

Abstract
Copalyl diphosphate synthase from Penicillium verruculosum (PvCPS) is a bifunctional diterpene synthase with both prenyltransferase and class II cyclase activities. The prenyltransferase α domain catalyzes the condensation of C5 dimethylallyl diphosphate with three successively added C5 isopentenyl diphosphates to form C20 geranylgeranyl diphosphate, which then undergoes a class II cyclization reaction at the βγ domain interface to generate copalyl diphosphate. The prenyltransferase α domain mediates oligomerization to form a 648-kD (αβγ)6 hexamer. In the current study, we explore prenyltransferase structure-function relationships in this oligomeric assembly-line platform with the goal of generating alternative linear isoprenoid products. Specifically, we report steady-state enzyme kinetics, product analysis, and crystal structures of various site-specific variants of the prenyltransferase α domain. Crystal structures of the H786A, F760A, S723Y, S723F, and S723T variants have been determined at resolutions of 2.8 Å, 3.10 Å, 3.15 Å, 2.65 Å, and 2.00 Å, respectively. The substitution of S723 with bulky aromatic amino acids in the S723Y and S723F variants constricts the active site, thereby directing the formation of the shorter C15 isoprenoid, farnesyl diphosphate. While the S723T substitution only subtly alters enzyme kinetics and does not compromise GGPP biosynthesis, the crystal structure of this variant reveals a nonproductive binding mode for isopentenyl diphosphate that likely accounts for substrate inhibition at high concentrations. Finally, mutagenesis of the catalytic general acid in the class II cyclase domain, D313A, significantly compromises prenyltransferase activity. This result suggests molecular communication between the prenyltransferase and cyclase domains despite their distant connection by a flexible polypeptide linker.
Graphical Abstract

Introduction
Terpenoids comprise the largest class of natural products, with more than 95,000 currently catalogued in the Dictionary of Natural Products (www.dnp.chemnetbase.com).1 Terpenoids are present in all domains of life and their incredible chemodiversity is rooted in just two 5-carbon precursors, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP).2–4 Prenyltransferases catalyze the Mg2+-dependent head-to-tail condensation of one or more molecules of IPP with DMAPP in processive chain-elongation reactions yielding increasingly longer isoprenoid diphosphates such as C10 geranyl diphosphate (GPP), C15 farnesyl diphosphate (FPP), or C20 geranylgeranyl diphosphate (GGPP). Final chain length is determined by the depth of the active site pocket.
A separate class of enzymes, the terpene cyclases, utilize linear isoprenoid diphosphates as substrates in multi-step cyclization reactions generating complex products that most often contain multiple rings and stereocenters.5–11 Many cyclic terpenoids exhibit potent anti-cancer, anti-inflammatory, and anti-microbial properties, and accordingly serve as leads in the development of novel therapeutics.12 Many terpenes are produced in small quantities in nature, so a large amount of natural source material must be harvested for terpene extraction and study. Alternatively, terpenoids can be synthesized in the laboratory, but they are often very challenging targets for synthetic organic chemistry.13 Thus, metabolic engineers, synthetic biologists, and biochemists are studying the development of systems and technologies for sustainable terpenoid biosynthesis.12–14
Prenyltransferases and terpene cyclases are most often found as two distinct proteins in bacteria, fungi, and plants.5–11 However, several bifunctional terpene synthases have been discovered in certain fungi in which the prenyltransferase and terpene cyclase are found as distinct and separate domains in a single protein.15–17 These bifunctional terpene synthases are regarded as assembly-line enzymes since they catalyze the first two consecutive steps of terpene biosynthesis starting with the universal C5 precursors DMAPP and IPP.18 Bifunctional terpene synthases exhibit either class I or class II cyclase activity and are found as oligomers with (αα)6,8 or (αβγ)6 architecture, respectively.18 A class I terpene synthase initiates prenyltransferase or cyclization activity by the Mg2+3-triggered ionization of the substrate diphosphate group,19 and a class II terpene synthase initiates cyclization by protonation of an isoprenoid C=C bond using an Asp general acid.20 The co-localization of catalytic domains in a single polypeptide chain, as well as their concentration through oligomerization, may confer a catalytic advantage due to active site proximity.21–24
Consider the bifunctional diterpene synthase, fusicoccadiene synthase, from Phomopsis amygdali (PaFS), which generates fusicoccadiene in the biosynthetic pathway leading to Fusicoccin A.15 PaFS adopts αα architecture with a C-terminal prenyltransferase that generates GGPP and an N-terminal class I cyclase that utilizes GGPP to form fusicoccadiene. Although full-length PaFS is refractory to crystallization, crystal structures of individual prenyltransferase and cyclase domains facilitate analysis of the hexameric structure formed by N333A/Q336A PaFS determined by small-angle X-ray scattering (SAXS).25 More recently, the structure of full-length wild-type PaFS determined by cryo-electron microscopy (cryo-EM) reveals a central octameric core of C-terminal prenyltransferase domains, with eight corresponding N-terminal cyclase domains splayed out in random positions; however, cyclase domains can also associate more closely with the prenyltransferase core.26 Hexamers of the full-length wild-type enzyme are also observed, with an approximate octamer:hexamer ratio of 9:1.
In contrast, the bifunctional diterpene synthase copalyl diphosphate synthase from Penicillium verruculosum (PvCPS)16 adopts αβγ domain architecture. This bifunctional enzyme catalyzes the synthesis of GGPP in its C-terminal prenyltransferase domain which is then cyclized in the N-terminal class II cyclase domain to form copalyl diphosphate (CPP) (Figure 1). The crystal structure of the PvCPS-α prenyltransferase reveals hexameric quaternary structure, and hexamerization of the prenyltransferase directs the hexamerization of the full-length enzyme as determined in SAXS experiments.27
Figure 1. Biosynthesis of copalyl diphosphate by PvCPS.

(A) The model of the PvCPS hexamer fits in the ab initio SAXS envelope with χ2 = 2.12 and exhibits a starburst-like structure, as previously reported by Ronnebaum and colleagues.27 The crystal structure of the central prenyltransferase α6 core is blue, the largely-disordered linker is red, and the class II cyclase βγ domains are green and yellow, respectively. (B) The prenyltransferase α domain catalyzes the coupling of one DMAPP and three IPP molecules to generate GGPP, which is then cyclized to form CPP in the class II cyclase active site (OPP = diphosphate). (C) Stereoview of the prenyltransferase domain (chain A, blue; chain B, cyan) of wild-type PvCPS (PDB 6V0K) showing variants presented in the current study. The contour of the active site cavity is shown; Mg2+ ions (green) and GGPP (magenta) are modeled in place based on crystal structures of related prenyltransferases45 to provide prospective.
