

Abstract
The intracellular molecular mechanisms underlying the genotype of cortisol-producing adenoma (CPA) have not been fully determined. We analyzed gene expressions in CPA and the human adrenocortical cell line (HAC15 cells) with PRKACA mutation. Clustering analysis using a gene set associated with responses to cAMP revealed the possible differences between PRKACA mutant CPAs and GNAS and CTNNB1 mutant CPAs. The levels of STAR, CYP11A1, CYP17A1, CYP21A2, and FDX1 transcripts and cortisol levels per unit area in PRKACA mutant CPAs were significantly higher than those in GNAS mutant CPAs. PRKACA mutations led to an increase in steroidogenic enzyme expression and cortisol production in HAC15 cells. Transcriptome analysis revealed differences between PRKACA mutant CPAs and GNAS and CTNNB1 mutant CPAs. Cortisol production in PRKACA mutant CPAs is increased by the cAMP-PKA signaling pathway-mediated upregulation of steroidogenic enzymes transcription. The intracellular molecular mechanisms underlying these processes would be notably important in PRKACA mutant CPAs.
Keywords: Cushing’s syndrome, cortisol, gene expression, PRKACA, genotype
1. Introduction
Cushing’s syndrome is classified as adrenocorticotropic hormone (ACTH)-independent or ACTH-dependent cortisol overproduction. ACTH-independent Cushing’s syndrome is most often due to a unilateral cortisol-producing adenoma. Cortisol excess leads to Cushingoid features such as central obesity, moon face, purple striae, buffalo hump and others. Patients with Cushing’s syndrome often present with diabetes mellitus, hypertension, atherosclerosis, osteoporosis and mental disorders (Newell-Price, 2006,Nieman, 2008).
The etiology of isolated adrenal cortisol-producing adenoma (CPA) has long remained unclear, but recent studies have demonstrated that many harbors somatic mutations in PRKACA, GNAS, or CTNNB1 (Beuschlein, 2014,Calebiro, 2014,Cao, 2014,Goh, 2014,Sato, 2014). The PRKACA is the most commonly somatic mutated gene, and occurring in 35 to 65% of CPA (Beuschlein, 2014,Thiel, 2015,Zhou, 2016). PRKACA encodes a catalytic subunit of protein kinase A (PKA) with a hotspot mutation in the PRKACA gene at L206R. In the absence of cAMP, the PRKACA is bound to the regulatory subunit rendering inactive. The PRKACA mutant does not bind to the regulatory subunit of PKA, allowing the PKA’s catalytic activity to be constitutively active in cAMP-PKA signaling (Beuschlein, 2014,Calebiro, 2017,Calebiro, 2014,Cao, 2014,Goh, 2014,Sato, 2014). Additionally, the PRKACA mutation-mediated suppression of the cAMP-dependent PKA regulatory subunit IIβ (RIIβ) further contributes to the stimulation of cortisol secretion in adrenocortical cells (Calebiro, 2017,Weigand, 2017,Weigand, 2021). GNAS and CTNNB1 encode guanine nucleotide-binding protein subunit alpha (Gsα) and β-catenin, respectively. These mutations stimulate different intracellular signaling pathways, including mutation-mediated tumorigenesis and/or cortisol production. In this study, we focused on PRKACA mutation-mediated molecular mechanisms underlying the pathogenesis of CPAs, since PRKACA mutations are the most frequent somatic mutations in CPAs.
Recently, the relationship between genotype and clinical or pathological phenotypes was reported in patients with adrenal Cushing’s syndrome. The clinical features of patients with PRKACA mutant CPA presented at a younger age, with higher cortisol levels compared to the cases in patients with non-PRKACA mutant CPA (Di Dalmazi, 2014,Li, 2016,Thiel, 2015). Pathological features based on the CPA genotype demonstrated that PRKACA mutant cortisol-producing adenomas had higher CYP17A1 and 3βHSD immunoreactivities than wild-type cortisol-producing adenomas (Gao, 2020). PRKACA mutant CPAs have high STAR mRNA expression (Cao, 2014), and CPAs with mutations in PRKACA and GNAS exhibited strong immune-positive staining for STAR (Zhou, 2016). Clustering analysis using RNA sequencing data revealed that PRKACA mutant CPAs tended to have different gene expression profiles from adrenocortical carcinomas, subclinical Cushing’s adenomas, nonfunctioning adrenocortical adenomas (NFAs) and CPAs with CTNNB1 mutations (Di Dalmazi, 2020).
cAMP-PKA signaling activation potentiates the transcription of gene encoding steroidogenic enzymes that are needed in cortisol production, including STAR, CYP11A1, CYP17A1, CYP21A2, and CYP11B1 (Aumo, 2010). FDX1 which encodes ferredoxin 1 (also called adrenodoxin) is also stimulated by cAMP-PKA signaling (Imamichi, 2013). Ferredoxin 1 plays a pivotal role in transferring electrons in the mitochondria and is essential for the synthesis of adrenal steroid hormones (Muller, 2001,Sheftel, 2010). Upregulation of ferredoxin 1 facilitates the action of CYP11A1 and participates in the molecular mechanisms underlying cortisol overproduction. Although transcriptome data of CPAs with several mutants have been reported (Di Dalmazi, 2020), the differences in steroidogenic enzyme and FDX1 expressions between PRKACA mutant CPAs and other genotypes of CPAs has not yet been fully studied. Additionally, the effects of PRKACA mutations on steroidogenic enzymes and FDX1 have not been investigated in vitro.
