

Abstract
Myeloproliferative neoplasms (MPNs) are a group of malignant disorders of the bone marrow where a dysregulated balance between proliferation and differentiation gives rise to abnormal numbers of mature blood cells. MPNs encompass a spectrum of disease entities with progressively more severe clinical features, including complications with thrombosis and haemostasis and an increased propensity for transformation to AML. There is an unmet clinical need for markers of disease progression. Our understanding of the precise mechanisms that influence pathogenesis and disease progression has been limited by access to disease-specific cells as bio-sources. Here, we review the landscape of MPN pathology and present blood platelets as potential candidates for disease-specific understanding. We conclude with our recent work discovering progressive platelet heterogeneity by subtype in a large clinical cohort of MPN patients.
Myeloproliferative neoplasms (MPNs) are a group of chronic haematological malignancies characterised by clonal over-production of mature cells of myeloid lineages1. They are defined by a combination of clinical, laboratory, morphological and molecular genetic features2. Three phenotypic subtypes, essential thrombocythemia (ET), polycythaemia vera (PV), and primary myelofibrosis (MF) constitute the ‘classical MPNs’. All have shared molecular genetic pathogenesis-somatic mutations in one of three driver genes (JAK2, CALR, MPL) that lead to constitutive JAK/STAT signalling in haematopoietic stem cells (HSC)3–7. Clinically they have distinct features, but abnormalities of haemostasis and thrombosis, and risk of progression to acute myeloid leukaemia (AML) are common to all 1. The chronic nature of MPNs and the accessibility of peripheral blood cells implies that these disorders could potentially serve as a generalizable model for events in cancer progression8. Furthermore, recent discovery that the mutational event precedes diagnosis by many years9, 10 highlights the opportunity MPNs present to investigate early detection and intervention strategies in cancer.
Our early work11 studying platelets in patients with MPNs established a methodological foundation for exploring subtype-specific signatures, not only of relevance to bleeding and thrombosis outcomes in these patients, but also of underlying disease pathobiology. More recently12, extended additional data in over 100 MPN patient samples across all three MPN subtypes identify distinct platelet transcriptomic signatures associated with disease processes. Megakaryocytes, which produce platelets, are derived from HSCs, reside in the bone marrow and contribute to MPN pathogenesis13–16. Platelets therefore provide both a snapshot of the status of the megakaryocyte at the time of platelet release, and a window into the bone marrow microenvironment.
MYELOPROLIFERATIVE NEOPLASMS
Clinical features
The clinical phenotype of the three classical MPNs differ, although there are overlapping features1. ET is characterised by an elevated platelet count and PV by an elevated haematocrit. Myelofibrosis, the most severe disease in the group, is typified by cytopaenias and presence of reticulin fibrosis in the bone marrow2. All patients experience systemic symptoms such as fatigue that impair quality of life 17. In ET, life expectancy may be near normal, whereas in PV, median life expectancy is around 13 years. Median life expectancy in MF is around 5 years although this varies according to risk score, highlighting the heterogeneity within this disease 18. There is a risk of transformation to AML, which has a worse prognosis than de-novo AML19, 20. Significant problems in haemostasis and thrombosis occur in all three diseases.
There are challenges in gathering data about long term outcomes for patients with MPNs, and in designing trials in this patient group21. Firstly, the diseases are relatively rare, affecting the cohort size. Secondly, adverse outcomes in these patients occur over decades of chronic progressive disease3, 4, 22.
Genetic landscape
In around 90% of patients with MPNs, an acquired mutation that promotes JAK/STAT signalling is identified4, 23. The JAK/STAT pathway transduces signals from cytokines including erythropoietin, thrombopoietin and granulocyte colony stimulating factor 24. A point mutation that activates JAK2, JAK2V617F, is present in around 95% of patients with PV and 40–60% of patients with ET and MF 25–28. Many of the remaining patients with ET and MF have frameshift mutations in the endoplasmic reticulum chaperone protein calreticulin29, 30 which cause ligand-independent activation of the TPO receptor 31, 32.
In addition to driver mutations, recurrent mutations are seen in TET2, DNMT3A, IDH1 and 2, ASXL1 and EZH24, 5, which contribute to disease initiation and progression by affecting DNA methylation and histone modification33. Profiling these mutations is used to refine prognosis amongst patients with MF18. Genes which acquire mutations in progression to AML include TP53 and transcription factors such as RUNX1 19, 20, 34.
Thrombosis and bleeding in MPNs
Patients with MPNs are paradoxically at risk of both thrombotic and bleeding complications. The risk of arterial and venous thrombosis is increased in patients with MPNs, with the time shortly after diagnosis being the period of highest risk. In the first three months following diagnosis, arterial thrombotic events are around three times commoner in patients with MPNs than they are in matched controls, and venous events are around ten times commoner. Rates are similar across PV, ET and MF 35 . Arterial events are more common than venous events in MPN patients, and in PV and ET cardiovascular disease is the most common cause of death36. Table 1 provides an overview of recent literature documenting clinical outcomes of patients with ET and PV.
