

Abstract
Introduction
We have earlier shown that hyperoxia (HO)-induced sphingosine kinase 1 (SPHK1)/sphingosine-1-phosphate (S1P) signaling contribute to bronchopulmonary dysplasia (BPD). S1P acts through G protein-coupled receptors, S1P1 through S1P5. Further, we noted that heterozygous deletion of S1pr1 ameliorated the HO-induced BPD in the murine model. The mechanism by which S1P1 signaling contributes to HO-induced BPD was explored.
Methods
S1pr1+/+ and S1pr1+/− mice pups were exposed to either room air (RA) or HO (75% oxygen) for 7 days from PN 1–7. Lung injury and alveolar simplification was evaluated. Lung protein expression was determined by Western blotting and immunohistochemistry (IHC). In vitro experiments were performed using human lung microvascular endothelial cells (HLMVECs) with S1P1 inhibitor, NIBR0213 to interrogate the S1P1 signaling pathway.
Results
HO increased the expression of S1pr1 gene as well as S1P1 protein in both neonatal lungs and HLMVECs. The S1pr1+/− neonatal mice showed significant protection against HO-induced BPD which was accompanied by reduced inflammation markers in the bronchoalveolar lavage fluid. HO-induced reduction in ANG-1, TIE-2, and VEGF was rescued in S1pr1+/− mouse, accompanied by an improvement in the number of arterioles in the lung. HLMVECs exposed to HO increased the expression of KLF-2 accompanied by reduced expression of TIE-2, which was reversed with S1P1 inhibition.
Conclusion
HO induces S1P1 followed by reduced expression of angiogenic factors. Reduction of S1P1 signaling restores ANG-1/ TIE-2 signaling leading to improved angiogenesis and alveolarization thus protecting against HO-induced neonatal lung injury.
Keywords: Neonatal lung disease, Oxidative stress, Sphingosine 1 phosphate receptor, Angiogenesis
Introduction
BPD is a chronic lung disease afflicting the preterm newborn resulting from prolonged oxygen therapy and mechanical ventilation [1, 2]. Exposure of the preterm neonates to HO contributes to poor lung development leading to BPD, which is characterized by fewer and larger alveoli due to decreased alveolar formation and impaired vascular growth [3, 4]. The incidence of BPD has risen over the past 25 years [5–7]. Long-term sequelae of BPD extend into adulthood leading to serious pulmonary disorders [8–12]. Despite advances in the understanding of the pathophysiology of BPD, there has been no breakthrough in the therapeutics. This makes BPD a major inevitable morbidity of prematurity, resulting in significant burden to the society.
Impaired angiogenesis is a salient accompaniment of alveolar simplification that characterizes BPD. The exact pathophysiology of abnormal angiogenesis is unclear [13], however, alveolar epithelial, and lung capillary endothelial cells are known to contribute to angiogenesis and participate in the secondary alveolar septation [14].
Potent bioactive sphingolipids such as sphingosine, sphingosine-1-phosphate (S1P), and ceramide regulate various cell processes including cell survival, proliferation, migration, angiogenesis, and vascular integrity. S1P-mediated signaling differentially regulates lung inflammation and injury in different pathological conditions [15]. The role of sphingosine kinase (SPHK)1/S1P signaling and ROS generation via NADPH oxidase (NOX) proteins in the pathogenesis of BPD has been reported by our lab earlier [16, 17]. SPHK1 phosphorylates sphingosine to S1P. Extracellular signaling of S1P is through ligation to five G protein-coupled S1P receptors, S1P1–5 [18]. Studies from our laboratory have unraveled a role for SPHK1 in the pathogenesis of BPD and SPHK1 inhibitor PF543 enabled improved lung development along with reduction of adverse airway remodeling [5, 16, 17]. We have shown that HO augments S1P levels in murine neonatal lung tissues and human lung microvascular endothelial cells (HLMVECs) [16]. Also, HO mediated ROS generation is in part, S1P dependent and is mediated through receptor S1P1 [17]. The downstream effect of S1P depends upon the type (s) of receptor(s) activated. S1P receptor modulation using Fingolimod, a sphingosine analog finds its clinical use in the effective therapy of multiple sclerosis (MS). Very little is known regarding the role of S1P receptor(s) in BPD. In this context we intended to study the role of S1P/S1P1 signaling in BPD.
We noted an increased expression of S1P1 in the lung tissue of neonatal mice exposed to HO. An increased S1P1 expression is known to suppress angiogenesis and sprouting of new vessels [18–20]. Therefore, we hypothesized that enhanced S1P1 expression during HO disrupts angiogenesis and alveolarization resulting in BPD. Our results suggest a reduction in the angiogenic factor such as Angiopoietin-1 (ANG-1) and its receptor TIE-2 following HO, which was restored following disruption of S1P1 signaling.
Materials and Methods
Mouse experiments and animal care
All experiments using S1pr1+/+ (WT) and S1pr1+/− animals were approved by the Institutional Animal Care and Use Committee at the Institutional Review Board of the University of Illinois at Chicago (UIC) (Protocol # 2012–1018). Genetic deletion of both alleles of S1P1 (S1pr1−/−) is embryonically lethal [21]. Therefore, we used S1pr1 heterozygous mice, S1pr1+/− provided to Dr. V. Natarajan by Dr. Richard L. Proia (NIDDK, National Institutes of Health, Bethesda) to evaluate role of S1P1 in alveolarization and lung development. The S1pr1+/+ littermate pups were used as the wild type controls. The neonatal pups with their lactating dams used for the study were rotated between room air (RA) and hyperoxia (HO) every 24 h [16]. The neonates were exposed to RA or HO (75% O2) from postnatal day (PN) PN1 to PN7. Following exposure to RA or HO, the pups were humanely sacrificed on PN 7 to collect lungs for histology and bronchoalveolar lavage (BAL). About 6–10 mice (both male and female) were used in each of the experiments and representative data are shown.
BAL collection and analysis
BAL collection was based on our previous protocol [16, 17]. Briefly, the mice were euthanized, the trachea were exposed, the tubing were secured, and BAL collection with PBS was performed. Protein concentrations in BAL fluid were measured using Bio-Rad Protein Assay (Bio-Rad, Hercules, CA).
Lung preparation for histology
The animals were intubated via the trachea, the left lung lobe was inflated with 10% neutral buffered formalin (at a pressure of 20 cm H2O), followed by fixation for a minimum of 24 h, and processed for paraffin embedding and sectioning. Lung tissue was cut into 5 μm sections at three levels from the apex to the base of the lung for subsequent alveolar analyses after hematoxylin and eosin (HE) staining.
Immunohistochemistry (IHC) was done to assess the expression of various proteins of interest. The sections were pretreated for antigen retrieval (10 mM citrate buffer), endogenous peroxidase was quenched by using 3% H2O2 followed by blocking nonspecific binding with appropriate blocking serum. Sections were then incubated overnight at 4 °C with primary antibodies of von Willebrand Factor (1:250, cat # ab9378, Abcam, Cambridge, MA), ANG-1 (1:250, cat # 23302–1-AP, Proteintech, Rosemont, IL), TIE-2 (1:600, cat # PA585241, Thermo Fisher), and VEGF (1:600, cat # 19003–1-AP, Proteintech, Rosemont, IL) followed by application of biotinylated secondary antibodies goat anti-mouse or goat anti-rabbit as indicated (1:700 & 1:800, Jackson ImmunoResearch, West Grove, PA). The indirect ABC method (cat # PK 6100, Vector Laboratories, Burlingame, CA) was performed and diaminobenzidine chromogen (ImmPACT TMDAB, Vector Laboratories Burlingame, CA) was used to visualize antibodies. The sections were counterstained with methylene blue. Appropriate negative controls were run by omitting the primary antibodies to confirm nonspecific staining. Stained sections were dehydrated and cover slipped with permount mounting medium (Fisher Scientific International, Pittsburgh, PA). All images were obtained using a Leica DFC7000T camera mounted on a Leica DMLB microscope (Leica Microsystems, Germany). The stained sections were analyzed using Image J software and the intensity of staining measured per area.
Morphometric analysis of lung histology
The objective assessment of alveolarization was calculated by the mean linear intercept (MLI) method [16, 17]. A minimum of 50 alveoli were measured from images obtained with 10x magnification. At least two sections from each pup were used for analysis.
Exposure of cells to HO
HLMVECs in complete EGM-2 medium with passages between 5 and 8 (~90% confluence) were exposed to 95% HO as described earlier [17]. The concentration of O2 inside the Billups-Rothenberg modular incubator chamber was monitored using digital oxygen monitor. The buffering capacity of the cell culture medium did not change significantly during the period of HO exposure and was maintained at a pH ~7.4.