The available data regarding the structural differences between each gene mutation and the differences in clinical and pathological findings among various CPA genotypes support the hypothesis that PRKACA mutations are associated with different gene expression profiles compared to those in CPAs with other mutations such as those in GNAS and CTNNB1. We first investigated whether gene expression in PRKACA mutant CPAs was different compared to that in other CPA genotypes. Next, we focused on the association between the genotypes and expression of cAMP-PKA signaling-associated genes in CPAs with PRKACA mutations. In addition, we modulated a PRKACA mutation in HAC15 cells and investigated the effects of PRKACA mutations on the transcription of steroidogenic enzyme-encoding genes.
2. Material and Methods
2.1. Patients and tissue collection
Cushing’s syndrome was diagnosed based on the existing guidelines (Nieman, 2008). Briefly, the patients presenting with Cushingoid features and adrenal tumors were detected using computed tomography. Suppressed basal ACTH levels, unsuppressed serum cortisol levels by overnight 1-mg dexamethasone suppression test (DST), and high midnight serum cortisol levels were observed in all patients. The patients without Cushingoid features, who were diagnosed as having subclinical Cushing’s syndrome, were excluded. NFA was diagnosed based on radiological findings showing lipid-containing adenomas obtained by computed tomography or magnetic resonance imaging, and by endocrinological findings that did not show cortisol or aldosterone excess, as reported previously (Baba, 2018,Oki, 2012).
Twenty-five adrenal CPA and six NFA samples were obtained by adrenalectomy at Hiroshima University Hospital between August 2007 and May 2019. The details of tissue collection and storage were mentioned, as reported previously (Kishimoto, 2016). This study was approved by the ethics committee of Hiroshima University, and written informed consent was obtained from all the patients.
2.2. Clinical measurements
Serum cortisol levels in patients were determined using the ECLusys 2010 cortisol assay (Roche Diagnostics Co., Germany). Tumor sizes were measured as the major and minor axes in the computed tomography images. The tumor area was calculated as half the length of the major axis × that of the minor axis × π as an ellipse. Cortisol levels per unit area were obtained by dividing the cortisol levels, as determined by overnight 1-mg DST, by the tumor area.
2.3. Cell culture and materials
HAC15 human adrenocortical carcinoma cells were provided by Professor WE Rainey (University of Michigan). The cells were cultured in DMEM/F12 with 10% Cosmic Calf serum (HyClone, Logan, UT) as described previously (Kishimoto, 2016,Kobuke, 2018). Incubation with forskolin in HAC15 cells were performed as described previously (Itcho, 2019). Forskolin was purchased from Sigma Aldrich Co.Ltd. (St. Louis, MO, USA).
2.4. Lentiviral production and infection
The open reading frame of the PRKACA gene with or without the L206R (c.620T>G) mutant was obtained from GenScript (Piscataway, NJ). They were ligated into the multiple cloning site of the lentiviral plasmid pCDH-CMV-MCS-EF1-Puro (System Bioscience, Palo Alto, CA). The empty plasmid was used for control. Lentivirus production and infection in HAC15 cells were performed as described previously (Itcho, 2020,Kishimoto, 2016).
2.5. DNA and RNA extraction and DNA genotyping
Genomic DNA and total RNA from a piece of adrenal frozen tissues and HAC15 cells were isolated using the AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany). Genotyping of the adenomas for PRKACA, GNAS and CTNNB1 was performed using targeted next-generation sequencing (NGS). The primers for PRKACA were 5’-TGCCAACTGCCTGTTCTTGTGC-3’ and 5’-GGAGGCTCCTACTTTGCTCAGG-3’. The primers for GNAS were 5’-CTTTGGTGAGATCCATTGACCTC-3’ and 5’-ACTGGGGTGAATGTCAAGAAACC-3’. The primers for CTNNB1 were 5’-TGATTTGATGGAGTTGGACATGG-3’ and 5’-TTGGGAGGTATCCACATCCTCTTC-3’. The PCR products were created using the Multiplex PCR Assay Kit Version 2 (TaKaRa Bio Inc. Shiga, Japan). The libraries were prepared for sequencing using the Nextera XT DNA Library Preparation Kit (Illumina. San Diego, CA). Amplicon-based NGS was performed using MiSeq platform (Illumina, San Diego, CA).
2.6. RNA sequencing analysis
Total RNA extracted from the NFA and CPA specimens and HAC15 cells (control cells and cells with PRKACA mutations) were used for RNA sequencing analysis. RNA integrity and quantitation were checked by Agilent 2100 bioanalyzer (Agilent Technologies Inc., Santa Clara, CA). Preparation of a paired-end RNA library and sequencing analysis using the Illumina HiSeq 2500 platform were performed as described previously (Itcho, 2020).
For the clustering analysis of all genes among the various genotypes, the single linkage method was performed using the R software package (https://www.r-project.org/). Ninety-four genes related to the response to cAMP in humans were retrieved using the AmiGO 2 website (http://amigo2.geneontology.org/amigo). Genes not expressed in adrenal cells were excluded from the analysis. Fifty-eight genes were used for the heatmap and clustering analyses. Gene ontology (GO) and pathway analyses using RNA sequencing data were performed using R software package. Bioinformatics data, such as those obtained from the GO (http://geneontology.org/), Kyoto Encyclopedia of Genes and Genomes (https://www.genome.jp/kegg/), and REACTOME databases (https://reactome.org/), were collected. Transcription factors which may regulate genes in each genotype of CPA were analyzed using Enrichr website (https://maayanlab.cloud/Enrichr/) with TRRUST data base (https://www.grnpedia.org/trrust/).
2.7. Cortisol and protein assays in vitro study
Cortisol levels in the cell culture and cellular protein levels were measured using an ELISA kit and a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA), respectively, as described previously (Itcho, 2020). The cortisol levels were determined as arbitrary units normalized to protein levels.