Table 1:
Representative overview of published literature on clinical outcomes of patients with ET and PV
| MPN Subtype | Study population (n) | Follow up (time) | Survival (time) | Thrombosis (all) | Thrombosis (arterial) | Thrombosis (venous) | Bleeding | Transformation |
|---|---|---|---|---|---|---|---|---|
| ET118 | 891 | 0-27 years (median 6.2) | Median overall survival 14.7 years (similar to sex and age standardized European population) | 22% at 15 years | Fibrotic 9%, Leukemic 2% (at 15 years) | |||
| ET47 (high vascular risk) | 809 (404 HU plus aspirin, 405 anagrelide plus aspirin) | 12-72 months (median 39) | Death 27/404 (hydroxyurea plus aspirin) 31/405 (anagrelide plus aspirin) |
17/404 (hydroxyurea plus aspirin) 37/405 (anagrelide plus aspirin) |
14/404 (hydroxyurea plus aspirin) 3/405 (anagrelide plus aspirin) |
Serious bleeding 8/404 (hydroxyurea plus aspirin) 22/405 (anagrelide plus aspirin) |
Myelofibrosis 5/404 (hydroxyurea plus aspirin), 16/405 anagrelide plus aspirin) AML or myelodysplasia 6/404 (hydroxyurea plus aspirin), 4/405 (anagrelide plus aspirin) |
|
| ET1, 119 (lacking high risk features for vascular events) | 382 (final analysis included 176 aspirin alone and 192 aspirin plus hydroxycarbamide | 0-187 months (median 73 months) | Death 7/176 (aspirin alone), 10/182 (aspirin plus hydroxycarbamide) | 7/176 (aspirin alone) 5/182 (aspirin plus hydroxycarbamide) |
3/176 (aspirin alone) 4/182 (aspirin plus hydroxycarbamide) |
Serious bleeding 2/176 (aspirin alone)3/182 (aspirin plus hydroxycarbamide) | Myelofibrosis 5/176 aspirin alone, 1/182 hydroxycarbamide plus aspirin AML 2/176 aspirin alone, 3/182 hydroxycarbamide plus aspirin. | |
| PV120 | 1638 | Mean 2.7 years, STDEV 1.3 years (4393 person years) | 3.7 deaths/100 patients/year (45% cardiovascular mortality, 13% haematological transformation) | 5.5/100 patients/year | Major bleeding 0.8/100 patients/year, any bleeding 2.9/100 patients/year |
Haematologic transformation 1.3/100patients/year | ||
| PV62 | 518 (253 aspirin, 265 placebo) | 1-3 years (1478 person-years) | Death 9/253 aspirin, 18/265 placebo | 8 out of 253 aspirin, 21/265 placebo | 5/253 aspirin, 13/265 placebo | 17/253 aspirin, 41/265 aspirin | Major bleeding 3/253 aspirin, 2/265 placebo. Any bleeding 23/253 aspirin 14/265 placebo | |
| PV43 | 365 (182 low target haematocrit <0.45, 183 high target haematocrit 0.45 to 0.5) | 1.5-48.1 mos (median 31) | Death 3/182 low haematocrit, 6/183 high haematocrit | 8/182 low haematocrit, 20/183 high haematocrit | 4/182 low haematocrit, 14/183 high haematocrit | 1/182 low haematocrit, 6/183 high haematocrit | 2/182 low haematocrit, 5/183 high haematocrit | Myelofibrosis 6/182 low haematocrit, 2/183 high haematocrit. AML or myelodysplasia 2/182 low haematocrit, 1/183 high haematocrit |
The pathogenesis of thrombosis in MPN patients is complex. Contributory factors include cell counts, clonal haematopoiesis, specific effects of driver mutations, and effects of systemic inflammation on platelets, granulocytes and endothelial cells37–41. Evidence for the contribution of elevated haematocrit and platelet counts is mixed. Amongst patients treated for PV cardiovascular events are more frequent in those with less stringent haematocrit control 42, 43. Thrombosis occurs even in individuals with a normal haematocrit however 44. Studies in ET have not shown an association between platelet count and thrombosis45, 46, and differences in rates of thrombosis were observed in a trial where platelet count did not differ between arms47.
There are several indicators that platelet function is altered in MPNs. There is upregulation of genes associated with thrombosis in platelets of patients with PV and ET compared to controls48. Clinical platelet function tests show increased aggregation in response to several ligands in patients with ET compared to healthy individuals49. Examination of the specific effect of JAK/STAT activation by JAK2V617F in platelets has produced conflicting results in different models, that nonetheless corresponds to the thrombotic and bleeding risks in patients. There is evidence both for a pro-thrombotic tendency50 and for impaired haemostasis with reduced granule formation and impaired aggregation51.
Increased rates of cardiovascular disease are seen in individuals with clonal haematopoiesis of undetermined significance 52. Evidence for a specific role of the driver mutation influencing thrombotic risk comes from ET, where patients with CALR mutations have significantly lower rates of thrombosis than patients with JAK2 or MPL mutations, despite those with CALR mutations having higher platelet counts53,54. Controversy over whether allele burden influences thrombotic risk 45, 55 also arises because peripheral blood measurements only partially reflect the cell populations9 in the bone marrow.
Inflammation is a risk factor for thrombosis, and in PV and ET an association is seen between C-reactive protein levels and occurrence of thrombotic events56. In addition to endothelial activation caused by systemic inflammation, there are reports of JAK2V617F in endothelial cells and a suggestion this may promote thrombosis57. Increased platelet activation and platelet-leucocyte complexes are seen in patients with MPNs and are thought to contribute to thrombosis58.
Bleeding problems can arise in patients with MPNs with thrombocytosis or thrombocytopaenia, for example in ET the relationship between major haemorrhage and platelet count is a U-shaped curve46. Around 6% of patients will experience bleeding complications at the time of diagnosis59. Thrombocytosis can cause acquired von Willebrand disease, with selective depletion of large von Willebrand factor multimers60 although the bleeding risk associated with thrombocytsis may differ between MPN sub-types61. Bleeding related to anti-platelet drugs is an important consideration in this patient population.
Therapy
Controlling thrombotic risk is the main goal of therapy in PV and ET. In PV there is evidence that thrombotic risk reduction can be achieved with control of the haematocrit43, and aspirin62. In ET, treatment decisions are based on individualized thrombotic risk. The link between platelet count and thrombotic risk is less clear46, 47, although there is evidence that hydroxycarbamide reduces risk in high-risk patients63. Aspirin is recommended to reduce thrombotic risk64, although in ET there is a lack of prospective evidence for benefit. There is an unmet need for tools to personalise aspirin treatment in MPNs65 since some patients experience excessive bleeding related to aspirin, whereas others suffer thrombotic events despite taking standard doses of aspirin. In ET, higher platelet turnover is associated with increased rates of thrombosis66. There is evidence that rapid platelet turnover reduces the effectiveness of aspirin, and that this can be overcome by adjusting dose regimens67. The identification that platelet RNA is a biomarker for response to antiplatelet therapy is highly relevant68. Clopidogrel may be used as an alternative in patients who are intolerant of aspirin although evidence for this is lacking44. For patients with venous thrombotic events, anticoagulation treatment followed by long-term prophylaxis is recommended due to a high recurrence risk. Traditionally this is with heparins and vitamin K antagonists, although there is emerging evidence that direct oral anticoagulants may be effective and safe69, 70.
Drugs inhibiting JAK2, such as Ruxolitinib, have been designed to target the molecular cause of the disease and have been partially effective in MF71 and PV72, but less so in ET73. Interferons are also used, and there is evidence that they alter the balance between normal and malignant haematopoiesis74.
HAEMATOPOIETIC STEM CELLS AND THE BONE MARROW NICHE IN MPNS
The HSC population in patients with MPNs is heterogeneous, consisting of wild-type HSCs and sub-clones of HSCs with one or more mutations 8. Haematopoietic stem and progenitor cells reside within niches75, specialised microenvironments in the bone marrow that sustain the equilibrium between proliferation and differentiation76. Maintenance and progression of haematological malignancies is facilitated by reciprocal interactions between the malignant cells and surrounding niche cells 75. Inflammation in the microenvironment 77 and JAK/STAT activation in wild-type cells 78 are important contributors to the niche changes in MPNs79.
