

Abstract
Objective:
Statin treatment reduces the risk of atherosclerotic cardiovascular disease (ASCVD) but is associated with a modest increased risk of type 2 diabetes (T2D), especially in those with insulin resistance or prediabetes. Our objective was to determine the physiologic mechanism for the increased T2D risk.
Approach and Results:
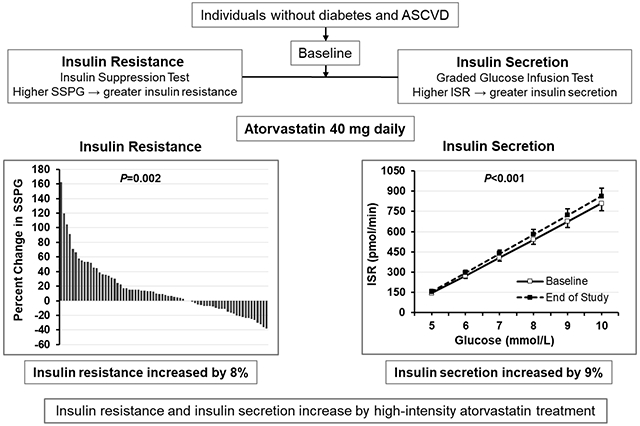
We conducted an open-label clinical trial of atorvastatin 40 mg daily in adults without known ASCVD or T2D at baseline. The co-primary outcomes were changes at 10 weeks versus baseline in insulin resistance as assessed by steady-state plasma glucose (SSPG) during the insulin suppression test and insulin secretion as assessed by insulin secretion rate area under the curve (ISRAUC) during the graded-glucose infusion test. Secondary outcomes included glucose and insulin, both fasting and during oral glucose tolerance test (OGTT). Of 75 participants who enrolled, 71 completed the study (median age 61 years, 37% women, 65% non-Hispanic white, median BMI 27.8 kg/m2). Atorvastatin reduced LDL-cholesterol (median decrease 53%, P<0.001) but did not change body weight. Compared to baseline, atorvastatin increased insulin resistance (SSPG) by a median of 8% (P=0.01) and insulin secretion (ISRAUC) by a median of 9% (P<0.001). There were small increases in OGTT glucoseAUC (median increase 0.05%, P=0.03) and fasting insulin (median increase 7%, P=0.01).
Conclusion:
In individuals without T2D, high-intensity atorvastatin for 10 weeks increases insulin resistance and insulin secretion. Over time, the risk of new-onset diabetes with statin use may increase in individuals who become more insulin resistant but are unable to maintain compensatory increases in insulin secretion.
Keywords: statin, insulin resistance, type 2 diabetes mellitus, insulin secretion, Lipids and cholesterol, Pathophysiology, Mechanism, Clinical Studies
Graphic abstract
INTRODUCTION
Statin treatment dramatically decreases the risk of atherosclerotic cardiovascular disease (ASCVD) by decreasing plasma levels of atherogenic lipoproteins (1–4). Statins are among the most prescribed medications in the world, and statin treatment is indicated for nearly half of the United States adult population either for primary or secondary prevention. (5) Statins are generally well tolerated but clinical trial data suggest that statin therapy is associated with an approximately 10% overall increased risk of incident type 2 diabetes (T2D) (6–10) over 5 years. This risk is increased in those with prediabetes and insulin resistance. (11,12)
The mechanisms for statin-related T2D are unclear. There is evidence that statins may adversely impact both insulin resistance and secretion. In that context, studies have shown that treatment with statins is associated with increase in fasting insulin (13–15) as well as increase in insulin resistance as assessed by measures obtained during the oral glucose tolerance test. (9,16) For example, Cederberg et al. (9) showed in a large prospective study (N=8749 men) that participants treated with statins (N=2142) had a 46% increase in incident T2D, associated with a 24% increase in insulin resistance and a 12% decrease in insulin secretion. These conclusions were based on surrogate estimates of insulin resistance and secretion which are only modestly correlated with direct measures of insulin action and secretion. (17–19) On the other hand, in small studies of 18 to 20 individuals that quantified insulin resistance by direct methods, insulin resistance did not significantly increase after treatment with pravastatin, simvastatin or rosuvastatin. (20–23) Furthermore, the effect of statin therapy on insulin secretion has not been evaluated with direct methods. Finally, different statins may also differently affect glucose and insulin metabolism (10,21–23) and direct assessments of the effect of atorvastatin (recommended as first line by current guidelines (4)) on insulin resistance and secretion are less available. Therefore, there is insufficient understanding of the effects that statins have on insulin resistance and insulin secretion, which limits our ability to identify the underlying mechanisms for this unwanted side effect and devise means to ameliorate it.
To investigate the mechanism of statin-related T2D, we conducted an open-label clinical trial to determine whether treatment with atorvastatin increases whole body insulin resistance and/or decreases insulin secretion as quantified by sensitive and reproducible direct methods.
MATERIALS AND METHODS
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request and summary statistics are available at ClinicalTrials.gov.
Study Design
This was an open-label, single group, prospective study to evaluate the effect of high-dose atorvastatin therapy on insulin resistance and insulin secretion rate. Each participant served as his or her own control. The study was performed between 2015 and 2019 at the Stanford Clinical and Translational Research Unit. The study was approved by Stanford University’s Institutional Review Board, and all participants gave written informed consent.
Study Participants
We recruited volunteers from the San Francisco Bay Area without T2D who were eligible for statin therapy for primary prevention of ASCVD. We excluded individuals with statin intolerance; marked kidney, liver, or heart disease; anemia or active malignancy (Figure 1). Detailed inclusion and exclusion criteria are presented in Supplementary Appendix. Our goal was to ensure recruitment across a broad range of insulin resistance. We and others have shown that high plasma triglyceride (TG) concentrations are associated with increased insulin resistance as assessed by gold-standard measures.(24,25) Therefore, we targeted advertisements to enrich for individuals with high TG levels (≥150 mg/dL) as a surrogate for increased insulin resistance. Individuals who were receiving statin therapy were included if they were able to undergo a 4-week statin washout with approval of their treating physician. The decision to include persons previously treated with statins was encouraged by the results of the study of Ahmadizar et al. showing that risk of incident diabetes is higher in current but not past users of statins.(15) Of the 71 subjects whose data was analyzed, 44 (62%) were statin naïve and 27 (38%) were receiving statin therapy at enrollment (statin exposed).
Figure 1. Study Participant Flow.
