

Abstract
Programmed cell death ligand 1 (PD-L1)/programmed cell death protein 1 (PD-1) cascade is an effective therapeutic target for immune checkpoint blockade (ICB) therapy. Targeting PD-L1/PD-1 axis by small-molecule drug is an attractive approach to enhance antitumor immunity. Using flow cytometry-based assay, we identify tubeimoside-1 (TBM-1) as a promising antitumor immune modulator that negatively regulates PD-L1 level. TBM-1 disrupts PD-1/PD-L1 interaction and enhances the cytotoxicity of T cells toward cancer cells through decreasing the abundance of PD-L1. Furthermore, TBM-1 exerts its antitumor effect in mice bearing Lewis lung carcinoma (LLC) and B16 melanoma tumor xenograft via activating tumor-infiltrating T-cell immunity. Mechanistically, TBM-1 triggers PD-L1 lysosomal degradation in a TFEB-dependent, autophagy-independent pathway. TBM-1 selectively binds to the mammalian target of rapamycin (mTOR) kinase and suppresses the activation of mTORC1, leading to the nuclear translocation of TFEB and lysosome biogenesis. Moreover, the combination of TBM-1 and anti-CTLA-4 effectively enhances antitumor T-cell immunity and reduces immunosuppressive infiltration of myeloid-derived suppressor cells (MDSCs) and regulatory T (Treg) cells. Our findings reveal a previously unrecognized antitumor mechanism of TBM-1 and represent an alternative ICB therapeutic strategy to enhance the efficacy of cancer immunotherapy.
KEY WORDS: PD-L1, Immune checkpoint blockade, Transcription factor EB, Lysosome, mTOR
Abbreviations: Baf, bafilomycin A1; CETSA, cellular thermal shift assay; CHX, cycloheximide; CQ, chloroquine; 4EBP1, eIF4E-binding protein 1; IB, immunoblotting; ICB, immune checkpoint blockade; IHC, immunohistochemistry; LLC, Lewis lung carcinoma; MDSCs, myeloid-derived suppressor cells; mTOR, mammalian target of rapamycin; NAG, β-N-acetylglucosaminidase; NSCLC, non-small cell lung cancer; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand- 1; p70S6K, phosphorylation of p70 S6 kinase; qRT-PCR, quantitative real-time polymerase chain reaction; SPR, surface plasmon resonance; TBM-1, tubeimoside-1; TFEB, nuclear transcriptional factor EB; TILs, tumor-infiltrating lymphocytes; Tregs, regulatory T-lymphocytes
Graphical abstract
This study reveals a previously unrecognized antitumor mechanism of tubeimoside-1 and represents an alternative immune checkpoint blockade therapeutic strategy to enhance the efficacy of cancer immunotherapy.
1. Introduction
Immune checkpoint blockade (ICB) therapy targeting programmed cell death protein 1 (PD-1) and programmed cell death ligand 1 (PD-L1) has provided clinical benefit in multiple cancer types involving non-small cell lung cancer (NSCLC), melanoma, and renal cell carcinoma1. As an immune checkpoint molecule, PD-L1 is frequently highly expressed on multiple cancers, protecting cancer cells from T cell-mediated immune surveillance2,3. PD-L1 on cancer cells binds to the T-cell-expressed PD-1 thereby suppressing the T cell receptor (TCR) pathway and inhibiting their effector responses, which is a key mechanism for tumor cells to evade immune surveillance4. Blocking the PD-1/PD-L1 axis has attracted considerable attention as a potential target for cancer immunotherapy. Unfortunately, a lot of patients do not obtain long-term clinical benefits from ICB5. Antibody-based drugs may efficiently disrupt the function of PD-L1 on the tumor cell surface, however, the compensatory upregulation of PD-L1 and its redistribution from the endoplasmic reticulum, Golgi apparatus, and endosomes to the cell membrane6,7 may counteract the efficacy of antibody-based drugs. Therefore, it is necessary to explore new combinatorial strategies to enhance the PD-1 or PD-L1 immunotherapies.
