

Abstract
Significant progress has been made in developing new treatments and refining the use of pre-existing ones against cancer. Their successful use and the longer survival of cancer patients have been associated with reports of new cardiotoxicities and the better characterization of the previously known cardiac complications. Immunotherapies with monoclonal antibodies against specific cancer-promoting genes, chimeric antigen receptor T cells (CAR-T-cells), and immune checkpoint inhibitors have been developed to fight cancer cells, but they can also show off-target effects on the heart. Some of these cardiotoxicities are thought to be due to non-specific immune activation and inflammatory damage. Unlike immunotherapy-associated cardiotoxicities which are relatively new entities, there is extensive literature on anthracycline-induced cardiomyopathy. Here, we provide a brief overview of the cardiotoxicities of immunotherapies for the purpose of distinguishing them from anthracycline cardiomyopathy. This is especially relevant as the expansion of oncologic treatments present greater diagnostic challenges in determining the etiology of cardiac dysfunction in cancer survivors with history of multiple cancer treatments including anthracyclines and immunotherapies administered concurrently or serially over time. We then provide a focused review of the mechanisms proposed to underlie the development of anthracycline cardiomyopathy based on experimental data mostly in mouse models. Insights into its pathogenesis may stimulate the development of new strategies to identify patients who are susceptible to anthracycline cardiomyopathy while permitting low cardiac risk patients to receive optimal treatment for their cancer.
Keywords: anthracycline, cardiomyopathy, immunotherapy, mitochondria, mitochondrial genome, p53
INTRODUCTION
Chemotherapy is the primary treatment for many types of cancers, and its improvement has contributed to the increased survival of cancer patients. Although chemotherapeutic agents generally target proliferating neoplastic cells, quiescent cardiomyocytes can also be damaged thereby leading to cardiomyopathy in cancer survivors. Treatment with one of the anthracycline class of chemotherapeutic agents such as daunorubicin, epirubicin, mitoxantrone, or doxorubicin can result in a progressive form of heart failure with a high mortality rate1–4. Among these anthracyclines, doxorubicin is an old but highly effective and commonly utilized agent. In a cumulative dose-dependent manner, it can cause myocardial dysfunction in a subset of patients for unclear reasons, and the onset of heart failure can occur many years after exposure to the chemotherapy. Despite extensive clinical characterization and a wealth of experimental data using model systems, there are still no preventive strategies or targeted treatment for anthracycline cardiomyopathy.
The introduction of immunotherapies against cancer has brought to the forefront the need to better understand the risk factors for anthracycline-induced cardiac damage. The use of trastuzamab, an anti- human epidermal growth factor receptor 2 (HER2) monoclonal antibody, in combination with an anthracycline resulted an unanticipated and significant increase in the risk of cardiac dysfunction5. More recently, immunotherapies using monoclonal antibodies against other cancer promoting genes and anti-cancer immune activating treatments such as immune checkpoint inhibitors (ICI) and chimeric antigen receptor T cells (CAR-T) have also turn out to have cardiovascular toxicities6, 7. Like the unexpected effect of anthracyclines on non-proliferating cardiomyocytes, eliciting cytotoxic effects using monoclonal antibodies for cancer cell recognition have also resulted in adverse side-effects. With these developments, the sub-specialty of cardio-oncology has coalesced with an emphasis on incorporating the advances in immune-oncology, such as CAR-T cell and ICI immunotherapy, for preventing and managing cancer treatment cardiotoxicities7.
Because over half of all cancer patients may be eligible for some form of immunotherapy7,8, it is useful to briefly summarize some of their cardiotoxicities in order to distinguish them from that of anthracyclines. Heart failure is the major cause of morbidity associated with anthracyclines, so we will review some of the immunotherapies and targeted therapies that have been associated primarily with adverse effects on myocardial contractility or the cardiovascular system. We will then focus on detailing the current understanding of the molecular mechanisms underlying anthracycline cardiotoxicity based mostly on in vivo mouse models with the goal of potentially translating these findings to assist in the management of cancer patients treated with anthracyclines. For the cardiotoxicities of chemotherapeutic agents such as proteasome inhibitors and tyrosine kinase inhibitors, the reader is referred to specialized reviews that more comprehensively discuss these and other chemotherapeutic agents.9,10,11, 12.
SOME CARDIOTOXICITIES ASSOCIATED WITH CANCER IMMUNOTHERAPIES AND TARGETED ANTI-ONCOGENIC SIGNALING ANTIBODIES
Immune Checkpoint Inhibitors (ICIs)
ICIs are therapeutic monoclonal antibodies that block T cell inhibitory regulators such as the cytotoxic T lymphocyte-associated protein-4 (CTLA-4), programmed cell death protein-1 (PD-1) and PD-1 ligand (PD-L1)6, 8. Disrupting these inhibitory activities with ICI treatment results in varying degrees of uncontrolled T cell activation and subsequent off-target immune-related adverse events that affect different organs such as skin, thyroid, intestine, liver, lung and heart. Examining mouse models with ablation of the immune checkpoint inhibitor genes support the concept of cardiac damage from T cell overactivation and infiltration into the myocardium (Table 1)13, 14. The PD-1 signaling pathway appears to play an important role in restraining CD4+ and CD8+ T cell activities in the myocardium, but the specific molecular events leading to their myocardial infiltration and subsequent pathogenesis remain to be elucidated15, 16. Human pathological studies show PD-L1 activation with inflammatory cell infiltrates while there are macrophage polarization changes observed in mouse models after ICI treatment17,18. PD-L1-expressed endothelial cells other than cardiomyocytes may be also targets of ICI-related cardiac injury19. Besides a direct effect on the heart, such interactions of the vasculature, cytokines or autoantibodies may explain the complex mechanism of ICI-related systemic adverse events which are more comprehensively reviewed elsewhere13, 16.
Table 1.
Cardiotoxicity mechanisms of some anti-cancer immunotherapies and targeted treatments
| Therapeutic agent | Molecular target | Proposed cardiotoxicity mechanism | References |
|---|---|---|---|
| Immune Checkpoint Inhibitors | |||
| Lipilimumab | CTLA-4 | Activation of CD4+ and CD8+ T cells with marked myocardial infiltration in mice | 8, 14, 119–122 |
| Pembrolizumab | PD-1 | Activated T cells in myocardium; cardiac troponin I autoantibodies and dilated cardiomyopathy observed in PD-1-deficient mice | |
| Nivolumab | |||
| Cemiplimab | |||
| Atezolizumab | PD-L1 | Activated T cells in myocardium | |
| Avelumab | |||
| Durvalumab | |||
| CAR-T cells | |||
| Axicabtagene ciloleucel | CD19 | Cytokine release syndrome; IL-6 implicated as key mediator | 24, 25 |
| Tisagenlecleucel | |||
| HER2 inhibitors | |||
| Trastuzumab | HER2 | Inhibition of HER2 signaling pathway involved in cell growth, survival, angiogenesis and migration | 33–36 |
| Pertuzumab | |||
| VEGF inhibitors | |||
| Bevacizumab | VEGF | VEGF inhibition alters blood vessel homeostasis and nitric oxide signaling which can both affect blood pressure and thrombosis | 28, 29 |
| Ramucirumab |
VEGF receptor | ||
Cardiotoxicity mechanisms of some anti-cancer immunotherapies and targeted treatments are summarized and referenced. CAR-T, chimeric antigen receptor T-cell; CTLA-4, cytotoxic T lymphocyte-associated protein-4; HER2, human epidermal growth factor receptor 2; PD-1, programmed cell death protein-1; PD-L1, PD-1 ligand; VEGF, vascular endothelial growth factor.
Although the incidence of clinically significant cardiotoxicities with a single ICI is low (range of 1% or less), the combined use of two different ICIs significantly increases the risk20, 21. Also, inhibitors of protein kinases BRAF and MEK can cause cardiovascular toxicities4, and their combined therapy with ICIs have been reported to worsen ICI-related myocarditis22. Distinct from the late onset cardiomyopathy caused by anthracyclines, ICI cardiotoxicity can occur within days after treatment but the median time is about 4 wk with most occurring within the first 6 wk after treatment. Of the different cardiotoxicity manifestations which include arrhythmias and non-inflammatory myocardial dysfunction such as stress cardiomyopathy, myocarditis poses the greatest risk with ~50% of the severe cases being fatal in some studies8, 21. A combination of clinical presentation, cardiac injury biomarkers and cardiac imaging (echocardiogram or magnetic resonance imaging) are used to diagnose ICI associated myocarditis, but endomyocardial biopsy, if available, is considered the gold standard criterion. In addition to heart failure treatments, immune suppression with corticosteroids is the mainstay of ICI myocarditis management21. There is general agreement to permanently withhold ICI treatment if a patient develops moderate to severe cardiotoxicity.