PLATELETS IN MPNs
Platelet origin and function
Blood platelets are critical in multiple processes and diseases, from their traditional role in haemostasis and wound healing to inflammation, immunity, cancer metastasis and angiogenesis80–82. Platelets originate from bone marrow precursor megakaryocytes, which themselves are differentiated from HSCs and the two cell types together play an important role in our understanding of MPN pathology. Megakaryocyte expansion is a diagnostic criterion in all three MPNs2. Populations of HSCs primed for megakaryopoiesis can be identified at an early stage of differentiation15, 83, 84. Megakaryocytes from patients with MF proliferate more than those from healthy controls and aberrantly overexpress myeloid transcription factors85. Megakaryocytes are a cellular component of the HSC niche75 and modulate HSC quiescence86, 87. In mice, expression of Jak2V617F in megakaryocyte lineage-committed cells triggers cell non-autonomous increased erythropoiesis 14. Megakaryocytes produce cytokines that promote fibrosis including IL6, CXCL4 and TGF-β14, 88, 89. Treatment with alisertib, an Aurora Kinase A inhibitor, that results in decreased megakaryocyte numbers and increased maturation led to a reduction in fibrosis in a mouse MF model16 and in five out of seven patients in a phase 1 clinical trial90.
The platelet transcriptome
Platelets contain a complex transcriptional landscape of messenger RNAs, unspliced pre-mRNAs, ribosomal RNAs, transfer RNAs and microRNAs13, 91–93. Most platelet RNA expression results from the transcription of nuclear DNA in the megakaryocyte, and thus reflects the status of the megakaryocyte at the time of platelet release into the circulation13, 91. This is overlaid with further complexities arising from splicing events triggered by receptor activation at the platelet surface94, 95 and inter-cellular transfer of RNA into megakaryocytes96 or circulating platelets97. The molecular signature of platelets is therefore changed in disease conditions80, 97, 98.
It is important to examine the transcriptome of megakaryocytes as well, given their centrality in the pathogenesis of MPNs; and identify the differential contributions to the platelet transcriptome from the parent megakaryocytes versus the peripheral disease environment. However, there are challenges to studying megakaryocytes from patient cohorts. Bone marrow biopsy is a required procedure and presents significantly more discomfort and risk to patients than a peripheral blood draw. Megakaryocytes are a relatively rare cell in healthy bone marrow though their transcriptomes have been examined99, 100 by enriching with density gradient centrifugation followed by positive selection for CD61 and identification in single cell analysis based on high expression of megakaryocyte-specific genes. For MF there is the added complication that fibrosis often prevents liquid bone marrow from being extracted by aspiration. The large size and fragility of megakaryocytes are also a concern. Single cell technologies can be used to identify and study megakaryocyte progenitors amongst haematopoietic stem and progenitor cells15, but it is unclear to what extent these reflect the transcriptomic profiles of mature megakaryocytes. In-vitro differentiation of megakaryocytes from stem cells or patient-derived HSCs, which can now be performed in large enough numbers to perform transcriptomic profiling101, 102, may be complicated by signatures reflecting the in-vitro differentiation process, and lack features conferred by the bone marrow microenvironment. Taken together, these challenges to studying megakaryocyte transcriptomes highlight the potential for exploring the platelet transcriptome as a feasible and viable alternative.
Transcriptomic heterogeneity in MPNs
In the context of MPNs therefore, the platelet transcriptome represents a biomarker of megakaryocyte activity, thus capturing information on the HSC niche and providing a snapshot of the underlying thrombotic, haemostatic and inflammatory derangements. Figure 1 illustrates the different potential influences on the platelet transcriptome in MPNs.
Figure 1. The transcriptomic signature of circulating platelets provides a window into disease biology in MPNs.
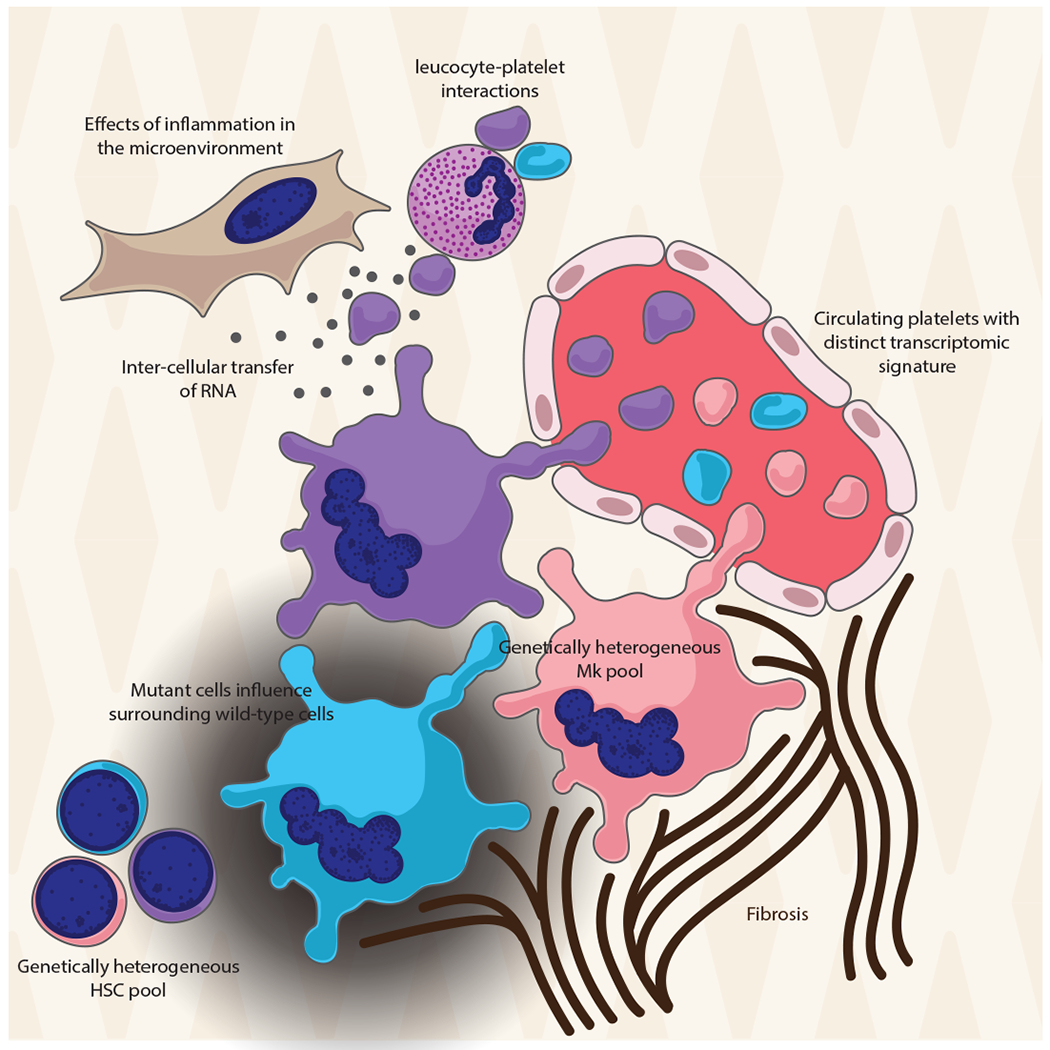
Megakaryocytes are expanded in MPNs and comprise a mixture of wild-type cells and cells with driver mutations and sub-clones with additional mutations, reflecting the heterogeneity within the HSC pool from which they are derived. Megakaryocytes influence surrounding cells in the bone marrow niche through non-cell-autonomous effects and are themselves influenced by environmental factors including inflammation and fibrosis. As well as reflecting the status of the megakaryocyte at the time of platelet release into the circulation, the platelet transcriptome is further influenced by inter-cellular transfer of RNA and by splicing events triggered by receptor activation. The transcriptome therefore gives insights into disease biology and carries signatures that distinguish between disease types and stages of disease progression.
