‐specific pathology isolates the hippocampal formation. Science, 225, 1168–1170. [DOI] [PubMed] [Google Scholar]
- Ichimiya, Y. , Emson, P. C. , Mountjoy, C. Q. , Lawson, D. E. , & Heizmann, C. W. (1988). Loss of calbindin‐28K immunoreactive neurons from the cortex in Alzheimer‐type dementia. Brain Research, 475, 156–159. [DOI] [PubMed] [Google Scholar]
- Janelidze, S. , Berron, D. , Smith, R. , Strandberg, O. , Proctor, N. K. , Dage, J. L. , Stomrud, E. , Palmqvist, S. , Mattsson‐Carlgren, N. , & Hansson, O. (2021). Associations of plasma phospho‐Tau217 levels with tau positron emission tomography in early Alzheimer Disease. JAMA Neurol, 78(2), 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze, S. , Stomrud, E. , Smith, R. , Palmqvist, S. , Mattsson, N. , Airey, D. C. , & Hansson, O. (2020). Cerebrospinal fluid p‐tau217 performs better than p‐tau181 as a biomarker of Alzheimer's disease. medRxiv. 10.1101/2020.01.15.20017236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha, G. A. , Weaver, C. , Lane, E. , Vianna, C. , Kress, Y. , Rockwood, J. , & Davies, P. (1999). cAMP‐dependent protein kinase phosphorylations on tau in Alzheimer's disease. Journal of Neuroscience, 19, 7486–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, L. E. , Wang, M. , Galvin, V. C. , Lightbourne, T. C. , Conn, P. J. , Arnsten, A. F. T. , & Paspalas, C. D. (2018). mGluR2 vs. mGluR3 in primate prefrontal cortex: Postsynaptic mGluR3 strengthen cognitive networks. Cerebral Cortex, 28, 974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, N. , Yin, X. , Yu, D. , Cao, M. , Gong, C. X. , Iqbal, K. , Ding, F. , Gu, X. , & Liu, F. (2015). Truncation and activation of GSK‐3β by calpain I: A molecular mechanism links to tau hyperphosphorylation in Alzheimer's disease. Scientific Reports, 5, 8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, S. K. , Del Tredici, K. , Thomas, T. L. , Braak, H. , & Diamond, M. I. (2018). Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho‐tau pathology in Alzheimer's disease and PART. Acta Neuropathologica, 136, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian, Z. S. (1991). Overview of basic research on Alzheimer disease: Implications for cognition. Alzheimer Disease and Associated Disorders, 5(S1), S1–S6. [DOI] [PubMed] [Google Scholar]
- King, A. (2018). The search for better animal models of Alzheimer's disease. Nature, 559, S13–S15. [DOI] [PubMed] [Google Scholar]
- Kondo, H. , Tanaka, K. , Hashikawa, T. , & Jones, E. G. (1999). Neurochemical gradients along monkey sensory cortical pathways: Calbindin‐immunoreactive pyramidal neurons in layers II and III. European Journal of Neuroscience, 11, 4197–4203. [DOI] [PubMed] [Google Scholar]
- Konopka, G. , Friedrich, T. , Davis‐Turak, J. , Winden, K. , Oldham, M. C. , Gao, F. , Chen, L. , Wang, G. Z. , Luo, R. , Preuss, T. M. , & Geschwind, D. H. (2012). Human‐specific transcriptional networks in the brain. Neuron, 75, 601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurbatskaya, K. , Phillips, E. C. , Croft, C. L. , Dentoni, G. , Hughes, M. M. , Wade, M. A. , Al‐Sarraj, S. , Troakes, C. , O'Neill, M. J. , Perez‐Nievas, B. G. , Hanger, D. P. , & Noble, W. (2016). Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer's disease brain. Acta Neuropathologica Communications, 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacampagne, A. , Liu, X. , Reiken, S. , Bussiere, R. , Meli, A. C. , Lauritzen, I. , Teich, A. F. , Zalk, R. , Saint, N. , Arancio, O. , Bauer, C. , Duprat, F. , Briggs, C. A. , Chakroborty, S. , Stutzmann, G. E. , Shelanski, M. L. , Checler, F. , Chami, M. , & Marks, A. R. (2017). Post‐translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease‐like pathologies and cognitive deficits. Acta Neuropathologica, 134, 749–767. [DOI] [PubMed] [Google Scholar]