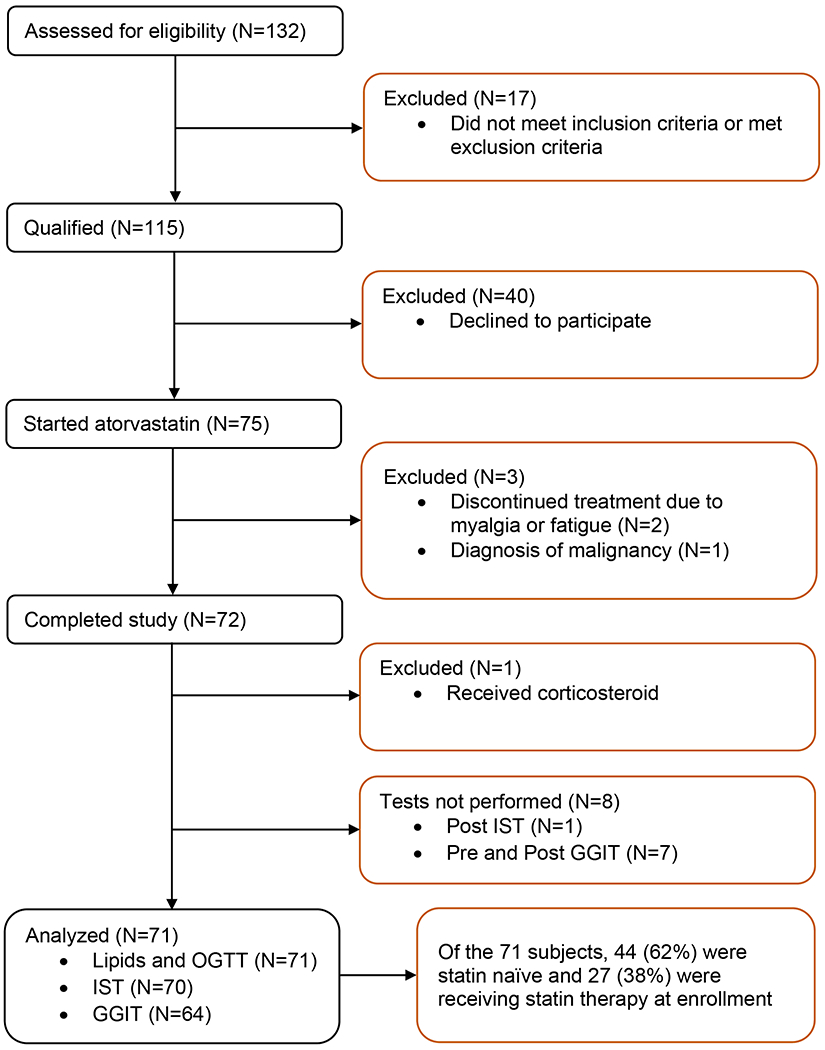
GGIT indicates graded glucose infusion test; OGTT, oral glucose tolerance test; and IST, insulin suppression test.
Study Visits
Participants gave written informed consent and were screened for the study on visit 1. Those who qualified underwent an oral glucose tolerance test (OGTT) on visit 2. Within two weeks, they were scheduled for the baseline graded-glucose infusion test (GGIT) and insulin suppression test (IST) which were generally performed about a week apart from one another. On the day of their last baseline test, a lipid panel with calculated LDL-C was performed and this was considered their baseline lipid panel.
Once participants completed the last baseline test (IST or GGIT) they were started on atorvastatin 40 mg daily (week zero). Study participants were asked to make no changes to their diet, weight or physical activity during the study.
While on atorvastatin, study participants were seen every 2 weeks from week zero to week 10 (Supplementary Table I). The OGTT was repeated at week 8, the GGIT at week 9 and the IST at week 10 while participants remained on atorvastatin. The lipid panel completed on the day of the final test was considered their statin-treated lipid panel. Body mass index (BMI) and blood pressure were assessed using standard techniques (Supplementary Appendix).
Oral Glucose Tolerance Test (OGTT)
Following a 12-hour fast, participants had a 75-gm OGTT (Supplementary Appendix). Blood was collected at baseline and 30, 60, 90, 120 minutes following an oral glucose load. Prediabetes (abnormal glucose tolerance) was defined as presence of having fasting glucose ≥100 mg/dL, 2-hour OGTT glucose ≥140 mg/dL, or both.
Graded Glucose Infusion Test
Insulin secretion was quantified during the graded-glucose infusion test (GGIT) as previously reported (26) after an overnight fast (Supplementary Appendix). The GGIT measures insulin secretion rate by deconvolution of peripheral C-peptide concentrations in response to increases in intravenous glucose.(27) The primary metric of insulin secretion during the GGIT is the insulin secretion rate area-under-the-curve (ISRAUC), where a higher ISRAUC indicates greater insulin secretion rate than a lower ISRAUC. Hereafter, for ease of use, we refer to ISRAUC (in pmol/min x 4 h) as “insulin secretion” except in the statistical analysis section.
Insulin Suppression Test
Insulin resistance was quantified by a modified version of the insulin suppression test (IST) (28) after an overnight fast (Supplementary Appendix). The IST measures peripheral insulin-stimulated glucose uptake, which primarily occurs in skeletal muscle. The primary metric of insulin resistance during the IST is the steady-state plasma glucose (SSPG) concentration, where a higher SSPG concentration indicates greater insulin resistance than a lower SSPG concentration. Insulin resistance measured by the IST highly correlates with that measured by the euglycemic hyperinsulinemic clamp. (29) Hereafter, for ease of use, we refer to SSPG concentration (in mg/dL) as “insulin resistance” except in the statistical analysis section.
Laboratory Measurements
Insulin, glucose and C-peptide measurements were performed at the Core Laboratory for Clinical Studies at Washington University School of Medicine. Lipids were measured at the Stanford Health Care Clinical Laboratory (Supplementary Appendix).
Outcomes
The co-primary outcomes were changes in insulin resistance during the IST and insulin secretion during the GGIT. Secondary outcomes included glucose and insulin concentrations measured fasting and during the OGTT. A prespecified subgroup analysis compared results between insulin sensitive versus insulin resistant participants. Additional analyses were performed by glucose tolerance status and by the diagnosis of the metabolic syndrome. (30)
Statistical Analysis
Based on our prior work (31), we calculated that with 60 subjects, we would be able to detect an 8% change in insulin resistance (SSPG concentration) and an 8% change in insulin secretion (ISRAUC) after atorvastatin therapy with 80% power and two-side significance level of 5% using a paired sample t test. Thus, we estimated needing to enroll 75 subjects with an anticipated dropout rate of 20%.
Summary statistics are reported as median (interquartile range) or number (percent) of participants unless otherwise specified. Shapiro-Wilk tests were used to assess normality of data, and variables that were not normally distributed were log-transformed, including: C-peptide, Homeostasis Model Assessment of Insulin Resistance (HOMA-IR), high sensitivity C-reactive protein (hs-CRP), insulin, ISRAUC, SSPG, steady-state plasma insulin (SSPI) and TG. Percent changes in variables were calculated by the formula: [(end-of study value) - (baseline value) / baseline value] x 100. Paired sample t tests were used to compare baseline and end-of-study means. One sample t tests were employed to evaluate whether percent changes in variables were significantly different from zero (no change). Pearson correlation coefficients were calculated to determine the strength of association between variables of interest. Prespecified subgroup analyses were carried out by stratifying for insulin resistant versus insulin sensitive subjects. Baseline SSPG concentration median (138 mg/dL) was used to define subjects as being insulin resistant (SSPG >138 mg/dL) or insulin sensitive (SSPG ≤138 mg/dL). As insulin resistance is a continuous trait, the median SSPG cutpoint (138 mg/dL) was informed by a prospective study of apparently healthy individuals, those with similar SSPG concentration (≥143 mg/dL) developed more cardiovascular disease (hypertension, coronary heart disease, stroke, or peripheral vascular disease) than those with SSPG below that cutpoint. (32) Subgroup means were compared by independent samples t tests and proportions by chi-square tests or Fisher’s exact tests. Statistical analyses were performed by using statistical software IBM SPSS version 26.0.