Our recent work103, 104 using RNA sequencing of purified platelets from two cohorts of patients with ET, PV, and MF (contrasted with healthy donors) confirms the intra-disease heterogeneity and identifies novel therapeutic targets. The platelet transcriptome is significantly reprogrammed in the MPN setting, with a wealth of transcript associations that may be missed in using conventional tissue sources such as serum, plasma, whole blood, or bulk bone marrow (Table 2 summarizes MPN studies across these bio-sources and suggests potential for future investigations on assessing intersecting signatures/pathways). Our data also closely overlaps with two other recent platelet transcriptomic studies on select MPN subtypes: thrombo-inflammatory signatures in PV from Gangaraju and Prchal et al105 and fibrosis-associated signatures in MF from Guo and Erber et al106.
Table 2.
Summary of cell types for transcriptomic study in MPNs, study findings, advantages and disadvantages of each source.
| Cell type | MPN subtypes | Findings |
|---|---|---|
| Whole blood121 | Healthy donors, ET, PV, MF | • Dysregulation of genes involved in inflammation • Detects signatures that might be missed by profiling isolated cell types |
| Granulocytes122 | Healthy donors, ET, PV, MF | • JAK-STAT signature common across disease sub-types and driver mutations • TET2 mutation is associated with a distinct signature • Extent of clonal predominance may influence signatures |
| Circulating stem and progenitor cells123 | JAK2V617F mutated PV | • Reveals heterogeneity, separates patients into distinct groups with different rates of disease complications • Rare cell type, may not be sufficiently abundant in all MPN subtypes |
| Circulating stem and progenitor cells15 | MF, mobilised cells in healthy donors | • Heterogeneity of Mk progenitors in MF cell type, cost if performing single cell sequencing |
| Platelets124 | ET, reactive thrombocytosis | • Transcript profiles distinguish ET from reactive thrombocytosis |
| Platelets106 | Healthy donors, MF | • Distinguishes MF from controls, separates patients into groups with and without fibrosis |
| Platelets12, 104 | Healthy donors, ET, PV, MF | • Two mutually validating MPN patient RNA-seq cohorts discriminate each clinical phenotype; and identify progressive transcriptomic markers that also enable predictive signatures for MF. |
Distinctive signatures of over and under-expressed genes are seen for each disease entity, with the greatest differences from normal seen in MF. Of note, these are independent of driver mutation, highlighting the significance of other factors in disease pathogenesis and progression. The data corroborates the role of inflammation in MPN pathogenesis and identifies gene expression signatures reflecting activation of inflammatory signalling pathways. Characteristic changes are noted in MF patients treated with the JAK1/2 inhibitor ruxolitinib, both confirming known mechanisms of action as well as identifying potential new or combinatorial targets for MPN therapy.
Molecular pathways differentially activated between MPN subtypes identified expected immune modulatory responses (e.g. a consistent interferon alpha/gamma, and IL2 STAT5) in addition to robust (FDR<0.05) signaling in oxidative phosphorylation (OXPHOS), mTORC1, and reactive oxygen species (ROS) production pathways. Coagulation- and complement-associated gene sets were also expectedly enriched across ET, PV, and MF. Particularly in MF, cycle progression and proliferation pathways around c-MYC and E2F targets, and G2M checkpoint pathways emerged as highly significant (FDR < 0.001) and altogether pointing to a strong unfolded protein response as a key factor, likely attributed to a chronic integrated stress response (ISR)107. Together, these data demonstrate that in addition to immune factors such as type I/II interferons and dysregulation of interleukin-dependent inflammatory responses, which have been linked to MPNs, platelet transcriptional signatures of proliferation, metabolic, and proteostasis signaling are a feature of MPN pathogenesis.
Most importantly, platelet gene expression profiling in MPN offers directions for prediction of myelofibrosis. Applying machine-learning algorithms of LASSO penalized regression under two conditions of external validation 108: temporal (using our two cohort design) and geographical (independently published datasets on healthy donors106, 109 and MF 106, we uniquely discriminate MPN subtypes from each other, and healthy controls using three model types and predict MF at high accuracy. The highest performing model used a set of progressively differentiated MPN genes at an area under the (ROC) curve of 0.96 (temporal) and 0.97 (geographical); and rendered a core signature of <5 candidate markers as top predictors of disease progression.
Collectively, our work presents the platelet transcriptome as a proof-of-concept not only in understanding disease progression but also in deciphering mechanistic insights and developing predictive machine learning algorithms.
Single-cell technologies
Despite the wealth of insight gained from bulk RNA sequencing of purified platelets, this approach does not answer questions arising from heterogeneity of sub-populations of HSC and their progeny (Figure 1). Single-cell approaches applied to stem and progenitor cells are starting to offer this granular perspective.
Our understanding of normal and malignant haematopoiesis is being transformed by single-cell transcriptomics, where expression of thousands of genes measured from individual cells is used to infer differentiation trajectories110–112. In MPNs, this has revealed heterogeneity in megakaryocyte progenitors in MF, where patients have a population resembling healthy controls, but also eight other distinct populations15. Methods that simultaneously determine the mutation status and transcript profile of single cells bring further resolution, including the potential to examine sub-clones with additional mutations113, 114. Surprisingly, driver-mutated cells do not form novel clusters with unique gene expression profiles but are found across all the stem and progenitor cell populations114, 115. Emerging technologies that combine single cell and spatial information116 have the potential to enrich this with information on influences of the marrow microenvironment.