The prenyltransferase domain of PvCPS is a type-III GGPP synthase.27 Structural and mechanistic analyses of other type-III GGPP synthases highlight the importance of residues lining the active site pocket, the depth of which determines final product chain length.28 Site-directed mutagenesis experiments show that product chain length can be altered by engineering the depth of the active site pocket.28 In essence, the active site pocket of a prenyltransferase is a “molecular ruler” that can be recontoured to confer new catalytic capabilities.28–31 For example, the S71Y mutation in the type-III GGPP synthase from Saccharomyces cerevisiae introduces a large bulky aromatic amino acid that occludes the middle portion of the active site pocket, resulting in production of the shorter C15 isoprenoid diphosphate product FPP when incubated with C10 GPP and C5 IPP.28 In this variant, the pocket is too shallow to allow for the generation of C20 GGPP. In contrast, the F108A mutation at the base of the active site pocket opens up the floor of the pocket to enable the generation of longer C30 and C35 isoprenoid diphosphate products.
Here, we present steady-state kinetics and X-ray crystal structures of PvCPS prenyltransferase domain variants H786A, F760A, S723Y, S723F, and S723T (Figure 1C) in which the active site pocket is recontoured. While the H786A and F760A variants still generate C20 GGPP, the S723Y and S723F variants establish the prenyltransferase of the PvCPS hexamer as a platform for generating the shorter C15 isoprenoid, farnesyl diphosphate (FPP). Interestingly, S723T PvCPS exhibits near-normal activity, but the crystal structure of the S723T prenyltransferase reveals a nonproductive binding mode for IPP that accounts for substrate inhibition. Finally, we present the steady-state kinetics of prenyltransferase activity for full-length D313A-PvCPS, in which the catalytically obligatory general acid in the class II cyclase domain is deleted. Even though this amino acid substitution is made in a distant domain, it influences catalytic activity in the prenyltransferase domain. This surprising result is suggestive of molecular communication between catalytic domains despite their connection through a flexible, 170-residue linker.
Materials and Methods
Reagents.
The chemicals used in buffers and crystallization conditions were purchased from Fisher Scientific, Millipore Sigma, or Hampton Research and used without further purification unless otherwise specified. Isoprenoids were purchased from Isoprenoids, LC.
Overexpression and purification of full length PvCPS variants.
Overexpression plasmids of codon-optimized mutant PvCPS genes were supplied by GenScript. PvCPS protein variants were expressed and purified as previously described.27 Briefly, BL21 (DE3) Escherichia coli containing the pvcps-variant overexpression plasmid were grown in Terrific Broth (TB) containing 50 μg/mL kanamycin at 37°C with shaking (250 rpm). Protein expression was induced when the OD600 reached ~0.6 by the addition of isopropyl β-d-thiogalactopyranoside (IPTG) to a final concentration of 500 μM. The temperature was reduced to 16°C for 24 h before cells were harvested by centrifugation (6,000 × g, 20 minutes, 4°C); cell pellets were stored at −80°C until purification. Protein purification was performed using a HisTrap™ (GE Healthcare) column for affinity gel filtration chromatography followed by size-exclusion chromatography. Enzymes were concentrated to 10 mg/mL and stored at −80°C until further use.
Preparation and purification of the prenyltransferase domain of PvCPS variants (PvCPS-α).
The preparation and purification of PvCPS-α variants with desired amino acid substitutions were previously described.27 Briefly, the specific variant of full-length PvCPS (1 mL, 10 mg/mL) was incubated with Endoproteinase Glu-C (Hampton) at a 1:1000 mg ratio for 1 h at room temperature. The mixture was loaded onto a 26/60 Superdex 200 size-exclusion column pre-equilibrated with Sup200 buffer [25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)•NaOH (pH 7.5), 150 mM NaCl, 1 mM TCEP, 10% glycerol]. The first peak containing the prenyltransferase domain, designated PvCPS-α, was collected and concentrated to 10 mg/mL, and stored at −80°C until further use. All variants were expressed and purified with similar levels of yield showing similar overall stability.
Crystallization.
Crystals of PvCPS-α variants were grown by the hanging drop vapor diffusion method in which a 1 μL drop of 4–10 mg/mL PvCPS-α protein solution was mixed with 1 μL of precipitant buffer and equilibrated against 500 μL of precipitant buffer at 21°C. Rectangular rod-shaped crystals generally appeared in 1–7 days. Crystals were equilibrated with mother liquor augmented with 20% (v/v) glycerol and flash-cooled32 in liquid nitrogen. Protein and precipitant solutions are listed in Table S1.
Crystallographic data collection and structure determination.
Diffraction data were collected remotely on beamline 17-ID-1 AMX at the National Synchrotron Light Source II (NSLS-II, Brookhaven, NY) at Brookhaven National Laboratory; on beamline 24-ID-C at the Northeastern Collaborative Access Team facility (NE-CAT) at the Advanced Photon Source at Argonne National Laboratory (Lemont, IL); and on beamline 9–2 at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA) at SLAC National Accelerator Laboratory. Data sets were indexed, integrated, and scaled using iMosflm33 and processed using AIMLESS.34
Molecular replacement solutions were determined with PHASER35 using wild-type PvCPS-α (PDB 6V0K)27 as a search probe for rotation and translation functions. For each structure determination, manual model building was achieved with COOT36 and refinement was performed with PHENIX.REFINE.37 Non-crystallographic symmetry restraints were not employed in refinement. Water molecules were iteratively placed during refinement and edited using the 2mFo-DFc electron density map contoured at 1.5σ after each round of refinement.
Atomic coordinates of non-proteogenic molecules (e.g., glycerol, IPP) were added to electron density maps during the later stages of refinement. Electron density for IPP was present in the IPP binding pocket when IPP was present in the crystallization condition. In crystallization conditions lacking IPP, electron density representative of a glycerol molecule was bound in the IPP binding pocket. C-terminal residues and some residues lacking complete electron density for side chains on the surface of the protein, likely due to disorder, were not fully modeled. Additionally, in some chains, helix J is not resolved, also likely due to disorder. Validation of final structures was performed with MolProbity.38,39 Disordered polypeptide segments and amino acid side chains not modeled in each final structure are indicated in Table S2. The Rwork values ranged from 0.175–0.210 and the Rfree values40 ranged from 0.195–0.269 for final refined structures. Data collection and refinement statistics for each structure are recorded in Table 1.
Table 1.