Chimeric Antigen Receptor T-cell Therapy (CAR-T)
CAR-T cell is another form of anti-cancer immunotherapy in which T cells are genetically modified to express a chimeric protein comprised of the cancer antigen binding receptor and T cell activating domains23. This directs both the innate and adaptive immune systems to target, for example, diffuse large B-cell lymphoma that express CD19 antigen using the axicabtagene ciloleucel or tisagenlecleucel CAR-T cells. The cardiac complications observed in patients undergoing CAR-T cell therapy include cardiac troponin elevation, heart failure, cardiogenic shock and arrhythmias which appear to stem from the phenomenon of cytokine release syndrome (CRS)24. CRS is characterized by a constellation of inflammatory signs and symptoms with hemodynamic instability that can be treated using tocilizumab, an anti-IL-6 receptor antagonist23, 25. Although some specific mechanisms for these side effects have been proposed such as the shared expression of B cell CD19 antigen by vascular mural cells in the brain, the full spectrum of CAR-T cell-induced toxicities of the heart and other tissues remain to be further elucidated26. Unlike some other cancer treatment toxicities, the side effects of CAR-T therapy are actually “on-target” and may be reversible when the specific cancer cells are eliminated. The interaction between CAR-T and other anti-cancer treatments also continue to be clarified. Although prior treatment with anthracyclines was thought to increase the risk of cardiovascular events associated with CAR-T cell therapy, this does not appear to be significant according to a retrospective analysis25. On the other hand, CAR-T therapy could be directed towards providing beneficial cardiac effects. An innovative pre-clinical study has proposed the use of CAR-T cells directed against a cardiac fibroblast-specific protein (fibroblast activation protein, FAP) to prevent myocardial fibrosis, a characteristic finding of anthracycline cardiomyopathy27.
Vascular Endothelial Growth Factor (VEGF) Inhibitors
Cancer cells require adequate blood supply for their growth, thus inhibiting the angiogenesis promoting activity of VEGF has been shown to have efficacy in cancer treatment, especially in suppressing metastatic cancer cell growth28. The monoclonal antibodies bevazicumab and ramucirumab can inactivate VEGF-A and block the signaling of its receptor VEGFR2, respectively. However, disrupting VEGF signaling can result in adverse cardiovascular effects such as systemic and pulmonary hypertension, cardiac ischemia, thromboembolism, and cardiac dysfunction29. Systemic hypertension is the most striking adverse effect of inhibiting VEGF while the least prominent cardiovascular effect is myocardial dysfunction, suggested to be in part secondary to hypertension exacerbation28, 29. VEGF receptors can also be blocked by small molecule kinase inhibitors such as sorafenib and sunitinib but their inhibition of other important receptor tyrosine kinases complicate the interpretation of their adverse cardiovascular effects28, 30.
Human Epidermal Growth Factor Receptor 2 (HER2) Inhibitor
The overexpression of human epidermal growth factor receptor 2, associated with aggressive disease, is observed in a significant fraction of breast cancers, and its inhibition using the monoclonal antibody trastuzumab improves the prognosis of patients with HER2-positive breast cancer5. However, this therapeutic benefit can be associated with symptomatic heart failure and declines in left ventricular ejection fraction initially reported in 2–3% of patients, usually within weeks of starting the treatment although it generally recovers in the months following therapy interruption10, 31. In a more recent clinical trial, about 30% of patients exhibited cardiotoxicity after HER2 inhibitor treatment while those concurrently or serially treated with anthracyclines were at even higher risk32. Unlike anthracyclines, HER2 cardiotoxicity does not show a cumulative dose dependent increase in risk10. These clinical observations were confirmed in the laboratory by the cardiac specific deletion of ErbB2, the mouse homolog of the human HER2 gene, which resulted in dilated cardiomyopathy and increased susceptibility to anthracycline cardiotoxicity while ErbB2 overexpression was protective33–35. These findings are also consistent with the involvement of HER2 in diverse cellular processes related to cell growth, survival, and energy metabolism10, 36.
CARDIOTOXICITY ASSOCIATED WITH ANTHRACYCLINES
Clinical Considerations of Anthracycline Cardiomyopathy
Although doxorubicin is the most commonly used anthracycline, there are 5 other class members available for the FDA- and non-FDA approved treatment of 14 and 7 different types of cancer, respectively37. Annually, an estimated one million patients received anthracyclines in North America38 so even a relatively low incidence of severe anthracycline cardiotoxicity translates to a large number of affected individuals. The recommended lifetime cumulative dose of doxorubicin is 550 mg/m2 or less, but the incidence of clinically significant decline (>10%) in left ventricular ejection fraction (LVEF) can be lowered further from ~65% at this dose to ~7% and ~18% at 150 mg/m2 and 350 mg/m2, respectively. This risk continuum is also sensitive to patient factors, such as age (>65 or <18 yr), female gender, race, and pre-existing cardiac conditions3.
Acute cardiotoxicity (during or immediately after infusion) of anthracycline can manifest as transient arrhythmias and LVEF depression, but it is rare (<1%) and usually reversible11. The timing of the cardiotoxicity has been further divided into early (≤1 yr) and late (median 7 yr after treatment) cardiotoxicity although the pathogenesis of anthracycline cardiomyopathy is most likely continuous, progressive, and irreversible unless detected early and intervened11. It is important to note here that the development of more sensitive imaging modalities such as cardiac magnetic resonance imaging has enabled the earlier detection of subclinical cardiac damage induced by anthracyclines39, 40. All 4 chambers of the heart may be dilated but not as severely as in some other forms of dilated cardiomyopathy, and the left ventricular wall may not be thinned significantly1. The affected left ventricle displays both systolic and diastolic contractile dysfunction. Other than careful risk stratification and monitoring of patients in order to intervene early with standard heart failure therapies if needed, the only approved preventive medication is dexrazoxane which appears to provide cardioprotective effects as measured by cardiac troponin and echocardiographic parameters3, 41, 42. Notably, the protective effects of dexrazoxane appear to be mediated in part by its activities on topoisomerases and mitochondrial iron homeostasis as discussed later in this review43, 44.
Pathogenesis Considerations in Human Anthracycline Cardiomyopathy
Although a number of different molecular mechanisms have been proposed to mediate anthracycline cardiomyopathy, there is general consensus on the histopathologic changes observed upon endomyocardial biopsy of the right ventricle, considered to be the most sensitive and specific diagnostic test for this condition45. The biopsy may show patchy interstitial fibrosis, fibroblast proliferation, histiocytic (monocytic origin) infiltration, loss of myofibrils, distension of the sarcoplasmic reticulum, and vacuolated cardiomyocytes next to the areas of fibrosis1. These histopathologic features are used to grade the degree of anthracycline-induced damage, but the availability of this diagnostic test is limited to specialized medical centers.
Relevance of Experimental Anthracycline Cardiotoxicity Models to Human Cardiomyopathy
From a mechanistic perspective, there have been many studies examining anthracycline cardiotoxicity using both in vitro and in vivo models which contrasts with the relative paucity of experimental data on immunotherapy associated cardiotoxicity albeit it is a newer treatment. There are many interesting in vitro studies, but in this review the mechanistic insights derived mostly from in vivo mouse models of anthracycline cardiotoxicity are discussed in order to associate them with cardiac function.
It is important to distinguish between the acute cardiotoxicity of doxorubicin, the most commonly used anthracycline in animal studies, and the late onset cardiomyopathy generally observed in patients. In fact, when apoptosis studies in doxorubicin cardiotoxicity were initially being performed, the important distinction between acute and chronic administration of doxorubicin in animal models and concerns about the relevance of supra-clinical doses and disease timing to the human condition were raised by some investigators46, 47. It is notable that in the representative anthracycline cardiotoxicity studies listed (Table 2), those that involve cell death mechanisms or early onset cardiac dysfunction tend to utilize an acute model and examine the cardiac response within days (versus weeks in chronic models) of anthracycline treatment (either bolus- or multi-injection dosing). In contrast, doxorubicin is usually given in low doses weekly in the clinics to prevent acute drug toxicity, now a rare complication in patients.
Table 2.