Pretreatment of cells with S1P1 inhibitor
HLMVECs grown to ~90% confluence was serum starved for 1 h and preincubated with NIBR-0213 (1 µM) in serum-free or media containing 1% FBS as indicated for 1 h prior to stimulation with HO (95% O2, 5%CO2) for 24 h.
Luciferase reporter assay
DNA fragments of 2 kb human S1PR1 gene promoter were amplified by polymerase chain reaction (PCR) using the human genomic DNA as described [22]. The fragments were fused to a pGL3-basic reporter vector (Promega, Madison, WI, USA) and transfected into HLMVECs. A plasmid with renilla luciferase gene (phRL-TK) was co-transfected by Fugene HD (Promega Co., Fitchburg, WI, USA) as a control. Transfected EC were cultured in growth medium, exposed to RA or HO for 6 h and lysed in passive lysis buffer. HLMVECs cultured in VEGF containing media was used as the positive control. Luciferase activity was measured by Dual-Luciferase Assay Kits and GloMax-Multi Detection System (Promega). The relative activities were expressed as the ratio of firefly luciferase in pGL3 to renilla luciferase in phRL-TK (RLU).
Western blotting
The protein expression was detected in the mouse lung and the HLMVECs as described earlier [16, 17]. The RA and HO-exposed lungs/HLMVECs were lysed for protein extraction, quantitated using BCA protein assay (Pierce, 23225) and subjected to immunoblotting. The antibodies against S1P1 (1:500), TIE-2 (1:500), KLF-2 (1:1000), KLF-4 (1:1000), ANG-1 (1:500), and GAPDH (1:20000, cat # 10494–1-AP, Proteintech) were used.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The gene expression was detected in the mouse lung and the HLMVECs after RA or HO exposure. Briefly, total RNA was extracted using Trizol reagent (Sigma, USA) according to the manufacturer’s instructions and quantified using a Nanodrop (Thermoscientific, USA). One microgram of RNA was converted to cDNA using the Superscript III reverse transcriptase enzyme (Invitrogen). About 250 ng of cDNA was mixed with respective forward and reverse primers using the specific primers and added to the SYBR-Green I master mix-No ROX (Roche, USA). Thirty-five cycles of reaction were performed on the iCycler (BioRad). For each gene, an assessment of quality was performed by examining PCR melt curves after real-time PCR to ensure specificity. The cycle threshold (Ct) value of the target gene was analyzed after normalization to the Ct value of GAPDH. Fold change (2−ΔΔCt) was calculated by comparing the expression levels of mRNA extracted from RA and HO samples.
Determination of HO-induced production of ROS
Total ROS production in HLMVECs, exposed to either RA or HO, was determined by the DCFDA fluorescence method. Briefly, HLMVECs (~90% confluent) were loaded with 10 μM DCFDA in EGM-2 basal medium and incubated at 37 °C for 30 min. The medium containing DCFDA was then aspirated; cells were rinsed once with EGM-2 complete medium, and cells were preincubated with vehicle and S1P1 inhibitor treated agents for the indicated time periods, followed by exposure to either RA (95% air and 5% CO2) or HO (95% O2 and 5% CO2) for 3 h. The cells were washed and were examined under a Nikon Eclipse TE 2000-S fluorescence microscope (Tokyo, Japan) with a Hamamatsu digital charge-coupled device camera (Hamamatsu, Japan) using a 20x objective lens.
Statistical analysis
Histological and morphometric data were analyzed by GraphPad Prism (GraphPad software, La Jolla, CA). Student’s t-test and analysis of variance (ANOVA) were used to compare means of two or more different treatment groups. The level of significance was set to ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. Results were expressed as mean ± SEM.
Results
HO increases expression of S1P1 in neonatal lungs
Exposure of wild-type (WT) mouse neonatal pups (n = 5–8 in each group) to HO (75% O2) for 7 days stimulated both mRNA (3.3 ± 0.2-fold) (Fig. 1A) and protein expression (3.4 ± 0.1-fold) of S1P1 in the lungs (Fig. 1B, C).
Fig. 1.
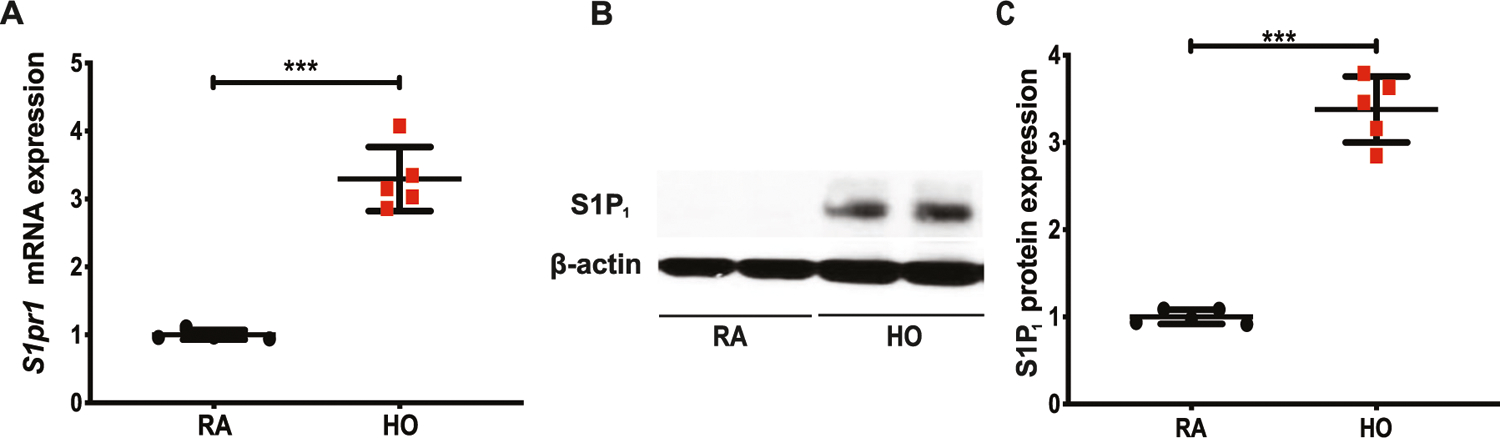
Hyperoxia (HO) increases the expression of S1P1. WT neonatal mice were exposed to HO (75% O2) or room air (RA) from postnatal day 1 (PN1) for 7 days. Quantitative real time-PCR showed increased expression of S1pr1 following HO (A). Western blot analysis of whole lung tissue lysates showed increased expression of S1P1 following HO in WT compared to RA controls (B, C). Statistical analysis was done with ANOVA test. *** indicates p < 0.0001, n = 5–8/group
Partial deletion of S1P1 protected neonatal mice from HO-mediated BPD
Following 7 days of HO exposure, impaired alveolar formation was noted in the S1pr1+/+ neonatal mice compared to RA controls. S1pr1+/− neonatal mice were resistant to HO-induced lung injury showing improved alveolarization (Fig. 2A, B). Quantitative analysis confirmed a reduction in alveolar size in the S1pr1+/− group exposed to HO (MLI of 60.8 ± 1.2 µm) compared with S1pr1+/+ HO-exposed mice (MLI of 86.2 ± 1.4 µm) and the RA controls, S1pr1+/− (MLI of 33.2 ± 1.1 µm) and S1pr+/+ (MLI of 34.4 ± 1.3 µm). This was accompanied by reduced inflammation, as evidenced by reduced protein concentration in BAL collected from the lungs of S1pr1+/− neonatal mice exposed to HO (0.079 ± 0.002 mg/mL) compared with their S1pr1+/+ litter mate controls exposed to HO (0.1 ± 0.009 mg/mL) (Fig. 2C). The protein levels in BAL of RA vehicle group were not statistically different (0.05 ± 0.004 mg/ml). We compared neonatal S1pr3−/− mice (also provided by Dr. Proia, NIH) exposed to HO to their WT counterparts (S1pr3+/+). Neonatal S1pr3−/− mice did not show protection against HO-induced lung injury compared to S1pr3+/+ (Fig. 2D, E, F).
Fig. 2.