Crystallographic data collection and refinement statistics for PvCPS-α variants
| H786A-PvCPSα | F760A-PvCPSα | S723Y-PvCPSα | S723F-PvCPSα | S723T-PvCPSα IPP | |
|---|---|---|---|---|---|
| Unit Cell | |||||
| Space Group | P 3 2 1 | P 3 2 1 | P 3 2 1 | P 3 2 1 | P 3 2 1 |
| a, b, c (Å) | 189.9, 189.9, 56.7 | 187.7, 187.7, 56.4 | 188.5, 188.5, 56.4 | 189.3, 189.3, 56.4 | 189.8, 189.8, 56.5 |
| Data Collection | |||||
| Laboratory, beamline | NSLS-II, 17-ID-1 | SSRL, 9–2 | NE-CAT, 24-ID-E | NE-CAT, 24-ID-C | NE-CAT, 24-ID-C |
| Detector | Dectris Eiger 9M |
Dectris Pilatus 6M PAD |
Dectris EIGER 16M |
Dectris Pilatus 6M-F |
Dectris Pilatus 6M-F |
| Resolution (Å) | 2.80 | 3.10 | 3.15 | 2.65 | 2.00 |
| Total/unique no. of reflections | 281,047/39,267 | 373,438/69,339 | 143,593/24,037 | 323,109/41,686 | 762,371/45,129 |
| R merge a,b | 0.384 (1.375) | 0.805 (2.116) | 0.418 (1.240) | 0.385 (1.497) | 0.165 (1.831) |
| R pim a,c | 0.130 (0.475) | 0.194 (0.516) | 0.168 (0.519) | 0.186 (0.730) | 0.056 (0.602) |
| CC 1/2 a,d | 0.970 (0.624) | 0.924 (0.544) | 0.947 (0.509) | 0.986 (0.521) | 0.998 (0.505) |
| I/σ(I) a | 6.6 (2.1) | 6.7 (1.50) | 5.3 (2.0) | 7.6 (2.3) | 10.8 (2.0) |
| Redundancy a | 9.6 (9.3) | 17.8 (18.4) | 7.1 (6.7) | 9.5 (9.3) | 9.7 (10.1) |
| Completeness (%) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (99.9) | 99.9 (99.9) | 100.0 (100.0) |
| Refinement | |||||
| Reflections used in refinement/test set | 29,152/2,876 | 20,936/2,068 | 20,131/1,983 | 33,846/3,360 | 78,736/7,786 |
| R work a,e | 0.191 (0.264) | 0.210 (0.247) | 0.187 (0.310) | 0.190 (0.297) | 0.175 (0.313) |
| R free a,e | 0.240 (0.321) | 0.269 (0.393) | 0.235 (0.345) | 0.229 (0.331) | 0.195 (0.346) |
| Number of nonhydrogen atoms | |||||
| Protein | 4658 | 4706 | 4781 | 4830 | 4867 |
| Solvent | - | - | - | - | 533 |
| Average B factors (Å2) | |||||
| Protein | 24 | 20 | 29 | 31 | 32 |
| Solvent | - | - | - | - | 42 |
| RMS deviations from ideal geometry | |||||
| Bonds (Å) | 0.009 | 0.009 | 0.012 | 0.009 | 0.007 |
| Ramachandran plot (%) f | |||||
| Favored | 97.77 | 90.15 | 95.04 | 99.01 | 99.34 |
| Outliers | 0.34 | 0.68 | 0.17 | 0.00 | 0.17 |
| PDB Accession Codes | |||||
| PDB ID: | 7S0A | 7S09 | 7S0H | 7S0L | 7S0M |
Values in parentheses refer to data in the highest shell.
, where 〈I〉hkl is the average intensity calculated for reflection hkl from replicate measurements.
where 〈I〉hkl is the average intensity calculated for reflection hkl from replicate measurements and N is the number of reflections.
Pearson correlation coefficient between random half-datasets.
for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively. Rfree is calculated using the same expression for reflections contained in the test set held aside during refinement.
Calculated with MolProbity.
Structure superpositions and active site cavity determination.
Superpositions of PvCPS-α variant structures were calculated using MatchMaker in Chimera.41 Each structure was also aligned in PyMOL (PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC) and active site contours were generated using GetCleft from the NRGsuite plugin.42
Steady-state enzyme kinetics.
The EnzChek Pyrophosphate Assay Kit (Molecular Probes) was used to determine the steady-state kinetic parameters for prenyltransferase activity as previously described.27 Briefly, reaction mixtures contained 50 mM Tris HCl pH (7.5), 1 mM MgCl2, 0.1 mM NaN3, 200 μM 2-amino-6-mercapto-7-methylpurine ribonucleoside (MesGR), 0.002 U purine nucleoside phosphorylase, and 0.00006 U inorganic pyrophosphatase. For steady-state kinetics, a final concentration of 500 nM PvCPS variants and 1 mM DMAPP were held constant while IPP concentration ranged from 10–1000 μM. A parent mixture containing all reaction components except IPP was mixed and incubated at room temperature for 15 min. The reaction was initiated by the addition of the parent mixture (95 μL) to varied IPP concentrations (5 μL) for a 100-μL total reaction mixture. Generation of 2-amino-6-mercapto-7-methylpurine (MesG) was monitored at A360 on a Tecan Infinite M1000 multi-mode plate reader. Negative controls (without enzyme, without substrate) were carried out for each PvCPS variant and the largest rate from the negative control was subtracted as background from all other determined rates. Trials for each PvCPS variant were performed in triplicate. Initial rates were fit to the substrate inhibition enzyme kinetics equation in the Prism 9 program suite since IPP exhibited substrate inhibition.27
PvCPS variant product formation assay.
Reaction assays performed with full-length PvCPS variants and different isoprenoid substrates were analyzed using a Shimadzu HPLC. Each reaction mixture (100 μL) contained 50 mM Tris HCl pH (7.5), 1 mM MgCl2, 0.1 mM NaN3, 1 mM IPP, 1 mM DMAPP, and 1– 10 μg PvCPS variant. Reactions were left overnight at room temperature and quenched 24 h later by adding an equivalent volume (100 μL) of methanol. After centrifugation (13 000 rpm, 5 mins), 50 μL of the soluble fraction was injected onto a Nucleosil® 300 C18 analytical column (4.6 × 150 mm, 5.0 μm) pre-equilibrated in 80% 10 mM NH4HCO3 and 20% acetonitrile flowing at a rate of 1 mL/min. A linear gradient of 20–70% acetonitrile from 2.5 to 15 minutes was used in accordance with a previously determined separation method for ent-copalyl diphosphate and GGPP.43 Analytical chromatography runs were monitored at A210 and prenyltransferase or cyclase products were confirmed by internal standards or high resolution mass spectrometry: copalyl diphosphate (CPP) HRMS: [M]− 449.19 (calcd), 449.507 (found). FPP elutes between 10.75–11.25 minutes, CPP elutes between 12.25–12.75 minutes, and GGPP elutes between 13.35–13.85 minutes. Chromatograms were subtracted by a negative control comprised of reaction mixture without enzyme.
Results
Steady-state kinetics of PvCPS variants.
