

Abstract
Airborne biohazards are risk factors in the development and severity of rheumatoid arthritis (RA) and RA-associated lung disease, yet the mechanisms explaining this relationship remain unclear. Lipopolysaccharide (LPS, endotoxin) is a ubiquitous inflammatory agent in numerous environmental and occupational air pollutant settings recognized to induce airway inflammation. Combining repetitive LPS inhalation exposures with the collagen induced arthritis (CIA) model, DBA1/J mice were assigned to either: sham (saline injection/saline inhalation), CIA (CIA/saline), LPS (saline/LPS 100 ng inhalation), or CIA+LPS for 5 weeks. Serum anti-citrullinated (CIT) protein antibody (ACPA) and anti-malondialdehyde-acetaldehyde (MAA) antibodies were strikingly potentiated with co-exposure (CIA+LPS). CIT- and MAA-modified lung proteins were increased with co-exposure and co-localized across treatment groups. Inhaled LPS exacerbated arthritis with CIA+LPS>LPS >CIA versus sham. Periarticular bone loss was demonstrated in CIA and CIA+LPS but not in LPS alone. LPS induced airway inflammation and neutrophil infiltrates were reduced with co-exposure (CIA+LPS). Potentially signaling transition to pro-fibrotic processes, there were increased infiltrates of activated CD11c+CD11b+ macrophages and transitioning CD11c+CD11bint monocyte-macrophage populations with CIA+LPS. Moreover, several lung remodeling proteins including fibronectin and matrix metalloproteinases as well as complement C5a were potentiated with CIA+LPS compared to other treatment groups. IL-33 concentrations in lung homogenates were enhanced with CIA+LPS with IL-33 lung staining driven by LPS. IL-33 expression was also significantly increased in lung tissues from patients with RA-associated lung disease (N=8) versus controls (N=7). These findings suggest that patients with RA may be more susceptible to developing interstitial lung disease following airborne biohazard exposures enriched in LPS.
Keywords: collagen induced arthritis, IL-33, rheumatoid arthritis, interstitial lung disease
1. INTRODUCTION
Rheumatoid arthritis (RA) is a systemic autoimmune disease affecting up to 1% of the population worldwide. Although its primary manifestations relate to inflammatory arthritis, respiratory-related deaths are the most over-represented cause of death in men and women with rheumatoid arthritis (RA) (1–3).Various airborne biohazard exposures have been implicated as risk factors in RA and in RA-associated lung disease (3–8), and the risk associated with these exposures appears to be disproportionately higher in men even though RA is more common among women (4). These biohazards include not only cigarette smoke exposure (3, 8), but also other exposures related to ambient air pollution, military duties (burn pits, organic dusts, military waste disposal), and various workplace environments (agriculture organic dusts, textiles, silica, construction, mining) (3–6, 8, 9). More common in men with RA than women with RA, pulmonary manifestations such as interstitial lung disease (ILD) are observed in up to 40% of RA patients. These epidemiologic observations also align with emerging evidence linking the generation of autoantigens and RA-specific autoimmunity to extraarticular sites, particularly the respiratory mucosa (10–13). Correspondingly, patients with RA and chronic lung diseases have increased articular disease activity and higher circulating disease-specific autoantibody concentrations (14, 15). However, the precise mechanisms underpinning the relationship of airborne exposure induced airway inflammation with the development and progression of RA are unknown.
Utilizing animal modeling strategies, it has been demonstrated that repetitive inhaled exposure to agriculture-related organic dust extracts (complex mixtures comprised of an abundance of bacterial products and particulates) in the setting of arthritis induction resulted in a potentiation of inflammatory arthritis and increases in pro-fibrotic indices in lung tissues (16). Sex differences were recapitulated in this pre-clinical mouse model whereby male mice were profoundly more susceptible to most effects of co-exposure. Repetitive inhaled exposures to these dust extracts has also resulted in increased expression of autoantigens in lung tissues, including both citrullinated (CIT)- and malondialdehyde acetaldehyde (MAA)- modified lung proteins (17), with associated increases in anti-MAA autoantibodies (16). In the setting of collagen-induced arthritis (CIA), repetitive inhalant exposure to these dust extracts induced higher circulating concentrations of anti-citrullinated protein antibody (ACPA) (16). These findings are highly relevant recognizing that ACPA responses are highly-specific and pathogenesis in RA, while anti-MAA autoantibodies have been identified as novel biomarkers of RA-associated lung disease (18).
Lipopolysaccharides (LPS) or endotoxin are common in various environmental settings (air pollution, agriculture production, military waste, textiles, silica, construction) that are associated with RA, RA-lung disease, and other chronic lung diseases (4–9, 19–22). For instance, LPS accounts for textile workplace-associated airway disease or Byssinosis (21). Likewise, occupational silica and sand dusts are contaminated with LPS that exacerbates silica-induced pulmonary fibrosis (23–25). Beyond these observations, innate immune responses, including those mediated by the LPS-binding Toll-like receptor (TLR) 4, appear to be central to RA. TLR4 is highly expressed in RA synovial tissue (26) and implicated as a potential target in the treatment of not only RA, but also pulmonary fibrosis (27, 28). Lastly, it is recognized that a number of endogenous TLR4 ligands are also likely to play a critical role in RA progression (29–34).
The objective of this study was therefore to investigate the combination of repetitive inhalant LPS exposure with the CIA model in arthritis-susceptible mice (i.e. DBA/1J strain) to understand the mechanisms and potential biomarkers underlying the dynamic interplay of lung inflammation with the arthritis and systemic autoimmunity that characterize RA.
2. MATERIALS AND METHODS
2.1. Study design for animal studies
We used an established 5-week exposure model (16) modified to replace organic dust extract with LPS. Mice were randomized to 1 of 4 treatment groups including Sham (saline injection/saline inhalation); collagen-induced arthritis (CIA) alone (CIA injection/saline inhalation), LPS alone (saline injection/LPS inhalation), and CIA+LPS (CIA injection/LPS inhalation) (Figure 1A, overview schematic). Male mice were utilized for all studies because we have previously demonstrated that female DBA/1J mice are strikingly less susceptible to LPS-enriched organic dust extract-associated lung disease, autoimmunity, arthritis, and periarticular bone loss in the setting of arthritis induction (16). Arthritis-prone DBA/1J mice between 6–8 weeks of age were purchased from The Jackson Laboratory (Bar Harbor, ME) and allowed to acclimate for one week prior to initiation of experiments. CIA was induced as per the Chondrex protocol (Chondrex, Inc, Redmond, WA) (16). Airway inflammation was induced using an established intranasal inhalation repetitive LPS exposure animal model (35) whereby mice were lightly sedated under isoflurane and received daily treatment with either 50 μL of sterile saline or LPS (100 ng in 50 μL sterile saline) from Escherichia coli (O55:B5; Sigma, St. Louis, MO) daily for up to 5 weeks (weekends excluded). Animals were euthanized 5 hours following the final exposure. No respiratory distress, signs of stress, or weight loss were observed throughout the exposure period. Animal procedures were approved by the Institutional Animal Care and Use Committee and were in accordance with the NIH guidelines for the use of rodents.
Fig. 1. Combined modeling of repetitive lipopolysaccharide (LPS)-induced airway inflammation with collagen-induced arthritis (CIA) with arthritis, bone deterioration, and serum pentraxin-2 and autoantibodies.
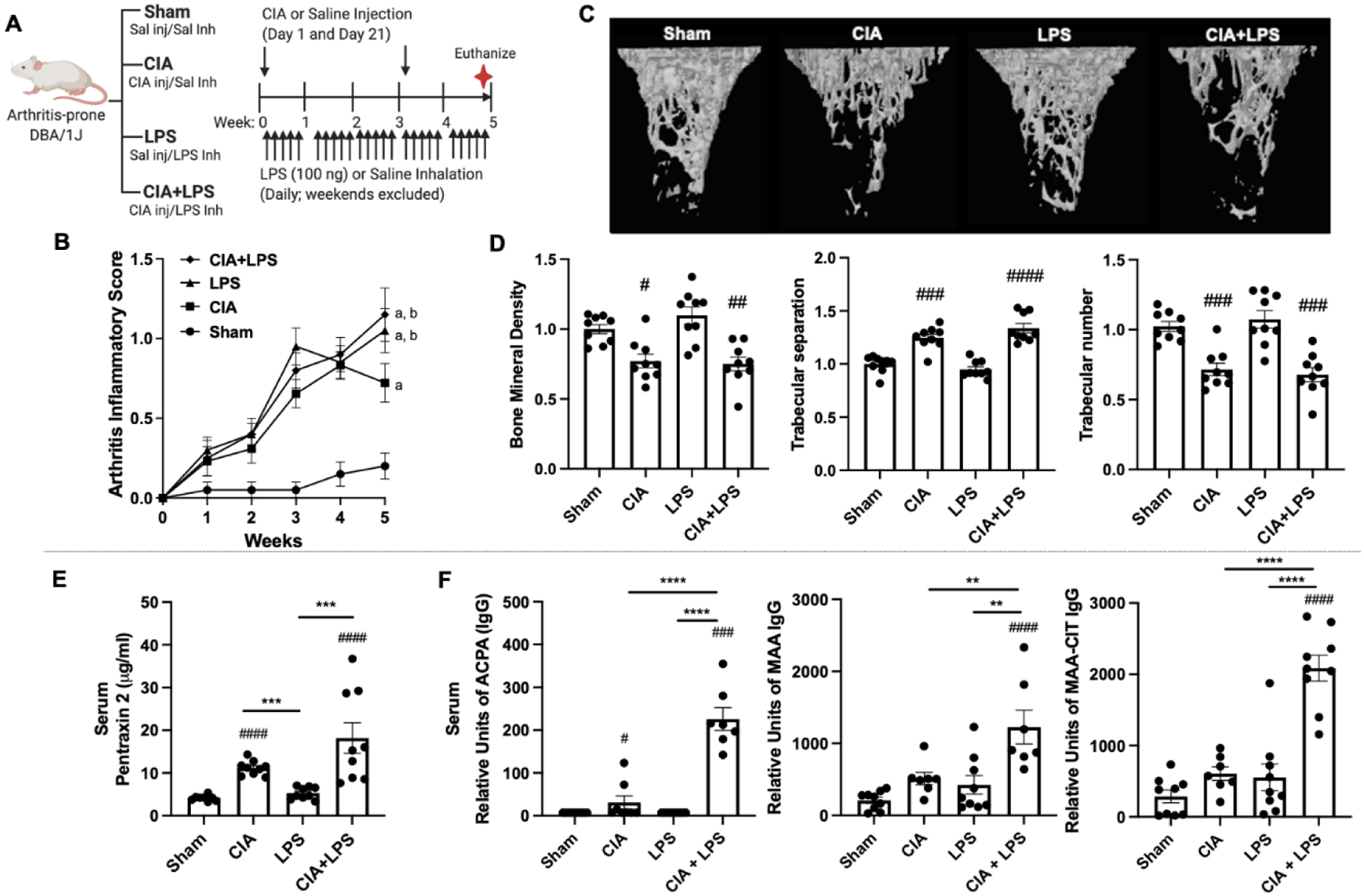
(A) Schematic of experimental design. (B) Line graph depicts mean with SE bars of arthritis inflammatory scores. Statistical difference versus sham denoted as “a” (p<0.0001); versus CIA denoted as “b” (p<0.05) with 9–10 mice/group/time point. (C) A representative 3D reconstructed image for region of interest of proximal tibia. Note the loss of trabecular bone in the CIA and CIA+LPS, but not LPS treatment groups as compared with Sham. Scatter plots depict mean with SE bars of (D) bone mineral density, trabecular separation and trabecular number with data representing the difference induced by treatments divided by sham. Serum levels of the (E) acute-phase reactant protein pentraxin 2 levels and (F) levels of IgG anti-citrullinated protein antibody (ACPA) levels to citrullinated (CIT) peptides, IgG antibody to malondialdehyde-acetaldehyde (MAA)-modified proteins, and combined anti-MAA-CIT autoantibodies. Statistical difference versus sham (#p<0.05, ##p<0.01, ###p<0.001, ####p<0.0001) and between groups as noted by lines (**p<0.01, ***p<0.001, ****p<0.0001).