Mechanisms of doxorubicin-induced cardiotoxicity in mouse models
| Mechanisms | Pathway/Genes | Mouse genotypes | Dosage | Ref |
|---|---|---|---|---|
| DNA damage | TOP2B | Cardiac specific-TOP2B−/− | Acute: 25 mg/kg once Chronic: 5 mg/kg/wk for 5 wk |
53 |
| Oxidative stress | MnSOD | MnSOD Tg | 10, 20 or 25 mg/kg once | 55 |
| NOX subunit | gp91 −/− | 3 mg/kg/wk for 3 wk then once at wk 5 |
56 | |
| iNOS | iNOS−/−; MnSOD Tg | 20 mg/kg once | 57, 58 | |
| NQO1 SIRT1 |
NQO1 −/− | 4 mg/kg/d × 3 d | 59 | |
| Apoptosis | p53 | p53 −/− | 20 mg/kg once | 64 |
| p53 ATM Bcl2 |
p53+/−; Bcl2 Tg | 6 mg/kg/wk for 4 wk | 65 | |
| p53 | Cardiac specific-p53−/− | 20 mg/kg once | 67 | |
| FAK p21 |
Muscle specific-FAK−/−; cardiac specific-superFAK Tg | 20 mg/kg once | 68 | |
| BAX | BAX −/− | Acute: 20 mg/kg once Chronic: 3 mg/kg × 8 over 2 wk |
71 | |
| Necrosis | BNIP3 | BNIP3 −/− | 20 mg/kg once | 70 |
| Necroptosis | RIP3 CaMKII |
RIP3 −/− | Acute: 20 mg/kg once Chronic: 5 mg/kg/wk for 4 wk |
48 |
| Ferroptosis | HFE | HFE −/− | Acute: 20 mg/kg once Chronic: 5 mg/kg × 6 at 2 wk intervals |
73 |
| ABCB8 | Cardiac specific-ABCB8−/−; cardiac specific-ABCB8 Tg | 10 mg/kg × 3 over 5 d; 6 mg/kg × 4 over 10 d |
44 | |
| NRF2 HO1 |
NRF2−/− | 20 mg/kg once | 49 | |
| GPX4 | GPX4+/−; GPX4 Tg | 6 mg/kg × 3 at 2 d intervals | 72 | |
| Autophagy | p53 mTOR |
Cardiac specific-CB7 (dominant interfering p53) Tg; cardiac specific-mTOR Tg | 10 mg/kg × 2 at 3 d interval | 66 |
| p53 Parkin |
p53−/−; Parkin−/−; Parkin Tg | 2.5 mg/kg × 5 over 2 wk | 75 | |
| Beclin1 | Beclin1+/−; cardiac specific-Beclin1 Tg | 5 mg/kg/wk for 4 wk | 79 | |
| Inflammation, Coagulation | COX-2 | Mice treated with COX-2 inhibitor | 4 mg/kg/wk for 6 wk | 81 |
| TLR2 TLR4 |
Mice treated with TLR2 or TLR4 neutralizing antibody | Acute: 10 mg/kg once Chronic: 3.5 mg/kg/wk for 8 wk |
82 | |
| PAR1 | PAR1−/− | Acute: 20 mg/kg once Chronic: 5 mg/kg/wk for 5 wk |
83 | |
| Mitochondrial damage | p53 mtDNA | p53 −/− | 20 mg/kg once | 107 |
| TOP1MT | TOP1MT −/− | 4 mg/kg/wk for 8 wk | 100 | |
| p53 STAT3 |
Cardiac specific-CB7 (dominant interfering p53); cardiac specific-STAT3−/− | 5 mg/kg/wk for 5 wk | 108 | |
| SIRT3 OGG1 |
SIRT3−/−; SIRT3 Tg | 5 mg/kg every 15 d × 3 | 103 | |
| p53 TFAM p53R2 |
p53−/−; p53 R172H knockin | 5 mg/kg/wk for 5 wk | 109 |
The genes/pathways involved in mediating anthracycline cardiotoxicity, the mouse model genotypes, and the corresponding references are shown. The dose and schedule of doxorubicin treatment used in each of the studies are also shown to better assess the chronicity of the cardiotoxicity model. ABCB8, ATP binding cassette subfamily B member 8; ATM, ataxia telangiectasia mutated kinase; BAX, Bcl2 associated x protein; Bcl2, B-cell lymphoma 2; BNIP3, Bcl2 interacting protein 3; CaMKII, calcium/calmodulin-dependent protein kinase II; COX-2, cyclooxygenase 2; FAK, focal adhesion kinase; GPX4, glutathione peroxidase 4; HFE, homeostatic iron regulator protein; HO1, heme oxygenase 1; iNOS, inducible nitric oxide synthase; MnSOD, manganese superoxide dismutase; mTOR, mammalian target of rapamycin; NOX, NAD(P)H oxidase; NRF2, nuclear factor erythroid 2-related factor 2; NQO1, NAD(P)H quinone oxidoreductase 1; OGG1, 8-oxoguanine DNA glycosylase; PAR1, protease-activated receptor 1; p53R2, p53-inducible ribonucleotide reductase; RIP3, receptor-interacting protein 3; SIRT, sirtuin; STAT3, signal transducer and activator of transcription 3; TFAM, transcription factor A, mitochondrial; TLR, toll-like receptor; TOP1MT, topoisomerase I mitochondrial; TOP2B, topoisomerase IIβ.
Generally, the acute cardiotoxicity models show cardiac dysfunction shortly after treatment with mortality rates sometimes exceeding 50%48, 49. This does not reflect anthracycline cardiomyopathy in the clinics where the heart failure presents only in a subset of patients and often occurs years after treatment exposure. Highlighting the importance of drug administration mode on cardiotoxicity outcome, the dosing of doxorubicin (2 mg/kg/wk over 7 wk) in a chronic cardiotoxicity model permitted the detection of rat heart mitochondrial dysfunction while this was not observed with an acute toxicity bolus dosing regimen (20 mg/kg once)50. Thus, in this review we have listed the available information on the dosing protocol of doxorubicin in various mouse models to provide context for interpreting the proposed mechanisms although it should be noted that in some instances both acute and chronic dosing schedules were used within the same study (Table 2).
Anthracyclines Can Induce DNA Damage via Topoisomerases
Anthracyclines such as doxorubicin can induce DNA strand breaks by interacting with cleavable topoisomerase IIα (TOP2A)-DNA complexes, thereby inhibiting the proliferation of cancer cells51. Non-proliferating cardiomyocytes express TOP2B rather than TOP2A (Figure 1)52, so an elegant study took advantage of this differential expression of topoisomerase II subtypes and showed that doxorubicin cardiotoxicity could be prevented by the cardiac-specific knockout of TOP2B gene in mice53. TOP2B involvement in mediating anthracycline cardiotoxicity was previously suggested by decreases in doxorubicin-induced DNA damage response upon disruption of TOP2B in mouse embryonic fibroblasts or by treatment with the cardio-protective agent dexrazoxane in rat H9C2 cardiomyoblasts43.
Figure 1.
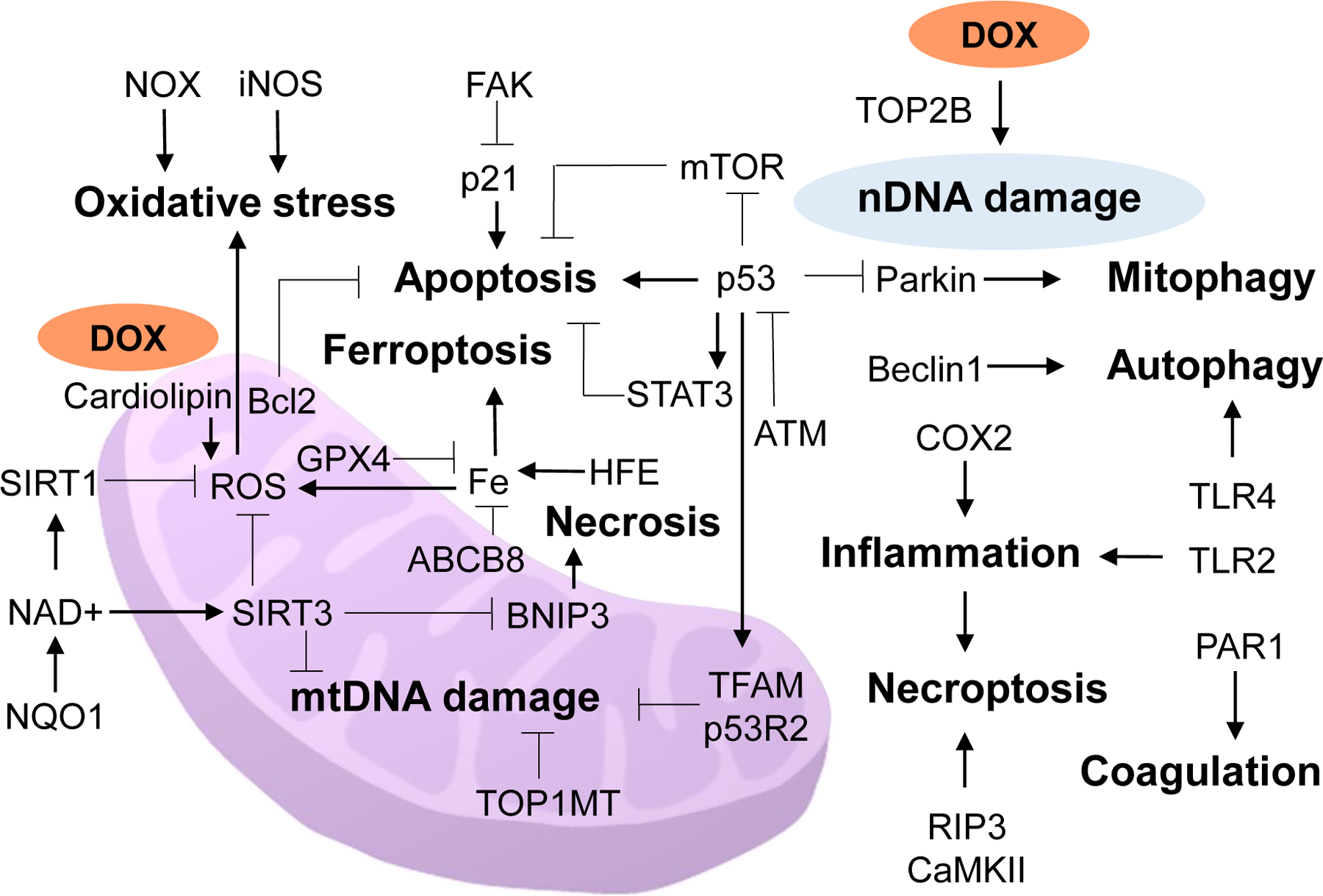
Schematic diagram of the major cellular processes (bold print) and their mediators/pathways of anthracycline cardiotoxicity. ABCB8, ATP binding cassette subfamily B member 8; ATM, ataxia telangiectasia mutated kinase; Bcl2, B-cell lymphoma 2; BNIP3, Bcl2 interacting protein 3; CaMKII, calcium/calmodulin-dependent protein kinase II; COX-2, cyclooxygenase 2; DOX, doxorubicin; FAK, focal adhesion kinase; GPX4, glutathione peroxidase 4; HFE, human homeostatic iron regulator protein; iNOS, inducible nitric oxide synthase; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; NOX, NAD(P)H oxidase; NQO1, NAD(P)H quinone oxidoreductase 1; nDNA, nuclear DNA; PAR1, protease-activated receptor 1; p53R2, p53-inducible ribonucleotide reductase; RIP3, receptor-interacting protein 3; SIRT, sirtuin; STAT3, signal transducer and activator of transcription 3; TFAM, transcription factor A, mitochondrial; TLR, toll-like receptor; TOP1MT, topoisomerase I mitochondrial; TOP2B, topoisomerase 2-beta.