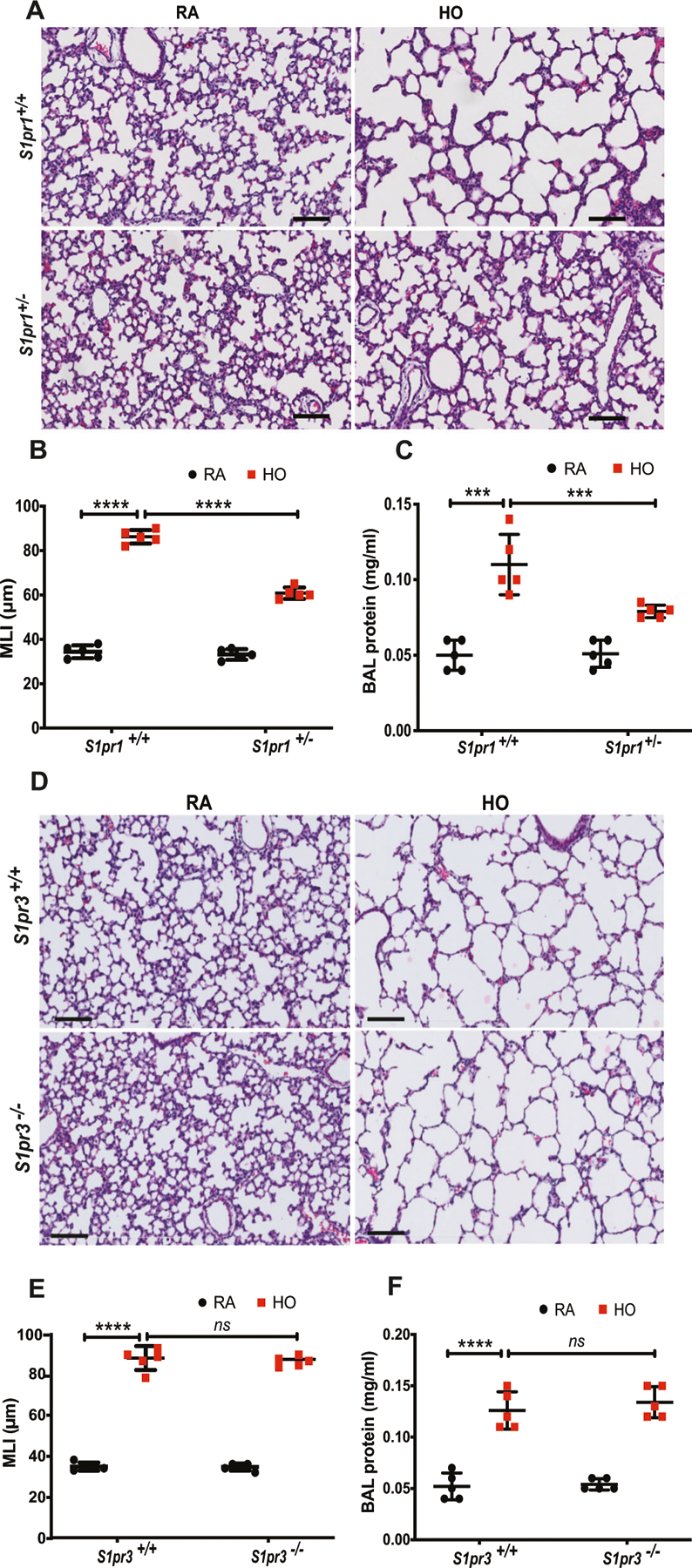
S1pr1 heterozygous (S1pr1+/−) newborn mice showed protection against hyperoxia (HO) induced lung injury whereas S1pr3−/− did not. Representative H&E photomicrographs of lung sections obtained from WT or S1pr1+/− neonatal mice exposed to HO (75% O2) from postnatal day 1 (PN1) for 7 days showed improved alveolarization as compared to S1pr1+/+ controls (A). Objective assessment of alveolarization demonstrated smaller alveoli with shorter mean linear intercept (MLI) in the S1pr1+/− group as compared to controls (B) and less inflammatory exudate in bronchoalveolar lavage fluid (C). Representative H&E photomicrographs of lung sections obtained from WT (S1pr3+/+) or S1pr3−/− neonatal mice exposed to HO (75% O2) from PN1 for 7 days showed no protection against HO-induced BPD as compared to WT controls (D). Objective assessment of alveolarization demonstrated larger alveoli with longer mean linear intercept (MLI) in both the HO-exposed group as compared to RA controls (E) and increased inflammatory exudate in bronchoalveolar lavage fluid (F). Statistical analyses were done with ANOVA. ***p < 0.001, n = 5–8/group
Partial deletion of S1P1 rescued TIE-2 and ANG-1 expression
mRNA analysis by qRT-PCR of lung tissue harvested from WT neonatal mouse exposed to HO showed a significant reduction in the Tie2 gene expression (0.3 ± 0.03-fold) as compared to its RA control (Fig. 3A). Similarly, there was a significant reduction in TIE-2 protein expression (Fig. 3B) in the HO-exposed WT mouse lung tissue (0.3 ± 0.04-fold). Upon performing IHC, the lung tissue from WT (S1pr1+/+ mice) showed a significant decrease in TIE-2 (0.58 ± 0.03-fold) compared to RA (1 ± 0.01) (Fig. 3C). There was also a concomitant reduction in ANG-1 in the WT pups exposed to HO (0.5 ± 0.04-fold) (Fig. 3E). This reduction was rescued in S1pr1+/− mice exposed to HO with a fold change of 0.74 ± 0.02 for TIE-2 (Fig. 3D) and 0.82 ± 0.03 for ANG-1 (Fig. 3F). Quantitative analysis by Image J confirmed this observation (Fig. 3D, F).
Fig. 3.
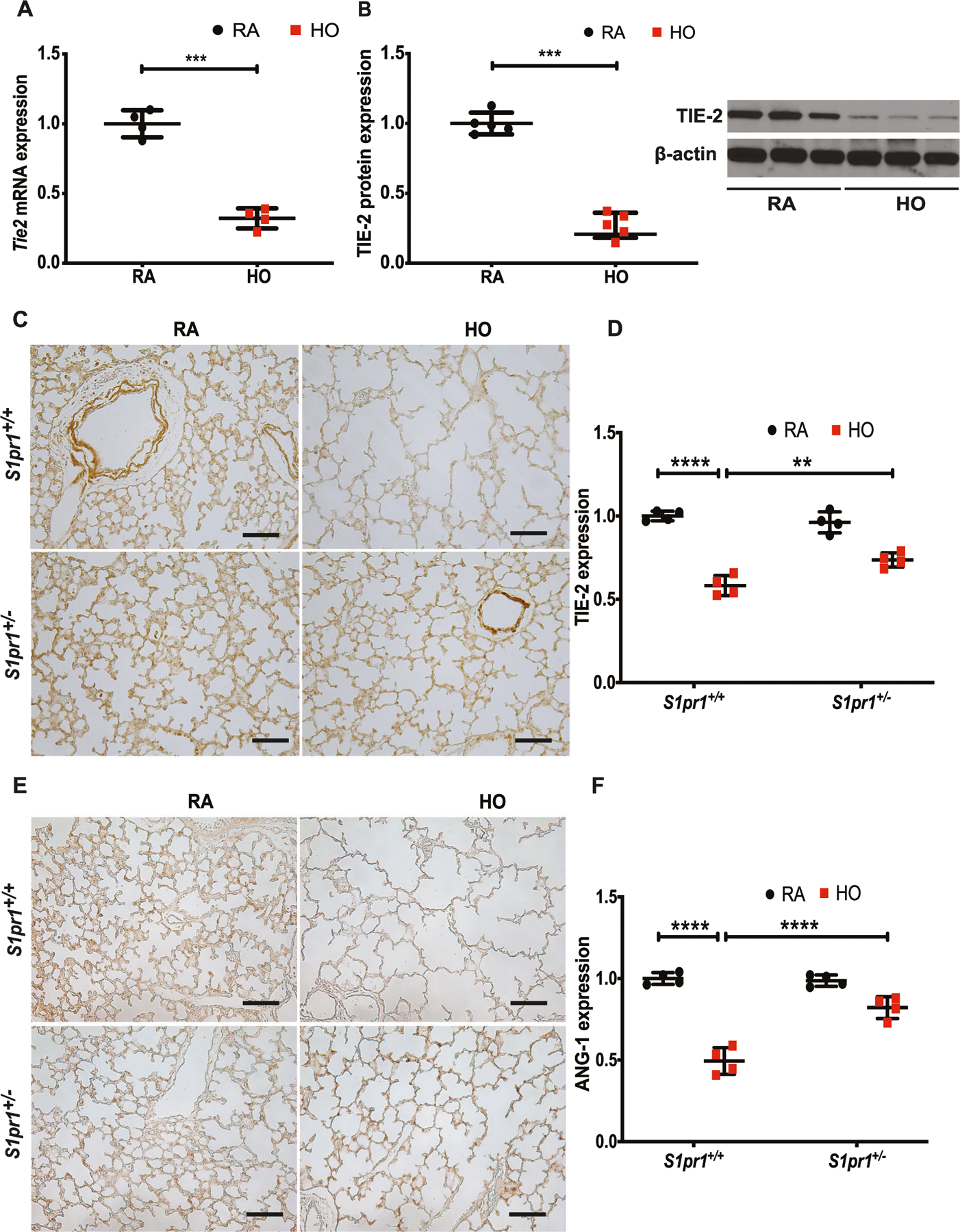
Hyperoxia (HO) reduced the expression of TIE-2 and ANG-1. Western blot analysis of mouse lung tissue subjected to HO showed decreased expression of Tie2 at RNA (A) and TIE-2 at protein levels (B) in WT mice. Immunohistochemistry (IHC) of lung tissue showed HO induced decrease in expression of TIE-2 in S1pr1+/+ mice which recovered in S1pr1+/− mice (C, D). IHC showed a decreased expression of ANG-1 in the HO exposed S1pr1+/+ mice which was rescued in the S1pr1+/− mice exposed to HO (E, F). The statistical analysis was carried out using ANOVA where ** indicate p < 0.01, ***p < 0.001, ****p < 0.0001, n = 5–8/group
Partial deletion of S1P1 rescued VEGF expression
Immunohistochemical analysis of the mouse lung tissue subjected to HO showed a decrease in VEGF expression (0.68 ± 0.02-fold) in the S1pr1+/+ group, which significantly improved (0.83 ± 0.01-fold) in the S1pr1+/− HO group (Fig. 4A, B). The number of order-1 arterioles (less than 20 µm diameter) decreased in the S1pr1+/+ group exposed to HO (8.4 ± 0.6) and was relatively more in the lungs of the S1pr1+/− partial knock out mice (12.4 ± 0.3) (Fig. 4C) compared to the RA controls (17 ± 0.2-fold and 15 ± 0.1-fold for S1pr1+/+ and S1pr1+/− RA groups, respectively).