Acting as a degradative and recycling acidic organelle in the cytosol, lysosomes play essential roles in maintaining cellular function such as energy metabolism, plasma membrane repair, and cell secretion8. Recent studies indicated that lysosomes control the fate of PD-L1. The CKLF-like MARVEL transmembrane domain containing protein 6 (CMTM6) stabilizes PD-L1 via lysosome-dependent proteolysis6. Huntingtin-interacting protein 1-related (HIP1R) binds with PD-L1 and targets it to lysosomal degradation to alter T cell-mediated cytotoxicity9. Moreover, our recent research also demonstrated that PD-L1 undergoes MITF-dependent lysosomal degradation10. Therefore, extending our knowledge of lysosomal degradation of checkpoint proteins would help develop new approaches for enhancing PD-L1 immunotherapies.
The main regulator of lysosomal function is the nuclear transcriptional factor EB (TFEB)11. The cellular localization and activity of TFEB are primarily regulated by the mammalian target of rapamycin (mTOR). mTOR is an evolutionarily conserved serine/threonine kinase and functions as a key regulator of cellular metabolism and growth in two complexes: mTOR complex 1 (mTORC1) and mTORC212. Under nutrient-rich condition, the active mTORC1 directly phosphorylates TFEB at the lysosomal membrane and sequesters TFEB in the cytosol11. Upon starvation or lysosomal stress, mTORC1 becomes inactive, dephosphorylated TFEB then translocates to nuclei and promotes the transcription of its target genes11. Moreover, mTORC1 increases the expression of PD-L1, thereby protecting cancer cells from being killed by immune cells13. Genetic or pharmacological abrogation of mTOR signaling also induces cytotoxic CD8+ T lymphocytes infiltration due to decreased secretion of inhibitory cytokines by cancer cells14, switches tumor-associated macrophages (TAMs) to a more pro-inflammatory M1 profile by suppressing the ACLY expression and regulation of histone acetylation15, and restrains myeloid-derived suppressor cells (MDSCs) accumulation via reducing G-CSF level16, thereby affecting tumor immunity17. Since mTOR signaling acts a pivotal part in promoting carcinogenesis and modulating tumor microenvironment18, it is possible to speculate that some mTOR inhibitors may play antitumor role through immune modulation.
Agents derived from natural products are the best sources for novel drug discovery19. Tubeimoside-1 (TBM-1) is the active ingredient of natural herb Bolbostemma paniculatum (Maxim) Franquet (Cucurbitaceae)20. It has been proved that TBM-1 could suppress the growth of various tumors and increase the responsiveness of tumors to chemotherapeutic agents21. The majority of these researches have concentrated on TBM-1's direct toxicity on tumor cells. Nevertheless, it remains unclear whether TBM-1 may act as an immunomodulator to mediate its antitumor effect. In this study, we carried out flow cytometry-based screening to identify small molecules that can reduce PD-L1 abundance in cancer cells. We provided evidences that TBM-1 is one of the most promising agents in decreasing the abundance of PD-L1 in cancer cells and enhancing antitumor immunity in tumor xenografts mice. Our findings show that TBM-1 induces nuclear translocation of TFEB by directly binding to and inactivating mTOR, therefore triggers lysosomal degradation of PD-L1.
2. Materials and methods
2.1. Cell culture
Human NSCLC cell lines A549, H157, H1299, H460, melanoma cell lines A375, A2058, and mouse Lewis lung carcinoma (LLC) cells, and B16 melanoma cells were obtained from Institute of Basic Medicine, Chinese Academy of Medical Sciences (Beijing, China). A375 cells were cultured in DMEM medium, A549 cells were cultured in DMEM/F-12 medium, and all the other cells were cultured in RPMI1640 medium (Gibco, Pittsburgh, PA, USA). The culture medium was supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were cultivated in a humidified atmosphere with 5% carbon dioxide at 37 °C. DAPI staining was used to confirm that no mycoplasma was contaminated in all cell lines. All study-used cell lines’ passages were limited in twenty after receipt or resuscitation.