Role of Oxidative Stress
In addition to the mechanisms by which it is known to damage DNA, doxorubicin accumulates in mitochondria to induce cytotoxic free radicals (Table 2)2. The redox metabolism of anthracyclines by the respiratory complex I NADH dehydrogenase can produce cytotoxic reactive oxygen species (ROS) in purified mitochondria54. Paralleling this in vitro study, mitochondrial manganese superoxide dismutase (MnSOD) overexpressing transgenic mice were protected from acute doxorubicin cardiotoxicity55. Other subcellular sources of ROS could also contribute to pathogenesis. For example, the disruption of the plasma membrane NAD(P)H oxidase gp91 has been shown to be protective against chronic doxorubicin cardiotoxicity in a mouse model56. This study also showed that specific polymorphisms of NAD(P)H oxidase subunits conferred differential sensitivity to anthracycline-induced cardiotoxicity in cancer patients.
Doxorubicin has also been suggested to cause myocardial injury through another free radical nitric oxide (NO) generated by inducible nitric oxide synthase (iNOS) enzyme in macrophages, but the role of NO in pathogenesis has been controversial57, 58. Another study which also examined acute toxicity within days after a bolus injection (20 mg/kg) of doxorubicin reported that iNOS deficient mice actually had worse cardiac outcome after treatment, suggesting an antioxidant role for NO57. Furthermore, the mitochondrial damage caused by doxorubicin in these iNOS deficient mice could be rescued by crossing them into transgenic mice overexpressing the antioxidant enzyme manganese superoxide dismutase (MnSOD)57. Another oxidative stress response enzyme NAD(P)H quinone oxidoreductase 1 (NQO1) has been shown to attenuate doxorubicin cardiotoxicity in association with reduced oxidative stress and inflammation via the NAD+/SIRT1 pathway59. Despite these and other pre-clinical studies showing their benefits in mouse models, the available clinical studies examining the protective effect of antioxidants against anthracycline toxicity in the heart have not been conclusive, except for dexrazoxane, an iron chelator which can mitigate mitochondrial oxidative stress42, 44, 60, 61.
Cell Death and Other Cellular Processes in Anthracycline Cardiotoxicity
Various types of cell death such apoptosis, necrosis, necroptosis, and ferroptosis with or without the involvement of p53, a well-established mediator of apoptosis62, have been reported to contribute to doxorubicin cardiotoxicity (Table 2). The apoptosis studies cited in Table 2 represent only a fraction of many that have been reported by various investigators over the years, but due to space limitations we provide only some representative in vivo studies. The apoptosis caused by oxidative DNA damage sensors ATM and p53 has been associated with doxorubicin cardiac injury and dysfunction, however in another study, p53 inhibition of mTOR signaling, not apoptosis, was proposed to be the major contributor of acute cardiotoxicity63–65,66. In certain cellular contexts, p53 can even prevent apoptosis via p21 mediated cell cycle arrest62, so perhaps it is not unexpected that the cardiac-specific knockout of p53 showed increased apoptosis with bolus dosing of doxorubicin albeit the homozygous cardiac-specific Cre mouse was embryonic lethal so that the heterozygous state had to be utilized67. The survival of cardiomyocytes in acute doxorubicin cardiotoxicity has also been shown to be promoted by tyrosine kinase FAK mediated upregulation of p2168. An additional factor to consider is the relative resistance of adult cardiomyocytes to apoptosis compared with young cardiomyocytes due to decreased expression of the apoptotic machinery, suggesting potentially different mechanisms of anthracycline cardiotoxicity depending on age69. Both necrosis (unprogrammed inflammatory cell death) and necroptosis (programmed inflammatory cell death) through BNIP3 and RIP3-CaMKII, respectively, have also been reported to play a role in the single bolus dosing model of acute doxorubicin cardiotoxicity48, 70. The importance of these cell death mechanisms in doxorubicin cardiotoxicity have been supported by its prevention in BAX deficient mice as well as by the innovative use of a small molecule inhibitor of BAX71.
Consistent with the essential role of iron in oxidative stress homeostasis, doxorubicin cardiotoxicity has been associated with mitochondrial iron overload and cell death by ferroptosis44, 49, 72. Reducing mitochondrial iron stores either by promoting its efflux out of the mitochondria or decreasing the pool of iron available for uptake into the mitochondria was cardioprotective while higher cellular iron content in a hemochromatosis mouse model was associated with increased cardiac sensitivity to doxorubicin73. Intriguingly, the results of various iron chelator studies in anthracycline cardiotoxicity have indicated that iron binding capacity may not be the sole determinant of their cardioprotective activity although the beneficial effect of dexrazoxane does appear to be mediated mainly by decreasing mitochondrial iron levels44, 60. Further studies may help resolve some of these differences observed in model systems.
There is also evidence that autophagy and mitophagy are involved in anthracycline cardiotoxicity. Cytoplasmic p53 has been reported to interact with parkin and impair mitophagy causing cardiac dysfunction in doxorubicin-treated mice, and to inhibit autophagy via mTOR and decrease cell survival74, 75. In addition to these inhibitory activities in the cytoplasm through protein-protein interactions, an in vitro study showed that p53 overexpression can increase BNIP3 mRNA and protein expression in association with increased autophagy and cell death76. It should be noted that p53 can also induce apoptosis via its nuclear transactivation of autophagy regulator gene DRAM77. Further linking autophagy to doxorubicin cardiotoxicity78, another group used Beclin1 mouse models to show that doxorubicin blocks autophagic flux, resulting in the accumulation of autolysosomes which can increase ROS and cause cardiac damage79.
Inflammation has been implicated in anthracycline cardiomyopathy80, and this is evidenced by studies, for example, showing the attenuation of cardiac dysfunction with the inhibition of either cyclooxygenase 2 (COX-2) or toll-like receptor 2 (TLR 2)81, 82. Although the antibody-mediated neutralization of TLR2 ameliorated inflammation, fibrosis, and cardiac dysfunction caused by doxorubicin treatment, neutralizing TLR4 worsened these parameters and suppressed autophagy indicating the complementary and distinct roles of TLRs in pathogenesis82. Related to inflammation, aberrant coagulation caused by the activation of the G-protein coupled protease-activated receptor 1 (PAR1) has also been shown to contribute to doxorubicin cardiotoxicity83. Additionally, the inhibition of matrix metalloproteinase inhibitors have been shown to ameliorate cardiac dysfunction and prevent myocardial fibrosis in a mouse model with chronic weekly administration of doxorubicin84. These representative in vivo mouse model studies of anthracycline cardiotoxicity, although not comprehensive, show mechanistic complexity and the need for further delineation as translation into the clinics is considered.
A Central Role for the Mitochondrion in Anthracycline Cardiomyopathy
Many different lines of investigations have led to the conclusion that mitochondrial structure and function are altered in anthracycline cardiomyopathy as well detailed in a comprehensive review85. As a positively charge hydrophobic molecule at physiologic pH, doxorubicin is known to interact with the negatively charged inner mitochondrial membrane lipid cardiolipin, potentially disrupting cytochrome c and other essential components of the respiratory chain86–89. Early studies in rat models showed that there was cumulative and irreversible cardiac mitochondrial dysfunction including abnormalities of calcium homeostasis that may affect cell death signaling upon chronic treatment with doxorubicin90, 91. Subsequent studies have continued to support a central role of the mitochondria in anthracycline cardiotoxicity ranging from the finding of preventing BNIP3 mediated necrosis by mitochondrially localized SIRT3 to a study demonstrating impaired mitochondria function/content, calcium handling, and antioxidant activity in induced pluripotent stem cell-derived cardiomyocytes of patients with doxorubicin cardiotoxicity70, 92, 93.