Fig. 4.
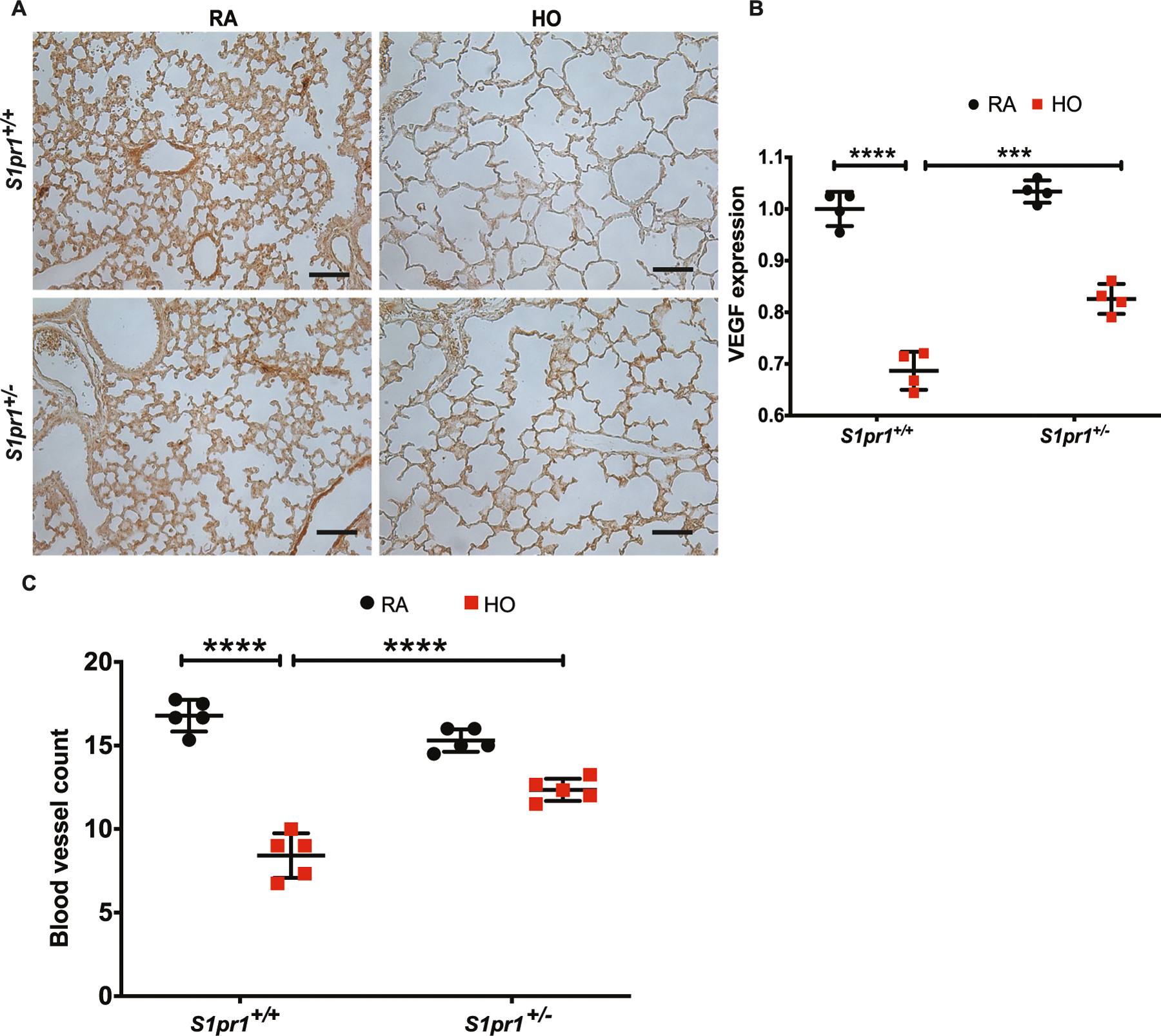
Hyperoxia (HO) exposure reduced the expression of VEGF and blood vessel count. HO exposure resulted in reduced VEGF in lung tissue of S1pr1+/+ mice as shown by immunohistochemistry (A, B). This reduction was ameliorated in S1pr1+/− mice which showed higher expression of VEGF. HO was accompanied by a significant reduction in the number of order-1 arterioles in the WT mice which was improved in the S1pr+/− mice (C). The statistical analysis was carried out using ANOVA where *** indicate p < 0.001, n = 5–8/group
HO enhances expression of Kruppel like factor (KLF)-2 and 4 and NF-κB in neonatal mouse lungs
In silico analysis of S1P1 promoter by Genomatix software revealed binding sites for NF-κB and KLF-2 upstream of transcription start site (TSS) for S1P1 (Fig. 5A–C). The expressions of Klf2 (3.8 ± 0.3) and Klf4 mRNA (4.9 ± 0.4) (Fig. 5D, E) in neonatal mouse lungs exposed to HO (75% for 7 days) were increased. The KLF-4 protein expression also increased (Fig. 5F) to 3.3 ± 0.2-fold in HO compared to RA (Fig. 5G). Similarly, short term exposure of HLMVECs to HO (60 min) stimulated NF-κB activation, as evidenced by translocation of p65 to the nucleus (Fig. 5H) which is quantified and represented in Fig. 5I (1 ± 0.03 for RA vs 1.6 ± 0.04 for HO treated cells).
Fig. 5.