- Lally, G. , Faull, R. L. , Waldvogel, H. J. , Ferrari, S. , & Emson, P. C. (1997). Calcium homeostasis in ageing: Studies on the calcium binding protein calbindin D28K. The Journal of Neural Transmission, 104, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Latimer, C. S. , Shively, C. A. , Keene, C. D. , Jorgensen, M. J. , Andrews, R. N. , Register, T. C. , Montine, T. J. , Wilson, A. M. , Neth, B. J. , Mintz, A. , Maldjian, J. A. , Whitlow, C. T. , Kaplan, J. R. , & Craft, S. (2019). A nonhuman primate model of early Alzheimer's disease pathologic change: Implications for disease pathogenesis. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 15, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie, S. N. , Datta, D. , Christensen, K. R. , van Dyck, C. H. , Arnsten, A. F. T. , & Nairn, A. C. (2020). Phosphodiesterase PDE4D is decreased in frontal cortex of aged rats and positively correlated with working memory performance and inversely correlated with PKA phosphorylation of tau. Frontiers in Aging Neuroscience, 14, 578483. 10.3389/fnagi.2020.576723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, D. A. , Campbell, M. J. , Terry, R. D. , & Morrison, J. H. (1987). Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer's disease: A quantitative study of visual and auditory cortices. Journal of Neuroscience, 7, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. T. , Xie, X. M. , Yu, J. Y. , Sun, Y. X. , Liao, X. M. , Wang, X. X. , Su, Y. A. , Liu, Y. J. , Schmidt, M. V. , Wang, X. D. , & Si, T. M. (2017). Suppressed calbindin levels in hippocampal excitatory neurons mediate stress‐induced memory loss. Cell Reports, 21, 891–900. [DOI] [PubMed] [Google Scholar]
- Liu, F. , Liang, Z. , Shi, J. , Yin, D. , El‐Akkad, E. , Grundke‐Iqbal, I. , Iqbal, K. , & Gong, C. X. (2006). PKA modulates GSK‐3β‐ and cdk5‐catalyzed phosphorylation of tau in site‐ and kinase‐specific manners. FEBS Letters, 580, 6269–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. J. , Zhang, J. Y. , Li, H. L. , Fang, Z. Y. , Wang, Q. , Deng, H. M. , Gong, C. X. , Grundke‐Iqbal, I. , Iqbal, K. , & Wang, J. Z. (2004). Tau becomes a more favorable substrate for GSK‐3 when it is prephosphorylated by PKA in rat brain. Journal of Biological Chemistry, 279, 50078–50088. [DOI] [PubMed] [Google Scholar]
- López‐Armada, M. J. , Riveiro‐Naveira, R. R. , Vaamonde‐García, C. , & Valcárcel‐Ares, M. N. (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion, 13, 106–118. [DOI] [PubMed] [Google Scholar]
- Lu, T. , Pan, Y. , Kao, S. Y. , Li, C. , Kohane, I. , Chan, J. , & Yankner, B. A. (2004). Gene regulation and DNA damage in the ageing human brain. Nature, 429, 883–891. [DOI] [PubMed] [Google Scholar]
- MacKenzie, K. F. , Wallace, D. A. , Hill, E. V. , Anthony, D. F. , Henderson, D. J. , Houslay, D. M. , & Houslay, M. D. (2011). Phosphorylation of cAMP‐specific PDE4A5 (phosphodiesterase‐4A5) by MK2 (MAPKAPK2) attenuates its activation through protein kinase A phosphorylation. Biochemical Journal, 435, 755–769. [DOI] [PubMed] [Google Scholar]
- Mattson, M. P. (2007). Calcium and neurodegeneration. Aging Cell, 6, 337–350. [DOI] [PubMed] [Google Scholar]
- Melov, S. , Adlard, P. A. , Morten, K. , Johnson, F. , Golden, T. R. , Hinerfeld, D. , Schilling, B. , Mavros, C. , Masters, C. L. , Volitakis, I. , Li, Q. X. , Laughton, K. , Hubbard, A. , Cherny, R. A. , Gibson, B. , & Bush, A. I. (2007). Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLOS One, 2, e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal, I. , Berto, S. , Usui, N. , Toriumi, K. , Chatterjee, P. , Douglas, C. , Huh, I. , Jeong, H. , Layman, T. , Tamminga, C. A. , Preuss, T. M. , Konopka, G. , & Yi, S. V. (2019). Cell type‐specific epigenetic links to schizophrenia risk in the brain. Genome Biology, 20, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monosov, I. , Haber, S. N. , Leuthardt, E. C. , & Jezzini, A. (2020). Anterior cingulate cortex and the control of dynamic behavior in primates. Current Biology, 30, R1442–R1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov, Y. M. , Datta, D. , Paspalas, C. D. , & Arnsten, A. F. (2017). Ultrastructural