RESULTS
Participants
Of the 132 volunteers who were screened, 115 qualified for the study and 75 participants completed baseline studies and began statin therapy (Figure 1). For the primary outcomes of insulin resistance and insulin secretion, complete data were available for 70 and 64 participants, respectively. This included 18 participants with a baseline TG concentration ≥150 mg/dL (enriched for insulin resistance). The median length of time between the first and last IST was 77 days (interquartile range, IQR, 70 to 84).
The median age of the study participants was 61 years; 37% were women; and 65% were non-Hispanic white (Supplementary Table II). At baseline, the overall study group was overweight (median BMI 27.8 kg/m2) with elevations in total and LDL-cholesterol concentrations (237 and 156 mg/dL, respectively) (Supplementary Table IV). There were 45 (63.4%) participants with abnormal glucose tolerance (AGT) and 26 (36.6%) with normal glucose tolerance (NGT) (Supplementary Table VIII). Participant with AGT had higher BMI, were more insulin resistant, and had higher insulin secretion than those with NGT. There were 29 (40.8%) participants with the metabolic syndrome and 42 (59.2%) without the metabolic syndrome (Supplementary Table IX). Participants with the metabolic syndrome were more insulin resistant and had higher insulin secretion than those without the metabolic syndrome. Correlations between baseline variables are shown in Supplementary Table III and are consistent with prior findings. Baseline fasting insulin strongly correlated with baseline insulin resistance (r=0.74; P<0.001) and insulin secretion (r=0.80; P<0.001).
Effect of atorvastatin on body weight and concentrations of lipids, glucose and insulin
Statin therapy reduced total cholesterol by 37%, LDL-C by 53% and triglycerides by 28% (Supplementary Table IV). There was no change in body weight.
Effect of atorvastatin on insulin resistance
Statin treatment significantly increased insulin resistance, a co-primary outcome. Across the entire study population, the median insulin resistance (i.e., SSPG) increased from 130 to 139 mg/dL (P=0.01) while the median percent increase in insulin resistance was 8% (IQR, −10 to 32%) (Table 1) (Figure 2A, Supplementary Figure I). SSPI decreased by 5% but there was no significant correlation between the change in SSPI and the change in insulin resistance (data not shown).
Table 1:
Effect of Atorvastatin on Primary and Secondary Outcomes of Insulin Resistance and Insulin Secretion (N=71*)
| Variable | Baseline | End of Study | P value† |
|---|---|---|---|
| Primary Outcomes | |||
| Insulin Suppression Test | |||
| Insulin Resistance (by SSPG, mg/dL) | 130 (85 – 193) | 139 (93 – 211) | 0.01 |
|
| |||
| SSPI, mU/L | 64.7 (54.6 – 75. 4) | 61.3 (55.7 – 71.4) | 0.02 |
|
| |||
| Graded Glucose Infusion Test | |||
| Insulin secretion (by ISRAUC, pmol/min x 4 h) | 1824 (1385 – 2549) | 1942 (1480 – 2755) | <0.001 |
|
| |||
| GlucoseAUC, mmol/L x 4 h | 31.2 (28.0 – 34.5) | 31.8 (28.4 – 34.7) | 0.10 |
|
| |||
| InsulinAUC, pmol/L x 4 h | 652 (430 – 1108) | 712 (458 – 1263) | 0.001 |
|
| |||
| C-peptideAUC, nmol/L x 4 h | 6.5 (4.9 – 8.3) | 6.5 (5.3 – 8.9) | <0.001 |
|
| |||
| Secondary Outcomes | |||
|
| |||
| Fasting glucose, mg/dL | 99 (92 – 108) | 100 (94 – 107) | 0.10 |
|
| |||
| Fasting insulin, mU/L | 10.1 (7.3 – 14.8) | 10.6 (7.6 – 15.1) | 0.01 |
|
| |||
| HOMA-IR | 2.44 (1.71 – 3.73) | 2.68 (1.81 – 4.07) | 0.01 |
|
| |||
| OGTT GlucoseAUC, mg/dL x 2 h | 295 (241 – 336) | 299 (254 – 339) | 0.03 |
|
| |||
| OGTT InsulinAUC, mU/L x 2 h | 127 (74 – 217) | 133 (88 – 218) | 0.27 |
Data are median (interquartile range).
SSPG (N=70); SSPI (N=66), ISRAUC, GlucoseAUC, InsulinAUC, and C-peptideAUC (N=64); fasting insulin and HOMA-IR (N=69); and OGTT InsulinAUC (N=64).
All variables were log-transformed except fasting glucose and glucoseAUC.
Paired sample t tests were used to compare baseline and end-of-study means.
AUC indicates area under the curve; HOMA-IR, homeostasis model assessment of insulin resistance; ISR, insulin secretion rate; OGTT, oral glucose tolerance test; SSPG, steady-state plasma glucose; and SSPI, steady-state plasma insulin.
Figure 2. Effect of Atorvastatin Treatment on Insulin Resistance and Insulin Secretion.
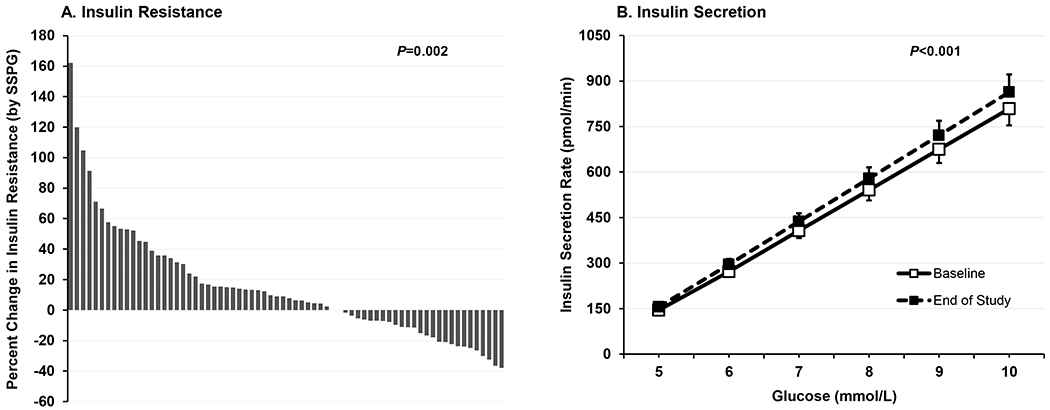
In panel A, waterfall plot depicts percent change in insulin resistance, measured by steady-state plasma glucose (SSPG), during the insulin suppression test (N=70). The mean (95% CI) percent change in SSPG concentration was 15 (6 – 23) mg/dL. One sample t test was used to compare mean percent change in SSPG to zero (no change). In Panel B, baseline and end-of-study insulin secretion rate (ISR) mean (SEM) values are plotted against the incremental increase in plasma glucose during the graded glucose infusion test (N=64). Baseline and end-of-study ISR area under the curve (ISRAUC) means were compared by paired sample t test after log-transformation of ISRAUC values.