2.8. Statistical analyses
Results of the clinical characteristics and basic research are expressed as the mean ± SD and mean ± SE, respectively. Differences between two groups were analyzed by the t-test, and those between multiple groups were analyzed by one-way ANOVA followed by Bonferroni comparisons. Differences were considered significant at P < 0.05. Analyses were performed using SPSS for Windows (version 27.0; SPSS Inc., Chicago, IL, USA).
3. Results
3.1. Clinical characteristics
Sequencing analysis using targeted NGS demonstrated that p.L206R (c.620T>G) in PRKACA gene was detected in 15 samples of CPA. In the GNAS gene, two samples were found to have p.R201H (c.602G>A) and two other samples were found to have p.R201C (c.601C>T) (Table 1). p.S45P (c.133T>C) in the CTNNB1 gene was found in one sample. One sample had a multiple mutations, R201S (c.601C>A) mutation in the GNAS gene and P44A (c.130C>G) and p.S45P (c.133T>C) in the CTNNB1 gene (Table 1). No mutations were detected in four CPA or the six NFA samples. Basal and midnight serum cortisol levels or serum cortisol levels by 1-mg DST were not different among Cushing’s syndrome patients separated based on the genotype (Table 1). Basal plasma ACTH levels were undetectable in all patients with CPAs.
Table 1.
Clinical characteristics of the patients with Cushing’s syndrome and NFA
| Diagnosis (number of patients) | Cushing’s syndrome (n = 25) | NFA (n = 6) | ||||
|---|---|---|---|---|---|---|
| Mutation (number of patients) | No mutation (n = 4) |
PRKACA (n = 15) |
GNAS (n = 4) |
CTNNB1 (n = 1) |
GNAS / CTNNB1 (n = 1) |
— |
| Male / Female | 2 / 2 | 3 / 12 | 0 / 4 | 1 / 0 | 0 / 1 | 1 / 5 |
| Age (years) | 54.8 ± 4.1 | 44.7 ± 11.5 | 51.5 ± 15.9 | 71 | 69 | 47.0 ± 11.5 |
| Body mass index | 22.0 ± 2.1 | 24.1 ± 2.9 | 23.6 ± 4.6 | 26.1 | 29.4 | 29.3 ± 7.5 |
| Basal serum cortisol level (μg/dl) | 16.9 ± 9.9 | 17.9 ± 4.2 | 14.8 ± 5.6 | 12.6 | 10.0 | 13.1 ± 6.6 |
| Midnight serum cortisol level (μg/dl) | 17.4 ± 10.1 | 16.7 ± 4.1 | 16.6 ± 6.0 | 13.2 | 9.9 | 2.2 ± 1.2 |
| Serum cortisol level by 1-mg DST (μg/dl) | 13.7 ± 8.3 | 17.2 ± 3.9 | 17.3 ± 3.1 | 10.5 | 16.0 | 0.7 ± 0.2 |
| Tumor area (cm2) | 4.4 ± 1.5 | 4.9 ± 1.6 | 8.0 ± 3.6 | 4.9 | 7.3 | 3.4 ± 1.7 |
NFA, nonfunctioning adenoma; DST, dexamethasone suppression test. Data presented as mean ± standard deviation or number.
3.2. RNA sequencing and clustering analysis
RNA sequencing analysis detected 40,600 genes in each sample. Genes whose expression levels in CPAs and NFAs showed significant and more than 2-fold differences were enrolled in the clustering analysis. As shown in Figure 1A, NFAs were markedly differentiated from CPAs. Furthermore, the gene expression in CPAs with PRKACA mutations differed from that of CPAs with GNAS mutations.
Figure 1.

Clustering analysis of cortisol-producing adenomas (CPAs) and nonfunctioning adrenocortical adenomas (NFAs). (A) Genes whose expression levels show significant and more than 2-fold differences between NFAs and CPAs were applied for clustering analysis using the R software package. The single-linkage statistical method was used. The figure shows the single-linkage dendrogram and the sequence of combinations of the clusters. The distances of merging between clusters, called heights, are illustrated on the y-axis. (B) Fifty-eight genes from the gene associated with responses to cAMP were used for clustering analysis using the R software package. Hierarchical clustering was performed for both the rows and columns of the data matrix. The columns of the data matrix, such as those showing gene expression, are re-ordered according to the hierarchical clustering results and are visualized by a heatmap with a color scheme.
Both PRKACA mutations and GNAS mutations stimulate the cAMP-PKA signaling pathway (Beuschlein, 2014), and thus the gene set associated with responses to cAMP was used for subsequent comparison. The analysis revealed that the gene expression profiles of PRKACA mutant CPAs were different from those of GNAS mutant CPAs and that the gene expression profiles of CTNNB1 mutant CPAs were similar to those of GNAS mutant CPAs (Figure 1B and Table S1). Genes related to the response to cAMP were listed in Table S1, and the highest expression in PRKACA mutant CPAs was STAR, followed by FDX1.
3.3. Expression of steroidogenic enzymes and FDX1 among various genotype of CPAs
For the analyses of gene expressions, one case of CPA with CTNNB1 mutation was excluded, and one case of CPA with GNAS and CTNNB1 mutation was included in GNAS mutation group. Thus, we compared the gene expression profiles between CPAs with PRKACA mutations and CPAs with GNAS mutations. PRKACA mutant CPAs had higher FDX1 expression than GNAS mutant CPAs (Figure 2). The expression levels of STAR, CYP11A1, CYP17A1, and CYP21A2 in PRKACA mutant CPAs were higher than those in GNAS mutant CPAs, whereas the expression of CYP11B1 in PRKACA mutant CPAs was lower than that in GNAS mutant CPAs (Figure 2).
Figure 2.