2.2. Arthritis evaluation
Mice were assessed weekly for the development of arthritis using the semi-quantitative, mouse arthritis scoring system provided by Chondrex (www.chondrex.com). This protocol is based on hind-foot examination with a range of 0 (no inflammation) to 4 (erythema and severe swelling encompassing ankle, foot, and digits).
2.3. Micro-CT analysis of the bone
Tibias were isolated and processed for micro-CT scanning and analysis (16, 36). Periarticular bone was scanned using high-resolution micro-CT (Skyscan 1172; Bruker, Kontich, Belgium) with bone position corrected using Dataviewer (Skyscan) to assure proper orientation (16, 36). The scanning parameters were set as the following: acquisition resolution at 8.66 μm, with X-ray source voltage and current set at 48 kV and 187μA, respectively, scanning rotation step at 0.4°, and frame averaging at 4. NRECON (Sky-scan) software was used to reconstruct scanned images. Analysis of the reconstructed tibia images was performed using CTAn (Skyscan) software, with final analysis conducted on a volume of interest of trabecular bone. Specifically, the region of interest (ROI; periarticular region) was defined as the region starting at the 0.98 mm away from epiphyseal plate and continued for 1.00 mm. Bone mineral density (g/cm3), trabecular number, and trabecular separation were calculated with data presented as the difference induced by treatments divided by sham compiled from independent studies (36, 37).
2.4. Serum
Whole blood was collected from mice at the time of euthanasia from the axillary artery (16). Pentraxin-2 (murine acute phase reactant protein) levels were quantified by Quantikine ELISA according to manufacturer’s instructions (R&D Systems, Minneapolis, MN). Anti-MAA antibodies (IgG) were quantified as previously described (38). Anti-citrullinated protein antibody (ACPA) and anti-MAA-CIT (IgG) antibodies were quantified using an ELISA and human serum albumin as the substrate antigen (39).
2.5. Lung autoantigen, extracellular protein staining and antigen capture assay
Paraffin embedded lung sections from mice were rehydrated and subjected to antigen retrieval using citrate buffer and steam. Sections were then blocked with goat serum and incubated with a Zenon Alex Fluor™ 594 label (Invitrogen, Carlsbad, CA) rabbit polyclonal IgG antibody to MAA and a mouse monoclonal anti-peptidyl-citrulline, clone F95 IgMκ (Millipore Sigma, Burlington, MA). Detection of the F95 antibody was done using an Alexa Fluor® 488 AffiniPure donkey anti-mouse IgM, μ chain specific antibody (Jackson Immunoresearch, West Grove, PA). DAPI (4′,6-diamidino-2-phenylindole; to identify nuclei) was added and samples were sealed with Fluormount-G (Southern Biotech, Birmingham, AL). Fluorochrome detections were done using a Revolve fluorescent microscope (ECHO, San Diego, CA). Images were quantified using ImageJ (NIH, Bethesda, MD) and colocalization done using the Image J FIJI plugin Coloc 2 as previously described (40).
To determine whether proteins co-modified with MAA and CIT were present in the lungs, a capture assay was developed. Briefly, rabbit IgG polyclonal anti-MAA antibody was coated onto ELISA plates and incubated overnight at 4°C. Plates were then washed, blocked in 2% casein, washed, and incubated with lung tissue homogenates overnight at 4°C. Samples were rewashed and the mouse monoclonal anti-peptidyl-citrulline, clone F95 IgMκ incubated on the plate for 1 hour. Plates were washed and incubated with an HRP donkey anti-mouse IgM, μ chain specific antibody (Jackson Immunoresearch). Following a washing step, the complex was developed using TMB substrate, stopped with 2N H2SO4 and reactivity detected at 450nm using Epoch plate reader (BioTek, Winooski, VT) and analyzed using Gene 5 Software (BioTek).
2.6. Bronchoalveolar lavage fluid cell analysis
Bronchoalveolar lavage fluid (BALF) was collected using 3 × 1 mL PBS. Total cell numbers from the combined recovered lavage were enumerated and differential cell counts determined from cytospin-prepared slides (cytopro cytocentrifuge, ELITech Group, Logan, UT) stained with DiffQuick (Siemens, Newark, DE). From cell-free supernate of the first lavage fraction, tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6) and murine neutrophil chemokines, CXCL1 and CXCL2, were quantitated by ELISA (R&D Systems, Minneapolis, MN; limits of detectability of 7.2, 1.8, 2.0, and 1.5 pg/ml, respectively), as these analytes have been implicated in airborne biohazard-induced lung inflammation (41–43).
2.7. Lung cell staining and flow cytometry
Following vascular perfusion and removal of BALF, harvested lungs (right lung) were subjected to an automated dissociation procedure using a gentleMACS Dissociator instrument (Miltenyi Biotech, Auburn, CA) (16). Viability of total lung cells was assessed by trypan blue exclusion and a LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen, Carlsbad, CA). Less than one percent of gated cells were not viable, with no differences by treatment group (data not shown). Lung cells from each animal were incubated with CD16/32 (Fc Block, BD Biosciences, San Jose, CA) to minimize non-specific antibody staining, and then stained with monoclonal antibodies (mAb) directed against rat anti-mouse CD45 (clone: 30-F11; eBiosciences), CD11b (clone: M1/70; Biolegend), Ly6G (clone 1A8; Biolegend), and CD11c (clone: N418; Invitrogen). Gating strategies for CD11c+CD11blo alveolar macrophages, CD11c+CD11bhi activated macrophages, CD11cintCD11bhi transitioning monocytes-macrophages, CD11c−CD11bhi monocytes, and Ly6G+ neutrophils were as previously reported (16). The percentage of all respective cell populations was determined from live CD45+ lung leukocytes after excluding debris and doublets. This percentage was multiplied by the respective total lung cell numbers to determine specific cell population numbers for each animal.
2.8. Lung histopathology
Following lavage, left lungs were excised and inflated to 15 cm H2O pressure with 10% formalin (Fisher Scientific, Fair Lawn, NJ) for 24 hours to preserve pulmonary architecture (16). Sectioned (4–5 μm) lungs were H&E stained and semi-quantitatively assessed for the degree and distribution of lung inflammation utilizing a previously published scoring system (44). This scoring system evaluates the spectrum of inflammatory changes for alveolar and bronchiolar compartments whereby each parameter is independently assigned a value from 0 to 4, with a higher score indicating greater inflammation. Lung sections were also stained for collagen with modified Masson’s Trichrome (collagen: blue) and Sirius Red/Fast Green Collagen Staining kit (Chondrex, Inc, Woodinville, WA) and quantified using Image J FIJI plug in with color thresholding set for blue for Masson’s Trichrome (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, https://imagej.nih.gov/ij/1997-2016) (16).
2.9. Lung homogenate mediator analysis
To facilitate analyte detection, tissue homogenates were prepared by homogenizing lung samples in 500 μl of sterile PBS. Matrix metalloproteinases (MMPs) and metallopeptidase inhibitor (TIMP-1) levels were determined by Magnetic Luminex Assay (R&D), with lower levels of detection of 4.8 pg/ml for MMP-3; 1276.5 pg/ml for MMP-8, 33.9 pg/ml for MMP-9, 24.7 pg/ml for TIMP-1. Complement component/anaphylatoxin C5a (R&D, lowest limit of detection of 15.6 pg/ml), alarmin IL-25 (Invitrogen, lowest limit of detection of 3.9 pg/ml) and alarmin IL-33 (R&D, with sensitivity of 14.3 pg/ml) were determined by individual ELISAs.
2.10. Human lung studies
Human lung sections of control subjects and patients with RA-associated lung disease were obtained from the University of Nebraska Medical Center institutional IRB-approved lung and/or rheumatology bio-banks. De-identified samples from normal donor human lungs (“controls”) (all with limited clinical information and deemed unsuitable for transplant) were obtained from lungs free from disease, or current smoking were accepted from the local organ procurement service, Live On Nebraska. All donors were de-identified and deceased. This protocol was reviewed by the University of Nebraska Medical Center IRB and was determined to be “non-human subjects research.”
2.11. Murine and human lung staining for IL-33 and vimentin
Lung tissue sections from mice (one section from each mouse/experimental group) as well as healthy human controls (n=7) and patients with RA-associated lung disease (n=9) were stained with mouse anti-IL-33 (AF3626, 2.5 μg/100 μl; R&D Systems) or human anti-IL-33 (AF3625, 2.5 μg/100 μl) and anti-vimentin (Abcam, AB 92547, 1:200). Donkey anti-goat (AlexaFluorPlus555, A32816) and anti-rabbit (AlexaFluor488, A21206; Thermo Fisher, Grand Island, NY) at 1:100 dilutions were used as secondary antibodies. DAPI (4’6-diamidino-2-phenylindole) was used to identify cell nuclei. Photographs (10 per lung section per patient/mouse) of lung parenchyma were taken from the entire section under a Zeiss fluorescent microscope at 20X for humans or 40X for mice. Sections (1 per patient, one from each mouse/experimental group) were then analyzed. Integrated densities (the product of area and mean gray value) of each protein were measured as single color on black background with color threshold determined by Image J FIJI plug in (version: 2.1.0/1.53c).
2.12. Statistical analysis
Data are presented as the mean ± standard error of mean (SEM). To detect significant changes among 3 or more groups, a one-way analysis of variance (ANOVA) was utilized and a post-hoc test (Tukey/LSD) was performed to account for multiple comparisons if the p value was < 0.05 or nonparametric Mann-Whitney test to detect significant changes among 2 groups. A two-way ANOVA for 2 variables (i.e., treatment groups vs. time point) was applied to determine differences in arthritis severity score. In confocal studies, the FIJI plugin, Coloc 2 in ImageJ was used to quantify the magnitude of co-localization of MAA or CIT using five regions of interest from each image. Pearson’s correlation coefficients were calculated based on the overlap of two colors to generate an R2-value. R2-values from the five regions of interest were averaged for each mouse and compared using a one-way ANOVA with Tukey’s post hoc test to account for multiple comparisons. All statistical analyses were performed using GraphPad Prism (version: 9.0.0) software and statistical significance accepted at p < 0.05.