The prenyltransferase domains of wild-type (WT)-PvCPS catalyze consecutive condensation reactions with DMAPP and 3 successive IPP molecules to generate the C20 isoprenoid GGPP, which then transits to the class-II βγ cyclase domains to generate CPP. For each PvCPS variant, Michaelis-Menten plots are presented in Figure 2A and steady-state kinetic parameters are recorded in Table 2.
Figure 2. Steady-state kinetics and products generated by full-length PvCPS variants.
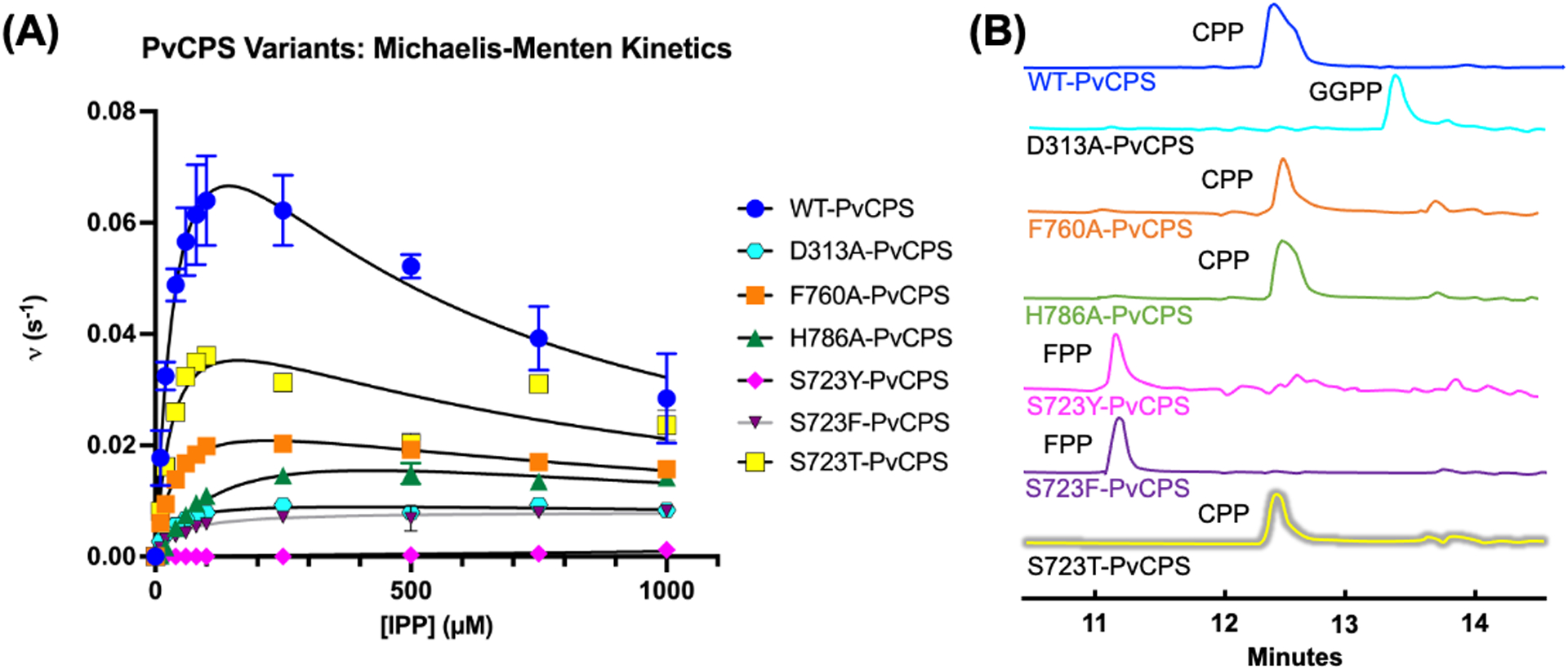
(A) Steady-state kinetics of PvCPS variants measured at [DMAPP] = 1.0 mM while varying IPP concentration. Steady-state kinetics for WT-PvCPS were previously reported by Ronnebaum and colleagues.27 Kinetic parameters for each variant are recorded in Table 2. (B) Product formation by each PvCPS variant was determined using HPLC. Each variant was incubated with 1 mM DMAPP and 1 mM IPP overnight before the reaction mixture was analyzed to determine the product formed. WT-PvCPS (blue), F760A-PvCPS (orange), H786A-PvCPS (green) and S723T-PvCPS (yellow) generated CPP as the major product, while S723Y-PvCPS (magenta) and S723F-PvCPS (violet) produced FPP. D313A-PvCPS (cyan), lacking the catalytic general acid in the cyclase domain, generated GGPP.
Table 2.
Prenyltransferase activity of PvCPS variants
| WTa | D313A | H786A | F760A | S723Yb | S723F | S723T | |
|---|---|---|---|---|---|---|---|
| kcat (s−1) | 0.11 ± 0.02 | 0.0102 ± 0.0002 | 0.027 ± 0.001 | 0.0279 ± 0.0008 | NA | 0.008 ± 0.001 | 0.051 ± 0.002 |
| KM (μM) | 50 ± 10 | 30 ± 2 | 160 ± 10 | 38 ± 1 | NA | 50 ± 10 | 36 ± 3 |
| kcat/KM (M−1s−1) | 2200 ± 200 | 340 ± 20 | 170 ± 10 | 740 ± 50 | NA | 180 ± 10 | 1400 ± 80 |
| Ki (μM) | 400 ± 40 | 5500 ± 900 | 1100 ± 100 | 1300 ± 100 | NA | NA | 700 ± 100 |
From ref. 27.
Only weakly active; steady-state kinetic parameters could not be determined.
Reaction kinetics for WT-PvCPS were previously reported by Ronnebaum and colleagues.27 As for other prenyltransferases, the steady-state kinetics of WT-PvCPS exhibit substrate inhibition at high IPP concentrations with Ki = 400 ± 40 μM. However, substrate inhibition is attenuated in site-specific variants. Additionally, each variant exhibits a lower turnover number (kcat) and lower catalytic efficiency (kcat/KM) compared to WT-PvCPS. The KM of IPP for each variant is equal to or lower than that recorded for WT-PvCPS (50 ± 10 μM), with the exception of H786A-PvCPS (160 ± 10 μM). The S723Y-PvCPS variant exhibits very low activity, thus preventing the measurement of steady-state kinetics. Surprisingly, mutation of the catalytic general acid in the class II cyclase domain, D313A-PvCPS, results in a 10-fold loss in catalytic turnover, a 1.7-fold decrease in KM, and 6.5-fold loss in overall catalytic efficiency for the prenyltransferase reaction.
Isoprenoid diphosphate products generated by PvCPS variants.