The mRNA expression levels of FDX1, STAR, and adrenal steroidogenic enzymes in nonfunctioning adrenocortical adenomas (NFAs) and cortisol-producing adenomas (CPAs) with PRKACA and GNAS mutations. The mRNA expression levels were detected by RNA-sequencing analysis. *, P < 0.01 vs. NFAs; #, P < 0.01 vs. CPAs with PRKACA mutation.
Presumed transcription factors in CPAs with PRKACA mutations and CPAs with GNAS mutations were detected by Enrichr using genes whose expression levels showed significant and more than 2-fold differences compared to NFAs (Table 2). Two transcription factors, ATF3 and CREB1, related with cAMP-PKA signaling were found in PRKACA mutant CPAs, but not in GNAS mutant CPAs (Table 2).
Table 2.
Top 10 transcription factors were extracted by TRRUST data base using RNA sequencing results in each genotype of Cushing’s adenoma.
| Rank | PRKACA | GNAS |
|---|---|---|
| 1 | GATA6 | MSX1 |
| 2 | SP1 | HOXA9 |
| 3 | ATF3 # | POU2F2 |
| 4 | PPARG | ZBTB16 |
| 5 | NR5A1 | HOXAIO |
| 6 | CREB1 # | TP63 |
| 7 | ERG | NFE2L2 |
| 8 | SMAD3 | FOXM1 |
| 9 | CDX2 | ELK1 |
| 10 | REST | POUF5F1 |
Transcription factors which may regulate genes in each genotype of cortisol-producing adenomas (CPAs) were analyzed by Enrichr website (https://maayanlab.cloud/Enrichr/) using TRRUST data base (https://www.grnpedia.org/trrust/). Genes with more than 2-fold and significant difference between NFAs and genotypes with CPAs were enrolled.
indicates transcription factors related with cAMP-protein kinase A (PKA) signaling.
3.4. Cortisol levels in patients with CPA
PRKACA mutant CPAs had higher levels of some steroidogenic enzymes. Thus, cortisol levels in patients with PRKACA mutant CPAs were compared with those in patients with GNAS mutant CPAs (Table S2). Two patients with bilateral CPAs were excluded for the analysis. Basal and midnight serum cortisol levels or serum cortisol levels by 1-mg DST were not different between them, whereas the tumor areas of CPAs with GNAS mutations were significantly larger than those of CPAs with PRKACA mutations (Table S2). Cortisol levels per unit area were significantly higher in PRKACA mutant CPAs than in GNAS mutant CPAs (Figure 3).
Figure 3.

Cortisol levels per unit area in cortisol-producing adenomas (CPAs) with PRKACA and GNAS mutations. The tumor area was calculated as half the length of the major axis × that of the minor axis × π as ellipse. Cortisol levels per unit area were obtained by dividing the cortisol levels of overnight 1-mg dexamethasone suppression test by the tumor area. *, P < 0.01 vs. PRKACA mutation group.
3.5. Effects of PRKACA mutation in HAC15 cells
We first compared cortisol levels in the media among control, PRKACA wild type and PRKACA mutation in HAC15 cells. Cortisol levels in PRKACA mutation cells was significantly higher than the others (Figure S1), and thus subsequent comparisons for gene expression were performed in control and PRKACA mutation cells. Cortisol levels in the media of HAC15 cells transduced with the PRKACA mutant were 5.5-fold higher than those in the control cells (P < 0.01) (Figure 4). Forskolin (10μM) had no effects on cortisol production in HAC15 cells with PRKACA mutation (Figure 4).
Figure 4.

Basal cortisol production in control HAC15 cells or cells with PRKACA mutations. Forty-eight hours after PRKACA mutation transduction using lentivirus, the cells were serum-starved by treatment with DMEM-F12 containing 0.1% cosmic calf serum for 24 hours. The cells were then incubated in fresh medium containing 0.1% serum with or without forskolin (10μM). Media were collected to measure the cortisol levels. Relative cortisol levels were compared between the control HAC15 cells and those with PRKACA mutations. *, P < 0.05 vs. control; **, P < 0.01 vs. control (n=3).
According to the results of RNA sequencing, average transduction efficacy was 26.0% in PRKACA mutant cells (Figure S2). GO and pathway analyses using RNA sequencing data revealed that steroid hormone biosynthesis was the most enriched pathway in PRKACA mutant cells (Table S3). The FDX1 expression in PRKACA mutant cells was significantly increased by 2.7-fold compared to that in control cells (P < 0.01) (Figure 5). The expression of all genes encoding steroidogenic enzymes associated with cortisol production was markedly higher in PRKACA mutant cells than in the control cells.
Figure 5.