3. RESULTS
3.1. Arthritis severity and periarticular bone loss
Throughout the 5-week experiment, arthritis scores increased in mice treated with CIA+LPS, LPS alone, and CIA alone vs. Sham (Fig. 1B). At 5-weeks, the highest arthritis score was for CIA+LPS, but this score did not differ significantly vs. LPS alone. Arthritis scores were significantly increased for both CIA+LPS (p<0.05) and LPS (p<0.05) vs. CIA alone at 5 weeks. Micro-CT analysis of periarticular tibia demonstrated that CIA and CIA+LPS resulted in significant trabecular bone loss (Fig. 1C). Compared with Sham, CIA and CIA+LPS resulted in significant declines that were similar in magnitude for bone mineral density (−16% and −27%) and trabecular number (−22% and −33%) and associated with commensurate increases in trabecular separation (+17% and +31%) (Fig. 1D). LPS alone did not result in significant bone loss vs. Sham.
3.2. Serum pentraxin-2 and autoantibodies
Serum concentrations of the murine acute phase reactant protein, pentraxin-2, were increased in CIA and CIA+LPS, but not LPS, with the highest concentrations observed in CIA+LPS (Fig. 1E). While serum ACPA were modestly increased in CIA compared to Sham, these levels were strikingly potentiated (~15-fold vs other groups) in the combined CIA+LPS mice (Fig. 1F). Serum anti-MAA IgG antibody responses as well as combined anti-CIT/MAA IgG antibody responses were only increased with combined CIA+LPS treatments (Fig. 1F).
3.3. Lung autoantigen expression
Lung tissues were stained and analyzed for CIT- and MAA-modified antigens. By confocal microscopy, both CIT and MAA modified proteins were significantly increased in CIA, LPS, and CIA+LPS vs. Sham, with the highest expression demonstrated with combined CIA+LPS administration (p<0.01 vs. all other groups) (Fig. 2A–C). Pearson correlation coefficients and R2 values were calculated as measures of autoantigen co-localization in lung tissues. CIT and MAA co-localization were similarly increased in each treatment group compared to Sham (Fig. 2D). Similarly, in separate studies using lung tissue homogenates, there was an increase in the co-capture of protein co-modified with MAA-CIT using sandwich ELISA, with increases demonstrated for CIA, LPS, and CIA+LPS as compared to Sham (Fig. 2E). Corresponding to confocal microscopy findings, the degree of co-capture of lung proteins co-modified with MAA-CIT was most significant with the CIA+LPS treatment group. There was no difference among treatment groups for capturing MAA alone or CIT alone (data not shown).
Fig. 2. Combined CIA+LPS modeling modulates lung CIT and MAA autoantigen expression.
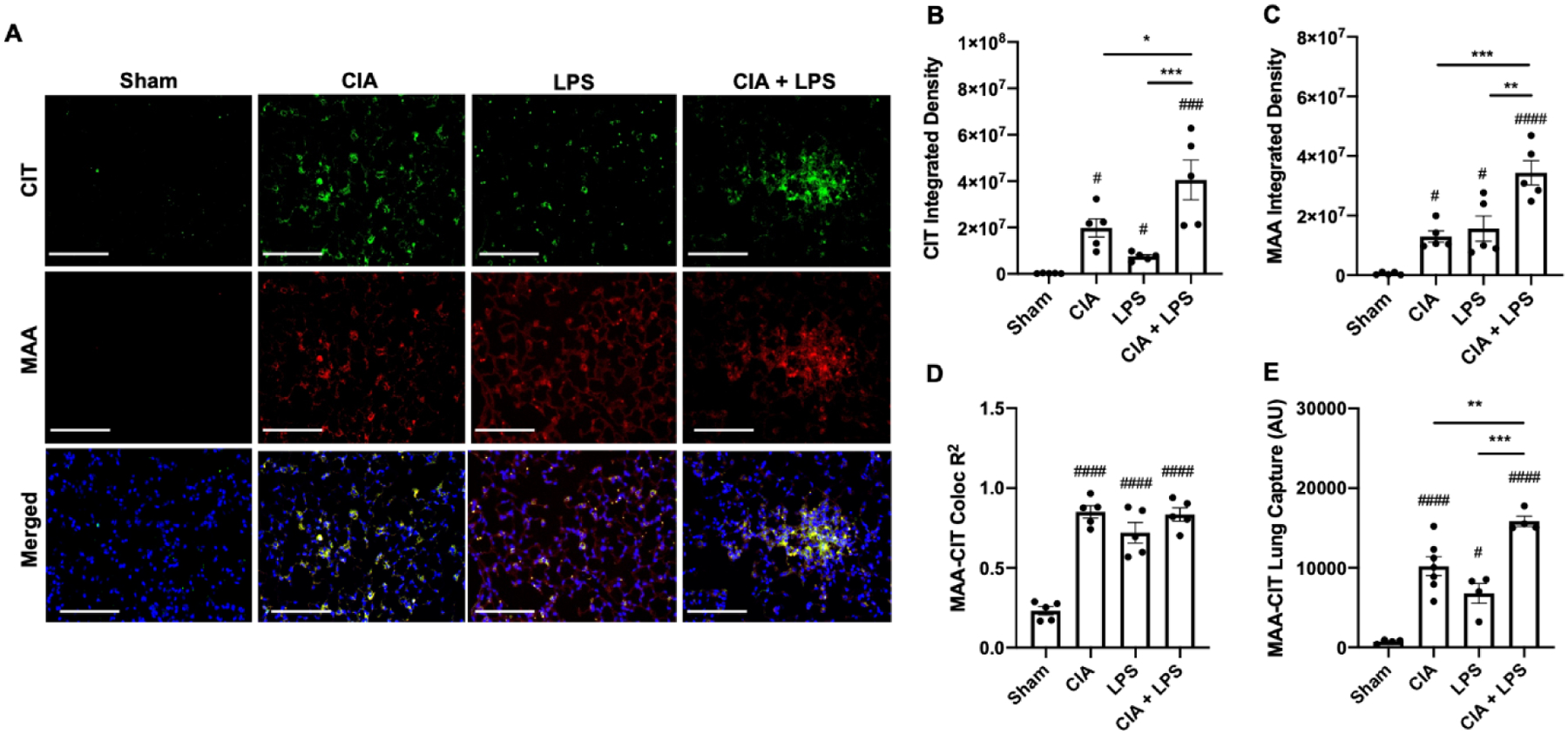
(A) Confocal images of lung tissue from treatment groups stained for citrulline (CIT; green) and malondialdehyde-acetaldehyde (MAA; red) modified proteins and merged with nuclei staining (DAPI, blue) shown at 40x magnification. Images were analyzed using Zen 2012 software (Zeiss). Scatter plots depict integrated density with SE bars of (B) CIT and (C) MAA-modified proteins quantified per each mouse. (D) R2-values demonstrating co-localization of MAA-CIT within each treatment group. (E) Co-capture of MAA-CIT from lung tissue homogenates by sandwich ELISA (AU; arbitrary units). Statistical difference versus sham (#p<0.05, ###p<0.001, ####p<0.0001) and between groups as noted by lines (*p<0.05, **p<0.01, ***p<0.001). Line scale denotes 100 μm.
3.4. Lung histopathology and extracellular matrix deposition
Lung histology demonstrated increased semi-quantitative inflammatory scores as well as increased bronchiolar and alveolar inflammation with LPS and CIA+LPS vs. Sham (Fig. 3A, 3C). There were no significant differences between LPS and CIA+LPS treatment groups and CIA did not induce significant lung inflammation vs. Sham. Correspondingly, there was also increased collagen staining (blue; Masson’s Trichrome and red/magenta; Sirius Red) in CIA, LPS and CIA+LPS vs. Sham (Fig. 3A); with the greatest collagen expression with LPS and CIA+LPS (Fig. 3D).
Figure 3. Combined CIA+LPS exposure modulates lung histopathology and extracellular matrix deposition.
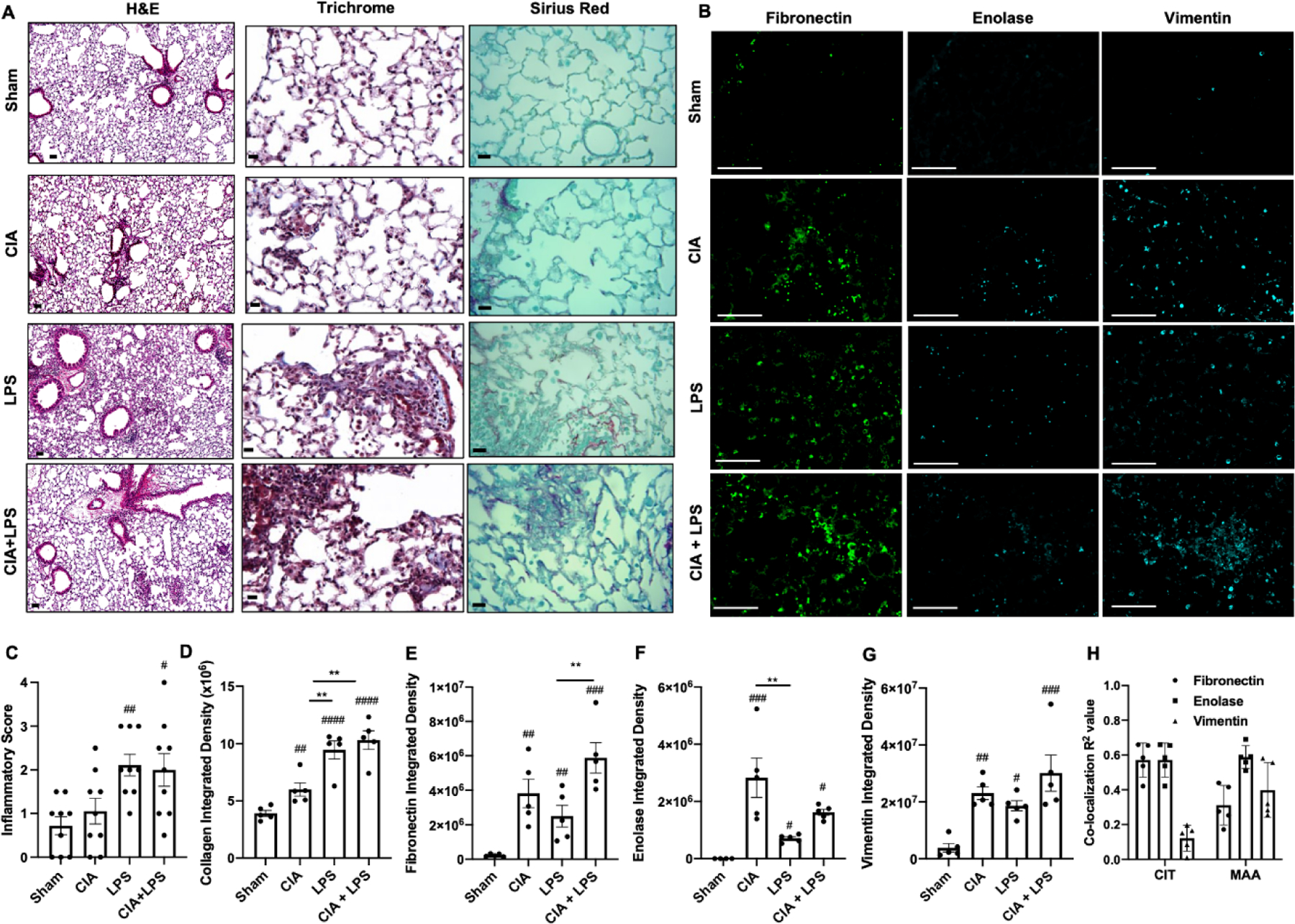
(A) Representative H&E and collagen (Trichrome; blue and Sirius Red; magenta)-stained lung section images from each treatment group. (B) Confocal microscopy images of fibronectin, enolase, and vimentin from each treatment group. Scatter plots depict with mean and SEM of (C) semiquantitative inflammatory score from H&E-stained sections and integrated density of (D) collagen (blue), (E) fibronectin, (F) enolase, and (G) vimentin staining. (H) Co-localization coefficient (R2) of fibronectin, enolase, and vimentin with CIT or MAA in the CIA+LPS treatment group. Statistical difference (#p<0.05, ##p<0.01, ###p<0.001, ####p<0.0001) versus saline and between groups denoted by line (**p<0.01). Line scale denotes 100 μm.