There was no significant relationship between the change in LDL-C and the change in insulin resistance (Pearson correlation, −0.06, P=0.65). In addition, the small changes in weight that did occur were not significantly associated with changes in insulin resistance (Pearson correlation, 0.08, P = 0.50). There was no difference in effect of statin treatment on insulin resistance in those who were statin naïve versus those who were statin exposed (median percent change 7%, IQR −10 to 36% versus 11%, IQR −10 to 26%, respectively; P=0.81) (Figure 1).
In terms of secondary outcomes, there was a small but significant increase in glucoseAUC during the OGTT (0.05%, IQR −0.1 to 0.2%). Fasting insulin levels were increased by 7% (IQR −4 to 27%) and HOMA-IR by 11% (IQR −4 to 23%) after statin treatment (Table 1).
Effect of atorvastatin on insulin secretion
During the graded-glucose infusion test, glucoseAUC was similar but insulinAUC and C-peptideAUC significantly increased following statin treatment (Table 1). Thus, the dose-response relationship between glucose and rate of insulin secretion was shifted higher by statin treatment, resulting in a significant increase in insulin secretion (i.e., ISRAUC) (P<0.001), a co-primary outcome (Figure 2B). The median percent increase in insulin secretion was 9% (IQR, −2 to 19%) (Supplementary Figure II). There was no significant relationship between the change in LDL-C and the change in insulin secretion (Pearson correlation, 0.12, P=0.35). There was no difference in effect of statin treatment on insulin secretion in those who were statin naïve versus statin exposed at the time of enrollment (median percent increase 11%, IQR 1 to 29% versus 8%, IQR −4 to 16%, respectively; P=0.28).
Effect of atorvastatin on the relationship between insulin resistance and insulin secretion
At baseline, there was a positive (r=0.68; P<0.001) relationship between insulin resistance and insulin secretion, confirming that insulin secretion increases linearly with increase in insulin resistance (Supplementary Figure III). To explore this interaction, we plotted the percent change in insulin secretion against the percent change in insulin resistance for each participant where full data were available (N=63) (Figure 3). There was an enrichment of participants in the upper right quadrant (N=29, 46%), indicating an increase in insulin resistance accompanied by an increase in insulin secretion. However, there remained a fraction of the study participants (N=9, 14%) who experienced an increase in insulin resistance and a decrease in insulin secretion.
Figure 3. Relationship between Change in Insulin Resistance and Change in Insulin Secretion after Atorvastatin Treatment.
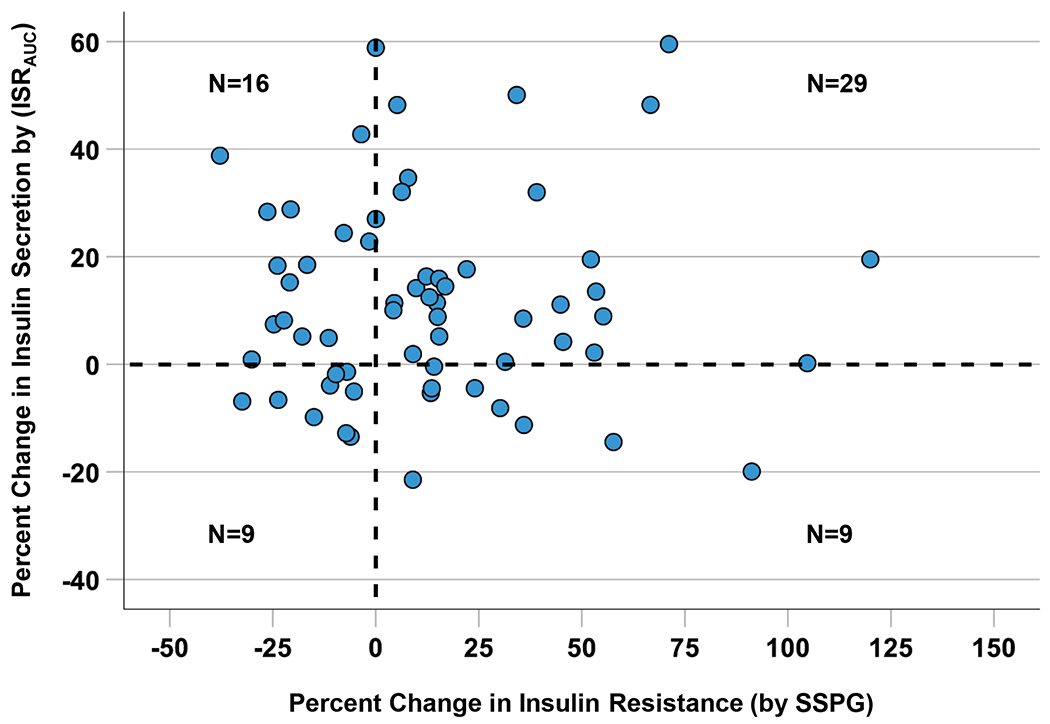
The four-quadrant scatterplot shows data on 63 individuals who underwent both the insulin suppression test and the graded glucose infusion test. Number of subjects in each quadrant is shown. Percent change was calculated by using the formula: [(end-of-study value) - (baseline value) / baseline value] x 100. ISRAUC indicates insulin secretion rate area-under-the-curve and SSPG, steady-state plasma glucose.
Effect of atorvastatin in insulin resistant versus insulin sensitive participants
Compared to insulin sensitive participants, those with insulin resistance at baseline (as defined in the statistical section) were heavier, were more likely to have dyslipidemia with higher median TG (122 vs 90 mg/dL) and lower HDL-cholesterol (46 vs 61 mg/dL) levels and had higher median fasting glucose (103 vs 97 mg/dL) and insulin concentrations (14.8 vs 7.9 mU/L). Among the insulin sensitive and insulin resistant participants, there were no difference in the distribution of individuals who were statin naïve versus statin exposed (P=0.74) (Supplementary Tables V, VI, VII).
Statins were equally effective in lowering LDL-C in insulin resistant versus insulin sensitive participants and changes in weight, glucose, and insulin were also similar between the two groups (Supplementary Tables VI, VII).
Insulin resistance increased in both insulin sensitive and insulin resistant participants (baseline values of 86 and 194 mg/dl which increased to 93 and 208 mg/dl respectively) though only the difference in insulin sensitive participants was significant (Supplementary Table VII and Supplementary Figure V). The median percent change in insulin resistance was a 16% increase (IQR: −3 to 50%) in insulin sensitive participants versus a 1% increase in insulin resistant participants (IQR: −21 to 14%) (Supplementary Figure V). The changes in insulin resistance also paralleled changes in fasting insulin in both the insulin sensitive and insulin resistant participants. To further examine the effect of baseline insulin resistance on statin-related increases in insulin resistance, a regression analysis was performed to determine if baseline insulin resistance predicted changes in insulin resistance after atorvastatin treatment. The results showed that baseline insulin resistance was a significant predictor of percent change in insulin resistance where lower baseline insulin resistance was associated with greater increase in insulin resistance (standardized coefficient = −0.39, P <0.001). For example, an individual with a baseline insulin resistance (i.e., SSPG) of 100 mg/dL would have a 23% increase in insulin resistance after atorvastatin therapy, whereas a person with baseline insulin resistance of 200 mg/dL would have a 3% increase in insulin resistance.