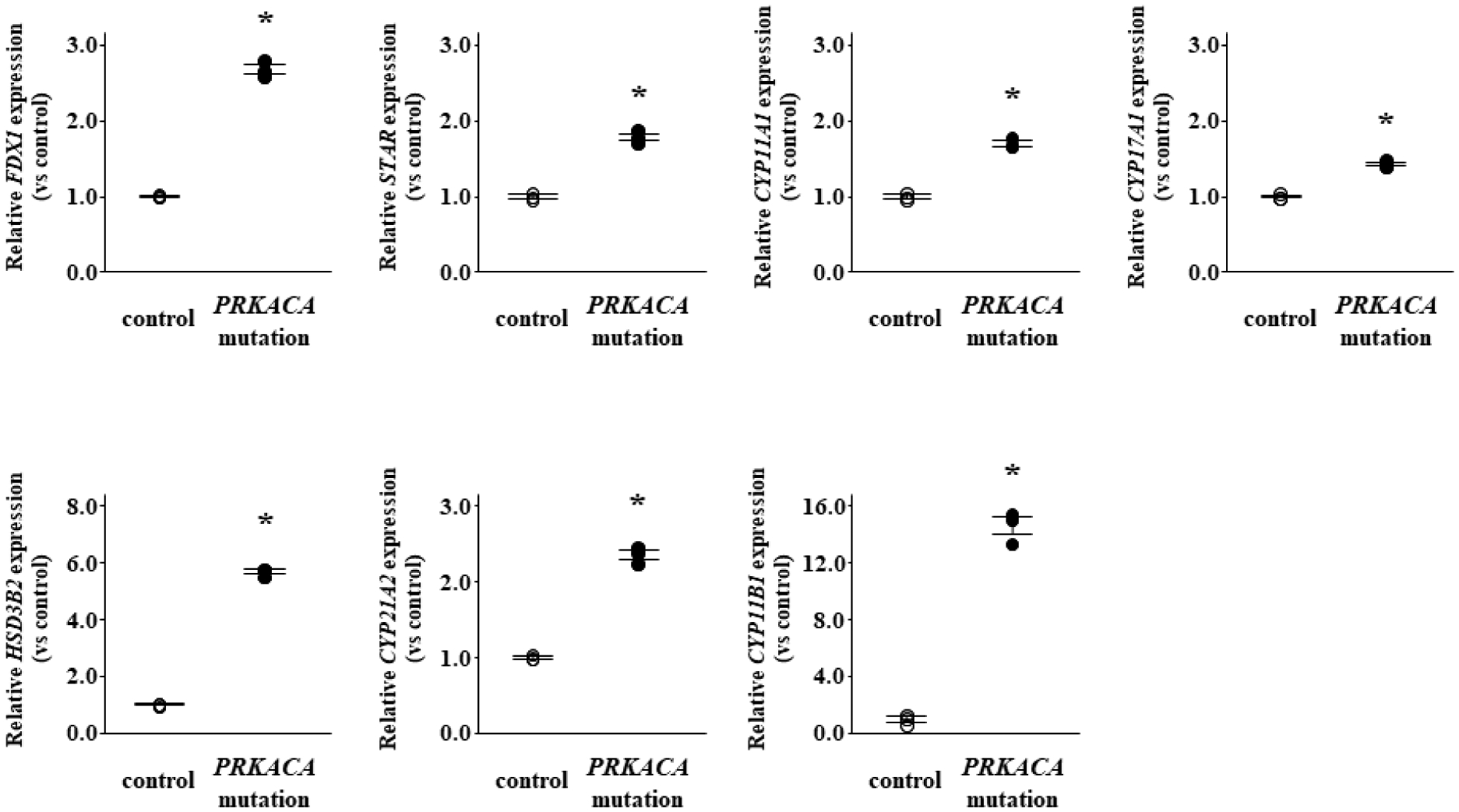
The effects of PRKACA mutation on the mRNA expression of FDX1, STAR, and adrenal steroidogenic enzymes in HAC15 cells. Forty-eight hours after PRKACA mutation transduction using lentivirus, the cells were serum starved by treatment with DMEM-F12 containing 0.1% cosmic calf serum for 24 hours, and then incubated with fresh medium containing 0.1% serum for 24 hours. After aspiration of medium, the cells were collected for RNA extraction and RNA sequencing. *, P < 0.01 vs. control (n=3).
4. Discussion
We demonstrated that PRKACA mutant CPAs differed from GNAS mutant CPAs, as shown by the clustering analysis using a gene set associated with responses to cAMP. FDX1 was the second most active gene among the gene set associated with responses to cAMP in PRKACA mutant CPAs. The levels of STAR, CYP11A1, CYP17A1, and CYP21A2 transcripts in PRKACA mutant CPAs were higher than those in CPAs with other mutations. Cortisol levels per unit area were higher in PRKACA mutant CPAs than in GNAS mutant CPAs. In vitro, HAC15 cells transduced with the PRKACA mutation showed increased expression levels of steroidogenic enzymes and cortisol production.
The gene expression profiles of CPAs with PRKACA mutations were different from those of CPAs with other mutations, as demonstrated by the clustering analysis using RNA sequencing. Although the clustering analysis of adrenal tumors including CPAs has already showed that PRKACA mutant CPAs tended to have different gene expression profiles from others (Di Dalmazi, 2020), especially when utilizing a gene set associated with responses to cAMP, gene expression in the PRKACA mutant CPAs was clearly distinguished from that in the GNAS and/or CTNNB1 mutant CPAs. Remarkably, the expression of adrenal steroidogenic enzyme-associated genes in the PRKACA mutant CPAs was higher than that in the GNAS mutant CPAs. Gao X, et al. reported that CYP17A1 immunoreactivities were higher in the PRKACA mutant CPAs than in the other CPAs (Gao, 2020); our results are consistent with the reports. ATF3 and CREB1, which are known to be activated by cAMP-PKA signaling (Tao, 2019), were candidate transcription factors to regulate genes in PRKACA mutant CPAs, but not in GNAS mutant CPAs (Table 2). Taken together, the cAMP-PKA signaling pathway is activated to a greater degree in PRKACA mutant CPAs among the various types of CPAs.
This is the first study to show the transcriptome data including entire steroidogenic enzymes for the transduction of the PRKACA mutation in HAC15 cells in vitro. Bioinformatics analysis revealed that the pathway whose activity was enhanced to the greatest degree by the PRKACA mutation was steroid biosynthesis (Table S3). The expression of steroidogenic enzymes and cortisol production were potentiated by PRKACA mutation in HAC15 cells (Figure 4 and 5). These results support prior findings that PRKACA mutations drive cortisol production in CPAs and cAMP-PKA signaling activation stimulated steroidogenic enzymes in H295R cells (Beuschlein, 2014,Cao, 2014,Di Dalmazi, 2014,Goh, 2014,Rizk-Rabin, 2020,Sato, 2014,Weigand, 2021). Calcium signaling pathway activation also induced STAR expression and cortisol production in vitro (Nishikawa, 1996), however PRKACA mutation mediated calcium signal activation was not found by our bioinformatics analysis (Table 2 and Table S3). Previous reports demonstrated that patients with PRKACA mutant CPA had higher cortisol levels and smaller adenoma size compared with those without any mutations, and higher cortisol levels compared with those without PRKACA mutation (Beuschlein, 2014,Di Dalmazi, 2014). These evidences also support our clinical results that the cortisol levels per unit area in CPAs with PRKACA mutation were higher than those with GNAS mutation.