evidence for impaired mitochondrial fission in the aged rhesus monkey dorsolateral prefrontal cortex. Neurobiology of Aging, 51, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, J. H. , & Baxter, M. G. (2012). The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nature Reviews Neuroscience, 13, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson, E. J. , Benzing, W. C. , Cole, G. M. , Wang, H. , Emerich, D. F. , Sladek, J. R. , Morrison, J. H. , & Kordower, J. H. (1994). Apolipoprotein E‐immunoreactivity in aged rhesus monkey cortex: Colocalization with amyloid plaques. Neurobiology of Aging, 15, 621–627. [DOI] [PubMed] [Google Scholar]
- Muntané, G. , Horvath, J. E. , Hof, P. R. , Ely, J. J. , Hopkins, W. D. , Raghanti, M. A. , Lewandowski, A. H. , Wray, G. A. , & Sherwood, C. C. (2015). Analysis of synaptic gene expression in the neocortex of primates reveals evolutionary changes in glutamatergic neurotransmission. Cerebral Cortex, 25, 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, J. D. , Bernacchia, A. , Freedman, D. J. , Romo, R. , Wallis, J. D. , Cai, X. , Padoa‐Schioppa, C. , Pasternak, T. , Seo, H. , Lee, D. , & Wang, X. J. (2014). A hierarchy of intrinsic timescales across primate cortex. Nature Neuroscience, 17, 1661–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, P. T. , Alafuzoff, I. , Bigio, E. H. , Bouras, C. , Braak, H. , Cairns, N. J. , Castellani, R. J. , Crain, B. J. , Davies, P. , Tredici, K. D. , Duyckaerts, C. , Frosch, M. P. , Haroutunian, V. , Hof, P. R. , Hulette, C. M. , Hyman, B. T. , Iwatsubo, T. , Jellinger, K. A. , Jicha, G. A. , … Beach, T. G. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. Journal of Neuropathology and Experimental Neurology, 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima, K. , Saitoh, R. , Tanaka, S. , Ohsato‐Suzuki, M. , Ohno, T. , & Kitajima, S. (2012). Life span of common marmoset (Callithrix jacchus) at CLEA Japan breeding colony. Biogerontology, 13, 439–443. [DOI] [PubMed] [Google Scholar]
- Palmqvist, S. , Janelidze, S. , Quiroz, Y. T. , Zetterberg, H. , Lopera, F. , Stomrud, E. , Su, Y. , Chen, Y. , Serrano, G. E. , Leuzy, A. , Mattsson‐Carlgren, N. , Strandberg, O. , Smith, R. , Villegas, A. , Sepulveda‐Falla, D. , Chai, X. , Proctor, N. K. , Beach, T. G. , Blennow, K. , … Hansson, O. (2020). Discriminative accuracy of plasma phospho‐tau217 for Alzheimer disease vs other neurodegenerative disorders. Journal of the American Medical Association, 324, 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas, C. D. , Carlyle, B. C. , Leslie, S. , Preuss, T. M. , Crimins, J. L. , Huttner, A. J. , van Dyck, C. H. , Rosene, D. L. , Nairn, A. C. , & Arnsten, A. F. T. (2018). The aged rhesus macaque manifests Braak‐stage III/IV Alzheimer's‐like pathology. Alzheimer's & Dementia, 14, 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passingham, R. E. , & Smaers, J. B. (2014). Is the prefrontal cortex especially enlarged in the human brain allometric relations and remapping factors. Brain, Behavior and Evolution, 84, 156–166. [DOI] [PubMed] [Google Scholar]
- Pearson, R. C. A. , Esiri, M. M. , Hiorns, R. W. , Wilcock, G. K. , & Powell, T. P. S. (1985). Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America, 82, 4531–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss, T. (2019). Critique of pure marmoset. Brain Behavior and Evolution, 93, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Callejas, J. D. , Fuchs, E. , & Perez‐Cruz, C. (2016). Evidence of tau hyperphosphorylation and dystrophic microglia in the common marmoset. Frontiers in Aging Neuroscience, 8, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, K. , Elce, J. S. , Hamos, J. E. , & Nixon, R. A. (1993). Widespread activation of calcium‐activated neutral proteinase (calpain) in the brain in Alzheimer disease: A potential molecular basis for neuronal degeneration. Proceedings of the National Academy of Sciences of the United States of America, 90, 2628–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, C. , Dehghani, F. , Hubbard, G. B. , Thal, D. R. , Struckhoff, G. , Braak, E. , & Braak, H. (2000). Filamentous tau pathology in nerve cells, astrocytes, and oligodendrocytes of aged baboons. Journal of Neuropathology and Experimental Neurology, 59, 39–52. [DOI] [PubMed] [Google Scholar]