Lung tissues were also stained for other extracellular matrix proteins implicated in tissue fibrosis, RA autoimmunity, and/or RA-associated lung disease including fibronectin, enolase, and vimentin (Fig. 3B). Fibronectin was increased with CIA and LPS and its expression was enhanced with combined CIA+LPS treatments (Fig. 3E). Enolase staining was increased with CIA, LPS, and CIA+LPS vs. Sham, with greatest expression demonstrated for CIA alone (Fig. 3F). Vimentin expression was increased in CIA, LPS, and CIA+LPS vs. Sham, but there was no difference among the treated groups (Fig. 3G). In CIA+LPS tissues, CIT co-localized with fibronectin and enolase (both R2 values ~0.6) but not vimentin (R2 value of 0.12), whereas MAA co-localized (R2 values of ~0.3, 0.6, and 0.4) with fibronectin, enolase, and vimentin.
3.5. Airway inflammation
LPS exposure induced increases in bronchoalveolar lavage fluid (BALF) total cells, neutrophils, and lymphocytes (Fig. 4A) as well as release of TNF-α, IL-6, and murine neutrophil chemoattractants (CXCL1 and CXCL2) (Fig. 4B) compared with Sham. CIA+LPS exposure led to increases in total cells, neutrophils, lymphocytes, and macrophages vs. Sham, with no difference vs. LPS alone. Eosinophils were rarely detected and did not differ across treatment groups (data not shown). However, a differential response was demonstrated with the inflammatory mediators given that CIA+LPS increased TNF-α and IL-6 release, but not neutrophil chemoattractants relative to Sham. Levels of IL-6, CXCL1, and CXCL2 were significantly lower in CIA+LPS vs. LPS alone. There were no increases in airway cell influx and cytokine/chemokine levels in CIA vs. Sham treatment groups.
Fig. 4. Combined CIA+LPS induced lung inflammatory cell influx and mediator release.
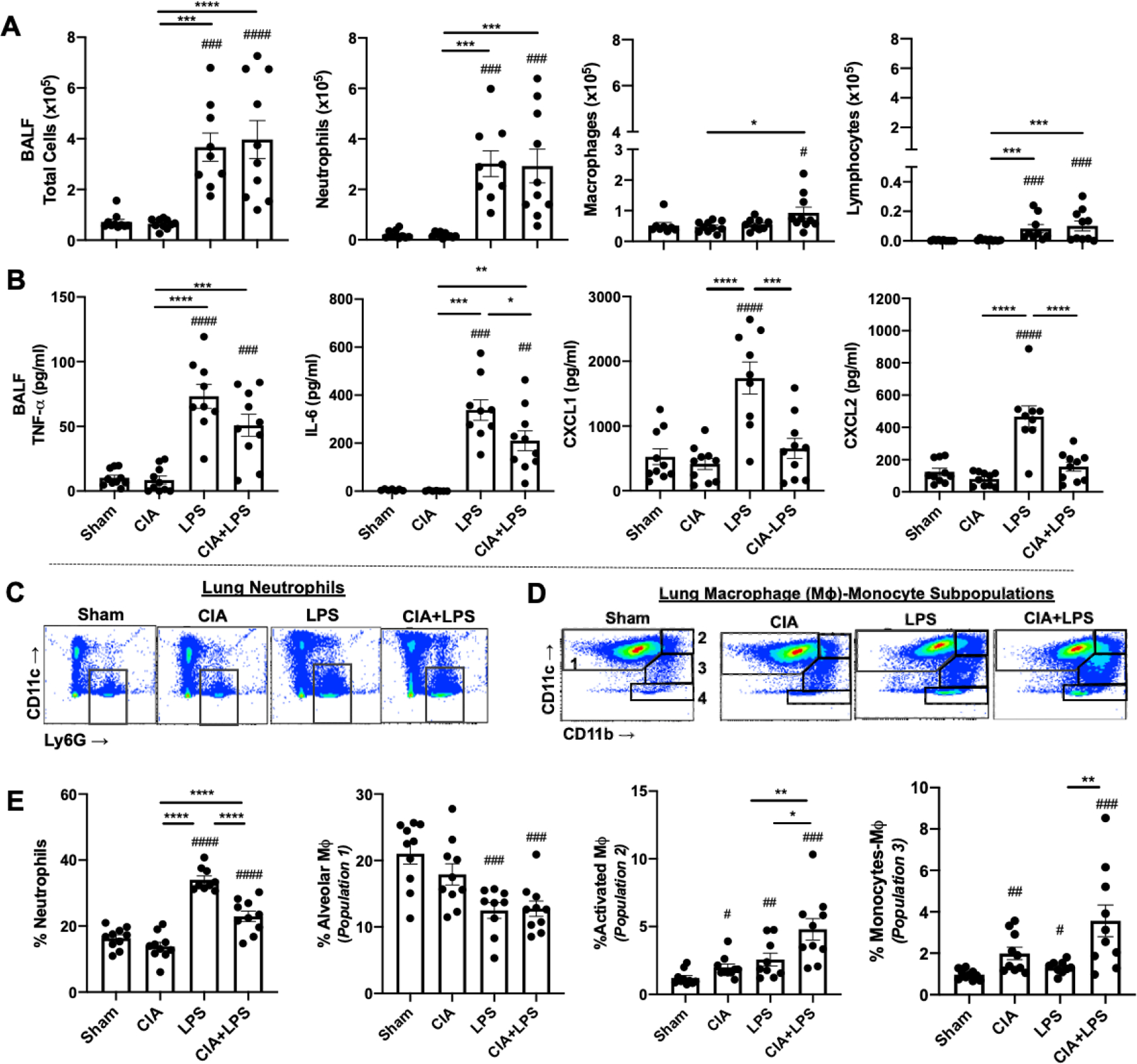
Scatter plots with bars depict mean with SEM. (A) Total cells, neutrophils, macrophages, and lymphocytes enumerated from bronchoalveolar lavage fluid (BALF). (B) BALF levels of pro-inflammatory cytokines (TNF-α, IL-6) and neutrophil chemoattractants (CXCL1 and CXCL2). A representative contour plot of lung neutrophils (CD11c−Ly6G+) (C) and the four macrophage-monocyte populations based upon CD11c and CD11b expression after removal of neutrophils and lymphocytes (D), all gated from live CD45+ cells after excluding debris and doublets. (E) Lung cell % populations of CD45+ cells. No difference among groups for monocytes (Population 4). Statistical difference (#p<0.05, ##p<0.01, ###p<0.001, ####p<0.0001) versus sham and between groups denoted by line (*p<0.05, **p<0.01, ***p<0.001, ***p<0.0001).
Lung neutrophil and monocyte/macrophage infiltrates were quantified by flow cytometry based on the gating strategies depicted in Fig. 4C–D. Lung neutrophil infiltrates were increased with LPS and CIA+LPS vs. Sham, but the percentage of neutrophils was reduced with CIA+LPS vs. LPS alone (Fig. 4E). There was no increase in neutrophil infiltrates with CIA vs. Sham. Whereas resting alveolar macrophages (CD11c+CD11b−, population 1) were reduced, activated lung macrophages (CD11c+CD11b+, population 2) and recruited/transitional monocyte-macrophages (CD11cintermediateCD11b+, population 3) were increased with CIA, LPS, and CIA+LPS, with the highest percentage demonstrated following combined CIA+LPS exposure (Fig. 4E). There was no difference in lung monocytes (CD11c−CD11b+, population 4) or lymphocytes including CD3+CD4+ T cells, CD3+CD8+ T cells, CD3−CD19+ B cells, or CD3−CD19−NK1.1+ Natural killer cells across the treatment groups (data not shown).
3.6. Lung matrix metalloproteinases, complement, and alarmins
Compared to Sham, CIA+LPS increased levels of MMP-2, -3, -8, -9, and TIMP-1 in lung tissue homogenates whereas LPS alone increased MMP-8, -9, and TIMP-1 and CIA increased only MMP-9 (Fig. 5A–B). TIMP-1 levels were potentiated with CIA+LPS as compared to LPS alone (Fig. 5B). Complement component C5a is a proinflammatory mediator that has been implicated in RA (45, 46), playing a role in neutrophil chemotaxis and neutrophil adhesion. C5a levels were significantly increased only in the CIA+LPS treatment group (Fig. 5C). Alarmins such as IL-33, thymic stromal lymphopoietin (TSLP), and IL-25 are released in tissues in response to cellular damage and stress, and they have also been suggested to be involved in pro-fibrotic responses in idiopathic pulmonary fibrosis (47, 48), a condition that resembles RA-ILD. IL-25 levels from lung homogenates were decreased in the LPS and CIA+LPS treatment groups relative to Sham, with significantly lower IL-25 levels in the CIA+LPS as compared to LPS alone (p<0.05) (Fig. 5D). In contrast, IL-33 concentrations were increased in lung homogenates from CIA, LPS, and CIA+LPS vs. Sham treatment groups (Fig. 5E). Levels of IL-33 were also significantly increased with CIA+LPS vs. LPS alone or CIA alone. TSLP levels were not detected across treatment groups (data not shown).
Figure 5. Combined CIA+LPS effects with MMPs, complement, and alarmins.