There was little difference in the change in insulin secretion between insulin sensitive and insulin resistant participants. The median percent change in insulin secretion was 11% in insulin sensitive participants (IQR: −3 to 23%) versus 9% in insulin resistant participants (IQR: −2 to 18%) (Supplementary Table VII).
Effect of atorvastatin on insulin resistance and insulin secretion by glucose tolerance status
Insulin resistance did not significantly increase in persons with abnormal glucose tolerance (AGT), but it did increase in those with normal glucose tolerance (NGT). The median percent change in insulin resistance was 5% in participants with AGT (IQR: −11 to 17%; P=0.07) versus 13% in participants with NGT (IQR: −1 to 53%; P=0.004). Insulin secretion significantly increased both in individuals with AGT and in those with NGT. The median percent change in insulin secretion was 11% in participants with AGT (IQR: −4 to 19%; P<0.001) versus 9% in participants with NGT (IQR: −0.0002 to 23%; P=0.01).
Effect of atorvastatin on insulin resistance and insulin secretion by the diagnosis of the metabolic syndrome
Insulin resistance did not significantly increase in persons with the metabolic syndrome, whereas it did increase in those without the metabolic syndrome. The median percent change in insulin resistance was 4%, in participants with the metabolic syndrome (IQR: −11 to 16%; P=0.21) versus 11% in participants without the metabolic syndrome (IQR: −7 to 41%; P=0.004). Insulin secretion significantly increased both in persons with the metabolic syndrome and in those without the metabolic syndrome. The median percent change in insulin secretion was 8% in participants with the metabolic syndrome (IQR: −3 to 16%; P=0.01) versus 11% in participants without the metabolic syndrome (IQR: −1 to 24%; P<0.001).
Other subgroup exploratory analyses
Atorvastatin treatment was associated with similar increases in insulin resistance and insulin secretion in subgroup analyses stratified by sex, race/ethnicity, age, BMI, TG, glucose tolerance, and the metabolic syndrome (Figure 4).
Figure 4. Changes in Insulin Resistance and Insulin Secretion in Subgroups after Atorvastatin Treatment.
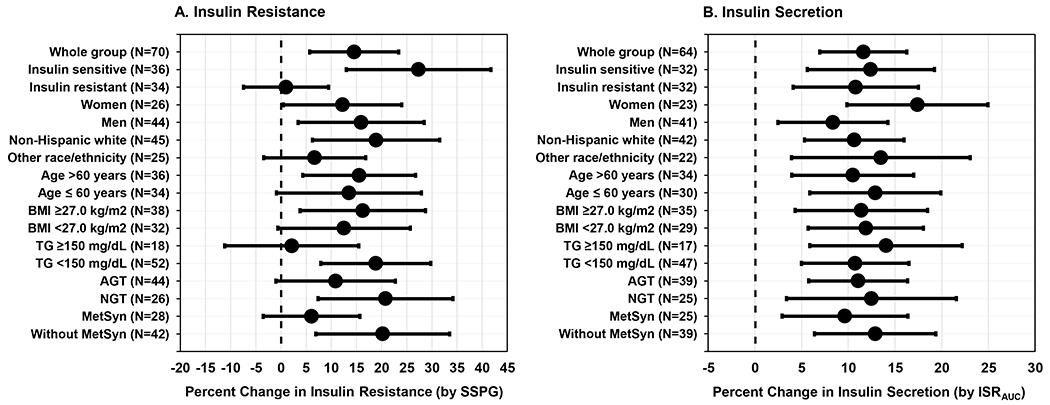
Forest plots depict changes in insulin resistance (Panel A) and insulin secretion (Panel B) in the whole group and in subgroups based on baseline characteristics: insulin resistance status, sex, race/ethnicity, age, BMI, TG, glucose tolerance, and diagnosis of the MetSyn. Data are means and 95% CIs.
Within each subgroup, means were compared by independent samples t tests. Only the differences in insulin resistance (Panel A) between insulin sensitive and insulin resistant subgroups were significantly different (P=0.002).
AGT indicates abnormal glucose tolerance; BMI, body mass index; ISRAUC, insulin secretion rate area under the curve; MetSyn, metabolic syndrome; NGT, normal glucose tolerance; NHW, Non-Hispanic white; SSPG, steady-state plasma glucose, and TG, triglycerides.
DISCUSSION
Statin therapy, a cornerstone of ASCVD prevention also increases the risk of developing T2D. (6–9,11,12,33,34) Individuals with T2D generally have defects in both insulin action and secretion as hyperglycemia only ensues when the insulin secretory response is inadequate. However, prior studies have not been able to determine whether statins increase the risk of T2D primarily by increasing insulin resistance or by decreasing insulin secretion. (35,36) Our results show that treatment with high-dose atorvastatin for 10 weeks increases insulin resistance and insulin secretion.
The modest increase we observed in insulin resistance (median 8% increase in SSPG) is likely the primary abnormality associated with statin treatment. Increase in insulin secretion is likely secondary and compensates for increase in insulin resistance and maintains glucose homeostasis in the short term. (37) Increases in insulin resistance have been reported in prior studies using surrogate estimates of insulin resistance. These studies showed that statin therapy was associated with increases in fasting insulin (13–15) as well as increases in insulin resistance assessed by OGTT-based measures (9,16). However, differences in insulin resistance were not seen in several prior smaller studies using gold-standard methods in nondiabetic individuals (20–23,38) treated with statins for 8 to 12 weeks. All of these trials enrolled 20 or fewer individuals and only two used what would be considered high intensity statin therapy (i.e., rosuvastatin 40 mg daily). (22,23) In contrast, in a study of 32 patients with type IIA and IIB dyslipidemia, treatment with pravastatin 10 to 20 mg per day for 3 months led to relatively small but significant increases in insulin resistance as measured by the IST. (24)
We did not observe obvious differences by sex, age or race/ethnicity. However, we saw proportionately greater increases in insulin resistance in those who were more insulin sensitive at baseline. This may represent a “ceiling effect” for some insulin resistant participants. In that vein, we have observed that those with marked insulin resistance at baseline may not get substantially more insulin resistant even with modest weight gain. (39)
The mechanism of increase in insulin resistance associated with statin therapy is unclear. Some literature suggests that long-term statin use could cause weight gain and thereby increase insulin resistance.(40) However, we detected an increase in insulin resistance without an increase in weight gain. Recent genetic observations have corroborated a potentially negative role of increased intracellular cholesterol for T2D and insulin resistance. Individuals with naturally occurring mutations that inhibit HMGCR have low plasma LDL-C levels, but increased intracellular cholesterol levels and a greater risk of T2D (41) while individuals with mutations in the low-density lipoprotein receptor (LDLR) have extreme elevations in plasma LDL-C levels but a decreased prevalence of T2D proportional to the severity of the LDLR mutation.(42) Other proposed mechanisms for how statins may increase insulin resistance include deregulation of intracellular or membrane bound cholesterol levels (43); suppression of intracellular levels of isoprenoids (44); perturbation of insulin signaling pathways (45–47); accumulation of free fatty acids (48); and mitochondrial dysfunction (49,50); all of which have been associated with increased insulin resistance. (51–56) Additional human studies are needed to determine if these or other mechanisms explain our results.