2.2. Antibodies, chemicals and plasmids
Natural product chemical monomers (Supporting Information Tables S1 and S2) were purchased from Shanghai Standard Technology Co., Ltd. (Shanghai, China). The antibodies used in this study are listed in Supporting Information Tables S3 and S4. Cycloheximide (CHX), MG132, bafilomycin A1 (Baf), chloroquine (CQ), and Hochest33342 were products of Sigma–Aldrich (St. Louis, MO, USA). The plasmid GFP tagged-PD-L1 (GFP-PD-L1) was described as before10. The pEGFP-MITF (#38131) and pEGFP-TFEB (#38119) plasmids were obtained from Addgene (Watertown, MA, USA). Human and mouse mTOR CRISPR/Cas9 double nickase plasmids obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
2.3. Flow cytometry-based screening of PD-L1 modulator and immunoblotting
5 × 105 H157 cells were seeded in the 6-well plates overnight. A set of 282 natural compounds (Table S1) were added to the culture medium at concentration of 5 μmol/L. After 36 h, cells were harvested and labeled with PE-conjugated anti-human PD-L1 or APC-conjugated anti-human MHC-I antibody (1:100) for 30 min at 4 °C. Fluorescence intensity was quantified by Novocyte flow cytometer with Novo Express software (Agilent, San Diego, CA, USA). The cytotoxicity effect of hit compounds that reduced the PD-L1 abundance more than 50% but did not affect the MHC-I level on H157 cells were determined by the xCELLigence system (ACEA Biosciences, San Diego, CA, USA) with RTCA Software10. Cell index values were measured by continuous impedance recordings every 15 min. Immunoblotting was conducted as described previously10.
2.4. PD-L1/PD-1 interaction assay and T cell-mediated tumor cell-killing assay
To measure the PD-1 and PD-L1 protein interaction in vitro, cells were seeded on 6-well plates and then incubated with human recombinant PD-1 Fc protein and anti-human Alexa Fluor 488 dye conjugated22. DAPI was used to stain the cell nuclei. The green fluorescence signal was captured by a Zeiss Axio Vert A1 microscope (Carl Zeiss, Thornwood, NY, USA) or quantified by Novocyte flow cytometer with Novo Express software (Agilent). Human peripheral blood mononuclear cells (PBMC) were obtained from StemEry Biotech (Fuzhou, China). PBMC-mediated tumor cell-killing assay was performed by the xCELLigence system (Agilent). Briefly, each well of E-plate 16 was added 50 μL of full medium for 30 min firstly. Additional 50 μL medium containing of 5 × 103 H157 or A375 cells was added in plate and the final volume reached 100 μL. Each treatment was conducted two times. After 24 h treatment with the indicated conditions, PBMC cells (activated by 100 ng/mL anti-CD3, 100 ng/mL anti-CD28 and 10 ng/mL IL-2) were incubated with tumor cells at the ratio of 10:1. The measurements for cell index values were performed by continuous impedance recordings every 10 min. The results were analyzed simultaneously by xCELLigence system (Agilent) with RTCA Software.
2.5. PD-L1/PD-1 blockade assay
Functional alterations in PD-1/PD-L1 interactions with TBM-1 modulation were inspected with the Promega J1250 PD-L1/PD-1 blockade assay kit (Promega, Madison, WI, USA)22. TBM-1 (0–5 μmol/L) was pretreatment with PD-L1-expressing H157 or A375 cells (1 × 104) for 16 h. The following day, the medium which contains drugs were removed, and each of the treated wells was supplement with 1 × 104 Jurkat T cells (stably transfected with NFAT-luciferase reporter and human PD-1). The NFAT-luciferase was activated through co-culturing of foresaid cells via TCR and MHC interaction. The NFAT-luciferase was downregulated by the interaction between PD-L1 and PD-1. Thus, the extent of decrease in luminescence is proportional to expression degree of PD-L1. Six hours later, each well was added with Bio-Glo reagent (Promega). After 5-min incubation, the wells were measured by a LB942 multimode microplate reader (Berthold, Bad Wildbad, Germany) and the data were analyzed by ICE software.