- Song, X. , Jensen, M. Ø. , Jogini, V. , Stein, R. A. , Lee, C. H. , Mchaourab, H. S. , Shaw, D. E. , & Gouaux, E. (2018). Mechanism of NMDA receptor channel block by MK‐801 and memantine. Nature, 556, 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spocter, M. A. , Hopkins, W. D. , Barks, S. K. , Bianchi, S. , Hehmeyer, A. E. , Anderson, S. M. , Stimpson, C. D. , Fobbs, A. J. , Hof, P. R. , & Sherwood, C. C. (2012). Neuropil distribution in the cerebral cortex differs between humans and chimpanzees. Journal of Comparative Neurology, 520, 2917–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner, K. N. , McGowen, M. R. , Chugani, H. T. , Tarca, A. L. , Sherwood, C. C. , Hof, P. R. , Kuzawa, C. W. , Boddy, A. M. , Raaum, R. L. , Weckle, A. , Lipovich, L. , Grossman, L. I. , Uddin, M. , Goodman, M. , & Wildman, D. E. (2013). Characterization of human cortical gene expression in relation to glucose utilization. American Journal of Human Biology, 25, 418–430. [DOI] [PubMed] [Google Scholar]
- Stutzmann, G. E. (2007). The pathogenesis of Alzheimers disease is it a lifelong "calciumopathy"? Neuroscientist, 13, 546–559. [DOI] [PubMed] [Google Scholar]
- Suzuki, W. A. , & Porteros, A. (2002). Distribution of calbindin D‐28k in the entorhinal, perirhinal, and parahippocampal cortices of the macaque monkey. Journal of Comparative Neurology, 451, 392–412. [DOI] [PubMed] [Google Scholar]
- Taniguchi, S. , Fujita, Y. , Hayashi, S. , Kakita, A. , Takahashi, H. , Murayama, S. , Saido, T. C. , Hisanaga, S. , Iwatsubo, T. , & Hasegawa, M. (2001). Calpain‐mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Letters, 489, 46–50. [DOI] [PubMed] [Google Scholar]
- Tardif, S. D. , Mansfield, K. G. , Ratnam, R. , Ross, C. N. , & Ziegler, T. E. (2011). The marmoset as a model of aging and age‐related diseases. Institute for Laboratory Animal Research Journal, 52, 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigges, J. , Gordon, T. P. , McClure, H. M. , Hall, E. C. , & Peters, A. (1988). Survival rate and lifespan of rhesus monkeys at the Yerkes Regional Primate Research Center. American Journal of Primatology, 15, 263–273. [DOI] [PubMed] [Google Scholar]
- Uddin, M. , Opazo, J. C. , Wildman, D. E. , Sherwood, C. C. , Hof, P. R. , Goodman, M. , & Grossman, L. I. (2008). Molecular evolution of the cytochrome c oxidase subunit 5A gene in primates. BMC Evolutionary Biology, 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um, J. W. , Kaufman, A. C. , Kostylev, M. , Heiss, J. K. , Stagi, M. , Takahashi, H. , Kerrisk, M. E. , Vortmeyer, A. , Wisniewski, T. , Koleske, A. J. , Gunther, E. C. , Nygaard, H. B. , & Strittmatter, S. M. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron, 79, 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno, H. , & Walker, L. C. (1993). The age of biosenescence and the incidence of cerebral beta‐amyloidosis in aged captive rhesus monkeys. Annals of the New York Academy of Sciences, 695, 232–235. [DOI] [PubMed] [Google Scholar]
- Vogels, T. , Leuzy, A. , Cicognola, C. , Ashton, N. J. , Smolek, T. , Novak, M. , Blennow, K. , Zetterberg, H. , Hromadka, T. , Zilka, N. , & Schöll, M. (2020). Propagation of tau pathology: Integrating insights from postmortem and in vivo studies. Biological Psychiatry, 87, 808–818. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Gamo, N. J. , Yang, Y. , Jin, L. E. , Wang, X. J. , Laubach, M. , Mazer, J. A. , Lee, D. , & Arnsten, A. F. T. (2011). Neuronal basis of age‐related working memory decline. Nature, 476, 210–213. 