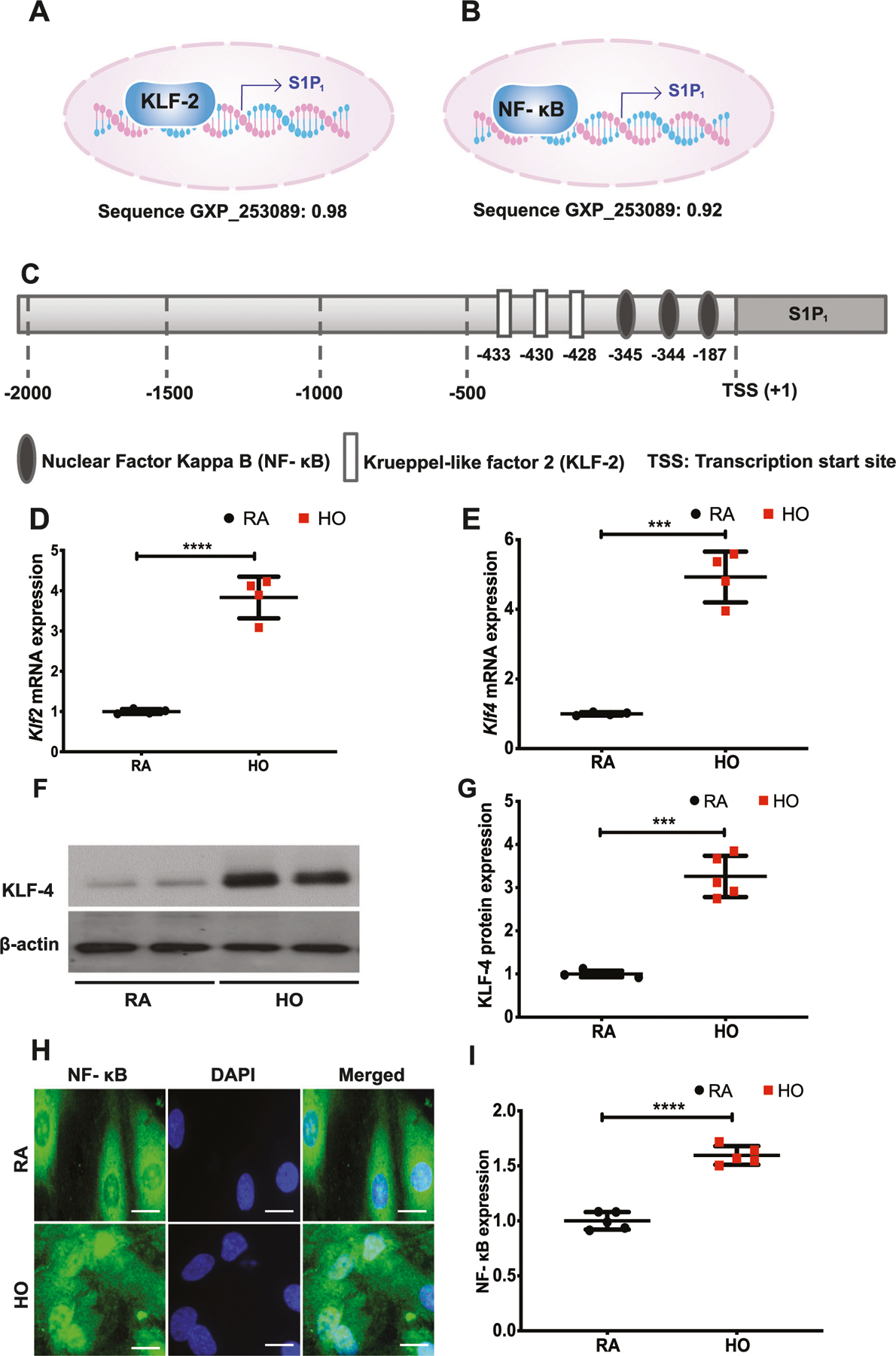
Hyperoxia (HO) induced the expression of Klf2 and Klf4 accompanied by a translocation of NF-κB to the nucleus. In silico analyses showed a strong probability for NF-κB and KLF-2 binding to the promoter site of S1P1 with a score of 0.98 and 0.92, respectively (A–C). Real time PCR analysis of lung tissues obtained from WT neonatal mice exposed to HO (75% O2) showed increased mRNA expression of Klf2 (D), and Klf4 (E). Western blot analysis showed increased expression of KLF-4 (F, G). Immunofluorescence showed an increased translocation of NF-κB to the nucleus (H, I). Statistical analysis was done using ANOVA test. *** for p < 0.001 and **** for p < 0.0001, HO vs RA and HO vs treatment. n = 6–8/group
HO induces S1P1 and ROS in HLMVECs and increase in ROS is attenuated by S1P1 inhibitor
HLMVECs exposed to 95% O2 for 24 h showed enhanced S1P1 protein expression (2.2 ± 0.3-fold) (Fig. 6A, B). The ability of HO to modulate S1P1 was further confirmed by luciferase reporter assay. As shown in Fig. 6C, HO enhanced luciferase reporter activity of S1PR1 to 7.0 ± 0.5-fold compared to RA (normalized to Renilla). HO stimulated ROS generation, as evidenced by DCFDA oxidation, and increased fluorescence. This HO-induced increase in ROS production was significantly decreased by S1P1 inhibition in cells (Fig. 6D). Quantitative data of fluorescence intensity by Image J (NIH, Bethesda, MD) showed a 4.4 ± 0.2-fold increase in HO as compared to NO controls. HO induced ROS generation decreased 2.0 ± 0.2-fold upon treatment with S1P1 inhibitor (Fig. 6E).
Fig. 6.
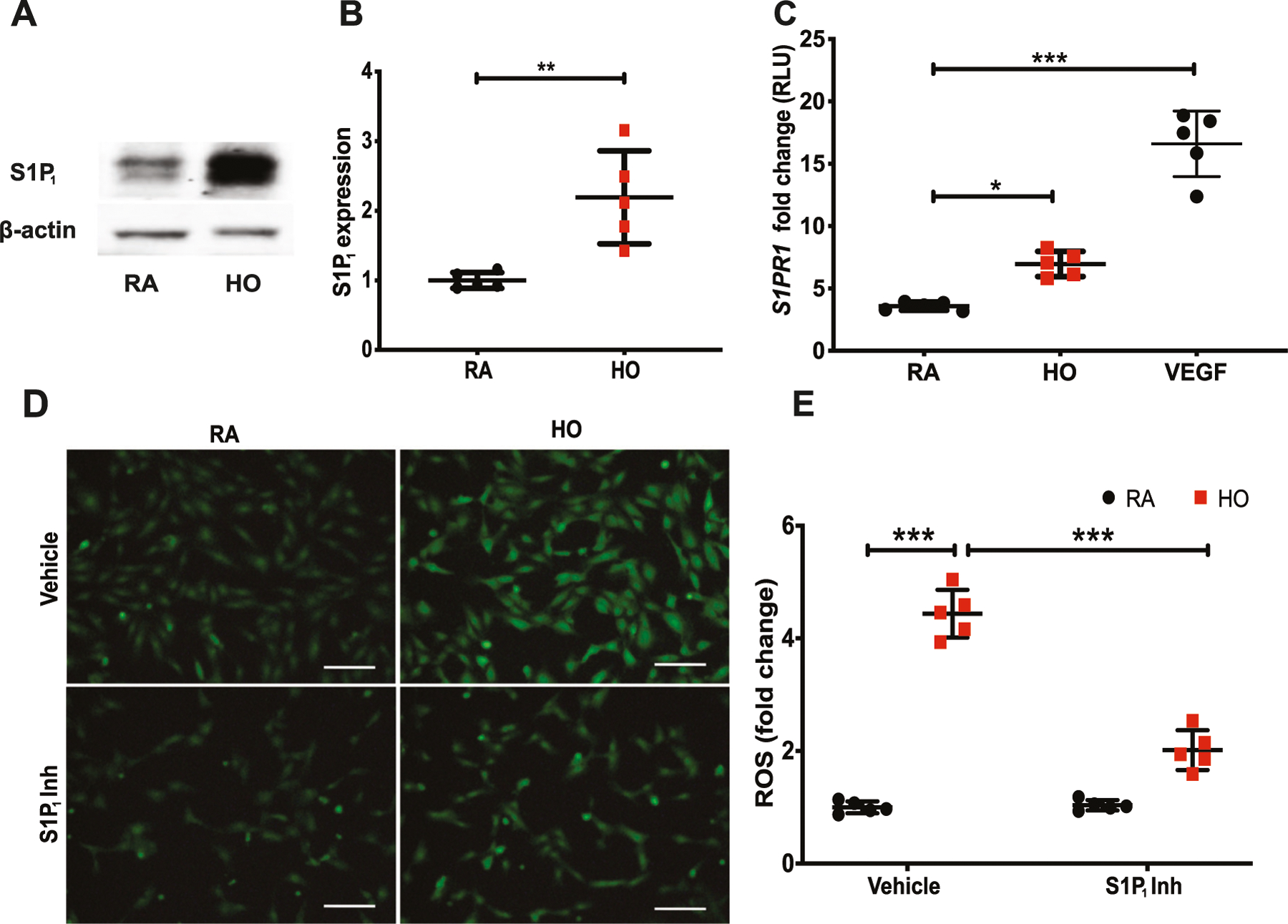
Hyperoxia (HO) increased the expression of S1P1 in human lung microvascular endothelial cells (HLMVECs) and S1P1 inhibition blocked HO-induced reactive oxygen species (ROS) generation. Western blot analysis of HLMVECs exposed to 95% HO or NO for 24 h showed increased expression of S1P1 (A, B). Luciferase reporter assay on HLMVECs exposed to 95% O2 or RA for 24 h showed increased S1PR1 gene transcription following HO (C). VEGF was used as a known promoter of S1PR1. HLMVECs treated with the vehicle or S1P1 inhibitor and then exposed to HO for 3 h showed increased production of ROS as determined by the DCFDA fluorescence method. The HO induced increase in the production of ROS was inhibited by the S1P1 inhibitor (D, E). The statistical analysis was carried out using ANOVA where * indicate p < 0.05, **p < 0.01, ***p < 0.001, n = 5
HO-induced decrease in TIE-2 expression is rescued by S1P1 inhibition in HLMVECs
S1P1 inhibitor treatment reversed the HO-induced reduction of TIE-2 in HLMVECs (1.08 ± 0.03-fold) (Fig. 7A, B). Exposure of HLMVECs to HO stimulated KLF-2 expression (Fig. 7C). Quantitatively, there was a 1.4 ± 0.05-fold increase in KLF-2 expression upon HO which reduced to 1.0 ± 0.09-fold upon inhibitor treatment and is statistically similar to that of RA control (Fig. 7D).