In addition to an increase in insulin resistance, we also show that short term statin treatment increases insulin secretion, a well-known compensatory response to increases in insulin resistance. (37) Prior studies examining the effect of statin therapy on insulin secretion used surrogate measures and reported disparate results. In a prospective, randomized, double-blind, placebo-controlled study of 28 women with polycystic ovarian syndrome, treatment with atorvastatin 20 mg per day for 6 months increased insulin secretion measured by the insulinogenic index. (16) In contrast, in a prospective study of 8749 men without diabetes, treatment with atorvastatin or simvastatin was associated with decreases in insulin secretion measured by OGTT-derived disposition index after a follow up of 5.9 years. (9) In contrast to the prior studies that employed surrogate estimates of insulin secretion, we quantified insulin secretion using a direct method to evaluate the effect of atorvastatin treatment on insulin secretion.
The short duration of our study did not allow us to determine the trajectory of insulin secretion with long-term statin therapy. Nevertheless, our results provide some insights into the potential course of beta cell function over time. As seen in Figure 3, insulin secretion increased with increase in insulin resistance in the majority of participants (upper right quadrant). This pattern indicates that the increase in insulin secretion was driven by change in insulin resistance to maintain glucose homeostasis. In other participants (lower right quadrant), insulin secretion decreased despite increase in insulin resistance. This pattern may indicate an inability to compensate for increase in insulin resistance and might be a harbinger of statin-related T2D. Lastly, in some participants (upper left quadrant), insulin secretion increased despite a decrease or no change in insulin resistance. This pattern may represent an independent effect of statins to increase insulin secretion.
The cellular mechanisms that could explain the increased insulin secretion are not completely understood though some evidence suggests that exposure of pancreatic beta cells to increased LDL-C levels may disrupt glucose homeostasis. First, incubating human and mouse pancreatic beta cells and islets with LDL-C decreases glucose-stimulated insulin secretion and increases cell death. (57–59) Second, statin treatment of mouse pancreatic islets in vitro reduced intracellular cholesterol levels and enhanced insulin secretion. (60) Finally, in vivo studies in high-fat fed mice suggests that atorvastatin treatment preserves beta cell function, increases proliferation, and reduces ER stress and apoptosis. (61) We cannot exclude the possibility that long-term statin treatment could adversely affect insulin secretion and note that lifelong deletion of Hmgcr in mouse beta cells causes a decrease in beta cell mass and insulin secretion.(62)
Persons with greater severity of the metabolic syndrome are at higher risk for developing incident T2D due to statin use (33,34). In that regard, individuals with the metabolic syndrome have features such as greater insulin resistance and higher fasting glucose that increase their risk of T2D. Consistent with that, in our study, participants with the metabolic syndrome were more insulin resistant than those without the metabolic syndrome (Supplemental Table IX). In addition, participants with more elements of the metabolic syndrome had greater insulin resistance and higher insulin secretion than those with fewer elements of the metabolic syndrome (Supplemental Table X). However, after statin therapy, insulin resistance did not increase in participants with the metabolic syndrome but insulin secretion did increase (Figure 4). Due to the short duration of our study, we could not determine the trajectories of insulin resistance and insulin secretion with statin use as a function of the severity of the metabolic syndrome. With long-term statin therapy, T2D likely occur in those individuals with the metabolic syndrome who develop additional increases in insulin resistance (due to statins, weight gain or physical inactivity) and are unable to maintain increase in insulin secretion to compensate for insulin resistance.
Adverse effects such as T2D are associated with a lack of acceptance and underutilization of statins.(63,64) Our study should not be interpreted as an argument against the use of statins. In primary prevention, modeling suggests that treatment of 10000 patients with an underlying ASCVD risk of 5-10% over 5 years with atorvastatin 40 mg daily for 5 years would be expected to cause an excess of about 100 new cases of T2D.(3) Among the subset of 100 patients with incident (excess) statin-related T2D, even if statin-related T2D doubles the risk of an ASCVD resulting in an excess of 5 to10 ASCVD events, this small number of excess events pales in comparison to the estimated 500 major ASCVD events that would be prevented(3). Furthermore, clinical trial data suggests that statin-related T2D may not markedly increase risk. (33)
The risk of statin-related T2D varies considerably depending on the population. In large statin trials both insulin resistance and prediabetes are independent risk factors for statin-related T2D and when they co-exist the risk of incident T2D over 7 years is 20% (11,12) versus 3% when neither insulin resistance nor prediabetes is present. However, those at risk for statin-related T2D also have higher ASCVD risk. Therefore, the net clinical benefit of statins remains substantial even if those benefits are nominally blunted by statin-related, incident T2D. An analysis of the primary prevention JUPITER trial limited to the 486 participants (out of 17,603) who developed T2D during a median follow-up of 1.9 years (N=270 on rosuvastatin 20 mg daily, N=216 on placebo), showed that the cardiovascular risk reduction associated with rosuvastatin (hazard ratio 0.63 versus placebo) was consistent with that observed for the entire trial (hazard ratio 0.56 versus placebo) and that rosuvastatin accelerated the diagnosis of T2D by only about 5.4 weeks (84.3 versus 89.7 weeks). (8) Given the unambiguous effects of statins in reducing the risk of ASCVD, it is imperative to emphasize healthy diet and lifestyle choices such as physical activity and maintenance of a healthy body weight to offset statin-related insulin resistance and T2D.
Limitations of our study include a single-center trial design with a single dose of atorvastatin without a placebo control group. However, we demonstrate adherence with statin treatment with resultant decreases in LDL-C accompanied by significant changes in insulin resistance and insulin secretion. Participants in our study were without diabetes and ASCVD and may not have been representative of the general population of patients taking statins. We are not able to determine the primary tissue responsible for the increase in insulin resistance. As the IST quantifies insulin-mediated glucose uptake primarily at skeletal muscle, it is likely that the increase in insulin resistance by atorvastatin occurs in skeletal muscle. In general, muscle insulin resistance parallels adipose tissue insulin resistance. We have shown that insulin-mediated glucose uptake, highly correlates with insulin-mediated suppression of lipolysis. (65) Our study was also short term and longer trials are needed to assess for potential decreases in insulin secretion or further increases in insulin resistance.
Strengths of our study include that we investigated the mechanism of statin-related T2D by using gold-standard methods for accurately measuring both insulin resistance and insulin secretion in a relatively large number of participants (N=71). The study subjects were treated with a commonly used high-intensity statin (atorvastatin 40 mg daily). Furthermore, baseline insulin resistance of our study participants varied several-fold.
In summary, we found that short-term treatment with high-intensity atorvastatin therapy results in increases in insulin resistance accompanied by increases in insulin secretion. Over time, the risk of new-onset T2D associated with statin therapy may increase in those individuals who become more insulin resistant but are unable to maintain compensatory increase in insulin secretion. All individuals on statin therapy should be encouraged to mitigate risk of diabetes through healthy lifestyle incorporating a nutrition and exercise plan. Finally, although our study provides insights into a potential mechanism of statin-related T2D, investigations are needed into the cellular mechanisms of increased insulin resistance and the trajectory of insulin secretion with long-term statin therapy.
Supplementary Material
Highlights.
Statin therapy decreases the risk of ASCVD but increases the risk of T2D.
It is unclear whether the increased T2D risk is caused by increased insulin resistance or decreased insulin secretion.