Besides the various mechanisms of damage to the myocardium depending on doxorubicin dosing, the manifestation of slowly progressive heart failure well after the chemotherapeutic agent has been eliminated from the body suggested a more persistent alteration of the mitochondrion in anthracycline cardiotoxicity85. Changes to the cardiac mitochondrial genomic DNA (mtDNA) could explain such a permanent alteration, and indeed an early investigation reported that the impaired synthesis of mtDNA was slower to recover after doxorubicin treatment compared with that of nuclear DNA94. Notably, the higher sensitivity of the heart than the liver to doxorubicin damage could be due to its slower recovery of mtDNA synthesis after being impaired by treatment compared with that of the liver. Furthermore, mtDNA of heart has a faster turnover rate than that of liver (T1/2 ~7 d versus ~9 d, respectively; nuclear DNA T1/2 >30 d), suggesting greater susceptibility to DNA damaging events94.
A study using chronic low dose doxorubicin treatment of mice reported increased mtDNA deletions which could be prevented by the antioxidant coenzyme Q10, invoking oxidative stress as the cause of damage95. In other studies, acute doxorubicin treatment of rats (single injection of 15 mg/kg) resulted in higher levels of oxidative mtDNA modifications (8-hydroxydeoxyguanosine) compared with nuclear DNA, again with cardiac tissue showed greater mtDNA modifications than liver96. Sub-chronic doxorubicin treatment (2 mg/kg/wk over 6 wk) caused the greatest cumulative damage in cardiac mtDNA that persisted even after the treatment was complete, suggesting the temporal and genetic bases of anthracycline cardiomyopathy in cancer patients97. Strong evidence for the role of mtDNA in anthracycline cardiomyopathy was provided by the finding of increased mtDNA deletions, decreased mtDNA content, and decreased mtDNA-encoded, but not nuclear DNA-encoded, respiratory subunits in autopsy cardiac tissues of patients treated with doxorubicin98. Similar reductions in mtDNA content and mtDNA-encoded respiratory subunit gene expression were reproduced in a rat model of chronic doxorubicin cardiomyopathy by these investigators99.
Mechanistically, a number of different genes have been linked to changes in mtDNA homeostasis after doxorubicin exposure. In addition to increased defects in cardiac cell myofibrils and mitochondrial ultrastructure by electron microscopy, mice that are deficient in mitochondrial topoisomerase I (TOP1MT) were observed to have greater loss of mtDNA content and respiratory complex subunit proteins upon doxorubicin exposure100. As mentioned earlier, doxorubicin cardiotoxicity was proposed to be mediated by TOP2B induced nuclear DNA strand breaks with resultant defective mitochondrial biogenesis53, but TOP2B is also known to be the only type II topoisomerase in the mitochondria and thus may affect mtDNA homeostasis as shown for TOP1MT100, 101. In fact, it has been suggested that TOP2B in the mitochondria may be the primary mediator of doxorubicin cardiotoxicity, because the gene program regulated by TOP2B in the nucleus is not typically related to mitochondrial biogenesis101, 102. Other genes that are well-established to promote mitochondrial biogenesis and activities such as SIRT1 and SIRT3 have been demonstrated to attenuate doxorubicin cardiotoxicity, further supporting the central role of the mitochondrion in its pathogenesis59, 103.
p53 as Guardian of mtDNA in Anthracycline Cardiomyopathy
p53 is best known as a mediator of cell death for cancer prevention, but it has a dual nature and is equally important for promoting cell survival through its DNA repair, cell cycle regulatory, antioxidant, metabolic, and other activities depending on factors such as the cellular context and level of p53 induction104, 105. In fact, there are evidence that the translocation of p53 into the mitochondria under physiological conditions may serve to maintain the integrity of the mitochondrial genome106. An early study reported increased p53 immunoreactivity in cardiomyocyte mitochondria after doxorubicin treatment, and increased oxidative mtDNA damage was observed in p53−/− (null) mouse hearts (Table 2)107. A subsequent insightful study using mice with cardiac overexpression of a dominant-interfering mutant p53 showed protection from acute doxorubicin cardiotoxicity, presumably due to loss of wild-type p53 cell death activity, but the cardiac function of these mice deteriorated in the chronic phase indicating a protective role for p53108. The blunting of doxorubicin-induced STAT3 by mutant p53 was associated with increased expression of DNA damage markers, portending the worse late cardiac outcome.
We examined chronic doxorubicin cardiotoxicity in p53 null mice compared with mice that had knockin of the p53 R172H mutation, the mouse homolog of the human hotspot R175H mutation that causes the inherited early onset cancer disorder Li-Fraumeni syndrome109. This mutation had previously been shown to retain the mitochondrial biogenesis activity of wild-type p53 while losing its other activities such as apoptosis and cell cycle arrest110,111. The mitochondrial activities of p53 could be mediated by its regulation of mitochondrial biogenesis genes such as TFAM and p53R2 in the nucleus or by its interactions with other proteins such as POLG in the mitochondria106. As in wild-type p53 mice, the hearts of p53 R172H mutant knockin mice were relatively protected from loss of cardiac and mitochondrial function in contrast to that of p53 null mice after doxorubicin treatment (Table 2)109. Furthermore, the selective decrease in the expression of mtDNA-encoded versus nuclear DNA-encoded mitochondrial respiratory subunit genes in p53 null hearts upon doxorubicin exposure was consistent with the previous reports of mtDNA depletion85, 107, 109. Taken together, the finding of p53-regulated mtDNA homeostasis playing a critical role in preventing anthracycline-induced cardiotoxicity correlates well the clinical observation of mtDNA damage observed in the heart samples of patients with anthracycline cardiomyopathy98. Thus, it could be interesting to examine whether the molecular components involved in regulating mtDNA in easily accessible patient samples such as blood provide diagnostic or prognostic information for the management of anthracycline treated patients.
CONCLUSIONS
The various cardiotoxicities associated with different types of cancer treatments are increasing with the advancement of cancer therapies. With the improved survival of cancer patients from their primary disease, a new specialty of cardio-oncology has emerged for managing the cardiovascular complications of anti-cancer therapies112. While it is important to recognize and understand the molecular bases of the cardiotoxicities associated with the newer oncologic treatments, the cardiomyopathy caused by one of the oldest classes of chemotherapeutic agents, the anthracyclines, still does not have a widely accepted and targeted prevention treatment despite the effort of many investigators4, 40, 112.
Because of the different mechanisms by which anthracyclines can cause cardiotoxicities, one challenge is focusing on the ones that are most relevant to human cardiomyopathy pathogenesis. One criterion may be the dosing of the chemotherapy in the in vivo model system which is summarized in Table 2 for some representative studies. As a DNA damaging agent that concentrates in the mitochondria, various studies from different laboratories point to damaging of the mitochondrial genome as a major contributor85. Because newer anti-cancer treatments utilizing immune checkpoint inhibition are likely to be used in combination with anthracyclines, interaction or augmentation of their specific cardiotoxicity mechanisms could occur with potentially worse outcome, making it imperative to understand pathogenesis. Besides bioenergetic deficiency secondary to mitochondrial dysfunction, the intracellular release of mtDNA damaged by doxorubicin may activate innate immune signaling113, known to be stimulated by immunotherapy114, and synergistically increase myocardial fibrosis in the setting of ICI therapy115, 116. Although based on an anecdotal case report, it is also tempting to consider underlying subclinical mitochondrial genomic DNA disease as a susceptibility factor for anthracycline cardiotoxicity117. In parallel, the development of a doxorubicin analog that uncouples its cardiotoxic DNA damaging effect from its cancer cell destroying chromatin modification activity would be of great benefit and timely needed for the care of cancer patients118.
Highlights.
With recent advances in oncology and improved survival of cancer patients, the cardiotoxicities of new treatments such as immunotherapy are becoming more evident and require further understanding for their management and prevention.
The cardiotoxicity of anthracyclines, one of the oldest but most effective class of chemotherapeutic agents, is the major limiting factor in its use, and various molecular mechanisms of this side effect have been proposed using experimental models.
Determining the mechanisms most relevant to human anthracycline cardiomyopathy pathogenesis remains a challenge, but one criterion may be the dosing protocol of the chemotherapy in the in vivo model.
Accumulation evidence points to damage of the cardiomyocyte mitochondrial genome as a major determinant of anthracycline cardiomyopathy, and its protection during treatment or better risk stratification based on a firm molecular understanding of pathogenesis may be key to minimizing this side effect while optimizing treatment of the cancer.
Acknowledgments
We wish to thanks current and past members of our laboratory who have contributed to the writing of this review by all their helpful discussions and research work over the years.
Sources of Funding
This work was supported by the Division of Intramural Research, National Heart, Lung and Blood Institute (NHLBI), NIH.