Fig. 7.
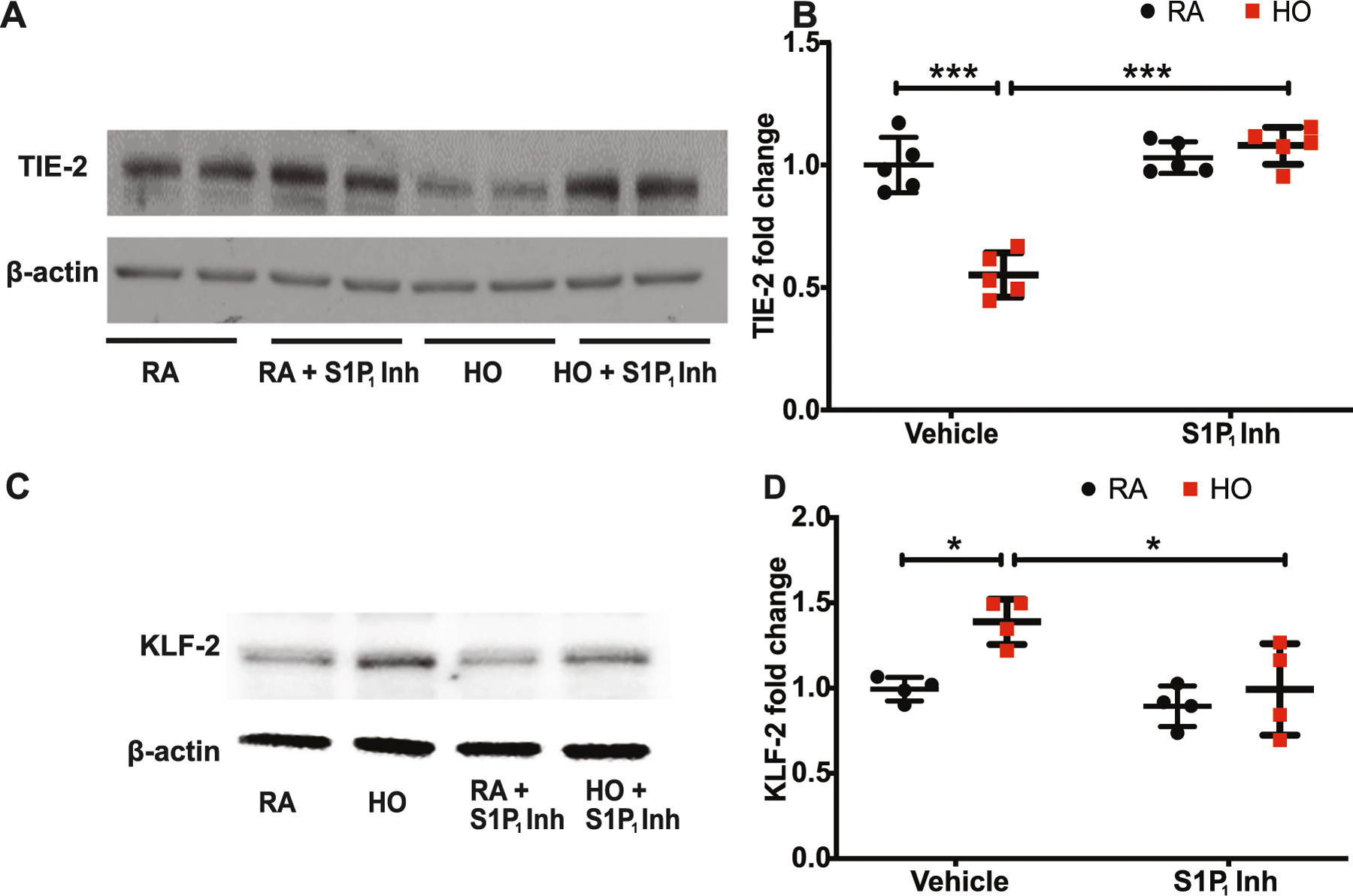
Hyperoxia (HO)-induced alteration in TIE-2 and KLF-2 were restored by S1P1 inhibition. Hyperoxia (HO)-reduced TIE-2 expression in human lung microvascular endothelial cells (HLMVECs) which was restored with the S1P1 inhibitor treatment (A, B). HLMVEC exposed to HO increased the expression of KLF-2 which was reduced by inhibitor treatment (C, D). Statistical analysis was done using ANOVA test. * for p < 0.05, *** for p < 0.001 and, HO vs RA and HO vs treatment
Discussion
BPD is a neonatal lung condition with long term pulmonary sequelae. The pathological role of sphingolipids in BPD has been recently identified [5, 16, 23, 24]. S1P is a potent bioactive sphingolipid with a diverse range of functions, which are mediated via S1P1 receptor [8]. However, the role of S1P1 in the pathogenesis of BPD is not well understood, and hence this was investigated in the current study. We observed an increased expression of S1P1 in the WT neonatal mice exposed to HO and partial genetic deletion of S1pr1 (S1pr1+/−) in mice conferred protection against developing experimental BPD.
Recent studies have shown that elevated S1P1 expression inhibits sprouting angiogenesis in developing blood vessels by affecting endothelial capillary formation [18, 20, 25]. BPD is characterized by suppression of sprouting angiogenesis and arrest of secondary septation [4]. The present study demonstrated that by genetically abrogating the expression of S1P1 receptor, angiogenesis is improved with rescue of impaired alveolarization seen in BPD. The current study also delineated the role of S1P/S1P1 signaling in HO-induced generation of ROS, and angiogenesis in the lungs thus identifying S1P1 signaling as therapeutic target for BPD. Genetic deletion of S1P1 (S1pr1+/−) conferred protection against HO-induced lung inflammation/injury and improved alveolarization. We also found that S1P1 expression was significantly elevated accompanied with altered expression of angiogenic factors including elevated Kruppel like factors (KLF)-2 and 4, reduced TIE-2, ANG-1, and VEGF expression in WT neonatal pups exposed to HO. This was reversed in the S1pr1+/− mice exposed to HO, suggesting suppression of angiogenesis by increased S1P1 (Figs. 3, 4, and 5).
KLF-2 and 4 have been shown to be key transcriptional regulators of normal lung development and angiogenesis [26]. KLF-2 is a transcription factor and a known promoter for S1P1 whereas KLF-4 regulates multiple pathways including that of NF-κB. We noted an increase in Klf2 gene expression in lung tissue and the protein expression in HLMVECs following HO, which could be a stress response to HO [27]. An increase in KLF-2 is interestingly accompanied by reduced HO-induced angiogenesis. This could be because KLF-2, as a promoter of S1P1, triggers a pathological increase in S1P1 inhibiting angiogenesis. In addition, NF-κB could be promoting S1P1 expression too inhibiting angiogenesis. ANG-1 is a ligand for the receptor tyrosine kinase TIE-2 and is expressed on lung endothelial and epithelial cells. ANG-1 secretion has been shown to be responsible for restoring epithelial protein permeability through suppression of NF-κB activity in human type II alveolar epithelial cells [28]. ANG-1/TIE-2 signaling has been shown to be mainly involved in angiogenic activity and promoting maturation of blood vessels, regulated by Akt and MAPK signaling [29]. A downregulation of ANG-1/TIE-2 signaling was observed in miR-34a induced HO model of BPD involving vascular and alveolar malformation. This downregulation was observed in type II alveolar epithelial cells isolated from human samples as well as MLE-12 cells and was rescued with the inhibition of the miRNA. Significant downregulation of ANG-1 and its receptor TIE-2 has been noted in pulmonary hypoplasia associated with animal models of congenital diaphragmatic hernia [30]. We noted that partial deletion of S1P1 (S1pr1+/−) in mice restored the ANG-1/TIE-2 expression, which was downregulated in HO [31, 32]. In in vitro studies using HLMVECs, a reduced expression of TIE-2 was noted following HO which was restored by S1P1 inhibition. TIE-2 plays a crucial role in the maintenance of normal vasculature and angiogenesis. Fluorescent co-labeling of TIE-2 with CD34 demonstrated that expression of the TIE-2 receptor is co-localized to the endothelium of the developing vasculature during early lung development [30]. It has been shown that TIE-2 expression progressively increases during lung development from the canalicular to saccular stages plateauing thereafter. TIE-2-mediated signaling has been shown to promote development of the perialveolar vasculature in the surrounding embryonic mesenchyme [33, 34]. Our data suggest that genetic abrogation of S1pr1 allele reduced increased expression of S1P1 under HO and restored ANG-1 and TIE-2.