Challenges remain in deciphering heterogeneity within platelet populations, for example to identify the contribution of wild-type and driver-mutated megakaryocytes to the circulating platelet population and establish whether there are transcriptomic differences between them. It will be of interest to see whether single-cell transcriptomic technologies can be adapted to study single platelets, although low RNA yields may be a potential limitation.
Early detection of MPNs
Novel work10, 117 using somatic mutations as molecular clocks to calculate the timing of driver mutation acquisition in patients with MPNs has revealed a very long latency between mutation acquisition and presentation with overt disease. Furthermore, it was inferred that the mutations would have been detectable many years before disease presentation10, 115. Notably, rates of clonal expansion varied between individuals. Platelets are potential sources to investigate the multiple influences including inflammation and the bone marrow microenvironment which could be contributing to this trajectory. Future directions for personalised medicine in MPNs therefore encompasses not only risk prediction for patients already diagnosed with an MPN, but also early detection, risk stratification for progression to disease and potentially early intervention for individuals with low burden mutant clones10.
CONCLUSIONS
Platelets play a central role in the haemostatic and thrombotic complication of MPNs and provide a window into the stem and progenitor cell populations and bone marrow microenvironment that are responsible for disease pathogenesis and progression. The platelet transcriptome offers markers of disease phenotype, disease progression and biological insights into potentially targetable aspects of disease biology - one step closer to personalising medicine in MPNs.
Highlights.
Myeloproliferative neoplasms (MPNs) arise when haematopoietic stem cells (HSCs) acquire driver mutations that cause abnormal blood counts.
MPNs are a model for cancer progression broadly relevant to bone marrow biology.
MPN disease phenotype is the consequence of a combination of factors including effects of driver mutations on HSCs and their progeny, non-cell-autonomous effects in the bone marrow microenvironment, and systemic inflammation.
Thrombosis and bleeding cause significant morbidity and mortality in MPN patients. Megakaryocyte populations are expanded in patients with MPNs and are particularly central to disease pathogenesis in myelofibrosis.
The platelet transcriptome integrates disease-specific information from the parent megakaryocytes as well as the bone marrow microenvironment and the peripheral circulation. There is an unmet need for markers of disease progression in MPNs and therefore, platelets are ideal peripherally accessible biosources that also reflect underlying disease.
Acknowledgements
a) Authors thank the patients at the Stanford Cancer Center for their generous participation in the research that led to this review article and Dr. Kellie Machlus for her inspirational leadership of the Blood and Bone seminar series over the 2020 COVID pandemic. b) This work was funded by US National Institutes of Health grants 1K08HG010061–01A1 and 3UL1TR001085–04S1 (research re-entry award) to A.K.
c) The authors declare no competing interests.
Abbreviations
- AML
acute myeloid leukaemia
- CALR
calreticulin
- ET
essential thrombocytosis
- HSC
haematopoietic stem cells
- JAK2
janus kinase 2
- MF
myelofibrosis
- MPN
myeloproliferative neoplasm
- PV
polycythaemia vera
Contributor Information
Sally Thomas, Department of Oncology and Metabolism, University of Sheffield and Department of Haematology, Royal Hallamshire Hospital, Sheffield, UK.
Anandi Krishnan, Department of Pathology, Stanford University School of Medicine, Stanford, CA.
Bibliography
- 1.Spivak JL. Myeloproliferative neoplasms. New England Journal of Medicine. 2017;376:2168–2181 [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405 [DOI] [PubMed] [Google Scholar]
- 3.Rumi E, Cazzola M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. 2017;129:680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129:667–679 [DOI] [PubMed] [Google Scholar]
- 5.Zoi K, Cross NC. Genomics of myeloproliferative neoplasms. J Clin Oncol. 2017;35:947–954 [DOI] [PubMed] [Google Scholar]
- 6.Bao EL, Nandakumar SK, Liao X, Bick AG, Karjalainen J, Tabaka M, et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature. 2020;586:769–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marneth AE, Mullally A. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb Perspect Med. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372:601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Egeren D, Escabi J, Nguyen M, Liu S, Reilly CR, Patel S, et al. Reconstructing the lineage histories and differentiation trajectories of individual cancer cells in myeloproliferative neoplasms. Cell Stem Cell. 2021;28:514–523 e519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams N, Lee J, Moore L, Baxter EJ, Hewinson J, Dawson KJ, et al. Phylogenetic reconstruction of myeloproliferative neoplasm reveals very early origins and lifelong evolution. BioRxiv. 2020:2020.2011.2009.374710 [Google Scholar]
- 11.Krishnan A, Zhang Y, Perkins C, Gotlib J, Zehnder JL. Platelet transcriptomic signatures in myeloproliferative neoplasms. Blood. 2017;130:5288–5288 [Google Scholar]
- 12.Shen Z, Du W, Perkins C, Fechter L, Natu V, Maecker H, et al. Platelet transcriptome yields progressive markers in chronic myeloproliferative neoplasms and identifies potential targets of therapy. BioRxiv. 2021: 10.1101/2021.1103.1112.435190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davizon-Castillo P, Rowley JW, Rondina MT. Megakaryocyte and platelet transcriptomics for discoveries in human health and disease. Arteriosclerosis, thrombosis, and vascular biology. 2020;40:1432–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woods B, Chen W, Chiu S, Marinaccio C, Fu C, Gu L, et al. Activation of jak/stat signaling in megakaryocytes sustains myeloproliferation in vivo. Clin Cancer Res. 2019;25:5901–5912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Psaila B, Wang G, Rodriguez-Meira A, Li R, Heuston EF, Murphy L, et al. Single-cell analyses reveal megakaryocyte-biased hematopoiesis in myelofibrosis and identify mutant clone-specific targets. Molecular cell. 2020;78:477–492.e478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen QJ, Yang Q, Goldenson B, Malinge S, Lasho T, Schneider RK, et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by aurka inhibition. Nature medicine. 2015;21:1473–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emanuel RM, Dueck AC, Geyer HL, Kiladjian JJ, Slot S, Zweegman S, et al. Myeloproliferative neoplasm (mpn) symptom assessment form total symptom score: Prospective international assessment of an abbreviated symptom burden scoring system among patients with mpns. J Clin Oncol. 2012;30:4098–4103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27:1861–1869 [DOI] [PubMed] [Google Scholar]