Nonstandard Abbreviations and Acronyms
- CAR-T
chimeric antigen receptor T-cell
- CRS
cytokine release syndrome
- HER2
human epidermal growth factor receptor 2
- ICI
immune checkpoint inhibitors
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- PD-1
programmed cell death protein-1
- PD-L1
PD-1 ligand
- VEGF
vascular endothelial growth factor
Footnotes
Disclosures
None.
REFERENCES
- 1.Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. 2010;115:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volkova M, Russell R 3rd. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Current cardiology reviews. 2011;7:214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henriksen PA. Anthracycline cardiotoxicity: An update on mechanisms, monitoring and prevention. Heart. 2018;104:971–977 [DOI] [PubMed] [Google Scholar]
- 4.Herrmann J Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat Rev Cardiol. 2020;17:474–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against her2 for metastatic breast cancer that overexpresses her2. N Engl J Med. 2001;344:783–792 [DOI] [PubMed] [Google Scholar]
- 6.Hu JR, Florido R, Lipson EJ, Naidoo J, Ardehali R, Tocchetti CG, Lyon AR, Padera RF, Johnson DB, Moslehi J. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc Res. 2019;115:854–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaha VG, Meijers WC, Moslehi J. Cardio-immuno-oncology. Circulation. 2020;141:87–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moslehi J, Lichtman AH, Sharpe AH, Galluzzi L, Kitsis RN. Immune checkpoint inhibitor-associated myocarditis: Manifestations and mechanisms. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10:111–126 [DOI] [PubMed] [Google Scholar]
- 10.Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol. 2015;12:547–558 [DOI] [PubMed] [Google Scholar]
- 11.Zamorano JL, Lancellotti P, Rodriguez Munoz D, Aboyans V, Asteggiano R, Galderisi M, Habib G, Lenihan DJ, Lip GYH, Lyon AR, Lopez Fernandez T, Mohty D, Piepoli MF, Tamargo J, Torbicki A, Suter TM, Group ESCSD. 2016 esc position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the esc committee for practice guidelines: The task force for cancer treatments and cardiovascular toxicity of the european society of cardiology (esc). Eur Heart J. 2016;37:2768–2801 [DOI] [PubMed] [Google Scholar]
- 12.Wu P, Oren O, Gertz MA, Yang EH. Proteasome inhibitor-related cardiotoxicity: Mechanisms, diagnosis, and management. Curr Oncol Rep. 2020;22:66. [DOI] [PubMed] [Google Scholar]
- 13.Sury K, Perazella MA, Shirali AC. Cardiorenal complications of immune checkpoint inhibitors. Nat Rev Nephrol. 2018;14:571–588 [DOI] [PubMed] [Google Scholar]
- 14.Wei SC, Meijers WC, Axelrod ML, Anang NAS, Screever EM, Wescott EC, Johnson DB, Whitley E, Lehmann L, Courand PY, Mancuso JJ, Himmel LE, Lebrun-Vignes B, Wleklinski MJ, Knollmann BC, Srinivasan J, Li Y, Atolagbe OT, Rao X, Zhao Y, Wang J, Ehrlich LIR, Sharma P, Salem JE, Balko JM, Moslehi JJ, Allison JP. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 2021;11:614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarrio ML, Grabie N, Bu DX, Sharpe AH, Lichtman AH. Pd-1 protects against inflammation and myocyte damage in t cell-mediated myocarditis. J Immunol. 2012;188:4876–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khunger A, Battel L, Wadhawan A, More A, Kapoor A, Agrawal N. New insights into mechanisms of immune checkpoint inhibitor-induced cardiovascular toxicity. Curr Oncol Rep. 2020;22:65. [DOI] [PubMed] [Google Scholar]
- 17.Palaskas N, Lopez-Mattei J, Durand JB, Iliescu C, Deswal A. Immune checkpoint inhibitor myocarditis: Pathophysiological characteristics, diagnosis, and treatment. J Am Heart Assoc. 2020;9:e013757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia W, Zou C, Chen H, Xie C, Hou M. Immune checkpoint inhibitor induces cardiac injury through polarizing macrophages via modulating microrna-34a/kruppel-like factor 4 signaling. Cell Death Dis. 2020;11:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, Keir ME, Freeman GJ, Sharpe AH, Lichtman AH. Endothelial programmed death-1 ligand 1 (pd-l1) regulates cd8+ t-cell mediated injury in the heart. Circulation. 2007;116:2062–2071 [DOI] [PubMed] [Google Scholar]
- 20.Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, Becker JR, Slosky DA, Phillips EJ, Pilkinton MA, Craig-Owens L, Kola N, Plautz G, Reshef DS, Deutsch JS, Deering RP, Olenchock BA, Lichtman AH, Roden DM, Seidman CE, Koralnik IJ, Seidman JG, Hoffman RD, Taube JM, Diaz LA Jr., Anders RA, Sosman JA, Moslehi JJ. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen DY, Huang WK, Chien-Chia Wu V, Chang WC, Chen JS, Chuang CK, Chu PH. Cardiovascular toxicity of immune checkpoint inhibitors in cancer patients: A review when cardiology meets immuno-oncology. J Formos Med Assoc. 2020;119:1461–1475 [DOI] [PubMed] [Google Scholar]
- 22.Guo CW, Alexander M, Dib Y, Lau PKH, Weppler AM, Au-Yeung G, Lee B, Khoo C, Mooney D, Joshi SB, Creati L, Sandhu S. A closer look at immune-mediated myocarditis in the era of combined checkpoint blockade and targeted therapies. Eur J Cancer. 2020;124:15–24 [DOI] [PubMed] [Google Scholar]
- 23.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganatra S, Carver JR, Hayek SS, Ky B, Leja MJ, Lenihan DJ, Lenneman C, Mousavi N, Park JH, Perales MA, Ryan TD, Scherrer-Crosbie M, Steingart RM, Yang EH, Zaha V, Barac A, Liu JE. Chimeric antigen receptor t-cell therapy for cancer and heart: Jacc council perspectives. J Am Coll Cardiol. 2019;74:3153–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alvi RM, Frigault MJ, Fradley MG, Jain MD, Mahmood SS, Awadalla M, Lee DH, Zlotoff DA, Zhang L, Drobni ZD, Hassan MZO, Bassily E, Rhea I, Ismail-Khan R, Mulligan CP, Banerji D, Lazaryan A, Shah BD, Rokicki A, Raje N, Chavez JC, Abramson J, Locke FL, Neilan TG. Cardiovascular events among adults treated with chimeric antigen receptor t-cells (car-t). J Am Coll Cardiol. 2019;74:3099–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, Haris M, Wilson NE, Liu F, Gabunia K, Scholler J, Montine TJ, Bhoj VG, Reddy R, Mohan S, Maillard I, Kriegstein AR, June CH, Chang HY, Posey AD Jr., Satpathy AT. Single-cell analyses identify brain mural cells expressing cd19 as potential off-tumor targets for car-t immunotherapies. Cell. 2020;183:126–142 e117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Pure E, Albelda SM, Epstein JA. Targeting cardiac fibrosis with engineered t cells. Nature. 2019;573:430–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Touyz RM, Herrmann J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis Oncol. 2018;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdel-Qadir H, Ethier JL, Lee DS, Thavendiranathan P, Amir E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat Rev. 2017;53:120–127 [DOI] [PubMed] [Google Scholar]
- 30.Maurea N, Coppola C, Piscopo G, Galletta F, Riccio G, Esposito E, De Lorenzo C, De Laurentiis M, Spallarossa P, Mercuro G. Pathophysiology of cardiotoxicity from target therapy and angiogenesis inhibitors. J Cardiovasc Med (Hagerstown). 2016;17 Suppl 1 Special issue on Cardiotoxicity from Antiblastic Drugs and Cardioprotection:e19–e26 [DOI] [PubMed] [Google Scholar]
- 31.Suter TM, Procter M, van Veldhuisen DJ, Muscholl M, Bergh J, Carlomagno C, Perren T, Passalacqua R, Bighin C, Klijn JG, Ageev FT, Hitre E, Groetz J, Iwata H, Knap M, Gnant M, Muehlbauer S, Spence A, Gelber RD, Piccart-Gebhart MJ. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol. 2007;25:3859–3865 [DOI] [PubMed] [Google Scholar]
- 32.Guglin M, Krischer J, Tamura R, Fink A, Bello-Matricaria L, McCaskill-Stevens W, Munster PN. Randomized trial of lisinopril versus carvedilol to prevent trastuzumab cardiotoxicity in patients with breast cancer. J Am Coll Cardiol. 2019;73:2859–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, Ross J Jr., Chien KR, Lee KF. Erbb2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459–465 [DOI] [PubMed] [Google Scholar]
- 34.Ozcelik C, Erdmann B, Pilz B, Wettschureck N, Britsch S, Hubner N, Chien KR, Birchmeier C, Garratt AN. Conditional mutation of the erbb2 (her2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci U S A. 2002;99:8880–8885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belmonte F, Das S, Sysa-Shah P, Sivakumaran V, Stanley B, Guo X, Paolocci N, Aon MA, Nagane M, Kuppusamy P, Steenbergen C, Gabrielson K. Erbb2 overexpression upregulates antioxidant enzymes, reduces basal levels of reactive oxygen species, and protects against doxorubicin cardiotoxicity. Am J Physiol Heart Circ Physiol. 2015;309:H1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitani T, Ong SG, Lam CK, Rhee JW, Zhang JZ, Oikonomopoulos A, Ma N, Tian L, Lee J, Telli ML, Witteles RM, Sharma A, Sayed N, Wu JC. Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation. 2019;139:2451–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venkatesh P, Kasi A. Anthracyclines. Statpearls. Treasure Island (FL); 2021. [Google Scholar]