We observed a decrease in VEGF in the HO exposed lung tissue which was restored upon S1P1 deletion. HO-induced lung damage is associated with decreased lung VEGF and VEGFR-2 expression [4, 35, 36]. Postnatal intratracheal adenovirus-mediated VEGF gene therapy improved survival, promoted lung capillary formation, and preserved alveolar structure. Combined VEGF and ANG-1 gene transfer, matured the new vasculature, reducing the vascular leakage seen in VEGF-induced capillaries [4]. Significantly decreased VEGF levels were found in the tracheal aspirates of infants born at 28–29 weeks gestation who later developed BPD [37]. Interestingly VEGF is a known inducer of S1P1 which we had used in our luciferase promoter experiment. HO triggered an increase in S1P1 accompanied by a reduction in VEGF. Going beyond an association the mechanism of this regulation is to be investigated in future. Another interesting aspect that merits further mechanistic studies include the mechanism by which S1P/S1P1 signaling modulates ANG-1/TIE-2 signaling regulating angiogenesis. The proposed mechanism is depicted in Fig. 8.
Fig. 8.
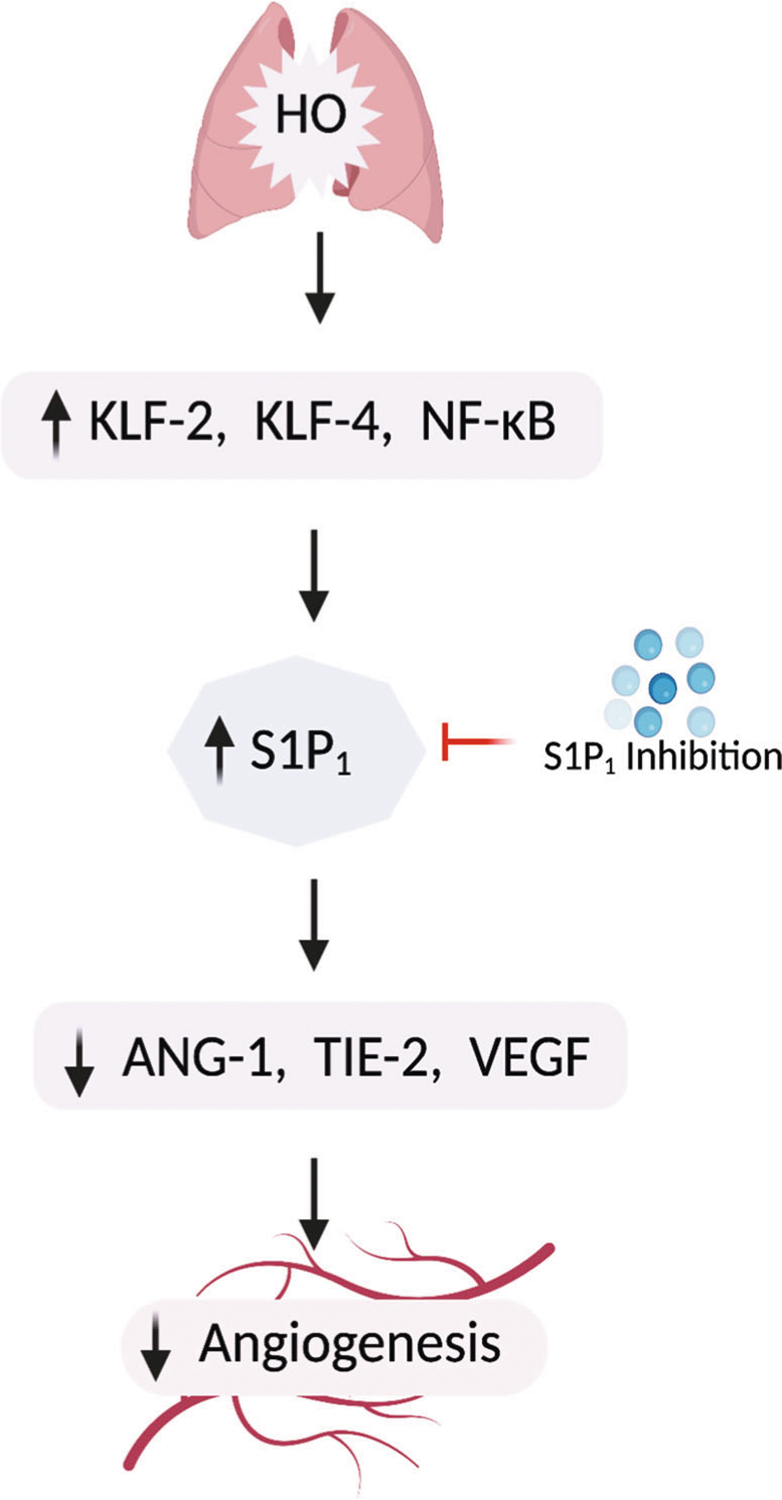
Proposed mechanism. Hyperoxia (HO) triggers an increase in KLF-2 and KLF-4 and activates NF-κB. These transcription factors putatively bind to S1P1 promoter resulting in increased transcription of S1P1. The increased S1P1 results in reduced ANG-1, TIE-2, and VEGF which are factors important for blood vessel maintenance and angiogenesis, there by inhibiting angiogenesis. S1P1 inhibition reversed the inhibition of angiogenesis by increased S1P1 ameliorating BPD. Thus, S1P1 could serve as a potential druggable target for treating BPD
In summary, our data demonstrates that S1P/S1P1 signaling axis plays a key role in the pathogenesis of BPD and suppression of S1P1 improves angiogenesis. In this study, we also identified the role of key downstream targets of S1P/S1P1 signaling pathway and defined the therapeutic effectiveness of S1P1 inhibition against HO-induced BPD in the preclinical model. The identification of the involvement of ANG 1/TIE 2 signaling as well as VEGF/KLF2 signaling has the potential to identify more druggable targets for BPD.
Acknowledgements
We gratefully acknowledge the assistance of Research Resources Center histology core of University of Illinois, Chicago, and that of University of Chicago in the processing of lung tissue, including IHC and image processing. We have used biorender.com to create the illustrations.
Funding
This work was supported in part by R01HD090887–01A1 from Eunice Kennedy Shriver National Institute of Child Health and Human Development and by Transitional Grant # 18TPA34230095 from American Heart Association to AH. No role was played by the funding body in the design of the study, collection, analysis and interpretation of data, or in writing the manuscript.
Footnotes
Competing interests The authors declare no competing interests.