- 19.Alvarez-Larrán A, Senín A, Fernández-Rodríguez C, Pereira A, Arellano-Rodrigo E, Gómez M, et al. Impact of genotype on leukaemic transformation in polycythaemia vera and essential thrombocythaemia. British Journal of Haematology. 2017;178:764–771 [DOI] [PubMed] [Google Scholar]
- 20.Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. 2020;136:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barosi G, Tefferi A, Besses C, Birgegard G, Cervantes F, Finazzi G, et al. Clinical end points for drug treatment trials in bcr-abl1-negative classic myeloproliferative neoplasms: Consensus statements from european leukemianet (eln) and internation working group-myeloproliferative neoplasms research and treatment (iwg-mrt). Leukemia. 2015;29:20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spivak JL. Myeloproliferative neoplasms. N Engl J Med. 2017;376:2168–2181 [DOI] [PubMed] [Google Scholar]
- 23.Nangalia J, Green TR. The evolving genomic landscape of myeloproliferative neoplasms. Hematology/the Education Program of the American Society of Hematology. American Society of Hematology. Education Program. 2014;2014:287–296 [DOI] [PubMed] [Google Scholar]
- 24.Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the jak/stat pathway. Protein Science. 2018;27:1984–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase jak2 in human myeloproliferative disorders. The Lancet.365:1054–1061 [DOI] [PubMed] [Google Scholar]
- 26.James C, Ugo V, Le Couedic J-P, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148 [DOI] [PubMed] [Google Scholar]
- 27.Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of jak2 in myeloproliferative disorders. New England Journal of Medicine. 2005;352:1779–1790 [DOI] [PubMed] [Google Scholar]
- 28.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, et al. Activating mutation in the tyrosine kinase jak2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer cell. 2005;7:387–397 [DOI] [PubMed] [Google Scholar]
- 29.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic calr mutations in myeloproliferative neoplasms with nonmutated jak2. N Engl J Med. 2013;369:2391–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–2390 [DOI] [PubMed] [Google Scholar]
- 31.Elf S, Abdelfattah NS, Baral AJ, Beeson D, Rivera JF, Ko A, et al. Defining the requirements for the pathogenic interaction between mutant calreticulin and mpl in mpn. Blood. 2018;131:782–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.How J, Hobbs GS, Mullally A. Mutant calreticulin in myeloproliferative neoplasms. Blood. 2019;134:2242–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tefferi A, Abdel-Wahab O, Cervantes F, Crispino JD, Finazzi G, Girodon F, et al. Mutations with epigenetic effects in myeloproliferative neoplasms and recent progress in treatment: Proceedings from the 5th international post-ash symposium. Blood cancer journal. 2011;1:e7–e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frédéric C, Nadine C, Séverine G, Arnaud G, José A, Nathalie C, et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica. 2017;102:e11–e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hultcrantz M, Björkholm M, Dickman PW, Landgren O, Derolf ÅR, Kristinsson SY, et al. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: A population-based cohort study. Ann Intern Med. 2018;168:317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hultcrantz M, Wilkes SR, Kristinsson SY, Andersson TML, Derolf ÅR, Eloranta S, et al. Risk and cause of death in patients diagnosed with myeloproliferative neoplasms in sweden between 1973 and 2005: A population-based study. Journal of Clinical Oncology. 2015;33:2288–2295 [DOI] [PubMed] [Google Scholar]
- 37.Moliterno A, Ginzburg Y, Hoffman R. Clinical insights into the origins of thrombosis in myeloproliferative neoplasms. Blood. 2020;Online ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finazzi G, De Stefano V, Barbui T. Are mpns vascular diseases? Current hematologic malignancy reports. 2013;8:307–316 [DOI] [PubMed] [Google Scholar]
- 39.Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood. 2013;122:2176–2184 [DOI] [PubMed] [Google Scholar]
- 40.Reeves BN, Beckman JD. Novel pathophysiological mechanisms of thrombosis in myeloproliferative neoplasms. Current hematologic malignancy reports. 2021;16:304–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuura S, Thompson CR, Belghasem ME, Bekendam RH, Piasecki A, Leiva O, et al. Platelet dysfunction and thrombosis in jak2(v617f)-mutated primary myelofibrotic mice. Arterioscler Thromb Vasc Biol. 2020;40:e262–e272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMullin MF, Harrison CN, Ali S, Cargo C, Chen F, Ewing J, et al. A guideline for the diagnosis and management of polycythaemia vera. A british society for haematology guideline. Br J Haematol. 2019;184:176–191 [DOI] [PubMed] [Google Scholar]
- 43.Marchioli R, Finazzi G, Specchia G, Cacciola R, Cavazzina R, Cilloni D, et al. Cardiovascular events and intensity of treatment in polycythemia vera. New England Journal of Medicine. 2012;368:22–33 [DOI] [PubMed] [Google Scholar]
- 44.McMullin MFF, Mead AJ, Ali S, Cargo C, Chen F, Ewing J, et al. A guideline for the management of specific situations in polycythaemia vera and secondary erythrocytosis. British Journal of Haematology. 2019;184:161–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carobbio A, Antonioli E, Guglielmelli P, Vannucchi AM, Delaini F, Guerini V, et al. Leukocytosis and risk stratification assessment in essential thrombocythemia. J Clin Oncol. 2008;26:2732–2736 [DOI] [PubMed] [Google Scholar]
- 46.Campbell PJ, MacLean C, Beer PA, Buck G, Wheatley K, Kiladjian J-J, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: Analysis of the prospective pt1 cohort. Blood. 2012;120:1409–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353:33–45 [DOI] [PubMed] [Google Scholar]
- 48.Gangaraju R, Song J, Kim SJ, Tashi T, Reeves BN, Sundar KM, et al. Thrombotic, inflammatory, and hif-regulated genes and thrombosis risk in polycythemia vera and essential thrombocythemia. Blood Advances. 2020;4:1115–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedersen OH, Larsen ML, Grove EL, van Kooten Niekerk PB, Bønløkke S, Nissen PH, et al. Platelet characteristics in patients with essential thrombocytosis. Cytometry. Part B, Clinical cytometry. 2018;94:918–927 [DOI] [PubMed] [Google Scholar]
- 50.Hobbs CM, Manning H, Bennett C, Vasquez L, Severin S, Brain L, et al. Jak2v617f leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood. 2013;122:3787–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuura S, Thompson CR, Belghasem ME, Bekendam RH, Piasecki A, Leiva O, et al. Platelet dysfunction and thrombosis in jak2(v617f)-mutated primary myelofibrotic mice. Arteriosclerosis, thrombosis, and vascular biology. 2020;40:e262–e272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rotunno G, Mannarelli C, Guglielmelli P, Pacilli A, Pancrazzi A, Pieri L, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014;123:1552–1555 [DOI] [PubMed] [Google Scholar]