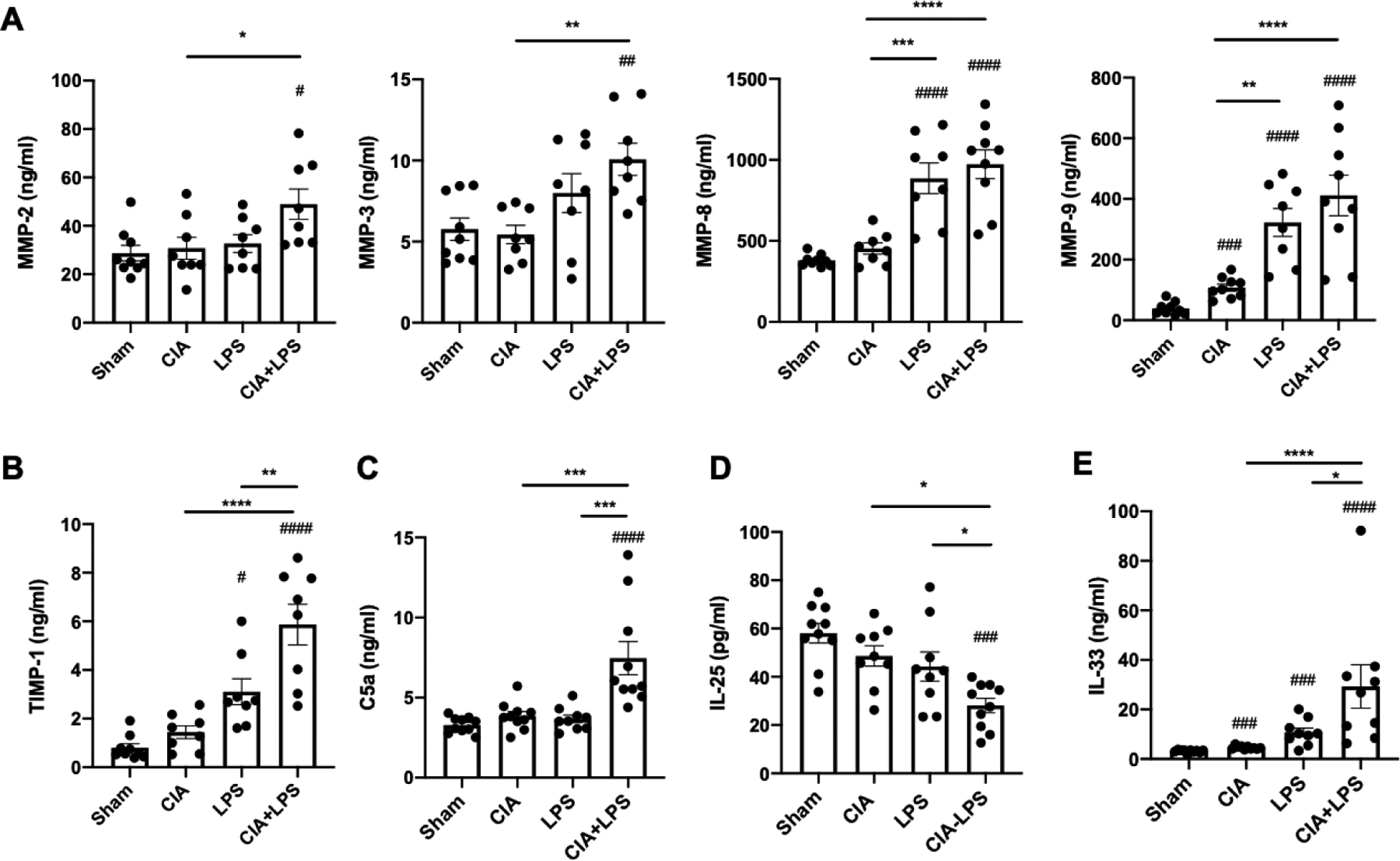
Scatter plots with bars depict mean with SEM. Lung tissue levels of matrix metalloproteinases (A, MMPs) and metallopeptidase inhibitor (B, TIMP-1) determined by multiplex analysis. Lung tissue levels of complement component C5a (C), IL-25 (D) and IL-33 (E) by ELISA. Statistical difference (#p<0.05, ##p<0.01, ###p<0.001, ####p<0.0001) versus sham and between groups denoted by line (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
3.7. Lung IL-33 and vimentin expression in mice
Based upon the increased lung expression of IL-33 with combined CIA+LPS exposure, lung sections were stained for IL-33 as well as vimentin to assess pro-fibrotic processes (49) given that vimentin expression is largely attributable to mesenchymal cells (i.e. fibroblasts, smooth muscle cells, and endothelial cells) and macrophages. Lung IL-33 was increased in LPS and CIA+LPS vs. Sham and vs. CIA alone (Fig. 6A), with no significant differences between LPS and CIA+LPS. Vimentin expression was increased across all treatment groups vs. Sham, but there was no significant difference between CIA, LPS, and CIA+LPS treatment groups.
Figure 6. Lung IL-33 and vimentin expression in murine co-exposure modeling and humans with RA-associated lung disease.
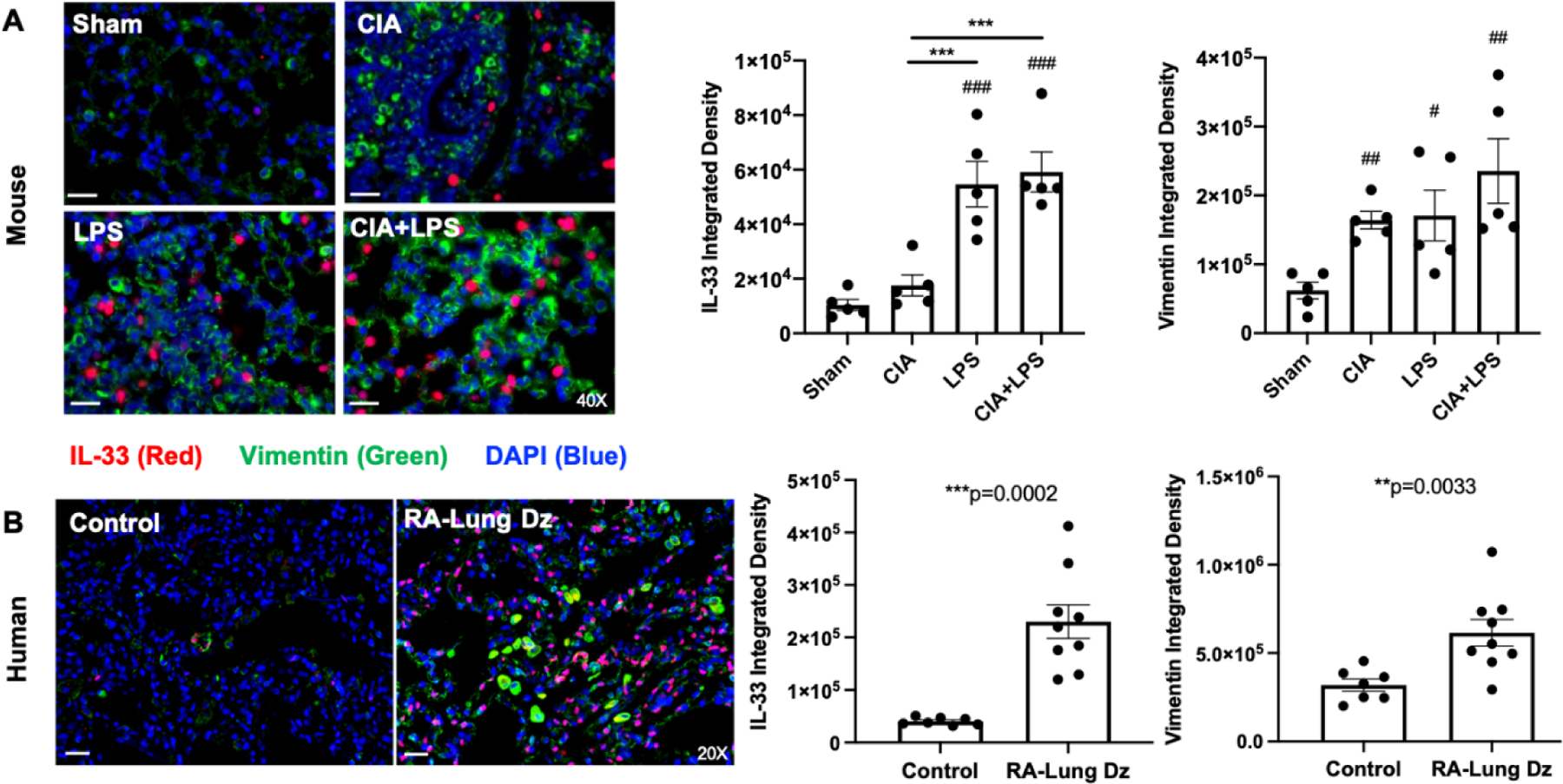
Photomicrographs were taken of entire lung section under a Zeiss fluorescent microscope. A representative merged image of all three stains of IL-33 (red), vimentin (green) and DAPI (blue; nuclei) from each murine treatment group (A) and human populations (B) of control subjects and patients with RA-associated lung disease (Dz). Scatter plots with bars depict mean + SEM per mouse (A) and human subjects (B) for IL-33 and vimentin. The individual values are averaged from 10 images per section per mouse or human for respective proteins. N=5 mice/group, N=7 control subjects, N=9 RA-associated lung disease subjects. Statistical difference versus sham (#p<0.05, ##p<0.01, ###p<0.001, ####p<0.0001) and between groups as noted by lines (**p<0.01, ***p<0.001). Line scale denotes 100 μm.
3.8. Lung IL-33 and vimentin expression in humans with RA-associated lung disease
To examine the translational significance of these findings, tissue sections from de-identified normal human lungs (“controls”) deemed unsuitable for transplant (N=7, age range: 19–59, 14.3% female) were stained for IL-33 as well as vimentin and compared to lung sections from patients with RA-associated lung disease (N=9, age range: 42–67 years, 55.6% female; pathology summarized in Table 1). As summarized in Figure 6, lung IL-33 expression was strikingly increased in the lung tissues of patients with RA-associated lung disease, with lesser (but still significant) increases in vimentin staining (Fig. 6B). Excluding 2 subjects with RA-related pulmonary nodules (in the absence of parenchymal disease) and 1 subject with mild lung inflammation, increases in IL-33 and vimentin expression remained significant, p=0.0012 and p=0.0023, respectively.
Table 1.
Characteristics of RA patients and corresponding lung tissue sample.
| # | Sex | Age | Disease | Pathology Report |
|---|---|---|---|---|
| 1 | M | 59 | RA-ILD | Patchy and non-specific foci of subpleural fibrosis, chronic cellular bronchiolitis associated fibrosis and chronic interstitial pneumonitis suggestive of usual interstitial pneumonia |
| 2 | M | 67 | RA-ILD | Organizing diffuse alveolar damage, chronic interstitial pneumonia, and patchy foci of significant interstitial fibrosis with honeycomb change and areas of chronic pleuritis |
| 3 | F | 62 | RA-mild disease | Bronchial mucosa with mild chronic inflammation and eosinophils |
| 4 | F | 49 | RA-ILD | Patchy fibrosis with subpleural, paraseptal and peribronchiolar distribution, bronchiectasis, focal lymphoid aggregates |
| 5 | F | 49 | RA-ILD | Fibrovascular pleural adhesion, mild diffuse interstitial lymphoid hyperplasia, patchy non-specific chronic interstitial pneumonitis, chronic cellular bronchiolitis |
| 6 | F | 60 | RA-nodules | Nodules with diffuse interstitial deposition of eosinophilic extracellular materials with multinucleated giant cells on background of chronic inflammation of plasma cells and lymphocytes. |
| 7 | M | 51 | RA-ILD | Focal areas of pleural fibrosis with necrotizing granuloma with peripheral palisading histiocytes1 |
| 8 | M | 65 | RA-nodules | Nodules of necrotizing granulomatous inflammation |
| 9 | F | 42 | RA-ILD | Mild ground-glass opacity, mild honeycombing, mild air trapping, features more consistent with usual interstitial pneumonia with fibrosis |
Evidence of both RA and RA-nodules.