10.1038/nature10243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Ramos, B. P. , Paspalas, C. D. , Shu, Y. , Simen, A. , Duque, A. , Vijayraghavan, S. , Brennan, A. , Dudley, A. , Nou, E. , Mazer, J. A. , McCormick, D. A. , & Arnsten, A. F. T. (2007). Alpha2A‐adrenoceptor stimulation strengthens working memory networks by inhibiting cAMP‐HCN channel signaling in prefrontal cortex. Cell, 129, 397–410. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Yang, Y. , Wang, C. J. , Gamo, N. J. , Jin, L. E. , Mazer, J. A. , Morrison, J. H. , Wang, X. J. , & Arnsten, A. F. T. (2013). NMDA receptors subserve working memory persistent neuronal firing In dorsolateral prefrontal cortex. Neuron, 77(4), 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. J. (2020). Macroscopic gradients of synaptic excitation and inhibition in the neocortex. Nature Reviews Neuroscience, 21, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Zhang, Y. , Hu, W. , Xie, S. , Gong, C. X. , Iqbal, K. , & Liu, F. (2015). Rapid alteration of protein phosphorylation during postmortem: Implication in the study of protein phosphorylation. Scientific Reports, 5, 15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Y. , de Lange, S. C. , Scholtens, L. H. , Watanabe, K. , Ardesch, D. J. , Jansen, P. R. , Savage, J. E. , Li, L. , Preuss, T. M. , Rilling, J. K. , Posthuma, D. , & van den Heuvel, M. P. (2019). Genetic mapping and evolutionary analysis of human‐expanded cognitive networks. Nature Communications, 10, 4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad, B. , Jones, R. W. , Wirth, Y. , Stöffler, A. , & Möbius, H. J. (2007). Memantine in moderate to severe Alzheimer's disease: A meta‐analysis of randomised clinical trials. Dementia and Geriatric Cognitive Disorders, 24, 20–27. [DOI] [PubMed] [Google Scholar]
- Yamashima, T. (2013). Reconsider Alzheimer's disease by the 'calpain‐cathepsin hypothesis'—A perspective review. Progress in Neurobiology, 105, 1–23. [DOI] [PubMed] [Google Scholar]
- Yang, S. T. , Wang, M. , Paspalas, C. P. , Crimins, J. L. , Altman, M. T. , Mazer, J. A. , & Arnsten, A. F. (2018). Core differences in synaptic signaling between primary visual and dorsolateral prefrontal cortex. Cerebral Cortex, 28, 1458–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Paspalas, C. D. , Jin, L. E. , Picciotto, M. R. , Arnsten, A. F. T. , & Wang, M. (2013). Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America, 110(29), 12078–12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, F. , Sancheti, H. , Patil, I. , & Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radical Biology and Medicine, 100, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, M. E. , Ohm, D. T. , Dumitriu, D. , Rapp, P. R. , & Morrison, J. H. (2014). Differential effects of aging on dendritic spines in visual cortex and prefrontal cortex of the rhesus monkey. Neuroscience, 274, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Bassam, B. , Thomas, A. G. , Williams, M. , Liu, J. , Nance, E. , Rojas, C. , Slusher, B. S. , & Kannan, S. (2016). Maternal inflammation leads to impaired glutamate homeostasis and up‐regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiology of Disease, 94, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , Raina, A. K. , Lee, H. G. , Chao, M. , Nunomura, A. , Tabaton, M. , Petersen, R. B. , Perry, G. , & Smith, M. A. (2003). Oxidative stress and neuronal adaptation in Alzheimer disease: The role of SAPK pathways. Antioxidants & Redox Signaling, 5, 571–576. [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Rottkamp, C. A. , Boux, H. , Takeda, A. , Perry, G. , & Smith, M. A. (2000). Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle‐related events in Alzheimer disease. Journal of Neuropathology and Experimental Neurology, 59, 880–888. [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Su, B. , Wang, X. , Smith, M. A. , & Perry, G. (2007). Causes of oxidative stress in Alzheimer disease. Cellular and Molecular Life Science, 64, 2202–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]