2.6. Animal experiments
All procedures with C57BL/6 mice and BALB/c-nu/nu nude mice (6- to 8-week-old females, Beijing Vital River Laboratory, Beijing, China) were conducted under guidelines approved by the animal ethics committee of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences (approval ethic number IMB-20180620D3, IMB-20181018D3 and IMB-20190416D3). C57BL/6 mice inoculated subcutaneously with LLC or B16 melanoma cells (5 × 106 cells/mice) in the right flank were intraperitoneally administered with 0, 1, 2, 4, 8 and 16 mg/kg of TBM-1 when the average tumor volume reached approximately 50 mm3 (n = 5). During the 14 days treatment, 2 and 3 mice died in the 8 and 16 mg/kg group, respectively, while 1 mg/kg of TBM-1 showed little antitumor effect. Therefore, 1, 2 and 4 mg/kg of TBM-1 were used in our animal study. When the average tumor volume reached round about 50 mm3, mice were divided randomly in each group and intraperitoneally treated with daily dose of 1, 2, 4 mg/kg TBM-1 and vehicle (PBS) for 14 days. For antibodies treatment in mice, on Days 6 and 12 after tumor cell inoculation, 50 μg of CTLA-4 antibody (Bio X cell, Lebanon, NH, USA), or control hamster IgG (Bio X cell) was injected intraperitoneally into mice. Body weight and tumor volume were measured every 2 days. The formula, π/6 × length × width2, was used to calculate tumor volume. The mice were sacrificed on Day 18, and the blood, major organs (Heart, liver, spleen, lung, and kidney) samples were collected to measure the drug safety. Serum samples were tested by automated biochemistry analyzer SYNCHRON CX4 PRO (Beckman Coulter, Brea, CA, USA). The separated organs (Heart, liver, spleen, lung, and kidney) were fixed in 4% paraformaldehyde, then the samples were captured by Axio Vert A1 microscope (Carl Zeiss) after staining with haematoxylin and eosin (H&E)22.
2.7. Tumor-infiltrating lymphocyte isolation and T cell profile analysis
Tumors tissues in mice treated with vehicle or TBM-1 (2 mg/kg) were collected and divided into small pieces in culture medium. Then type 4 collagenase (1 mg/mL, Sigma–Aldrich) and DNase I (0.1 mg/mL, Sigma–Aldrich) were used to digest tissues’ pieces to signal cells for 1 h at 37 °C. After blocking with anti-CD16/CD32 antibodies, suspension cells were stained with fixable viability dye for 15 min at 4 °C. Immunological surface marker antibodies CD45, CD3, CD8, CD69, CD137, CD25, FOXP3, CD11b, and Gr-1 were used to stained cells for half an hour at 4 °C. After that, cells were fixed and permeabilized after the stimulation with Cell Stimulation Cocktail (eBioscience, San Diego, CA, USA) at incubator for 6 h, and labeled with anti-mouse interferon-gamma (IFN-γ) or granzyme B (GzmB) for half an hour at 4 °C. Cells were washed three times with cell staining buffer (Biolegend, San Diego, CA, USA) and quantitatively analyzed by Cytoflex flow cytometer with CytExpert software (Beckman Coulter).
2.8. Small interfering RNA knockdown and generation of mTOR knock out cells
For siRNA-mediated silencing, cells were transfected with 100 nmol/L of target siRNA against TFEB, TFE3, or MITF and a control siRNA (Table S5) using Vigofect (Vigorous Biotechnology, Beijing, China) according to the manufacturer's recommendations. Forty-eight hours post-transfection, immunoblotting was used to analyze the protein expression. For mTOR knockout, H157, A375 or LLC, B16 cells were transfected with the human or mouse mTOR double nickase plasmid (Santa Cruz Biotechnology) using Lipofectamin transfection reagent. After 24 h of transfection, cells were expanded as single clones with 2–4 μg/mL puromycin (InvivoGen, San Diego, CA, USA).
2.9. Quantitative real-time PCR (qRT-PCR)
Quantitative real-time PCR was conducted as previously described10,23. The primers used are listed in Table S6.
2.10. Immunohistochemical staining
Immunohistochemical staining was conducted as described previously10. Briefly, tumor tissues were co-cultured with antibodies against PD-L1, Ki67 and cleaved caspase 3 and a biotin-labeled secondary antibody and then co-cultured with an avidin–biotin–peroxidase complex. Amino-ethylcarbazole chromogen was used for visualization. Images were captured through an Axio Vert A1 microscope (Carl Zeiss). The staining intensity was ranked as 0 (negative), 1 (low), 2 (medium) or 3 (high).