3.9. Combination exposure modeling at earlier time points
To determine earlier temporal events in this murine co-exposure modeling system, lung and serum specimens were collected and evaluated at 1 week (Table S1) and 3 weeks (Table S2), time points that fall between the two injections used in CIA induction. There was an early (starting at 1 week) and robust airway inflammatory cellular influx with an associated release in pro-inflammatory cytokines, chemokines and remodeling proteins including fibronectin, IL-33, MMP-8, MMP-9, and TIMP-1. Serum pentraxin-2 (acute phase protein) was strikingly elevated at 1 week in CIA, LPS, and most significantly, CIA+LPS treatment groups. Serum anti-MAA and anti-MAA-CIT antibodies were also significantly increased with CIA+LPS at 1 week as compared to Sham. Similar trends were demonstrated at 3 weeks, but the magnitude of the response was less. The systemic inflammatory and autoimmune response also dampened at 3 weeks, except for the CIA treatment group.
4. DISCUSSION
Airborne biohazard exposures are strongly associated with the risk of both RA and RA-associated lung diseases that together represent the most overrepresented cause of death in RA (1, 2, 50, 51). Knowledge of this dynamic interplay of environmental influences in the setting of RA is critical to disease management because current therapeutic options for RA-associated lung disease are quite limited and clinically relevant biomarkers for predicting the onset or progression of RA-associated lung disease are lacking (52). Here, we identify that repetitive inhaled exposure to a ubiquitous environmental and occupational air pollutant, LPS, in the setting of inflammatory arthritis leads to markedly increased levels of serum autoantibodies relevant to RA, increased lung expression of respective autoantigens, and key features of pulmonary inflammation and fibrosis. Of added translational significance, we also report the novel observation that increased IL-33 expression in lung tissue of mice exposed to CIA+LPS can be replicated in patients with RA-associated lung disease.
ACPA are a cardinal feature of RA, as they are highly disease specific and appear to render a direct pathogenic role in the progression of RA (53). Although ACPA have been shown to be higher in patients with established lung disease, concentrations do not appear to be predictive of incident lung disease (53, 54). We previously demonstrated that serum anti-MAA antibodies were independently associated with ILD in the context of RA and that MAA is highly expressed in RA-ILD lung tissues where it co-localizes with CIT and extracellular matrix proteins (18). Corresponding to these clinical observations (18), the combination of CIA+LPS resulted in synergistic increases in ACPA as well as antibodies to both MAA and MAA-CIT. To further underscore the significance of this observation, it is a known limitation that the CIA model alone yields only nominal ACPA responses (55, 56) and is unable to induce a robust anti-MAA antibody response (16). Moreover, we have shown in prior work that agricultural dust extracts also did not potentiate anti-MAA antibody in the setting of arthritis induction (16). Yet, in this co-exposure modeling, both ACPA and anti-MAA antibody were strikingly increased, yielding an autoantibody profile that more closely mimics human disease. In comparison, repetitive inhalant LPS alone did not induce systemic autoantibody responses, although this exposure did result in modest increases in the lung expression of both CIT- and MAA-modified proteins. Thus, these studies emphasize that an underlying predisposition to autoimmunity plus a second “hit” such as airborne biohazard exposure may be driving tolerance loss and disease manifestations. This model, using LPS, a ubiquitous airborne exposure that is commercially available and highly reproducible, could be further exploited to test various pre-clinical therapeutic interventions to reduce articular and lung disease burden.
Consistent with prior observations in patients with RA (18), CIT and MAA co-localized in the lungs of experimental mice. Furthermore, we were only able to significantly co-capture lung MAA-CIT together as opposed to capturing MAA or CIT alone, suggesting an important and perhaps “interdependent” relationship between these post-translational modifications. MAA is typically generated from lipid peroxidation during oxidative stress, and MAA adducts are highly immunogenic, induce tolerance loss (57), and upregulate pro-inflammatory and pro-fibrotic pathways (58, 59). LPS induces lung inflammation by signaling through TLR4 to activate NF-κB, leading to release of reactive oxidative species and pro-inflammatory cytokines (60) that could further promote MAA adduct formation. LPS is also recognized as a cofactor for MAA-mediated release by liver endothelial and Kupffer cells (61), and therefore, it is possible that MAA circulates to the lung or to the joint where it might combine with LPS to activate macrophages, other inflammatory processes, ultimately resulting in tissue damage. In the combined exposure modeling, there was co-localization of CIT with fibronectin and enolase, but not vimentin, whereas MAA modestly co-localized with fibronectin, enolase, and vimentin. Investigation using strategies to inhibit reactive aldehyde species to inhibit MAA formation (62) could further elucidate whether MAA is directly pathogenic, impacts citrullination of autoantigens and autoantibody formation, and/or represents a highly-relevant biomarker of lung disease in RA.
Repetitive LPS exposure activated lung neutrophil influx and pro-inflammatory cytokine/chemokine release. Intriguingly, this traditional airway inflammatory response remained but was blunted in the setting of inflammatory arthritis induced by CIA. Instead, with both exposures, the lung disease skewed towards pro-fibrotic inflammatory processes that parallel the interstitial fibrotic manifestations of RA-associated lung disease. These pro-fibrotic features in the combined model included increases in infiltrates of transitional monocyte-macrophages populations as well as increases in several MMPs, fibronectin, complement, and IL-33. Inflammatory monocytes and transitional monocyte-macrophages are implicated in promoting lung fibrosis (63–66) and have also been identified and profiled in animal studies combining CIA with inhalant agriculture organic dusts exposures (16, 46). Future studies involving genetically modified mice or inhibition of infiltrating inflammatory monocytes through systemic clodronate liposome-reduction techniques should clarify the role of this cell population in RA-associated lung disease development.
Interestingly, several lung MMPs including murine MMP-2, -3, -8, -9 and TIMP-1 were increased with combined exposures, recapitulating findings recently observed in U.S. Veterans with RA-ILD (67). Of note, the Veteran RA-ILD patient cohort examined in this recent study consisted primarily of men with history of various military and occupational exposures as well as tobacco use (8). Given sex-based differences that have been demonstrated in prior animal studies as well as epidemiological/clinical investigations, future efforts aimed at dissecting the specific role of sex hormones including testosterone, progesterone, and/or estrogen are needed. In support of this sex-based paradigm, MMPs appear to be important not only in male mice, but also in men with RA-ILD, suggesting a potential therapeutic role for agents targeting these tissue remodeling proteins.
Other mediators that were potentiated with combined CIA+LPS exposure were complement C5a and IL-33. Conversely, the alarmin IL-25 was slightly, albeit significantly, reduced with combined exposures, perhaps due to “consumption” by on-going inflammatory processes. Next, our finding that complement C5a increased over time with combined CIA+LPS exposure (but not in the other treatment groups) provide support for the potential role of complement in the initiation and progression of RA-associated lung disease (45, 46), suggesting that this mediator could be therapeutically targeted by available agents. Finally, IL-33 is released predominately in response to damage or stress, but is also suggested to be involved in pro-fibrotic responses and tissue remodeling in idiopathic pulmonary fibrosis (IPF) in its un-cleaved or full-length form (47, 48). In contrast, full-length IL-33 does not promote eosinophilic responses as those classic allergic responses are promoted by cleaved forms of IL-33 (68). Here, lung levels of IL-33 were increased over time in the setting of combined CIA+LPS exposure, though expression of this cytokine appears to be driven primarily by LPS exposure. Because the antibody utilized recognizes both full-length and cleaved forms through recognition of the Ser112-Thr270 amino acid sequences, we are unable to speculate on which IL-33 form was present. Moreover, our studies demonstrate for the first time that lung IL-33 expression is increased in patients with RA-associated lung disease, which has also been observed with other chronic lung diseases including IPF, chronic obstructive pulmonary disease (COPD), and asthma (68–71). Clinical studies are currently underway utilizing anti-IL-33/ST2 pathway antibodies in the treatment of COPD (clinicaltrials.gov; NCT04701983, NCT03615040), suggesting that this approach could be explored in RA-lung disease. Alternatively, IL-33 may be an important early identifier of patients with RA who are at risk of developing RA-associated lung disease.
Limitations of this study include the sole use of LPS as the airborne biohazard prototype, as other airborne biohazards relevant to RA-lung disease (such as silica and cigarette smoke) should be explored. Cigarette smoke, for example, accelerates arthritis and citrullination in mice (72, 73). Notably, cigarettes contain not only LPS (74), but also acetaldehydes that, when coupled with alcohol (source of acetaldehyde generation), robustly increase MAA adduct formation as detected in bronchioalveolar lavage fluid of patients (75). In addition, LPS has a short half-life, and it may not be the appropriate inflammatory agent to model former/past exposures, such as cigarette smoking, to the risk of RA-associated lung disease. Finally, it has been recently demonstrated that citrullinated vimentin is a TLR4 ligand that precipitates pulmonary fibrosis and that citrullinated vimentin is induced by cigarette component of carbon black and cadmium (76) to suggest that future studies are warranted to investigate the potential additive role of LPS plus cadmium and carbon black in the setting of arthritis induction.
In conclusion, we found that repetitive exposure to bacterial endotoxin in the setting of inflammatory arthritis leads to increases in lung expression of RA-associated antigens and correlating circulating autoantibodies targeting post-translationally modified proteins, ultimately shifting towards a pro-fibrotic, pro-inflammatory lung phenotype. Meaningful mitigation of airborne biohazard exposures that are enriched in endotoxin may therefore represent an important measure in reducing the disease burden posed by RA and RA-associated lung disease.
Supplementary Material
Highlights.
Airborne biohazards are risk factors in the development and progression of rheumatoid arthritis (RA) and RA-associated lung disease. However, mechanisms underpinning this relationship are unclear. Knowledge of this dynamic interplay is critical because current therapeutic options and biomarkers are limited.
A novel murine model combining repetitively inhaled endotoxin with inflammatory arthritis potentiated RA-related autoantibody responses, expression of relevant lung autoantigens, and pro-fibrotic/pro-inflammatory lung disease characterized by enhanced IL-33 with the latter findings also demonstrated in patients with RA. Meaningful mitigation of airborne exposures enriched in endotoxin may represent an important measure in reducing disease burden in RA.
Acknowledgments:
The authors acknowledge members of the Tissue Sciences Facility at the Department of Pathology and Microbiology (University of Nebraska Medical Center, Omaha, NE, USA) for assistance with tissue processing and staining. We acknowledge members of the Experimental Immunology Laboratory including Nozima Aripova and Karen C. Easterling for their technical assistance. We thank Victoria B. Smith, Samantha Wall, and Craig Semerad in the Flow Cytometry Research Core Facility at the University of Nebraska Medical Center for providing assistance with flow cytometry studies. We thank Xioayan Wang & Gang Zhao for assistance with the micro-CT scanning and analysis. We also thank Lisa Chudomelka for manuscript submission assistance.