Steroidogenic factor 1 (SF-1), also known as adrenal 4-binding protein (Ad4BP), is encoded by NR5A1, and regulates development of adrenal gland and the expression of steroidogenesis-related genes (Lala, 1992,Morohashi, 1992). According to the predicting transcription factor analysis, NR5A1 may participate the regulation of gene expressions in PRKACA mutant CPA (Table 2). NR5A1 expression was stimulated by cAMP-PKA signaling activation in Y1 cells (Kulcenty, 2015). mRNA expression of NR5A1 was correlated with CYP17A1 and CYP11B1 mRNA expression in CPA (Kubota-Nakayama, 2016). Taken together, NR5A1 would play a pivotal role in the regulation of gene expressions including steroidogenic enzymes in PRKACA mutant CPA.
Although PRKACA mutation stimulated the transcription of CYP11B1 in HAC15 cells, CYP11B1 expressions in CPAs with PRKACA and GNAS mutations was lower than that in NFAs. A previous study also showed that CYP11B1 transcription was not stimulated in CPAs (Sakuma, 2013). CYP11B1 immunoreactivity is detected in CPAs, although the expression was not compared between CPAs and NFAs (Gomez-Sanchez, 2014,Kubota-Nakayama, 2016). It has been suggested that CYP11B1 expression in PRKACA mutant adenomas is not a rate-limiting factor in the synthesis of cortisol.
Somatic mutations of CTNNB1 causes loss of β-catenin phosphorylation, and it prevents the ubiquitination of β-catenin and leads to the constitutive activation of the Wnt/β-catenin signaling pathway (Kikuchi, 2003). CTNNB1 mutations have been detected in several human cancers, and the molecular mechanisms support their role in the causation and progression of tumors (Kikuchi, 2003,Polakis, 2000). Clustering analysis using RNA sequencing has shown that CPAs with CTNNB1 mutations are similar to adrenocortical carcinomas (Di Dalmazi, 2020). It is suggested that CTNNB1 mutations induce the proliferation of zona fasciculata cells and lead to the development of CPAs.
In this study, the results obtained for the GNAS mutant CPAs following the clustering analysis of the gene set associated with responses to cAMP were similar to those obtained for the CTNNB1 mutants (Figure 1B). Previous reports regarding the intracellular molecular mechanisms underlying the role of GNAS mutations in several tumors or cancers state that these mutations activate not only the cAMP-PKA signaling pathway, but also the Wnt/β-catenin and RAS-MAPK pathways, to stimulate gene transcription (Goh, 2014,Lin, 2020). The cAMP-PKA signaling pathway may be less important in GNAS mutant CPAs than in PRKACA mutant CPAs. Both the Wnt/β-catenin and RAS-MAPK pathways were well known to stimulate cell proliferation and tumor progression (Kikuchi, 2003,Pereira, 2019,Polakis, 2000), and thus GNAS mutations as well as CTNNB1 mutations would induce the zona fasciculata cells proliferation and the development of CPAs. Further basic experiments are needed to clarify the molecular mechanisms underlying tumorigenesis and cortisol production in GNAS mutant CPAs.
A limitation of present study is small numbers of GNAS and CTNNB1 mutations, because the frequency of those mutations in CPAs is small. Although a large sample size is needed to investigate whether similar results would be obtained, the current study would be helpful to reveal gene expression profile and clinical findings in CPAs with PRKACA mutation.
In conclusion, it is suggested that the cAMP-PKA signaling pathway is activated mainly in PRKACA mutant CPAs, resulting in cortisol overproduction via the increase in the expression of FDX1 and steroidogenic enzymes. PRKACA mutations led to increased cortisol production and increased steroidogenic enzymes activities in HAC15 cells. Collectively, these findings indicate that cortisol production may be activated by the cAMP-PKA signaling pathway mainly in PRKACA mutant CPAs.
Supplementary Material
Highlights.
RNA sequencing analyses showed that PRKACA mutants were separated with others.
Cushing’s adenoma with PRKACA mutant had elevated steroidogenic enzyme expression.
Cortisol levels per unit area were high in Cushing’s adenoma with PRKACA mutant.
PRKACA mutation increased in steroidogenic enzyme expression in HAC15 cells.
PRKACA mutations led to an increase in cortisol production in HAC15 cells.
Acknowledgement
This work was partly carried out with the kind cooperation of the Analysis Center of Life Science, Hiroshima University.
Sources of Funding
This study was financially supported by JSPS KAKENHI Grant Number JP21K08557 (K.O.), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (K.O.), Takeda Science Foundation (K.O.), Suzuken Memorial Foundation (K.O.), JSPS KAKENHI Grant Number JP19K17964 (K.I.), JSPS KAKENHI Grant Number JP21K16058 (K.K.), National Heart, Lung and Blood Institute grant R01 HL144847 (C.-E.GS.), the National Institute of General Medical Sciences under Award Number 1U54GM115428 (C.-E.GS.), and the Department of Veteran Affairs I01 BX004681 (C.-E.GS.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statements
The authors have nothing to disclose.