2.11. NAG assay and LysoTracker Red staining
NAG assays were conducted by a kit from Sigma–Aldrich (CS0780) as described before24. Briefly, cells treatments with TBM-1 were lysed in RIPA buffer (250 μL). Ten micrograms of cell lysates were recorded in triplicate for NAG activity in compliance with the protocol from the supplier. 50 nmol/L of LysoTracker Red DND-99 dye (Thermo Fisher, Shanghai, China) was used to label the lysosomes at 37 °C for 30 min23. The medium was aspirated and washed two times with PBS quickly so that the unbound dye can be removed. Red fluorescence was detected using an Axio Vert A1 microscope (Carl Zeiss).
2.12. Coupling of TBM-1 to epoxy-activated sepharose beads
TBM-1 was coupled to Epoxy-activated Sepharose 6 B (GE Healthcare, Piscataway, NJ, USA) via stable ether linkages to hydroxyl groups. TBM-1 and Epoxy-activated Sepharose 6 B were mixed in PBS buffer for 24 h, and then washed with three cycles of alternating pH. Each cycle is comprised of a wash with 0.1 mol/L acetate pH 4.0 buffer including 0.5 mol/L NaCl followed by a wash with 0.1 mol/L Tris-HCl pH 8 buffer including 0.5 mol/L NaCl.
2.13. Surface plasmon resonance analysis
The BIAcore T200 biosensor system (GE Healthcare) was used to perform the surface plasmon resonance experiments at 25 °C22. TBM-1 was dissolved in 10 mmol/L sodium borate of pH 8.5 and immobilized on an activated CM5 dextran chip through amine-coupling chemistry (GE Healthcare). 1 mol/L pH 8.3 ethanolamine was used to block the remaining active sites. In 1 × PBS buffer containing 0.05% Tween-20 (Sigma–Aldrich), pH 7.4, the TBM-1 binding of rhmTOR at different concentrations was performed at a flow rate of 20 μL/min for 2 min. BIAevaluation software 2.0 was used to analyze the binding kinetics by using the 1:1 Langmuir binding model.
2.14. Cellular thermal shift assay
Cellular thermal shift assay (CETSA) was conducted to evaluate the stabilization of targets in cells by compound interaction as described previously25. Briefly, 80% confluency cells grown in 100-mm dishes were treated with 20 μmol/L TBM-1 or DMSO for 120 min. Cells were collected into PBS with protease inhibitors. The cell suspension was then added into 5 PCR tubes, heated for 10 min to 40, 43, 46, 49 or 52 °C followed by 3 cycles of freeze-thawing with liquid nitrogen and centrifugation at 4 °C, 17,000×g for 20 min. The soluble fractions were analyzed using SDS-PAGE and immunoblotted with anti-mTOR antibody.
2.15. Molecular modelling
The TBM-1 molecular docking with 3D structure of mTOR (PDB code: 4JT5) was performed by Discovery Studio 4.5 and UCSF chimera1.7. To detect the important amino acids in the predicted binding pocket, the regularized protein was used. Interactive docking for all the TBM-1 conformers to the selected active site was conducted by CDOCKER after energy minimization. A score was assigned to the docked compound based on its binding mode onto the binding site.
2.16. Statistics
Results were presented as mean ± standard deviation (SD) and examined by GraphPad Prism 6 software (GraphPad Software, La Jolla, CA, USA). The unpaired two-tailed Student's t-test and ANOVA with a Tukey's post hoc test were used to analyze the statistical significance between groups. The P-value <0.05 was considered statistically significant.
3. Results
3.1. Identification of TBM-1 as a potent negative modulator of PD-L1
To identify novel compounds that could reduce the PD-L1 abundance in cancer cells, we evaluated the ability for a set of 282 natural compounds to alter PD-L1 level in human lung cancer H157 cells by flow cytometry-based assay. The compounds (5 μmol/L) were added to the culture medium of H157 cells, and the PD-L1 levels were detected by flow cytometry analysis after 36 h of incubation. We found approximately 19 agents that reduced cell surface PD-L1 level (Tables S1 and S2). As MHC-I is indispensable for antigen presentation and T cell recognition, we further investigated whether these hit compounds affected MHC-I abundance. Flow cytometry results showed that tubeimoside-1 (TBM-1) and cinobufagin (Cbf) decreased PD-L1 abundance of cancer cells without reducing the level of MHC-I on the cell surface (Fig. 1A and B, Supporting Information Fig. S1A and Table S2). However, cinobufagin (5 μmol/L) treatment resulted in a high percentage of cell death (Fig. 1C), thus it was excluded from the following studies. In addition, TBM-1 (5 μmol/L) failed to affect H157 cell viability (Fig. S1B), and TBM-1 exhibited little or no direct toxicity towards LLC and B16 cells at concentrations lower than 5 μmol/L (Fig. S1C). Therefore, 0–5 μmol/L TBM-1 was used in the following experiments of this study.