References
- 1.Davis JM (2002). Role of oxidant injury in the pathogenesis of neonatal lung disease. Acta Paediatrica, 91(437), 23–25. 10.1111/j.1651-2227.2002.tb00156.x. [DOI] [PubMed] [Google Scholar]
- 2.Asikainen TM, & White CW (2004). Pulmonary antioxidant defenses in the preterm newborn with respiratory distress and bronchopulmonary dysplasia in evolution: Implications for antioxidant therapy. Antioxidants & Redox Signaling, 6(1), 155–167. 10.1089/152308604771978462. [DOI] [PubMed] [Google Scholar]
- 3.Domm W, Misra RS, & O’Reilly MA (2015). Affect of early life oxygen exposure on proper lung development and response to respiratory viral infections. Frontiers in Medicine, 2, 55. 10.3389/fmed.2015.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, & Archer SL (2005). Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation, 112(16), 2477–2486. 10.1161/CIRCULATIONAHA.105.541524. [DOI] [PubMed] [Google Scholar]
- 5.Ha AW, Sudhadevi T, Ebenezer DL, Fu P, Berdyshev EV, Ackerman SJ, & Harijith A (2020). Neonatal therapy with PF543, a sphingosine kinase 1 inhibitor, ameliorates hyperoxia-induced airway remodeling in a murine model of bronchopulmonary dysplasia. American Journal of Physiology Lung Cellular and Molecular Physiology, 319(3), L497–L512. 10.1152/ajplung.00169.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zysman-Colman Z, Tremblay GM, Bandeali S, & Landry JS (2013). Bronchopulmonary dysplasia - trends over three decades. Paediatrics & Child Health, 18(2), 86–90. 10.1093/pch/18.2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Reilly M, Sozo F, & Harding R (2013). Impact of preterm birth and bronchopulmonary dysplasia on the developing lung: long-term consequences for respiratory health. Clinical and Experimental Pharmacology & Physiology, 40(11), 765–773. 10.1111/1440-1681.12068. [DOI] [PubMed] [Google Scholar]
- 8.Sudhadevi T, Ha AW, Ebenezer DL, Fu P, Putherickal V, Natarajan V, & Harijith A (2020). Advancements in understanding the role of lysophospholipids and their receptors in lung disorders including bronchopulmonary dysplasia. Biochimica Et Biophysica Acta. Molecular and Cell Biology of Lipids, 1865(7), 158685. 10.1016/j.bbalip.2020.158685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoll BJ, Hansen NI, Bell EF, Shaκaran S, Laptook AR, & Walsh MC, Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. (2010). Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics, 126(3), 443–456. 10.1542/peds.2009-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landry JS, & Menzies D (2011). Occurrence and severity of bronchopulmonary dysplasia and respiratory distress syndrome after a preterm birth. Pediatrics & Child Health, 16(7), 399–403 https://pubmed.ncbi.nlm.nih.gov/22851893/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisman LE (2003). Populations at risk for developing respiratory syncytial virus and risk factors for respiratory syncytial virus severity: Infants with predisposing conditions. The Pediatric Infectious Disease Journal, 22(2 Suppl), S33–S37. 10.1097/01.inf.0000053883.08663.e5. discussion S37–S39. [DOI] [PubMed] [Google Scholar]
- 12.Ng DK, Lau WY, & Lee SL (2000). Pulmonary sequelae in long-term survivors of bronchopulmonary dysplasia. Pediatrics International: Official Journal of the Japan Pediatric Society, 42 (6), 603–607. 10.1046/j.1442-200x.2000.01314.x. [DOI] [PubMed] [Google Scholar]
- 13.Bourbon J, Boucherat O, Chailley-Heu B, & Delacourt C (2005). Control mechanisms of lung alveolar development and their disorders in bronchopulmonary dysplasia. Pediatric Research, 57(5 Pt 2), 38R–46R. 10.1203/01.PDR.0000159630.35883.BE. [DOI] [PubMed] [Google Scholar]
- 14.Ladha F, Bonnet S, Eaton F, Hashimoto K, Korbutt G, & Thébaud B (2005). Sildenafil improves alveolar growth and pulmonary hypertension in hyperoxia-induced lung injury. American Journal of Respiratory and Critical Care Medicine, 172 (6), 750–756. 10.1164/rccm.200503-510OC. [DOI] [PubMed] [Google Scholar]
- 15.Rosen H, Stevens RC, Hanson M, Roberts E, & Oldstone MBA (2013). Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annual Review of Biochemistry, 82, 637–662. 10.1146/annurev-biochem-062411-130916. [DOI] [PubMed] [Google Scholar]
- 16.Harijith A, Pendyala S, Reddy NM, Bai T, Usatyuk PV, Berdyshev E, & Natarajan V (2013). Sphingosine kinase 1 deficiency confers protection against hyperoxia-induced bronchopulmonary dysplasia in a murine model: Role of S1P signaling and Nox proteins. The American Journal of Pathology, 183(4), 1169–1182. 10.1016/j.ajpath.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harijith A, Pendyala S, Ebenezer DL, Ha AW, Fu P, Wang Y-T, & Natarajan V (2016). Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2, and S1P/S1P1&2 signaling axis in lung endothelium. American Journal of Physiology Lung Cellular and Molecular Physiology, 311(2), L337–L351. 10.1152/ajplung.00447.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung B, Obinata H, Galvani S, Mendelson K, Ding B, Skoura A, & Hla T (2012). Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Developmental Cell, 23(3), 600–610. 10.1016/j.devcel.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae J-S, & Rezaie AR (2010). Thrombin upregulates the angiopoietin-Tie2 Axis: endothelial protein C receptor occupancy prevents the thrombin mobilization of angiopoietin 2 and P-selectin from Weibel-Palade bodies. Journal of Thrombosis and Haemostasis, 8(5), 1107–1115. 10.1111/j.1538-7836.2010.03812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ben Shoham A, Malkinson G, Krief S, Shwartz Y, Ely Y, Ferrara N, & Zelzer E (2012). S1P1 inhibits sprouting angiogenesis during vascular development. Development, 139(20), 3859–3869. 10.1242/dev.078550. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, & Proia RL (2000). Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. The Journal of Clinical Investigation, 106(8), 951–961. 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun X, Ma S-F, Wade MS, Flores C, Pino-Yanes M, Moitra J, & Garcia JGN (2010). Functional variants of the sphingosine-1-phosphate receptor 1 gene associate with asthma susceptibility. The Journal of Allergy and Clinical Immunology, 126(2), 241–249. 10.1016/j.jaci.2010.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tibboel J, Reiss I, de Jongste JC, & Post M (2014). Sphingolipids in lung growth and repair. Chest, 145(1), 120–128. 10.1378/chest.13-0967. [DOI] [PubMed] [Google Scholar]
- 24.Hendricks-Muñoz KD, Xu J, & Voynow JA (2018). Tracheal aspirate VEGF and sphingolipid metabolites in the preterm infant with later development of bronchopulmonary dysplasia. Pediatric Pulmonology, 53(8), 1046–1052. 10.1002/ppul.24022. [DOI] [PubMed] [Google Scholar]
- 25.Yamakawa D, Kidoya H, Sakimoto S, Jia W, Naito H, & Takakura N (2013). Ligand-independent Tie2 dimers mediate kinase activity stimulated by high dose angiopoietin-1. The Journal of Biological Chemistry, 288(18), 12469–12477. 10.1074/jbc.M112.433979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, & Jameson SC (2006). Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature, 442(7100), 299–302. 10.1038/nature04882. [DOI] [PubMed] [Google Scholar]
- 27.Sun X, Mathew B, Sammani S, Jacobson JR, & Garcia JGN (2017). Simvastatin-induced sphingosine 1−phosphate receptor 1 expression is KLF2-dependent in human lung endothelial cells. Pulmonary Circulation, 7(1), 117–125. 10.1177/2045893217701162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang X, Neyrinck AP, Matthay MA, & Lee JW (2010). Allogeneic human mesenchymal stem cells restore epithelial protein permeability in cultured human alveolar type II cells by secretion of angiopoietin-1. The Journal of Biological Chemistry, 285(34), 26211–26222. 10.1074/jbc.M110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Syed M, Das P, Pawar A, Aghai ZH, Kaskinen A, Zhuang ZW, & Bhandari V (2017). Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nature Communications, 8(1), 1173. 10.1038/s41467-017-01349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grzenda A, Shannon J, Fisher J, & Arkovitz MS (2013). Timing and expression of the angiopoietin-1-Tie-2 pathway in murine lung development and congenital diaphragmatic hernia. Disease Models & Mechanisms, 6(1), 106–114. 10.1242/dmm.008821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah D, Sandhu K, Das P, Aghai ZH, Andersson S, Pryhuber G, & Bhandari V (2020). miR-184 mediates hyperoxia-induced injury by targeting cell death and angiogenesis signaling pathways in the developing lung. The European Respiratory Journal. 10.1183/13993003.01789-2019. [DOI] [PubMed]
- 32.Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, & Elias JA (2006). Hyperoxia causes angiopoietin 2–mediated acute lung injury and necrotic cell death. Nature Medicine, 12(11), 1286–1293. 10.1038/nm1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones N, Iljin K, Dumont DJ, & Alitalo K (2001). Tie receptors: New modulators of angiogenic and lymphangiogenic responses. Nature Reviews Molecular Cell Biology, 2(4), 257–267. 10.1038/35067005. [DOI] [PubMed] [Google Scholar]
- 34.Peters KG, Kontos CD, Lin PC, Wong AL, Rao P, Huang L, & Sankar S (2004). Functional significance of Tie2 signaling in the adult vasculature. Recent Progress in Hormone Research, 59, 51–71. 10.1210/rp.59.1.51. [DOI] [PubMed] [Google Scholar]
- 35.Fujinaga H, Baker CD, Ryan SL, Markham NE, Seedorf GJ, Balasubramaniam V, & Abman SH (2009). Hyperoxia disrupts vascular endothelial growth factor-nitric oxide signaling and decreases growth of endothelial colony-forming cells from preterm infants. American Journal of Physiology Lung Cellular and Molecular Physiology, 297(6), L1160–L1169. 10.1152/ajplung.00234.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosford GE, & Olson DM (2003). Effects of hyperoxia on VEGF, its receptors, and HIF-2alpha in the newborn rat lung. American Journal of Physiology Lung Cellular and Molecular Physiology, 285(1), L161–L168. 10.1152/ajplung.00285.2002. [DOI] [PubMed] [Google Scholar]
- 37.Been JV, Debeer A, van Iwaarden JF, Kloosterboer N, Passos VL, Naulaers G, & Zimmermann LJ (2010). Early alterations of growth factor patterns in bronchoalveolar lavage fluid from preterm infants developing bronchopulmonary dysplasia. Pediatric Research, 67(1), 83–89. 10.1203/PDR.0b013e3181c13276. [DOI] [PubMed] [Google Scholar]