References
- Aumo L, Rusten M, Mellgren G, Bakke M and Lewis AE, Functional roles of protein kinase A (PKA) and exchange protein directly activated by 3’,5’-cyclic adenosine 5’-monophosphate (cAMP) 2 (EPAC2) in cAMP-mediated actions in adrenocortical cells, Endocrinology. 151, 2010. 2151–2161. [DOI] [PubMed] [Google Scholar]
- Baba R, Oki K, Kobuke K, Itcho K, Okubo H, Ohno H, Yoneda M and Hattori N, Measurement of midnight ACTH levels is useful for the evaluation of midnight cortisol levels, Steroids. 140, 2018. 179–184. [DOI] [PubMed] [Google Scholar]
- Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, Schaak K, Schmittfull A, Schwarzmayr T, Barreau O, Vezzosi D, Rizk-Rabin M, Zabel U, Szarek E, Salpea P, Forlino A, Vetro A, Zuffardi O, Kisker C, Diener S, Meitinger T, Lohse MJ, Reincke M, Bertherat J, Strom TM and Allolio B, Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome, N Engl J Med. 370, 2014. 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Bathon K and Weigand I, Mechanisms of Aberrant PKA Activation by Calpha Subunit Mutations, Horm Metab Res. 49, 2017. 307–314. [DOI] [PubMed] [Google Scholar]
- Calebiro D, Hannawacker A, Lyga S, Bathon K, Zabel U, Ronchi C, Beuschlein F, Reincke M, Lorenz K, Allolio B, Kisker C, Fassnacht M and Lohse MJ, PKA catalytic subunit mutations in adrenocortical Cushing’s adenoma impair association with the regulatory subunit, Nat Commun. 5, 2014. 5680. [DOI] [PubMed] [Google Scholar]
- Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, Zhou W, Li X, Zhong X, Lei Y, Su T, Wang H, Jiang Y, Yang L, Wei W, Yang X, Jiang X, Liu L, He J, Ye J, Wei Q, Li Y, Wang W, Wang J and Ning G, Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome, Science. 344, 2014. 913–917. [DOI] [PubMed] [Google Scholar]
- Di Dalmazi G, Altieri B, Scholz C, Sbiera S, Luconi M, Waldman J, Kastelan D, Ceccato F, Chiodini I, Arnaldi G, Riester A, Osswald A, Beuschlein F, Sauer S, Fassnacht M, Appenzeller S and Ronchi CL, RNA Sequencing and Somatic Mutation Status of Adrenocortical Tumors: Novel Pathogenetic Insights, J Clin Endocrinol Metab. 105, 2020. [DOI] [PubMed] [Google Scholar]
- Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullie M, Mantero F, Rubin B, Waldmann J, Bartsch DK, Pasquali R, Lohse M, Allolio B, Fassnacht M, Beuschlein F and Reincke M, Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study, J Clin Endocrinol Metab. 99, 2014. E2093–2100. [DOI] [PubMed] [Google Scholar]
- Gao X, Yamazaki Y, Tezuka Y, Pieroni J, Ishii K, Atsumi N, Ono Y, Omata K, Morimoto R, Nakamura Y, Satoh F and Sasano H, Intratumoral heterogeneity of the tumor cells based on in situ cortisol excess in cortisol-producing adenomas; approximately An association among morphometry, genotype and cellular senescence approximately, J Steroid Biochem Mol Biol. 204, 2020. 105764. [DOI] [PubMed] [Google Scholar]
- Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kunstman JW, Korah R, Suttorp AC, Dietrich D, Haase M, Willenberg HS, Stalberg P, Hellman P, Akerstrom G, Bjorklund P, Carling T and Lifton RP, Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors, Nat Genet. 46, 2014. 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, Satoh F, Maekawa T, Nakamura Y, Sasano H and Gomez-Sanchez EP, Development of monoclonal antibodies against human CYP11B1 and CYP11B2, Mol Cell Endocrinol. 383, 2014. 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamichi Y, Mizutani T, Ju Y, Matsumura T, Kawabe S, Kanno M, Yazawa T and Miyamoto K, Transcriptional regulation of human ferredoxin 1 in ovarian granulosa cells, Mol Cell Endocrinol. 370, 2013. 1–10. [DOI] [PubMed] [Google Scholar]
- Itcho K, Oki K, Gomez-Sanchez CE, Gomez-Sanchez EP, Ohno H, Kobuke K, Nagano G, Yoshii Y, Baba R, Hattori N and Yoneda M, Endoplasmic Reticulum Chaperone Calmegin Is Upregulated in Aldosterone-Producing Adenoma and Associates With Aldosterone Production, Hypertension. 75, 2020. 492–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itcho K, Oki K, Kobuke K, Ohno H, Yoneda M and Hattori N, Angiotensin 1–7 suppresses angiotensin II mediated aldosterone production via JAK/STAT signaling inhibition, J Steroid Biochem Mol Biol. 185, 2019. 137–141. [DOI] [PubMed] [Google Scholar]
- Kikuchi A, Tumor formation by genetic mutations in the components of the Wnt signaling pathway, Cancer Sci. 94, 2003. 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto R, Oki K, Yoneda M, Gomez-Sanchez CE, Ohno H, Kobuke K, Itcho K and Kohno N, Gonadotropin-Releasing Hormone Stimulate Aldosterone Production in a Subset of Aldosterone-Producing Adenoma, Medicine (Baltimore). 95, 2016. e3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobuke K, Oki K, Gomez-Sanchez CE, Gomez-Sanchez EP, Ohno H, Itcho K, Yoshii Y, Yoneda M and Hattori N, Calneuron 1 Increased Ca(2+) in the Endoplasmic Reticulum and Aldosterone Production in Aldosterone-Producing Adenoma, Hypertension. 71, 2018. 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota-Nakayama F, Nakamura Y, Konosu-Fukaya S, Azmahani A, Ise K, Yamazaki Y, Kitawaki Y, Felizola SJ, Ono Y, Omata K, Morimoto R, Iwama N, Satoh F and Sasano H, Expression of steroidogenic enzymes and their transcription factors in cortisol-producing adrenocortical adenomas: immunohistochemical analysis and quantitative real-time polymerase chain reaction studies, Hum Pathol. 54, 2016. 165–173. [DOI] [PubMed] [Google Scholar]