- 54.Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res. 2016;140 Suppl 1:S71–75 [DOI] [PubMed] [Google Scholar]
- 55.Pemmaraju N, Moliterno AR, Williams DM, Rogers O, Spivak JL. The quantitative jak2 v617f neutrophil allele burden does not correlate with thrombotic risk in essential thrombocytosis. Leukemia. 2007;21:2210–2212 [DOI] [PubMed] [Google Scholar]
- 56.Barbui T, Carobbio A, Finazzi G, Vannucchi AM, Barosi G, Antonioli E, et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of c-reactive protein and pentraxin 3. Haematologica. 2011;96:315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teofili L, Martini M, Iachininoto MG, Capodimonti S, Nuzzolo ER, Torti L, et al. Endothelial progenitor cells are clonal and exhibit the jak2(v617f) mutation in a subset of thrombotic patients with ph-negative myeloproliferative neoplasms. Blood. 2011;117:2700–2707 [DOI] [PubMed] [Google Scholar]
- 58.Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Villamor N, Colomer D, Cervantes F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the jak2 mutational status. Haematologica. 2006;91:169–175 [PubMed] [Google Scholar]
- 59.Rungjirajittranon T, Owattanapanich W, Ungprasert P, Siritanaratkul N, Ruchutrakool T. A systematic review and meta-analysis of the prevalence of thrombosis and bleeding at diagnosis of philadelphia-negative myeloproliferative neoplasms. BMC Cancer. 2019;19:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Budde U, Scharf RE, Franke P, Hartmann-Budde K, Dent J, Ruggeri ZM. Elevated platelet count as a cause of abnormal von willebrand factor multimer distribution in plasma. Blood. 1993;82:1749–1757 [PubMed] [Google Scholar]
- 61.Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M, et al. Incidence and risk factors for bleeding in 1104 patients with essential thrombocythemia or prefibrotic myelofibrosis diagnosed according to the 2008 who criteria. Leukemia. 2012;26:716–719 [DOI] [PubMed] [Google Scholar]
- 62.Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350:114–124 [DOI] [PubMed] [Google Scholar]
- 63.Cortelazzo S, Finazzi G, Ruggeri M, Vestri O, Galli M, Rodeghiero F, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332:1132–1136 [DOI] [PubMed] [Google Scholar]
- 64.Rumi E, Cazzola M. How i treat essential thrombocythemia. Blood. 2016;128:2403–2414 [DOI] [PubMed] [Google Scholar]
- 65.Braunstein EM, Chaturvedi S. Aspirin in et: Will twice a day keep thrombosis away? Blood. 2020;136:151–153 [DOI] [PubMed] [Google Scholar]
- 66.Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Colomer D, Villamor N, Bellosillo B, et al. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelial activation in essential thrombocythemia: Relationship with thrombosis occurrence and jak2 v617f allele burden. American journal of hematology. 2009;84:102–108 [DOI] [PubMed] [Google Scholar]
- 67.Rocca B, Tosetto A, Betti S, Soldati D, Petrucci G, Rossi E, et al. A randomized double-blind trial of 3 aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood. 2020;136:171–182 [DOI] [PubMed] [Google Scholar]
- 68.Voora D, Ginsburg GS, Åkerblom A. Platelet rna as a novel biomarker for the response to antiplatelet therapy. Future Cardiology. 2014;10:9–12 [DOI] [PubMed] [Google Scholar]
- 69.Ianotto JC, Couturier MA, Galinat H, Mottier D, Berthou C, Guillerm G, et al. Administration of direct oral anticoagulants in patients with myeloproliferative neoplasms. International journal of hematology. 2017;106:517–521 [DOI] [PubMed] [Google Scholar]
- 70.Hamulyák EN, Daams JG, Leebeek FWG, Biemond BJ, Te Boekhorst PAW, Middeldorp S, et al. A systematic review of antithrombotic treatment of venous thromboembolism in patients with myeloproliferative neoplasms. Blood Adv. 2021;5:113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. Jak inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798 [DOI] [PubMed] [Google Scholar]
- 72.Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372:426–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harrison CN, Mead AJ, Panchal A, Fox S, Yap C, Gbandi E, et al. Ruxolitinib vs best available therapy for et intolerant or resistant to hydroxycarbamide. Blood. 2017;130:1889–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kiladjian JJ, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112:3065–3072 [DOI] [PubMed] [Google Scholar]
- 75.Méndez-Ferrer S, Bonnet D, Steensma DP, Hasserjian RP, Ghobrial IM, Gribben JG, et al. Bone marrow niches in haematological malignancies. Nature Reviews Cancer. 2020;20:285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu YC, Sanada C, Xavier-Ferrucio J, Wang L, Zhang PX, Grimes HL, et al. The molecular signature of megakaryocyte-erythroid progenitors reveals a role for the cell cycle in fate specification. Cell reports. 2018;25:2083–2093.e2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koschmieder S, Chatain N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood reviews. 2020;42:100711. [DOI] [PubMed] [Google Scholar]
- 78.Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. Jak-stat pathway activation in malignant and nonmalignant cells contributes to mpn pathogenesis and therapeutic response. Cancer Discov. 2015;5:316–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Curto-Garcia N, Harrison C, McLornan DP. Bone marrow niche dysregulation in myeloproliferative neoplasms. Haematologica. 2020;105:1189–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circulation research. 2013;112:1506–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weyrich AS. Platelets: More than a sack of glue. Hematology Am Soc Hematol Educ Program. 2014;2014:400–403 [DOI] [PubMed] [Google Scholar]
- 82.Portier I, Campbell RA. Role of platelets in detection and regulation of infection. Arterioscler Thromb Vasc Biol. 2021;41:70–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nature cell biology. 2017;19:271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, et al. Clonal analysis of lineage fate in native haematopoiesis. Nature. 2018;553:212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gilles L, Finke C, Lasho TL, Pardanani A, Tefferi A, Crispino J. Aberrant megakaryocyte gene expression contributes to primary myelofibrosis. Blood. 2012;120:2867–2867 [Google Scholar]
- 86.Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through cxcl4 secretion. Nature medicine. 2014;20:1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nature medicine. 2014;20:1321–1326 [DOI] [PubMed] [Google Scholar]
- 88.Gleitz HFE, Dugourd AJF, Leimkühler NB, Snoeren IAM, Fuchs SNR, Menzel S, et al. Increased cxcl4 expression in hematopoietic cells links inflammation and progression of bone marrow fibrosis in mpn. Blood. 2020;136:2051–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ciurea SO, Merchant D, Mahmud N, Ishii T, Zhao Y, Hu W, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007;110:986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gangat N, Marinaccio C, Swords R, Watts JM, Gurbuxani S, Rademaker A, et al. Aurora kinase a inhibition provides clinical benefit, normalizes megakaryocytes, and reduces bone marrow fibrosis in patients with myelofibrosis: A phase i trial. Clinical Cancer Research. 2019;25:4898–4906 [DOI] [PubMed] [Google Scholar]