We conducted an open-label clinical trial of atorvastatin 40 mg daily in 75 adults without known ASCVD or T2D.
High-intensity atorvastatin for 10 weeks increased insulin resistance and insulin secretion, measured by the insulin suppression test and the graded-glucose infusion test, respectively.
Investigations are needed into the cellular mechanisms of increased insulin resistance and the trajectory of insulin secretion with long-term statin therapy.
Acknowledgements
The authors thank all the individuals in the trial for their participation as well as the staff of the Stanford CTRU for their assistance. We thank David Maron, Fatima Rodriguez and David Waters for helpful comments.
Sources of Funding
The Doris Duke Charitable Foundation, grant #2016097 provided major funding for this trial. Dr. Knowles is supported by the NIH through grants: P30DK116074 (to the Stanford Diabetes Research Center), R01 DK116750, R01 DK120565, R01 DK106236; and by the American Diabetes Association (grant #1-19-JDF-108). Dr. Kim is supported by the Bose Family Foundation and P30DK116074. The Washington University at St. Louis Diabetes Research Center central lab is supported by NIH grant P30 DK020579. Dr. Snyder was supported by the Metabolic Health Center through Lucille Packard Children’s Hospital, the Stanford PHIND Initiative and the NIH R01 AT01023203.
Abbreviations
- ASCVD
atherosclerotic cardiovascular disease
- T2D
type 2 diabetes
- TG
plasma triglycerides
- LDL-C
low-density lipoprotein cholesterol
- IST
insulin suppression test
- GGIT
graded glucose infusion test
- OGTT
oral glucose tolerance test
- AUC
area under the curve
- ISR
insulin secretion rate
Footnotes
Disclosures
The authors declare that there is no conflict of interest.
References
- 1.Baigent C, Blackwell L, Emberson J et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010;376:1670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor F, Huffman MD, Macedo AF et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev 2013:CD004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collins R, Reith C, Emberson J et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016;388:2532–2561. [DOI] [PubMed] [Google Scholar]
- 4.Grundy SM, Stone NJ, Bailey AL et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2019;73:3168–3209. [DOI] [PubMed] [Google Scholar]
- 5.Pencina MJ, Navar-Boggan AM, D’Agostino RB Sr., et al. Application of new cholesterol guidelines to a population-based sample. N Engl J Med 2014;370:1422–31. [DOI] [PubMed] [Google Scholar]
- 6.Sattar N, Preiss D, Murray HM et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010;375:735–42. [DOI] [PubMed] [Google Scholar]
- 7.Preiss D, Seshasai SR, Welsh P et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA : the journal of the American Medical Association 2011;305:2556–64. [DOI] [PubMed] [Google Scholar]
- 8.Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012;380:565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cederberg H, Stancakova A, Yaluri N, Modi S, Kuusisto J, Laakso M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: a 6 year follow-up study of the METSIM cohort. Diabetologia 2015;58:1109–17. [DOI] [PubMed] [Google Scholar]
- 10.Olotu BS, Shepherd MD, Novak S et al. Use of Statins and the Risk of Incident Diabetes: A Retrospective Cohort Study. Am J Cardiovasc Drugs 2016;16:377–90. [DOI] [PubMed] [Google Scholar]
- 11.Kohli P, Waters DD, Nemr R et al. Risk of new-onset diabetes and cardiovascular risk reduction from high-dose statin therapy in pre-diabetics and non-pre-diabetics: an analysis from TNT and IDEAL. Journal of the American College of Cardiology 2015;65:402–4. [DOI] [PubMed] [Google Scholar]
- 12.Kohli P, Knowles JW, Sarraju A, Waters DD, Reaven G. Metabolic Markers to Predict Incident Diabetes Mellitus in Statin-Treated Patients (from the Treating to New Targets and the Stroke Prevention by Aggressive Reduction in Cholesterol Levels Trials). Am J Cardiol 2016;118:1275–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 2010;55:1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thongtang N, Ai M, Otokozawa S et al. Effects of maximal atorvastatin and rosuvastatin treatment on markers of glucose homeostasis and inflammation. Am J Cardiol 2011;107:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmadizar F, Ochoa-Rosales C, Glisic M, Franco OH, Muka T, Stricker BH. Associations of statin use with glycaemic traits and incident type 2 diabetes. Br J Clin Pharmacol 2019;85:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puurunen J, Piltonen T, Puukka K et al. Statin therapy worsens insulin sensitivity in women with polycystic ovary syndrome (PCOS): a prospective, randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab 2013;98:4798–807. [DOI] [PubMed] [Google Scholar]
- 17.Reaven GM. HOMA-beta in the UKPDS and ADOPT. Is the natural history of type 2 diabetes characterised by a progressive and inexorable loss of insulin secretory function? Maybe? Maybe not? Diab Vasc Dis Res 2009;6:133–8. [DOI] [PubMed] [Google Scholar]
- 18.Ingelsson E, Langenberg C, Hivert MF et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes 2010;59:1266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pisprasert V, Ingram KH, Lopez-Davila MF, Munoz AJ, Garvey WT. Limitations in the use of indices using glucose and insulin levels to predict insulin sensitivity: impact of race and gender and superiority of the indices derived from oral glucose tolerance test in African Americans. Diabetes Care 2013;36:845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galvan AQ, Natali A, Baldi S et al. Effect of a reduced-fat diet with or without pravastatin on glucose tolerance and insulin sensitivity in patients with primary hypercholesterolemia. J Cardiovasc Pharmacol 1996;28:595–602. [DOI] [PubMed] [Google Scholar]
- 21.Altunbas H, Balci MK, Karayalcin U. No effect of simvastatin treatment on insulin sensitivity in patients with primary hypercholesterolemia. Endocr Res 2003;29:265–75. [DOI] [PubMed] [Google Scholar]
- 22.ter Avest E, Abbink EJ, de Graaf J, Tack CJ, Stalenhoef AF. Effect of rosuvastatin on insulin sensitivity in patients with familial combined hyperlipidaemia. Eur J Clin Invest 2005;35:558–64. [DOI] [PubMed] [Google Scholar]
- 23.Lamendola C, Abbasi F, Chu JW et al. Comparative effects of rosuvastatin and gemfibrozil on glucose, insulin, and lipid metabolism in insulin-resistant, nondiabetic patients with combined dyslipidemia. Am J Cardiol 2005;95:189–93. [DOI] [PubMed] [Google Scholar]
- 24.Sheu WH, Shieh SM, Fuh MM et al. Insulin resistance, glucose intolerance, and hyperinsulinemia. Hypertriglyceridemia versus hypercholesterolemia. Arteriosclerosis and thrombosis : a journal of vascular biology / American Heart Association 1993;13:367–70. [DOI] [PubMed] [Google Scholar]
- 25.Armato J, Ruby R, Reaven G. Plasma triglyceride determination can identify increased risk of statin-induced type 2 diabetes: a hypothesis. Atherosclerosis 2015;239:401–4. [DOI] [PubMed] [Google Scholar]
- 26.Kim SH, Abbasi F, Chu JW et al. Rosiglitazone reduces glucose-stimulated insulin secretion rate and increases insulin clearance in nondiabetic, insulin-resistant individuals. Diabetes 2005;54:2447–52. [DOI] [PubMed] [Google Scholar]