Following the production of GGPP by the prenyltransferase domain, PvCPS catalyzes a class II cyclization reaction at the βγ domain interface. Here, general acid D313 initiates the GGPP cyclization cascade leading to formation of CPP.16 WT-PvCPS generates only CPP, whereas deletion of the catalytic general acid in D313A-PvCPS yields only GGPP (Figure 2B). Alanine substitutions for F760 and H786 were expected to generate longer isoprenoid chains, which would not be utilized for catalysis by the cyclase domain. However, F760A-PvCPS and H786A-PvCPS generate only CPP, indicating that these amino acid substitutions do not alter product length achieved by the prenyltransferase domain. Mutations to S723 using larger amino acids were hypothesized to generate shorter isoprenoids such as FPP or GPP. Pleasingly, large aromatic mutations as found in S723Y-PvCPS and S723F-PvCPS result in the generation of FPP. The S723T-PvCPS variant still generates CPP, indicating that GGPP biosynthesis is not compromised by the conservative S723T substitution.
Crystal structures of PvCPS prenyltransferase α domain variants.
The crystal structure of the C-terminal prenyltransferase domain of WT-PvCPS (residues 660–961, designated PvCPS-α) was determined by limited proteolysis of the full-length enzyme using Endoproteinase GluC and reported by Ronnebaum and colleagues.27 The WT-PvCPS-α enzyme crystallizes as a hexamer with D3 symmetry, such that the hexamer can be described as a trimer of isologous dimers. The asymmetric unit consists of one dimer, and a single glycerol molecule binds in the IPP binding site of each monomer; the active site is otherwise vacant, apart from a few ordered water molecules. The overall structure of the active site is informative with regard to structure-function relationships that can redirect enzyme activity.
The active site of WT-PvCPS-α contoured using GetCleft (via the PyMOL plugin NRGsuite) indicates an active site volume of 797 Å3 (Table S3). Residues responsible for ligand binding or for defining the active site contour are highlighted in Figure 3A (a wider view is shown in Figure S1). The current study focuses on S723 at the side of the active site pocket and F760 and H786 at the base of the pocket.
Figure 3. Active site contours of PvCPS-α variants.

In each figure plate, 3 Mg2+ ions (green spheres) and the isoprenoid product are modeled to illustrate possible product binding modes in the active site pocket. GGPP (cyan) is modeled in the IPP binding site of the active site pockets of WT-PvCPS-α and the F760A, H786A, and S723T variants, whereas FPP (aquamarine) is modeled in the active site pockets of S723Y-PvCPS-α and S723F-PvCPS-α. GGPP and FPP were modeled after the GGPP binding mode observed in the complex with S. cerevisiae GGPP synthase,45 with slight adjustments made as necessary to fit in each active site pocket. WT-PvCPS-α is overlayed in plates B–F. Residues from the adjacent monomer are denoted with a prime symbol. Active site volumes are recorded in Table S3. (A) WT-PvCPS-α (blue), (B) S723Y-PvCPS-α (magenta), (C) S723F-PvCPS-α (violet), (D) S723T-PvCPS-α (yellow), (E) F760A-PvCPS-α (orange), (F) H786A-PvCPS-α (green).
Three different amino acid substitutions for S723 were made to generate a catalytically competent FPP synthase. It was hypothesized that larger residues substituted for S723 would occlude the active site pocket so as to direct the biosynthesis of shorter isoprenoid diphosphates such as FPP or GPP. The crystal structures of S723Y-PvCPS-α, S723F-PvCPS-α, and S723T-PvCPS-α were determined at resolutions of 3.15 Å, 2.65 Å, and 2.00 Å, respectively. No major structural changes were observed in comparison to WT-PvCPS-α (root-mean-square deviations (RMSDs) are recorded in Table S4).
In the crystal structure of WT-PvCPS-α, S723 donates a hydrogen bond to the backbone carbonyl of H720. In S723Y-PvCPS-α, the phenolic hydroxyl group of Y723 points away from the active site and donates a hydrogen bond to the backbone carbonyl of G817 on helix H. This results in a shallower but wider active site pocket due to compensatory structural changes (Figure 3B); the active site volume increases to 871 Å3 (Table S3). This variant directs the formation of FPP instead of GGPP, although catalytic activity is significantly compromised. In S723F-PvCPS-α, the F723 side chain packs against the wall of the active site, resulting in slight conformational changes of L785 and Y759 (Figure 3C). While the active site volume of 785 Å3 is only marginally smaller than that of WT-PvCPS-α (797 Å3) and the active site pocket is only slightly shallower than that of the wild-type enzyme, this slight constriction is sufficient to direct the formation of FPP instead of GGPP (Table 2, Figure 2). In S723T-PvCPS-α, 723 donates a hydrogen bond to the backbone carbonyl of L719 and accepts a hydrogen bond from Y759. Due to compensatory structural changes, the active site pocket appears to be shallower (Figure 3D), yet the volume of the pocket actually increases to 850 Å3 (Table S3) and GGPP synthase activity is sustained (Table 2, Figure 2).
The S723 variant side chains generally do not, themselves, directly constrict the active site. Instead, in both S723Y-PvCPS-α and S723T-PvCPS-α (Figure 3B, 3D), D727 (the first residue of the characteristic aspartate-rich metal-binding motif DDXXD, protrudes into the active site and accepts a hydrogen bond from Q789. In S723Y-PvCPS-α, Q789 also forms a hydrogen bond with Q730. The position of D727 may result from the binding of IPP in a nonproductive conformation in the IPP binding site.
Each S723 variant was co-crystallized with the co-substrate IPP. However, clear electron density for IPP is found in the IPP binding pocket only in the crystal structure of S723T-PvCPS-α. While electron density in the IPP binding pocket is present in the lower resolution crystal structures of S723Y-PvCPS-α and S723F-PvCPS-α, the density is ambiguous; thus, no ligand was modeled in these structures. Even so, it is worthwhile to examine more closely the crystal structure of S723T-PvCPS-α, which depicts IPP bound nonproductively in the IPP binding pocket (Figure 4A). The productive binding conformation of IPP has been observed in crystal structures of GGPP synthase and FPP synthase (Figures 4B–D),44,45 in which the isoprene tail is oriented toward the middle of the active site and positioned adjacent to the DMAPP analogue. Surprisingly, the diphosphate group of IPP is flipped 180° in the complex with S723T-PvCPS-α, such that the isoprene tail folds underneath the diphosphate group and is sandwiched by the backbone of the B–C loop (Figure 4E). Hydrogen bonds with the nonproductively bound IPP molecule are similar to those observed with productively bound IPP in other prenyltransferases:44,45 R691 and K688 donate hydrogen bonds to the β-phosphate and H720 and R737 donate hydrogen bonds to the β-phosphate. The backbone NH group of M690 appears to engage with the C=C bond of IPP through a dipole-π interaction.