- 91.Rowley JW, Schwertz H, Weyrich AS. Platelet mrna: The meaning behind the message. Curr Opin Hematol. 2012;19:385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schubert S, Weyrich AS, Rowley JW. A tour through the transcriptional landscape of platelets. Blood. 2014;124:493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Simon LM, Edelstein LC, Nagalla S, Woodley AB, Chen ES, Kong X, et al. Human platelet microrna-mrna networks associated with age and gender revealed by integrated plateletomics. Blood. 2014;123:e37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the nuclear confines: Signal-dependent pre-mrna splicing in anucleate platelets. Cell. 2005;122:379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, et al. Signal-dependent splicing of tissue factor pre-mrna modulates the thrombogenicity of human platelets. J Exp Med. 2006;203:2433–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cunin P, Bouslama R, Machlus KR, Martínez-Bonet M, Lee PY, Wactor A, et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. Rna-seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. 2015;28:666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Middleton EA, Rowley JW, Campbell RA, Grissom CK, Brown SM, Beesley SJ, et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood. 2019;134:911–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Davizon-Castillo P, McMahon B, Aguila S, Bark D, Ashworth K, Allawzi A, et al. Tnf-α-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood. 2019;134:727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heazlewood SY, Williams B, Storan MJ, Nilsson SK. The prospective isolation of viable, high ploidy megakaryocytes from adult murine bone marrow by fluorescence activated cell sorting. Methods in molecular biology (Clifton, N.J.). 2013;1035:121–133 [DOI] [PubMed] [Google Scholar]
- 101.Shepherd JH, Howard D, Waller AK, Foster HR, Mueller A, Moreau T, et al. Structurally graduated collagen scaffolds applied to the ex vivo generation of platelets from human pluripotent stem cell-derived megakaryocytes: Enhancing production and purity. Biomaterials. 2018;182:135–144 [DOI] [PubMed] [Google Scholar]
- 102.Thon JN, Mazutis L, Wu S, Sylman JL, Ehrlicher A, Machlus KR, et al. Platelet bioreactor-on-a-chip. Blood. 2014;124:1857–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shen Z, Du W, Perkins C, Fechter L, Natu V, Maecker H, et al. Progressive and predictive markers of disease evolution: Platelet transcriptome in chronic myeloproliferative neoplasms. BioRxiv. 2021:2021.2003.2012.435190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Krishnan A, Zhang Y, Perkins C, Gotlib J, Zehnder JL. Platelet transcriptomic signatures in myeloproliferative neoplasms. Blood. 2017;130:5288 [Google Scholar]
- 105.Gangaraju R, Song J, Kim SJ, Tashi T, Reeves BN, Sundar KM, et al. Thrombotic, inflammatory, and hif-regulated genes and thrombosis risk in polycythemia vera and essential thrombocythemia. Blood Adv. 2020;4:1115–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guo BB, Linden MD, Fuller KA, Phillips M, Mirzai B, Wilson L, et al. Platelets in myeloproliferative neoplasms have a distinct transcript signature in the presence of marrow fibrosis. Br J Haematol. 2020;188:272–282 [DOI] [PubMed] [Google Scholar]
- 107.Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moons KG, Kengne AP, Grobbee DE, Royston P, Vergouwe Y, Altman DG, et al. Risk prediction models: Ii. External validation, model updating, and impact assessment. Heart. 2012;98:691–698 [DOI] [PubMed] [Google Scholar]
- 109.Rondina MT, Voora D, Simon LM, Schwertz H, Harper JF, Lee O, et al. Longitudinal rna-seq analysis of the repeatability of gene expression and splicing in human platelets identifies a platelet selp splice qtl. Circulation research. 2020;126:501–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Watcham S, Kucinski I, Gottgens B. New insights into hematopoietic differentiation landscapes from single-cell rna sequencing. Blood. 2019;133:1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Acosta J, Ssozi D, Galen Pv. Single-cell rna sequencing to disentangle the blood system. Arteriosclerosis, thrombosis, and vascular biology.0:ATVBAHA.120.314654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shepherd MS, Kent DG. Emerging single-cell tools are primed to reveal functional and molecular heterogeneity in malignant hematopoietic stem cells. Current opinion in hematology. 2019;26:214–221 [DOI] [PubMed] [Google Scholar]
- 113.Rodriguez-Meira A, Buck G, Clark SA, Povinelli BJ, Alcolea V, Louka E, et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel rna sequencing. Molecular cell. 2019;73:1292–1305.e1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nam AS, Kim K-T, Chaligne R, Izzo F, Ang C, Taylor J, et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature. 2019;571:355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Van Egeren D, Escabi J, Nguyen M, Liu S, Reilly CR, Patel S, et al. Reconstructing the lineage histories and differentiation trajectories of individual cancer cells in myeloproliferative neoplasms. Cell stem cell. 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nature cell biology. 2020;22:38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Williams N, Lee J, Moore L, Baxter JE, Hewinson J, Dawson KJ, et al. Driver mutation acquisition in utero and childhood followed by lifelong clonal evolution underlie myeloproliferative neoplasms. Blood. 2020;136:LBA-1–LBA-1 [Google Scholar]
- 118.Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. Journal of Clinical Oncology. 2011;29:3179–3184 [DOI] [PubMed] [Google Scholar]
- 119.Godfrey AL, Campbell PJ, MacLean C, Buck G, Cook J, Temple J, et al. Hydroxycarbamide plus aspirin versus aspirin alone in patients with essential thrombocythemia age 40 to 59 years without high-risk features. J Clin Oncol. 2018;36:3361–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. Journal of Clinical Oncology. 2005;23:2224–2232 [DOI] [PubMed] [Google Scholar]
- 121.Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, et al. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leukemia Research. 2012;36:1387–1392 [DOI] [PubMed] [Google Scholar]
- 122.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of jak-stat pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123:e123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spivak JL, Considine M, Williams DM, Talbot CC, Rogers O, Moliterno AR, et al. Two clinical phenotypes in polycythemia vera. New England Journal of Medicine. 2014;371:808–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gnatenko DV, Zhu W, Xu X, Samuel ET, Monaghan M, Zarrabi MH, et al. Class prediction models of thrombocytosis using genetic biomarkers. Blood. 2010;115:7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]