- 27.Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992;41:368–77. [DOI] [PubMed] [Google Scholar]
- 28.Pei D, Jones CN, Bhargava R, Chen YD, Reaven GM. Evaluation of octreotide to assess insulin-mediated glucose disposal by the insulin suppression test. Diabetologia 1994;37:843–5. [DOI] [PubMed] [Google Scholar]
- 29.Knowles JW, Assimes TL, Tsao PS et al. Measurement of insulin-mediated glucose uptake: direct comparison of the modified insulin suppression test and the euglycemic, hyperinsulinemic clamp. Metabolism 2013;62:548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alberti KG, Eckel RH, Grundy SM et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009;120:1640–5. [DOI] [PubMed] [Google Scholar]
- 31.Kim SH, Liu A, Ariel D et al. Pancreatic beta cell function following liraglutide-augmented weight loss in individuals with prediabetes: analysis of a randomised, placebo-controlled study. Diabetologia 2014;57:455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yip J, Facchini FS, Reaven GM. Resistance to insulin-mediated glucose disposal as a predictor of cardiovascular disease. The Journal of clinical endocrinology and metabolism 1998;83:2773–6. [DOI] [PubMed] [Google Scholar]
- 33.Waters DD, Ho JE, DeMicco DA et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. Journal of the American College of Cardiology 2011;57:1535–45. [DOI] [PubMed] [Google Scholar]
- 34.Waters DD, Ho JE, Boekholdt SM et al. Cardiovascular event reduction versus new-onset diabetes during atorvastatin therapy: effect of baseline risk factors for diabetes. J Am Coll Cardiol 2013;61:148–52. [DOI] [PubMed] [Google Scholar]
- 35.Betteridge DJ, Carmena R. The diabetogenic action of statins - mechanisms and clinical implications. Nat Rev Endocrinol 2016;12:99–110. [DOI] [PubMed] [Google Scholar]
- 36.Ward NC, Watts GF, Eckel RH. Statin Toxicity. Circ Res 2019;124:328–350. [DOI] [PubMed] [Google Scholar]
- 37.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988;37:1595–607. [DOI] [PubMed] [Google Scholar]
- 38.Baker WL, Talati R, White CM, Coleman CI. Differing effect of statins on insulin sensitivity in non-diabetics: a systematic review and meta-analysis. Diabetes Res Clin Pract 2010;87:98–107. [DOI] [PubMed] [Google Scholar]
- 39.McLaughlin T, Craig C, Liu LF et al. Adipose Cell Size and Regional Fat Deposition as Predictors of Metabolic Response to Overfeeding in Insulin-Resistant and Insulin-Sensitive Humans. Diabetes 2016;65:1245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swerdlow DI, Preiss D, Kuchenbaecker KB et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet 2015;385:351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ference BA, Robinson JG, Brook RD et al. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N Engl J Med 2016;375:2144–2153. [DOI] [PubMed] [Google Scholar]
- 42.Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA 2015;313:1029–36. [DOI] [PubMed] [Google Scholar]
- 43.Parpal S, Karlsson M, Thorn H, Stralfors P. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J Biol Chem 2001;276:9670–8. [DOI] [PubMed] [Google Scholar]
- 44.Chamberlain LH. Inhibition of isoprenoid biosynthesis causes insulin resistance in 3T3-L1 adipocytes. FEBS Lett 2001;507:357–61. [DOI] [PubMed] [Google Scholar]
- 45.Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 2006;49:1881–92. [DOI] [PubMed] [Google Scholar]
- 46.Takaguri A, Satoh K, Itagaki M, Tokumitsu Y, Ichihara K. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes. J Pharmacol Sci 2008;107:80–9. [DOI] [PubMed] [Google Scholar]
- 47.Li W, Liang X, Zeng Z et al. Simvastatin inhibits glucose uptake activity and GLUT4 translocation through suppression of the IR/IRS-1/Akt signaling in C2C12 myotubes. Biomed Pharmacother 2016;83:194–200. [DOI] [PubMed] [Google Scholar]
- 48.Kain V, Kapadia B, Misra P, Saxena U. Simvastatin may induce insulin resistance through a novel fatty acid mediated cholesterol independent mechanism. Sci Rep 2015;5:13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mullen PJ, Zahno A, Lindinger P et al. Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochim Biophys Acta 2011;1813:2079–87. [DOI] [PubMed] [Google Scholar]
- 50.Larsen S, Stride N, Hey-Mogensen M et al. Simvastatin effects on skeletal muscle: relation to decreased mitochondrial function and glucose intolerance. J Am Coll Cardiol 2013;61:44–53. [DOI] [PubMed] [Google Scholar]
- 51.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 2004;350:664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 2005;54:1392–9. [DOI] [PubMed] [Google Scholar]
- 53.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006;55 Suppl 2:S9–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patti M-E, Corvera S. The Role of Mitochondria in the Pathogenesis of Type 2 Diabetes. Endocrine Reviews 2010;31:364–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kleiner S, Mepani RJ, Laznik D et al. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc Natl Acad Sci U S A 2012;109:9635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson Andrew MF, Olefsky Jerrold M. The Origins and Drivers of Insulin Resistance. Cell 2013;152:673–684. [DOI] [PubMed] [Google Scholar]
- 57.Cnop M, Hannaert JC, Grupping AY, Pipeleers DG. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 2002;143:3449–53. [DOI] [PubMed] [Google Scholar]
- 58.Roehrich ME, Mooser V, Lenain V et al. Insulin-secreting beta-cell dysfunction induced by human lipoproteins. J Biol Chem 2003;278:18368–75. [DOI] [PubMed] [Google Scholar]
- 59.Rutti S, Ehses JA, Sibler RA et al. Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 2009;150:4521–30. [DOI] [PubMed] [Google Scholar]
- 60.Wijesekara N, Zhang LH, Kang MH et al. miR-33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes 2012;61:653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen ZY, Liu SN, Li CN et al. Atorvastatin helps preserve pancreatic beta cell function in obese C57BL/6 J mice and the effect is related to increased pancreas proliferation and amelioration of endoplasmic-reticulum stress. Lipids Health Dis 2014;13:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takei S, Nagashima S, Takei A et al. beta-Cell-Specific Deletion of HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A) Reductase Causes Overt Diabetes due to Reduction of beta-Cell Mass and Impaired Insulin Secretion. Diabetes 2020;69:2352–2363. [DOI] [PubMed] [Google Scholar]
- 63.Nielsen SF, Nordestgaard BG. Negative statin-related news stories decrease statin persistence and increase myocardial infarction and cardiovascular mortality: a nationwide prospective cohort study. Eur Heart J 2016;37:908–916. [DOI] [PubMed] [Google Scholar]
- 64.Fung V, Graetz I, Reed M, Jaffe MG. Patient-reported adherence to statin therapy, barriers to adherence, and perceptions of cardiovascular risk. PLoS One 2018;13:e0191817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abbasi F, McLaughlin T, Lamendola C, Reaven GM. The relationship between glucose disposal in response to physiological hyperinsulinemia and basal glucose and free fatty acid concentrations in healthy volunteers. J Clin Endocrinol Metab 2000;85:1251–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request and summary statistics are available at ClinicalTrials.gov.