Figure 4. Binding conformations of IPP in prenyltransferase active sites.

In plates A-D, metal ions are displayed as spheres and ligands as sticks. (A) Polder omit map (contoured at 4.0 σ) showing the nonproductive binding conformation of IPP in the active site of S723T-PvCPS-α. (B) Catalytically productive binding mode of IPP with farnesyl S-thiolodiphosphate in the active site of GGPP synthase from S. cerevisiae (marine blue, PDB 2E8T).45 (C) Catalytically productive binding mode of dimethylallyl S-thiolodiphosphate and IPP in FPP synthase from E. coli (firebrick red, PDB 1RQI).44 (D) Catalytically productive binding mode of geranyl S-thiolodiphosphate and IPP in the active site of FPP synthase from R. denitrificans (forest green, PDB 4LLS). (E) Stereoview showing intermolecular interactions of nonproductively bound IPP (cyan) in the active site of S723T-PvCPS-α (yellow).
While mutagenesis studies of S723 show that it is possible to generate a shorter isoprenoid diphosphate product by constricting the depth of the active site pocket, is it possible that longer isoprenoid diphosphates can be generated through mutagenesis intended to open up the base of the pocket? While other studies have shown that this is feasible,28 our studies with F760A-PvCPS and H786A-PvCPS show that the initial answer to this question is no, in part due to the oligomeric structure of the prenyltransferase. Residues from the opposing monomer of the dimer contribute to the active site contour and appear to stabilize the base of the active site. This structural feature makes it difficult to separate the contribution of one monomer from the other at the base of the active site pocket in the tightly-associated dimer.
The dimer interface in type-III GGPP synthases has been described as a “double floor”, in which the first floor is the base of the active site and the second floor is at the dimer interface.28 Specifically, N753’ (N753 of the opposing monomer) from helix F’ partially defines the bottom of each active site, while N756 and N756’ (from helices F and F’, respectively) largely comprise the second floor at the base of the active site pocket (Figure 3A). The side chain of H786 (from helix G) contributes to the base of the active site and makes a π-stacking interaction at the dimer interface with F760’ (from helix F’); F760’ also forms a π-stacking interaction with F760, which in turn forms a π-stacking interaction with H786’. Analysis of this π-stacking network led us to hypothesize that mutations to either H786 or F760 could deepen the active site pockets in both monomers simultaneously. However, product analysis assays conducted with the F760A and H786A variants do not reveal the generation of longer isoprenoid diphosphates, but at least GGPP generation is preserved based on the observation of the GGPP cyclization product CPP (Figure 2B). X-ray crystal structures reveal the consequences of the F760A and H786A substitutions.
The crystal structure of F760A-PvCPS-α is determined at 3.10 Å resolution and is structurally quite similar to WT-PvCPS-α (root-mean-square deviations between all pairs of structures are recorded in Table S4). The conformations of residues forming the active site contour are largely similar, although rotamer differences are evident for L785 and Y759 (Figure 3E). The F760A substitution slightly increases the active site volume from 797 Å3 to 832 Å3 (Table S3), but this is insufficient for the generation of longer isoprenoid diphosphate products based on the product analysis assay (Figure 2B).
The 2.80 Å resolution crystal structure of H786A-PvCPS-α is overall quite similar to that of WT-PvCPS-α (Table S4). The H786A substitution increases the active site volume from 797 Å3 to 892 Å3 (Table S3). However, this volume increase does not enable the generation of longer isoprenoid diphosphate products based on the product analysis assay (Figure 2B). Interestingly, the compensatory plasticity of the protein scaffolding allows for neighboring residues to shift to partially fill the void created by the H786A substitution (Figure 3F). Consequently, although the active site volume increases, its depth remains about the same. Thus, H786A-PvCPS-α remains a GGPP synthase, such that full-length H786A-PvCPS exclusively yields the cyclization product CPP (Figure 2B).
Discussion
The vast chemodiversity of terpenoid natural products is rooted in the first two steps of their biosynthesis catalyzed by prenyltransferases and then terpene cyclases. Genome mining experiments have led to the discovery of bifunctional terpene synthases in which these two activities are covalently linked in a single polypeptide chain.15–17, 46 Only recently have we begun to understand structure-function relationships in these catalytic assembly lines with regard to oligomerization and substrate channeling between active sites.25–27,47,48 Assembly-line terpene synthases adopt (αα)6 or (αα)8 architecture with class I cyclase activity, or (αβγ)6 architecture with class II cyclase activity.25–27 Importantly, two key functional properties have been observed in these bifunctional systems. First, as observed for PaFS, when incubated with DMAPP and IPP, most of the GGPP remains on the enzyme for cyclization;48 this feature remains to be demonstrated for other assembly-line terpene synthases. Second, when individual catalytic domains are expressed and purified separately, they retain functionality.15, 16, 25 These properties point toward new possibilities for engineering oligomeric assembly-line terpene synthases in which alternative prenyltransferase activities can be mixed and matched with different cyclase activities to generate new terpene products.
Protein engineering experiments demonstrate that a prenyltransferase can be covalently linked with a terpene cyclase to yield a two-fold advantage in cyclic terpene generation.49,50 The likely explanation for enhanced product generation may be rooted in the proximity of the prenyltransferase to the covalently linked cyclase. While the two-fold enhancement of product flux may appear modest, it is important to note that substrate channeling of any sort is desirable not necessarily to achieve kinetic superiority, but instead to achieve more efficient carbon management.51 In systems such as PaFS or PvCPS, it is possible that dynamic cluster channeling is operative, involving an agglomeration of multiple enzymes in a cluster that undergoes constant structural fluctuations.51,52
A strategy for product chain length determination in a type-III GGPP synthase from S. cerevisiae has been established by Chang and colleagues.28 Some elements of this strategy are transferable to PvCPS, and some elements are not. For example, a strategy that is effective with PvCPS is the substitution of active site residue S723 to convert the GGPP synthase into an FPP synthase. A naturally occurring assembly-line FPP synthase-cyclase has not yet been discovered in nature, but we show that the prenyltransferase domain of the PvCPS hexamer can be engineered to generate FPP with reasonable catalytic efficiency.
The conservative S723T substitution does not block GGPP biosynthesis and sustains near-normal catalytic activity (Figure 2, Table 2). Even so, the crystal structure of S723T-PvCPS-α (Figure 4A, 4E) yields important new information, namely, the nonproductive binding mode of IPP that likely accounts for the observed substrate inhibition (Figure 2A). At high IPP concentrations, prenyltransferases often exhibit substrate inhibition, and previous studies suggest that substrate inhibition by IPP arises from competitive binding to the homoallylic substrate region of the active site.53 Here, we observe that IPP can bind with a flipped orientation, thereby providing a plausible visualization for substrate inhibition.