Funding:
This work was supported by the National Institutes of Health (R01ES019325, JAP; R25AA020818, TRM, KLB; U54GM115458, TRM; R01AG053553, KLB), the National Institute for Occupational Safety and Health (U54OH010162, JAP, TAW, RG, DJR) and the Department of Veterans Affairs (BX004600, TRM, GMT; IK2 CX002203, BRE; IK6BX003781, TAW; CX001714, DJR). Additional support was provided by the Central States Center of Agricultural Safety and Health (CS-CASH) and Fred & Pamela Buffett Cancer Center [Flow Cytometry Research Facility and Tissue Sciences Facility] Shared Resource, supported by the National Cancer Institute award P30CA036727.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Decalaration of interest: BRE has received consulting fees from Boehringer-Ingelheim.
References
- 1.Kelly CA, Saravanan V, Nisar M, Arthanari S, Woodhead FA, Price-Forbes AN, Dawson J, Sathi N, Ahmad Y, Koduri G. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics—a large multicentre UK study. Rheumatology 2014; 53: 1676–1682. [DOI] [PubMed] [Google Scholar]
- 2.England BR, Sayles H, Michaud K, Caplan L, Davis LA, Cannon GW, Sauer BC, Solow EB, Reimold AM, Kerr GS, Schwab P, Baker JF, Mikuls TR. Cause-Specific Mortality in Male US Veterans With Rheumatoid Arthritis. Arthritis care & research 2016; 68: 36–45. [DOI] [PubMed] [Google Scholar]
- 3.England BR, Baker JF, Sayles H, Michaud K, Caplan L, Davis LA, Cannon GW, Sauer BC, Solow EB, Reimold AM, Kerr GS, Mikuls TR. Body Mass Index, Weight Loss, and Cause-Specific Mortality in Rheumatoid Arthritis. Arthritis care & research 2018; 70: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy D, Hutchinson D. Is Male Rheumatoid Arthritis an Occupational Disease? A Review. The open rheumatology journal 2017; 11: 88–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Too CL, Muhamad NA, Ilar A, Padyukov L, Alfredsson L, Klareskog L, Murad S, Bengtsson C, My ESG. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: results from a Malaysian population-based case-control study. Annals of the Rheumatic Diseases 2016; 75: 997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stolt P, Yahya A, Bengtsson C, Kallberg H, Ronnelid J, Lundberg I, Klareskog L, Alfredsson L, Group ES. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Annals of the Rheumatic Diseases 2010; 69: 1072–1076. [DOI] [PubMed] [Google Scholar]
- 7.Stolt P, Källberg H, Lundberg I, Sjögren B, Klareskog L, Alfredsson L, group Es. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis 2005; 64: 582–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebel AV, Lutt G, Poole JA, Thiele GM, Baker JF, Cannon GW, Gaffo A, Kerr GS, Reimold A, Schwab P, Singh N, Richards JS, Ascherman DP, Mikuls TR, England BR. Association of Agricultural, Occupational, and Military Inhalants With Autoantibodies and Disease Features in US Veterans With Rheumatoid Arthritis. Arthritis Rheumatol 2021; 73: 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yahya A, Bengtsson C, Larsson P, Too CL, Mustafa AN, Abdullah NA, Muhamad NA, Klareskog L, Murad S, Alfredsson L. Silica exposure is associated with an increased risk of developing ACPA-positive rheumatoid arthritis in an Asian population: evidence from the Malaysian MyEIRA case-control study. Mod Rheumatol 2014; 24: 271–274. [DOI] [PubMed] [Google Scholar]
- 10.Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. The Journal of clinical investigation 2006; 116: 3183–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gizinski AM, Mascolo M, Loucks JL, Kervitsky A, Meehan RT, Brown KK, Holers VM, Deane KD. Rheumatoid arthritis (RA)-specific autoantibodies in patients with interstitial lung disease and absence of clinically apparent articular RA. Clinical rheumatology 2009; 28: 611–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssen KM, de Smit MJ, Brouwer E, de Kok FA, Kraan J, Altenburg J, Verheul MK, Trouw LA, van Winkelhoff AJ, Vissink A, Westra J. Rheumatoid arthritis-associated autoantibodies in non-rheumatoid arthritis patients with mucosal inflammation: a case-control study. Arthritis research & therapy 2015; 17: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quirke AM, Perry E, Cartwright A, Kelly C, De Soyza A, Eggleton P, Hutchinson D, Venables PJ. Bronchiectasis is a Model for Chronic Bacterial Infection Inducing Autoimmunity in Rheumatoid Arthritis. Arthritis & rheumatology (Hoboken, NJ) 2015; 67: 2335–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry E, Eggleton P, De Soyza A, Hutchinson D, Kelly C. Increased disease activity, severity and autoantibody positivity in rheumatoid arthritis patients with co-existent bronchiectasis. International journal of rheumatic diseases 2017; 20: 2003–2011. [DOI] [PubMed] [Google Scholar]
- 15.Aubart F, Crestani B, Nicaise-Roland P, Tubach F, Bollet C, Dawidowicz K, Quintin E, Hayem G, Palazzo E, Meyer O, Chollet-Martin S, Dieude P. High levels of anti-cyclic citrullinated peptide autoantibodies are associated with co-occurrence of pulmonary diseases with rheumatoid arthritis. The Journal of rheumatology 2011; 38: 979–982. [DOI] [PubMed] [Google Scholar]
- 16.Poole JA, Thiele GM, Janike K, Nelson AJ, Duryee MJ, Rentfro K, England BR, Romberger DJ, Carrington JM, Wang D, Swanson BJ, Klassen LW, Mikuls TR. Combined Collagen-Induced Arthritis and Organic Dust-Induced Airway Inflammation to Model Inflammatory Lung Disease in Rheumatoid Arthritis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2019; 34: 1733–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poole JA, Mikuls TR, Duryee MJ, Warren KJ, Wyatt TA, Nelson AJ, Romberger DJ, West WW, Thiele GM. A role for B cells in organic dust induced lung inflammation. Respiratory research 2017; 18: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.England BR, Duryee MJ, Roul P, Mahajan TD, Singh N, Poole JA, Ascherman DP, Caplan L, Demoruelle MK, Deane KD, Klassen LW, Thiele GM, Mikuls TR. Malondialdehyde-Acetaldehyde Adducts and Antibody Responses in Rheumatoid Arthritis-Interstitial Lung Disease. Arthritis & rheumatology (Hoboken, NJ) 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebel AV, Lutt G, Poole JA, Thiele GM, Baker JF, Cannon GW, Gaffo A, Kerr GS, Reimold A, Schwab P, Singh N, Richards JS, Ascherman DP, Mikuls TR, England BR. Association of Agricultural, Occupational, and Military Inhalants with Autoantibodies and Disease Features in U.S. Veterans with Rheumatoid Arthritis. Arthritis Rheumatol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Matteis S, Heederik D, Burdorf A, C C, Cullinan P, Hennenberger P, A O, A R, J R, T S, J S, V S, vT M, S T. Current and new challenges in occupational lung diseases. Eur Respir Rev 2017; 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai PS, Hang JQ, Valeri L, Zhang FY, Zheng BY, Mehta AJ, Shi J, Su L, Brown D, Eisen EA, Christiani DC. Endotoxin and gender modify lung function recovery after occupational organic dust exposure: a 30-year study. Occup Environ Med 2015; 72: 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidson ME, Schaeffer J, Clark ML, Magzamen S, Brooks EJ, Keefe TJ, Bradford M, Roman-Muniz N, Mehaffy J, Dooley G, Poole JA, Mitloehner FM, Reed S, Schenker MB, Reynolds SJ. Personal exposure of dairy workers to dust, endotoxin, muramic acid, ergosterol and ammonia on large-scale dairies in the high plains western United States. Journal of Occupational and Environmental Hygiene 2017; 15: 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brass DM, Spencer JC, Li Z, Potts-Kant E, Reilly SM, Dunkel MK, Latoche JD, Auten RL, Hollingsworth JW, Fattman CL. Innate immune activation by inhaled lipopolysaccharide, independent of oxidative stress, exacerbates silica-induced pulmonary fibrosis in mice. PLoS One 2012; 7: e40789. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Chen S, Yuan J, Yao S, Jin Y, Chen G, Tian W, Xi J, Xu Z, Weng D, Chen J. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy 2015; 11: 2346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Y, Ichinose T, He M, Song Y, Yoshida Y, Yoshida S, Nishikawa M, Takano H, Sun G, Shibamoto T. Enhancement of OVA-induced murine lung eosinophilia by co-exposure to contamination levels of LPS in Asian sand dust and heated dust. Allergy Asthma Clin Immunol 2014; 10: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radstake TR, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PL, Barrera P, Joosten LA, van den Berg WB. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheum 2004; 50: 3856–3865. [DOI] [PubMed] [Google Scholar]
- 27.Samarpita S, Kim JY, Rasool MK, Kim KS. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res Ther 2020; 22: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karampitsakos T, Woolard T, Bouros D, Tzouvelekis A. Toll-like receptors in the pathogenesis of pulmonary fibrosis. Eur J Pharmacol 2017; 808: 35–43. [DOI] [PubMed] [Google Scholar]
- 29.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 2001; 276: 10229–10233. [DOI] [PubMed] [Google Scholar]
- 30.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 2009; 15: 774–780. [DOI] [PubMed] [Google Scholar]
- 31.Kuhns DB, Priel DA, Gallin JI. Induction of human monocyte interleukin (IL)-8 by fibrinogen through the toll-like receptor pathway. Inflammation 2007; 30: 178–188. [DOI] [PubMed] [Google Scholar]
- 32.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol 2001; 167: 2887–2894. [DOI] [PubMed] [Google Scholar]
- 33.Roelofs MF, Boelens WC, Joosten LA, Abdollahi-Roodsaz S, Geurts J, Wunderink LU, Schreurs BW, van den Berg WB, Radstake TR. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol 2006; 176: 7021–7027. [DOI] [PubMed] [Google Scholar]