- 38.Bernstein D Anthracycline cardiotoxicity: Worrisome enough to have you quaking? Circ Res. 2018;122:188–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jolly MP, Jordan JH, Melendez GC, McNeal GR, D’Agostino RB Jr., Hundley WG. Automated assessments of circumferential strain from cine cmr correlate with lvef declines in cancer patients early after receipt of cardio-toxic chemotherapy. J Cardiovasc Magn Reson. 2017;19:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexandre J, Cautela J, Ederhy S, Damaj GL, Salem JE, Barlesi F, Farnault L, Charbonnier A, Mirabel M, Champiat S, Cohen-Solal A, Cohen A, Dolladille C, Thuny F. Cardiovascular toxicity related to cancer treatment: A pragmatic approach to the american and european cardio-oncology guidelines. J Am Heart Assoc. 2020;9:e018403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipshultz SE, Rifai N, Dalton VM, Levy DE, Silverman LB, Lipsitz SR, Colan SD, Asselin BL, Barr RD, Clavell LA, Hurwitz CA, Moghrabi A, Samson Y, Schorin MA, Gelber RD, Sallan SE. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. 2004;351:145–153 [DOI] [PubMed] [Google Scholar]
- 42.Lipshultz SE, Karnik R, Sambatakos P, Franco VI, Ross SW, Miller TL. Anthracycline-related cardiotoxicity in childhood cancer survivors. Current opinion in cardiology. 2014;29:103–112 [DOI] [PubMed] [Google Scholar]
- 43.Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. Topoisomerase iibeta mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–8846 [DOI] [PubMed] [Google Scholar]
- 44.Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124:617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905 [DOI] [PubMed] [Google Scholar]
- 46.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–1792 [PubMed] [Google Scholar]
- 47.Auner HW, Tinchon C, Linkesch W, Halwachs-Baumann G, Sill H. Correspondence re: O. J. Arola et al., acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer res, 60: 1789–1792, 2000. Cancer Res. 2001;61:2335–2336 [PubMed] [Google Scholar]
- 48.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, Zhang M, Wu H, Guo J, Zhang X, Hu X, Cao CM, Xiao RP. Camkii is a rip3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–182 [DOI] [PubMed] [Google Scholar]
- 49.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pereira GC, Pereira SP, Pereira CV, Lumini JA, Magalhaes J, Ascensao A, Santos MS, Moreno AJ, Oliveira PJ. Mitochondrionopathy phenotype in doxorubicin-treated wistar rats depends on treatment protocol and is cardiac-specific. PLoS One. 2012;7:e38867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase ii. Science. 1984;226:466–468 [DOI] [PubMed] [Google Scholar]
- 52.Capranico G, Tinelli S, Austin CA, Fisher ML, Zunino F. Different patterns of gene expression of topoisomerase ii isoforms in differentiated tissues during murine development. Biochim Biophys Acta. 1992;1132:43–48 [DOI] [PubMed] [Google Scholar]
- 53.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–1642 [DOI] [PubMed] [Google Scholar]
- 54.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. Ii. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–3074 [PubMed] [Google Scholar]
- 55.Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996;98:1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wojnowski L, Kulle B, Schirmer M, Schluter G, Schmidt A, Rosenberger A, Vonhof S, Bickeboller H, Toliat MR, Suk EK, Tzvetkov M, Kruger A, Seifert S, Kloess M, Hahn H, Loeffler M, Nurnberg P, Pfreundschuh M, Trumper L, Brockmoller J, Hasenfuss G. Nad(p)h oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112:3754–3762 [DOI] [PubMed] [Google Scholar]
- 57.Cole MP, Chaiswing L, Oberley TD, Edelmann SE, Piascik MT, Lin SM, Kiningham KK, St Clair DK. The protective roles of nitric oxide and superoxide dismutase in adriamycin-induced cardiotoxicity. Cardiovasc Res. 2006;69:186–197 [DOI] [PubMed] [Google Scholar]
- 58.Mukhopadhyay P, Rajesh M, Batkai S, Kashiwaya Y, Hasko G, Liaudet L, Szabo C, Pacher P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol. 2009;296:H1466–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khadka D, Kim HJ, Oh GS, Shen A, Lee S, Lee SB, Sharma S, Kim SY, Pandit A, Choe SK, Kwak TH, Yang SH, Sim H, Eom GH, Park R, So HS. Augmentation of nad(+) levels by enzymatic action of nad(p)h quinone oxidoreductase 1 attenuates adriamycin-induced cardiac dysfunction in mice. J Mol Cell Cardiol. 2018;124:45–57 [DOI] [PubMed] [Google Scholar]
- 60.Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. 2009;61:154–171 [DOI] [PubMed] [Google Scholar]
- 61.Vincent DT, Ibrahim YF, Espey MG, Suzuki YJ. The role of antioxidants in the era of cardiooncology. Cancer Chemother Pharmacol. 2013;72:1157–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Green DR. The coming decade of cell death research: Five riddles. Cell. 2019;177:1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu X, Chua CC, Gao J, Chen Z, Landy CL, Hamdy R, Chua BH. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am J Physiol Heart Circ Physiol. 2004;286:H933–939 [DOI] [PubMed] [Google Scholar]
- 64.Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol Cell Biochem. 2005;273:25–32 [DOI] [PubMed] [Google Scholar]
- 65.Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-atm-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of rac1 activity. J Mol Cell Cardiol. 2009;47:698–705 [DOI] [PubMed] [Google Scholar]
- 66.Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W, Field LJ. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation. 2009;119:99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feridooni T, Hotchkiss A, Remley-Carr S, Saga Y, Pasumarthi KB. Cardiomyocyte specific ablation of p53 is not sufficient to block doxorubicin induced cardiac fibrosis and associated cytoskeletal changes. PLoS One. 2011;6:e22801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng Z, DiMichele LA, Rojas M, Vaziri C, Mack CP, Taylor JM. Focal adhesion kinase antagonizes doxorubicin cardiotoxicity via p21(cip1.). J Mol Cell Cardiol. 2014;67:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, Cantlon A, Fisch S, Golomb-Mello G, Ryan JA, Deng J, Jian B, Corbett C, Goldenberg M, Madsen JR, Liao R, Walsh D, Sedivy J, Murphy DJ, Carrasco DR, Robinson S, Moslehi J, Letai A. Developmental regulation of mitochondrial apoptosis by c-myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell. 2017;31:142–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, Dorn GW 2nd, Kirshenbaum LA. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci U S A. 2014;111:E5537–5544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amgalan D, Garner TP, Pekson R, Jia XF, Yanamandala M, Paulino V, Liang FG, Corbalan JJ, Lee J, Chen Y, Karagiannis GS, Sanchez LR, Liang H, Narayanagari SR, Mitchell K, Lopez A, Margulets V, Scarlata M, Santulli G, Asnani A, Peterson RT, Hazan RB, Condeelis JS, Oktay MH, Steidl U, Kirshenbaum LA, Gavathiotis E, Kitsis RN. A small-molecule allosteric inhibitor of bax protects against doxorubicin-induced cardiomyopathy. Nat Cancer. 2020;1:315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI, Imai H, Tsutsui H. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, Bertrand R, Andrews NC, Santos MM. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. 2003;102:2574–2580 [DOI] [PubMed] [Google Scholar]
- 74.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. [DOI] [PubMed] [Google Scholar]
- 76.Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V, Kirshenbaum LA. P53 mediates autophagy and cell death by a mechanism contingent on bnip3. Hypertension. 2013;62:70–77 [DOI] [PubMed] [Google Scholar]
- 77.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. Dram, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134 [DOI] [PubMed] [Google Scholar]
- 78.Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2017;104:1–8 [DOI] [PubMed] [Google Scholar]
- 79.Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, Schneider JW, Gillette TG, Hill JA. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fabiani I, Aimo A, Grigoratos C, Castiglione V, Gentile F, Saccaro LF, Arzilli C, Cardinale D, Passino C, Emdin M. Oxidative stress and inflammation: Determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail Rev. 2021;26:881–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delgado RM 3rd, Nawar MA, Zewail AM, Kar B, Vaughn WK, Wu KK, Aleksic N, Sivasubramanian N, McKay K, Mann DL, Willerson JT. Cyclooxygenase-2 inhibitor treatment improves left ventricular function and mortality in a murine model of doxorubicin-induced heart failure. Circulation. 2004;109:1428–1433 [DOI] [PubMed] [Google Scholar]