- Kulcenty K, Holysz M and Trzeciak WH, SF-1 (NR5A1) expression is stimulated by the PKA pathway and is essential for the PKA-induced activation of LIPE expression in Y-1 cells, Mol Cell Biochem. 408, 2015. 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lala DS, Rice DA and Parker KL, Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu-factor I, Mol Endocrinol. 6, 1992. 1249–1258. [DOI] [PubMed] [Google Scholar]
- Li X, Wang B, Tang L, Lang B, Zhang Y, Zhang F, Chen L, Ouyang J and Zhang X, Clinical characteristics of PRKACA mutations in Chinese patients with adrenal lesions: a single-centre study, Clin Endocrinol (Oxf). 85, 2016. 954–961. [DOI] [PubMed] [Google Scholar]
- Lin YL, Ma R and Li Y, The biological basis and function of GNAS mutation in pseudomyxoma peritonei: a review, J Cancer Res Clin Oncol. 146, 2020. 2179–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morohashi K, Honda S, Inomata Y, Handa H and Omura T, A common trans-acting factor, Ad4-binding protein, to the promoters of steroidogenic P-450s, J Biol Chem. 267, 1992. 17913–17919. [PubMed] [Google Scholar]
- Muller JJ, Lapko A, Bourenkov G, Ruckpaul K and Heinemann U, Adrenodoxin reductase-adrenodoxin complex structure suggests electron transfer path in steroid biosynthesis, J Biol Chem. 276, 2001. 2786–2789. [DOI] [PubMed] [Google Scholar]
- Newell-Price J, Bertagna X, Grossman AB and Nieman LK, Cushing’s syndrome, Lancet. 367, 2006. 1605–1617. [DOI] [PubMed] [Google Scholar]
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM and Montori VM, The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline, J Clin Endocrinol Metab. 93, 2008. 1526–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki K, Yamane K, Nakanishi S, Shiwa T and Kohno N, Influence of adrenal subclinical hypercortisolism on hypertension in patients with adrenal incidentaloma, Exp Clin Endocrinol Diabetes. 120, 2012. 244–247. [DOI] [PubMed] [Google Scholar]
- Pereira SS, Monteiro MP, Costa MM, Ferreira J, Alves MG, Oliveira PF, Jarak I and Pignatelli D, MAPK/ERK pathway inhibition is a promising treatment target for adrenocortical tumors, J Cell Biochem. 120, 2019. 894–906. [DOI] [PubMed] [Google Scholar]
- Polakis P, Wnt signaling and cancer, Genes Dev. 14, 2000. 1837–1851. [PubMed] [Google Scholar]
- Rizk-Rabin M, Chaoui-Ibadioune S, Vaczlavik A, Ribes C, Polak M, Ragazzon B and Bertherat J, Link between steroidogenesis, the cell cycle, and PKA in adrenocortical tumor cells, Mol Cell Endocrinol. 500, 2020. 110636. [DOI] [PubMed] [Google Scholar]
- Sakuma I, Suematsu S, Matsuzawa Y, Saito J, Omura M, Maekawa T, Nakamura Y, Sasano H and Nishikawa T, Characterization of steroidogenic enzyme expression in aldosterone-producing adenoma: a comparison with various human adrenal tumors, Endocr J. 60, 2013. 329–336. [DOI] [PubMed] [Google Scholar]
- Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, Suzuki H, Shiozawa Y, Kataoka K, Kon A, Aoki K, Chiba K, Tanaka H, Kume H, Miyano S, Fukayama M, Nureki O, Homma Y and Ogawa S, Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome, Science. 344, 2014. 917–920. [DOI] [PubMed] [Google Scholar]
- Sheftel AD, Stehling O, Pierik AJ, Elsasser HP, Muhlenhoff U, Webert H, Hobler A, Hannemann F, Bernhardt R and Lill R, Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis, Proc Natl Acad Sci U S A. 107, 2010. 11775–11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao T, Wei MY, Guo XW, Zhang J, Yang LY and Zheng H, Modulating cAMP responsive element binding protein 1 attenuates functional and behavioural deficits in rat model of neuropathic pain, Eur Rev Med Pharmacol Sci. 23, 2019. 2602–2611. [DOI] [PubMed] [Google Scholar]
- Thiel A, Reis AC, Haase M, Goh G, Schott M, Willenberg HS and Scholl UI, PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases, Eur J Endocrinol. 172, 2015. 677–685. [DOI] [PubMed] [Google Scholar]
- Weigand I, Ronchi CL, Rizk-Rabin M, Dalmazi GD, Wild V, Bathon K, Rubin B, Calebiro D, Beuschlein F, Bertherat J, Fassnacht M and Sbiera S, Differential expression of the protein kinase A subunits in normal adrenal glands and adrenocortical adenomas, Sci Rep. 7, 2017. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand I, Ronchi CL, Vanselow JT, Bathon K, Lenz K, Herterich S, Schlosser A, Kroiss M, Fassnacht M, Calebiro D and Sbiera S, PKA Calpha subunit mutation triggers caspase-dependent RIIbeta subunit degradation via Ser(114) phosphorylation, Sci Adv. 7, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Wu L, Xie J, Su T, Jiang L, Jiang Y, Cao Y, Liu J, Ning G and Wang W, Steroidogenic Acute Regulatory Protein Overexpression Correlates with Protein Kinase A Activation in Adrenocortical Adenoma, PLoS One. 11, 2016. e0162606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.