- 34.Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Götte M, Malle E, Schaefer RM, Gröne HJ. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest 2005; 115: 2223–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson AJ, Roy SK, Warren K, Janike K, Thiele GM, Mikuls TR, Romberger DJ, Wang D, Swanson B, Poole JA. Sex differences impact the lung-bone inflammatory response to repetitive inhalant lipopolysaccharide exposures in mice. Journal of immunotoxicology 2018; 15: 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dusad A, Thiele GM, Klassen LW, Gleason AM, Bauer C, Mikuls TR, Duryee MJ, West WW, Romberger DJ, Poole JA. Organic dust, lipopolysaccharide, and peptidoglycan inhalant exposures result in bone loss/disease. American journal of respiratory cell and molecular biology 2013; 49: 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2013; 28: 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thiele GM, Duryee MJ, Anderson DR, Klassen LW, Mohring SM, Young KA, Benissan-Messan D, Sayles H, Dusad A, Hunter CD, Sokolove J, Robinson WH, O’Dell JR, Nicholas AP, Tuma DJ, Mikuls TR. Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arthritis & rheumatology (Hoboken, NJ) 2015; 67: 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiele GM, Duryee MJ, Hunter CD, England BR, Fletcher BS, Daubach EC, Pospisil TP, Klassen LW, Mikuls TR. Immunogenic and inflammatory responses to citrullinated proteins are enhanced following modification with malondialdehyde-acetaldehyde adducts. Int Immunopharmacol 2020; 83: 106433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikuls TR, Duryee MJ, Rahman R, Anderson DR, Sayles HR, Hollins A, Michaud K, Wolfe F, Thiele GE, Sokolove J, Robinson WH, Lingampalli N, Nicholas AP, Talmon GA, Su K, Zimmerman MC, Klassen LW, Thiele GM. Enrichment of malondialdehyde-acetaldehyde antibody in the rheumatoid arthritis joint. Rheumatology (Oxford, England) 2017; 56: 1794–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poole JA, Wyatt TA, Oldenburg PJ, Elliott MK, West WW, Sisson JH, Von Essen SG, Romberger DJ. Intranasal Organic Dust Exposure-Induced Airway Adaptation Response Marked By Persistent Lung Inflammation and Pathology in Mice. Am J Physiol Lung Cell Mol Physiol 2009; 296: L1085–L1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Charavaryamath C, Juneau V, Suri SS, Janardhan KS, Townsend H, Singh B. Role of Toll-like receptor 4 in lung inflammation following exposure to swine barn air. Exp Lung Res 2008; 34: 19–35. [DOI] [PubMed] [Google Scholar]
- 43.Robbe P, Draijer C, Borg TR, Luinge M, Timens W, Wouters IM, Melgert BN, Hylkema MN. Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. American journal of physiologyLung cellular and molecular physiology 2015; 308: L358–367. [DOI] [PubMed] [Google Scholar]
- 44.Wyatt TA, Nemecek M, Chandra D, DeVasure JM, Nelson AJ, Romberger DJ, Poole JA. Organic dust-induced lung injury and repair: Bi-directional regulation by TNFalpha and IL-10. J Immunotoxicol 2020; 17: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holers VM, Banda NK. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front Immunol 2018; 9: 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaurav R, Mikuls TR, Thiele GM, Nelson AJ, Niu M, Guda C, Eudy JD, Barry AE, Wyatt TA, Romberger DJ, Duryee MJ, England BR, Poole JA. High-throughput analysis of lung immune cells in a combined murine model of agriculture dust-triggered airway inflammation with rheumatoid arthritis. PLoS One 2021; 16: e0240707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Majewski S, Tworek D, Szewczyk K, Kurmanowska Z, Antczak A, Górski P, Piotrowski WJ. Epithelial alarmin levels in exhaled breath condensate in patients with idiopathic pulmonary fibrosis: A pilot study. The Clinical Respiratory Journal 2019; 13: 652–656. [DOI] [PubMed] [Google Scholar]
- 48.Majewski S, Szewczyk K, Białas AJ, Miłkowska-Dymanowska J, Górski P, Piotrowski WJ. Epithelial Alarmins in Serum and Exhaled Breath in Patients with Idiopathic Pulmonary Fibrosis: A Prospective One-Year Follow-Up Cohort Study. Journal of clinical medicine 2019; 8: 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Surolia R, Li FJ, Wang Z, Li H, Dsouza K, Thomas V, Mirov S, Pérez-Sala D, Athar M, Thannickal VJ. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erickson Ar CACMTR. Clinical features of rheumatoid arthritis. In: Budd Rc GSBMIBODJR, editor. Kelley & Firestein’s Textbook of Rheumatology, II ed. Philadelphia: Elsevier; 2017. p. 1167-1167-1186. [Google Scholar]
- 51.England BR, Sayles H, Michaud K, Thiele GM, Poole JA, Caplan L, Sauer BC, Cannon GW, Reimold A, Kerr GS, Baker JF, Mikuls TR. Chronic lung disease in U.S. Veterans with rheumatoid arthritis and the impact on survival. Clinical rheumatology 2018; 37: 2907–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.England BR, Hershberger D. Management issues in rheumatoid arthritis-associated interstitial lung disease. Curr Opin Rheumatol 2020; 32: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang D, Zhang J, Lau J, Wang S, Taneja V, Matteson EL, Vassallo R. Mechanisms of lung disease development in rheumatoid arthritis. Nature Reviews Rheumatology 2019; 15: 581–596. [DOI] [PubMed] [Google Scholar]
- 54.Natalini JG, Baker JF, Singh N, Mahajan TD, Roul P, Thiele GM, Sauer BC, Johnson CR, Kawut SM, Mikuls TR, England BR. Autoantibody Seropositivity and Risk for Interstitial Lung Disease in a Prospective Male-Predominant Rheumatoid Arthritis Cohort of U.S. Veterans. Ann Am Thorac Soc 2021; 18: 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest 2006; 116: 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thiele GM, Duryee MJ, Dusad A, Hunter CD, Lacy JP, Anderson DR, Wang D, O’Dell JR, Mikuls TR, Klassen LW. Citrullinated mouse collagen administered to DBA/1J mice in the absence of adjuvant initiates arthritis. International immunopharmacology 2012; 13: 424–431. [DOI] [PubMed] [Google Scholar]
- 57.Thiele GM, Tuma DJ, Willis MS, Miller JA, McDonald TL, Sorrell MF, Klassen LW. Soluble proteins modified with acetaldehyde and malondialdehyde are immunogenic in the absence of adjuvant. Alcoholism, Clinical and Experimental Research 1998; 22: 1731–1739. [PubMed] [Google Scholar]
- 58.Wyatt TA, Kharbanda KK, McCaskill ML, Tuma DJ, Yanov D, Devasure J, Sisson JH. Malondialdehyde-acetaldehyde-adducted protein inhalation causes lung injury. Alcohol (Fayetteville, NY) 2012; 46: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wyatt TA, Kharbanda KK, Tuma DJ, Sisson JH. Malondialdehyde-acetaldehyde-adducted bovine serum albumin activates protein kinase C and stimulates interleukin-8 release in bovine bronchial epithelial cells. Alcohol 2001; 25: 159–166. [DOI] [PubMed] [Google Scholar]
- 60.Karki R, Igwe OJ. Toll-like receptor 4-mediated nuclear factor kappa B activation is essential for sensing exogenous oxidants to propagate and maintain oxidative/nitrosative cellular stress. PLoS One 2013; 8: e73840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duryee MJ, Klassen LW, Freeman TL, Willis MS, Tuma DJ, Thiele GM. Lipopolysaccharide is a cofactor for malondialdehyde-acetaldehyde adduct-mediated cytokine/chemokine release by rat sinusoidal liver endothelial and Kupffer cells. Alcoholism, Clinical and Experimental Research 2004; 28: 1931–1938. [DOI] [PubMed] [Google Scholar]
- 62.Clark D, Tauber J, Sheppard J, Brady TC. Early Onset and Broad Activity of Reproxalap in a Randomized, Double-Masked, Vehicle-Controlled Phase 2b Trial in Dry Eye Disease. Am J Ophthalmol 2021; 226: 22–31. [DOI] [PubMed] [Google Scholar]
- 63.Puttur F, Gregory LG, Lloyd CM. Airway macrophages as the guardians of tissue repair in the lung. Immunology and cell biology 2019. [DOI] [PubMed] [Google Scholar]
- 64.Gibbons MA, Mackinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, Hirani N, Gauldie J, Iredale JP, Sethi T, Forbes SJ. Ly6Chi Monocytes Direct Alternatively Activated Pro-fibrotic Macrophage Regulation of Lung Fibrosis. American journal of respiratory and critical care medicine 2011. [DOI] [PubMed] [Google Scholar]
- 65.Laskin DL, Malaviya R, Laskin JD. Role of Macrophages in Acute Lung Injury and Chronic Fibrosis Induced by Pulmonary Toxicants. Toxicological sciences : an official journal of the Society of Toxicology 2019; 168: 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aghasafari P, George U, Pidaparti R. A review of inflammatory mechanism in airway diseases. Inflammation Research 2018; 68: 59–74. [DOI] [PubMed] [Google Scholar]
- 67.Kass DJ, Nouraie M, Glassberg MK, Ramreddy N, Fernandez K, Harlow L, Zhang Y, Chen J, Kerr GS, Reimold AM, England BR, Mikuls TR, Gibson KF, Dellaripa PF, Rosas IO, Oddis CV, Ascherman DP. Comparative Profiling of Serum Protein Biomarkers in Rheumatoid Arthritis-Associated Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis. Arthritis Rheumatol 2020; 72: 409–419. [DOI] [PubMed] [Google Scholar]
- 68.Murdaca G, Greco M, Tonacci A, Negrini S, Borro M, Puppo F, Gangemi S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. International journal of molecular sciences 2019; 20: 5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, Wu K, You Y, Alevy Y, Girard JP, Stappenbeck TS, Patterson GA, Pierce RA, Brody SL, Holtzman MJ. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest 2013; 123: 3967–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luzina IG, Pickering EM, Kopach P, Kang PH, Lockatell V, Todd NW, Papadimitriou JC, McKenzie AN, Atamas SP. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol 2012; 189: 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gaurav R, Anderson DR, Radio SJ, Bailey KL, England BR, Mikuls TR, Thiele GM, Strah HM, Romberger DJ, Wyatt TA, Dickinson JD, Duryee MJ, Katafiasz DM, Nelson AJ, Poole JA. IL-33 Depletion in COVID-19 Lungs. Chest 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang J, Jeong SH, Lee K, Park N, Jung H, Lee K, Ju JH. Exacerbation of symptomatic arthritis by cigarette smoke in experimental arthritis. PLoS One 2020; 15: e0230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bidkar M, Vassallo R, Luckey D, Smart M, Mouapi K, Taneja V. Cigarette Smoke Induces Immune Responses to Vimentin in both, Arthritis-Susceptible and -Resistant Humanized Mice. PLoS One 2016; 11: e0162341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hasday JD, Bascom R, Costa JJ, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest 1999; 115: 829–835. [DOI] [PubMed] [Google Scholar]
- 75.Sapkota M, Burnham EL, DeVasure JM, Sweeter JM, Hunter CD, Duryee MJ, Klassen LW, Kharbanda KK, Sisson JH, Thiele GM, Wyatt TA. Malondialdehyde-Acetaldehyde (MAA) Protein Adducts Are Found Exclusively in the Lungs of Smokers with Alcohol Use Disorders and Are Associated with Systemic Anti-MAA Antibodies. Alcohol Clin Exp Res 2017; 41: 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li FJ, Surolia R, Li H, Wang Z, Liu G, Kulkarni T, Massicano AVF, Mobley JA, Mondal S, de Andrade JA, Coonrod SA, Thompson PR, Wille K, Lapi SE, Athar M, Thannickal VJ, Carter AB, Antony VB. Citrullinated vimentin mediates development and progression of lung fibrosis. Sci Transl Med 2021; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.