- 82.Ma Y, Zhang X, Bao H, Mi S, Cai W, Yan H, Wang Q, Wang Z, Yan J, Fan GC, Lindsey ML, Hu Z. Toll-like receptor (tlr) 2 and tlr4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS One. 2012;7:e40763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Antoniak S, Tatsumi K, Schmedes CM, Grover SP, Pawlinski R, Mackman N. Protease-activated receptor 1 activation enhances doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol. 2018;122:80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chan BYH, Roczkowsky A, Cho WJ, Poirier M, Sergi C, Keschrumrus V, Churko JM, Granzier H, Schulz R. Mmp inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc Res. 2021;117:188–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wallace KB, Sardao VA, Oliveira PJ. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. 2020;126:926–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duarte-Karim M, Ruysschaert JM, Hildebrand J. Affinity of adriamycin to phospholipids. A possible explanation for cardiac mitochondrial lesions. Biochem Biophys Res Commun. 1976;71:658–663 [DOI] [PubMed] [Google Scholar]
- 87.Goormaghtigh E, Brasseur R, Huart P, Ruysschaert JM. Study of the adriamycin-cardiolipin complex structure using attenuated total reflection infrared spectroscopy. Biochemistry. 1987;26:1789–1794 [DOI] [PubMed] [Google Scholar]
- 88.Sokolove PM. Interactions of adriamycin aglycones with mitochondria may mediate adriamycin cardiotoxicity. Int J Biochem. 1994;26:1341–1350 [DOI] [PubMed] [Google Scholar]
- 89.Pereira GC, Pereira SP, Tavares LC, Carvalho FS, Magalhaes-Novais S, Barbosa IA, Santos MS, Bjork J, Moreno AJ, Wallace KB, Oliveira PJ. Cardiac cytochrome c and cardiolipin depletion during anthracycline-induced chronic depression of mitochondrial function. Mitochondrion. 2016;30:95–104 [DOI] [PubMed] [Google Scholar]
- 90.Solem LE, Henry TR, Wallace KB. Disruption of mitochondrial calcium homeostasis following chronic doxorubicin administration. Toxicol Appl Pharmacol. 1994;129:214–222 [DOI] [PubMed] [Google Scholar]
- 91.Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001;61:771–777 [PubMed] [Google Scholar]
- 92.Du Q, Zhu B, Zhai Q, Yu B. Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of bnip3. Am J Transl Res. 2017;9:3360–3373 [PMC free article] [PubMed] [Google Scholar]
- 93.Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmstrom A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. 2016;22:547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hixon SC, Ellis CN, Daugherty JP. Heart mitochondrial DNA synthesis: Preferential inhibition by adriamycin. J Mol Cell Cardiol. 1981;13:855–860 [DOI] [PubMed] [Google Scholar]
- 95.Adachi K, Fujiura Y, Mayumi F, Nozuhara A, Sugiu Y, Sakanashi T, Hidaka T, Toshima H. A deletion of mitochondrial DNA in murine doxorubicin-induced cardiotoxicity. Biochem Biophys Res Commun. 1993;195:945–951 [DOI] [PubMed] [Google Scholar]
- 96.Palmeira CM, Serrano J, Kuehl DW, Wallace KB. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim Biophys Acta. 1997;1321:101–106 [DOI] [PubMed] [Google Scholar]
- 97.Serrano J, Palmeira CM, Kuehl DW, Wallace KB. Cardioselective and cumulative oxidation of mitochondrial DNA following subchronic doxorubicin administration. Biochim Biophys Acta. 1999;1411:201–205 [DOI] [PubMed] [Google Scholar]
- 98.Lebrecht D, Kokkori A, Ketelsen UP, Setzer B, Walker UA. Tissue-specific mtdna lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J Pathol. 2005;207:436–444 [DOI] [PubMed] [Google Scholar]
- 99.Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, Walker UA. Time-dependent and tissue-specific accumulation of mtdna and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation. 2003;108:2423–2429 [DOI] [PubMed] [Google Scholar]
- 100.Khiati S, Dalla Rosa I, Sourbier C, Ma X, Rao VA, Neckers LM, Zhang H, Pommier Y. Mitochondrial topoisomerase i (top1mt) is a novel limiting factor of doxorubicin cardiotoxicity. Clin Cancer Res. 2014;20:4873–4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sobek S, Boege F. DNA topoisomerases in mtdna maintenance and ageing. Exp Gerontol. 2014;56:135–141 [DOI] [PubMed] [Google Scholar]
- 102.Tiwari VK, Burger L, Nikoletopoulou V, Deogracias R, Thakurela S, Wirbelauer C, Kaut J, Terranova R, Hoerner L, Mielke C, Boege F, Murr R, Peters AH, Barde YA, Schubeler D. Target genes of topoisomerase iibeta regulate neuronal survival and are defined by their chromatin state. Proc Natl Acad Sci U S A. 2012;109:E934–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pillai VB, Bindu S, Sharp W, Fang YH, Kim G, Gupta M, Samant S, Gupta MP. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am J Physiol Heart Circ Physiol. 2016;310:H962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhuang J, Ma W, Lago CU, Hwang PM. Metabolic regulation of oxygen and redox homeostasis by p53: Lessons from evolutionary biology? Free Radic Biol Med. 2012;53:1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kruiswijk F, Labuschagne CF, Vousden KH. P53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405 [DOI] [PubMed] [Google Scholar]
- 106.Park JH, Zhuang J, Li J, Hwang PM. P53 as guardian of the mitochondrial genome. FEBS Lett. 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nithipongvanitch R, Ittarat W, Velez JM, Zhao R, St Clair DK, Oberley TD. Evidence for p53 as guardian of the cardiomyocyte mitochondrial genome following acute adriamycin treatment. J Histochem Cytochem. 2007;55:629–639 [DOI] [PubMed] [Google Scholar]
- 108.Zhu W, Zhang W, Shou W, Field LJ. P53 inhibition exacerbates late-stage anthracycline cardiotoxicity. Cardiovasc Res. 2014;103:81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li J, Wang PY, Long NA, Zhuang J, Springer DA, Zou J, Lin Y, Bleck CKE, Park JH, Kang JG, Hwang PM. P53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc Natl Acad Sci U S A. 2019;116:19626–19634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhuang J, Wang PY, Huang X, Chen X, Kang JG, Hwang PM. Mitochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc Natl Acad Sci U S A. 2013;110:17356–17361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, Ali QA, Tripodi DJ, Zhuang J, Lago CU, Strong LC, Talagala SL, Balaban RS, Kang JG, Hwang PM. Increased oxidative metabolism in the li-fraumeni syndrome. N Engl J Med. 2013;368:1027–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moslehi J, Amgalan D, Kitsis RN. Grounding cardio-oncology in basic and clinical science. Circulation. 2017;136:3–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1beta through activation of the nlrp3 inflammasome. Cancer Biol Ther. 2011;11:1008–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reislander T, Groelly FJ, Tarsounas M. DNA damage and cancer immunotherapy: A sting in the tale. Mol Cell. 2020;80:21–28 [DOI] [PubMed] [Google Scholar]
- 115.Meneghin A, Hogaboam CM. Infectious disease, the innate immune response, and fibrosis. J Clin Invest. 2007;117:530–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Frantz S, Falcao-Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M, Dawson D, de Windt LJ, Giacca M, Hamdani N, Hilfiker-Kleiner D, Hirsch E, Leite-Moreira A, Mayr M, Thum T, Tocchetti CG, van der Velden J, Varricchi G, Heymans S. The innate immune system in chronic cardiomyopathy: A european society of cardiology (esc) scientific statement from the working group on myocardial function of the esc. Eur J Heart Fail. 2018;20:445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yamasaki T, Yanishi K, Tateishi S, Nakanishi N, Zen K, Nakamura T, Yamano T, Shiraishi H, Shirayama T, Matoba S. Late-onset mitochondrial cardiomyopathy triggered by anticancer treatment. Intern Med. 2017;56:1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Qiao X, van der Zanden SY, Wander DPA, Borras DM, Song JY, Li X, van Duikeren S, van Gils N, Rutten A, van Herwaarden T, van Tellingen O, Giacomelli E, Bellin M, Orlova V, Tertoolen LGJ, Gerhardt S, Akkermans JJ, Bakker JM, Zuur CL, Pang B, Smits AM, Mummery CL, Smit L, Arens R, Li J, Overkleeft HS, Neefjes J. Uncoupling DNA damage from chromatin damage to detoxify doxorubicin. Proc Natl Acad Sci U S A. 2020;117:15182–15192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science. 1995;270:985–988 [DOI] [PubMed] [Google Scholar]
- 120.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in pd-1 receptor-deficient mice. Science. 2001;291:319–322 [DOI] [PubMed] [Google Scholar]
- 121.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, Ishida M, Hiai H, Matsumori A, Minato N, Honjo T. Autoantibodies against cardiac troponin i are responsible for dilated cardiomyopathy in pd-1-deficient mice. Nat Med. 2003;9:1477–1483 [DOI] [PubMed] [Google Scholar]
- 122.Grabie N, Lichtman AH, Padera R. T cell checkpoint regulators in the heart. Cardiovasc Res. 2019;115:869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]