A strategy developed with S. cerevisiae GGPP synthase that is ineffective with PvCPS is the substitution of residues at the base of the active site pocket with smaller amino acids to enable the generation of longer C25, C30, C35, and C40 isoprenoid diphosphate products. However, longer isoprenoid diphosphates are not generated by H786A-PvCPS or F760A-PvCPS (Figure 2B). Instead, these variants retain GGPP synthase activity, ultimately yielding CPP. Crystal structures of these variants reveal larger active site volumes, but the active site is only marginally deepened due in part to compensatory structural changes in the protein scaffolding (Figures 3D and 3F; Table S4). Such changes are not always anticipated in molecular modeling experiments, which highlights the importance of experimental structure determinations of site-specific variants to ascertain whether unanticipated structural changes are triggered by amino acid substitutions.
A question that remains unanswered in the study of bifunctional assembly-line terpene synthases is the degree to which one active site communicates with the other. While the two catalytic domains are typically connected by a flexible, disordered polypeptide linker, it is curious that the D313A substitution for the catalytic general acid in the class II cyclase domain not only obliterates cyclase activity, but also perturbs catalysis in the prenyltransferase domain (Figure 2, Table 2). This suggests the possibility of catalytic crosstalk between the prenyltransferase and cyclase domains despite the dynamic positioning of the cyclase domains surrounding the hexameric prenyltransferase core. The molecular envelope of full-length PvCPS derived from SAXS measurements (Figure 1A) suggests that the linker connecting the prenyltransferase and cyclase domains adopts a more compact globule-like conformation, which is consistent with a prediction based on amino acid sequence analysis.18 Since an active site mutation in the cyclase domain influences catalytic activity in the prenyltransferase domain, this implies that molecular communication between active sites, presumably transmitted by conformational changes, occurs in part through the flexible globule-like linker.
In conclusion, we have shown that the hexameric assembly-line diterpene synthase PvCPS can be engineered to be a sesquiterpene synthase, generating the linear isoprenoid diphosphate FPP instead of GGPP. While FPP is not utilized as a substrate by the diterpene cyclase domain of PvCPS, we envisage that the diterpene cyclase domain could be swapped with a sesquiterpene cyclase domain of choice to yield a bifunctional assembly-line sesquiterpene synthase. The structure of S723F-PvCPS-α is essentially that of the first hexameric FPP synthase; insofar that hexamerization of the prenyltransferase may enable substrate channeling between the prenyltransferase and the cyclase in an assembly-line synthase, PvCPS may serve as a platform in which desired prenyltransferase and cyclase activities can be introduced by mutagenesis to take advantage of enhanced efficiencies of carbon management. Future studies in this regard will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on beamline 24-ID-C is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This work is also based on research conducted at beamline 17-ID-1 (AMX) of the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health, NIGMS, through a Center Core P30 Grant (P30GM133893) and by the DOE Office of Biological and Environmental Research (KP1605010). Finally, this work is also based on research performed at beamline 9-2 of the Stanford Synchrotron Radiation Lightsource. SLAC National Accelerator Laboratory is supported by the DOE Office of Science by Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The Stanford Synchrotron Radiation Lightsource is supported by the US DOE of Biological and Environmental Research and by the National Institutes of Health, NIGMS, through NIH Grant (P41GM103393). Molecular analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. We would also like to thank the Biological Chemistry Resource Center at Penn Chemistry for use of the HPLC instrument and Tecan Plate Reader.
Funding
We thank the NIH for grant GM56838 to D.W.C. in support of this research. T.A.R. was supported by NIH postdoctoral fellowship F32-GM137461. S.A.E was supported by the Structural Biology and Molecular Biophysics NIH Training Grant T32 GM132039-03.
Footnotes
Accession Codes
The atomic coordinates and crystallographic structure factors of PvCPSα variants have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes as follows: 7S09, 7S0A, 7S0H, 7S0L, 7S0M.
The authors declare no competing financial interests.
References
- [1].Buckingham J (1993) Dictionary of Natural Products, Chapman & Hall, London, 8584 pp. [Google Scholar]
- [2].Ruzicka L (1953) The isoprene rule and the biogenesis of terpenic compounds. Experientia 9, 357–367. [DOI] [PubMed] [Google Scholar]
- [3].Sacchettini JC, and Poulter CD (1997) Creating isoprenoid diversity. Science 277, 1788–1789. [DOI] [PubMed] [Google Scholar]
- [4].Christianson DW (2007) Roots of biosynthetic diversity. Science 316, 60–61. [DOI] [PubMed] [Google Scholar]
- [5].Christianson DW (2006) Structural biology and chemistry of the terpenoid cyclases. Chem. Rev 106, 3412–3442. [DOI] [PubMed] [Google Scholar]
- [6].Christianson DW (2017) Structural and chemical biology of terpenoid cyclases. Chem. Rev 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pichersky E, Noel JP, and Dudareva N (2006) Biosynthesis of plant volatiles: nature’s diversity and ingenuity. Science 311, 808–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tholl D (2006) Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr. Opin. Plant. Biol 9, 297–304. [DOI] [PubMed] [Google Scholar]
- [9].Degenhardt J, Köllner TG, and Gershenzon J (2009) Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochem 70, 1621–1637. [DOI] [PubMed] [Google Scholar]
- [10].Cane DE, and Ikeda H (2012) Exploration and mining of the bacterial terpenome. Acc. Chem. Res 45, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Quin MB, Flynn CM, and Schmidt-Dannert C (2014) Traversing the fungal terpenome. Nat. Prod. Rep 31, 1449–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ajikumar PK, Tyo K, Carlsen S, Mucha O, Phon TH, and Stephanopoulos G (2008) Terpenoids: opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol. Pharm 5, 167–190. [DOI] [PubMed] [Google Scholar]
- [13].Jansen DJ, and Shenvi RA (2014) Synthesis of medicinally relevant terpenes: reducing the cost and time of drug discovery. Future Med. Chem 6, 1127–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Z, and Hui C (2021) Contemporary advancements in the semi-synthesis of bioactive terpenoids and steroids. Org. Biomol. Chem 19, 3791–3812. [DOI] [PubMed] [Google Scholar]
- [15].Toyomasu T, Tsukahara M, Kaneko A, Niida R, Mitsuhashi W, Dairi T, Kato N, and Sassa T (2007) Fusicoccins are biosynthesized by an unusual chimera diterpene synthase in fungi. Proc. Natl. Acad. Sci. USA 104, 3084–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mitsuhashi T, Okada M, and Abe I (2017) Identification of chimeric αβγ diterpene synthases possessing both type II terpene cyclase and prenyltransferase activities. Chembiochem 18, 2104–2109. [DOI] [PubMed] [Google Scholar]
- [17].Mitsuhashi T, and Abe I (2018) Chimeric terpene synthases possessing both terpene cyclization and prenyltransfer activities. ChemBioChem 19, 1106–1114. [DOI] [PubMed] [Google Scholar]
- [18].Faylo JL, Ronnebaum TA, and Christianson DW (2021) Assembly-line catalysis in bifunctional terpene synthases, Acc. Chem. Res, in press. DOI: 10.1021/acs.accounts.1c00296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aaron JA, and Christianson DW (2010) Trinuclear metal clusters in catalysis by terpenoid synthases. Pure Appl. Chem 82, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wendt KU (2005) Enzyme mechanisms for triterpene cyclization: new pieces of the puzzle. Angew. Chem. Int. Ed 44, 3966–3971. [DOI] [PubMed] [Google Scholar]
- [21].Srere PA (1987) Complexes of sequential metabolic enzyme. Annu. Rev. Biochem 56, 89–124. [DOI] [PubMed] [Google Scholar]
- [22].Miles EW, Rhee S, and Davies DR (1999) The molecular basis of substrate channeling. J. Biol. Chem 274, 12193–12196. [DOI] [PubMed] [Google Scholar]
- [23].Huang X, Holden HM, and Raushel FM (2001) Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem 70, 149–180. [DOI] [PubMed] [Google Scholar]
- [24].Wheeldon I, Minteer SD, Banta S, Barton SC, Atanassov P, and Sigman M (2016) Substrate channelling as an approach to cascade reactions. Nat. Chem 8, 299–309. [DOI] [PubMed] [Google Scholar]
- [25].Chen M, Chou W, Toyomasu T, Cane D, and Christianson D (2016) Structure and function of fusicoccadiene synthase, a hexameric bifunctional diterpene synthase. ACS Chem. Biol 11, 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Faylo JL, van Eeuwen T, Kim HJ, Gorbea Colón JJ, Garcia BA, Murakami K, and Christianson DW (2021) Structural insight on assembly-line catalysis in terpene biosynthesis. Nat. Commun 12, 3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ronnebaum TA, Gupta K, and Christianson DW (2020) Higher-order oligomerization of a chimeric αβγ bifunctional diterpene synthase with prenyltransferase and class II cyclase activities is concentration-dependent. J. Struct. Biol 210, 107463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chang TH, Guo RT, Ko TP, Wang AHJ, and Liang PH (2006) Crystal structure of type-III geranylgeranyl pyrophosphate synthase from Saccharomyces cerevisiae and the mechanism of product chain length determination. J. Biol. Chem 281, 14991–15000. [DOI] [PubMed] [Google Scholar]
- [29].Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, and Poulter CD (1996) Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl. Acad. Sci. USA 93, 15018–15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ohnuma S. i., Narita K, Nakazawa T, Ishida C, Takeuchi Y, Ohto C, and Nishino T (1996) A role of the amino acid residue located on the fifth position before the first aspartate-rich motif of farnesyl diphosphate synthase on determination of the final product. J. Biol. Chem 271, 30748–30754. [DOI] [PubMed] [Google Scholar]
- [31].Ohnuma SI, Nakazawa T, Hemmi H, Hallberg AM, Koyama T, Ogura K, and Nishino T (1996) Conversion from farnesyl diphosphate synthase to geranylgeranyl diphosphate synthase by random chemical mutagenesis. J. Biol. Chem 271, 10087–10095. [DOI] [PubMed] [Google Scholar]
- [32].Teng TY, and Moffat K (1998) Cooling rates during flash cooling. J. Appl. Cryst 31, 252–257. [Google Scholar]
- [33].Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Evans PR, and Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr D69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB, Snoeyink J, Adams PD, Lovell SC, Richardson JS, and Richardson DC (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brünger AT (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- [41].Meng EC, Pettersen EF, Couch GS, Huang CC, and Ferrin TE (2006) Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 7, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gaudreault F, Morency LP, and Najmanovich RJ (2015) NRGsuite: a PyMOL plugin to perform docking simulations in real time using FlexAID. Bioinformatics 31, 3856–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rudolf JD, Dong LB, Cao H, Hatzos-Skintges C, Osipiuk J, Endres M, Chang CY, Ma M, Babnigg G, Joachimiak A, Phillips GN, and Shen B (2016) Structure of the ent-copalyl diphosphate synthase PtmT2 from Streptomyces platensis CB00739, a bacterial type II diterpene synthase. J. Am. Chem. Soc 138, 10905–10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, and Finn J (2004) Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J. Biol. Chem 279, 8526–8529. [DOI] [PubMed] [Google Scholar]
- [45].Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Jeng WY, Chen CK, Zhang Y, Song Y, Kuo CJ, Yin F, Oldfield E, and Wang AH (2007) Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc. Natl. Acad. Sci. USA 104, 10022–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Qin B, Matsuda Y, Mori T, Okada M, Quan Z, Mitsuhashi T, Wakimoto T, and Abe I (2016) An unusual chimeric diterpene synthase from Emericella variecolor and its functional conversion into a sesterterpene synthase by domain swapping. Angew. Chem. Int. Ed 55, 1658–1661. [DOI] [PubMed] [Google Scholar]
- [47].Chen M, Harris GG, Pemberton TA, and Christianson DW (2016) Multi-domain terpenoid cyclase architecture and prospects for proximity in bifunctional catalysis. Curr. Opin. Struct. Biol 41, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pemberton TA, Chen M, Harris GG, Chou WK, Duan L, Köksal M, Genshaft AS, Cane DE, and Christianson DW (2017) Exploring the influence of domain architecture on the catalytic function of diterpene synthases. Biochemistry 56, 2010–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brodelius M, Lundgren A, Mercke P, and Brodelius PE (2002) Fusion of farnesyldiphosphate synthase and epi-aristolochene synthase, a sesquiterpene cyclase involved in capsidiol biosynthesis in Nicotiana tabacum. Eur. J. Biochem 269, 3570–3577. [DOI] [PubMed] [Google Scholar]
- [50].Camagna M, Grundmann A, Bär C, Koschmieder J, Beyer P, and Welsch R (2019) Enzyme fusion removes competition for geranylgeranyl diphosphate in carotenogenesis. Plant Physiol 179, 1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sweetlove LJ, and Fernie AR (2018) The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun 9, 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Castellana M, Wilson MZ, Xu Y, Joshi P, Cristea IM, Rabinowitz JD, Gitai Z, and Wingreen NS (2014) Enzyme clustering accelerates processing of intermediates through metabolic channeling Nature Biotechnol 32, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Laskovics FM, Krafcik JM, and Poulter CD (1979) Prenyltransferase. Kinetic studies of the 1’-4 coupling reaction with avian liver enzyme. J. Biol. Chem 254, 9458–9463. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.