Figure 1.

TBM-1 decreases PD-L1 abundance effectively in tumor cells. (A) PD-L1 and MHC-I levels in H157 cells treatment with 5 μmol/L of 19 selected compounds for 36 h were analyzed by flow cytometry. Results are shown as fold change by comparing mean fluorescence intensity (MFI) of PD-L1 and MHC-I to vehicle-treated cells. TBM-1, tubeimoside-1; Cbf, cinobufagin. (B) Chemical structure of TBM-1. (C) Cell death effect of TBM-1 (5 μmol/L) and cinobufagin (5 μmol/L) on H157 cells were determined by cell impedance assay. (D) Flow cytometry analyzing the membrane PD-L1 levels in H157 and A375 cells treatment with TBM-1 (5 μmol/L) for 24 h. Statistic of MFI of PD-L1 is shown at the right. (E) H460, H157, A375 and A2058 cells were treated with indicated concentrations of TBM-1 for 24 h, or treated with 5 μmol/L of TBM-1 for the indicated times, the PD-L1 levels were detected by immunoblotting. Quantifications of PD-L1 to GAPDH were shown at the right. (F) Cellular PD-L1 and IDO1 levels in A549 or H1299 cells pretreatment with TBM-1 (5 μmol/L, 2 h) and IFN-γ (5 ng/mL) stimulation for 24 h without removing TBM-1 were determined by immunoblotting. Quantifications of PD-L1 to GAPDH were shown on the bottom. (G) Membrane PD-L1 levels in A549 or H1299 cells pretreatment with TBM-1 (5 μmol/L, 2 h) and IFN-γ (5 ng/mL) stimulation for 24 h without removing TBM-1 were detected by flow cytometry. Statistic MFI of PD-L1 is shown. Data are presented as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ns, no significant.
We attempted to further validate the ability of TBM-1 to reduce PD-L1 abundance in cancer cells. As presented in Fig. 1D, the PD-L1 levels on the cell surface of H157 and A375 cells were concentration-dependently reduced by TBM-1. In addition, TBM-1 induced decrease of PD-L1 abundance in a both time-dependent and dose-dependent manner in NSCLC cells H460, H157 as well as melanoma cells A375 and A2058 (Fig. 1E). The abundance of other immune inhibitory ligands, for example, PD-L2 and Siglec-15, were not attenuated in cells treated with TBM-1 (Fig. S1D), indicating that TBM-1 selectively reduced PD-L1 abundance in tumor cells. Since IFN-γ has been identified to be a main inducer of PD-L1 upregulation in the tumor microenvironment26, we then examined whether TBM-1 alters inductive PD-L1 expression. Indeed, TBM-1 attenuated the abundance of PD-L1 induced by IFN-γ in both A549 and H1299 cells, while there were no significant differences in IFN-γ-induced IDO1 in cells treated with TBM-1 compared with control (Fig. 1F). In addition, flow cytometry assay showed that TBM-1 dramatically attenuated inductive PD-L1, but not CD155, on the cell surface of IFN-γ-stimulated A549 and H1299 cells (Fig. 1G and Fig. S1E). Altogether, these results indicate that TBM-1 may be a specific negative modulator of PD-L1 in various tumor cells.
3.2. The regulation of TBM-1 on PD-L1 affects PD-1 binding and T cell-dependent cytotoxicity
It has been verified that PD-L1 on cancer cells binds to PD-1, the homologous receptor on tumor-infiltrating T cells, and shuts down their antitumor activity27. To examine whether TBM-1-mediated PD-L1 inhibition would influence their ability to bind to PD-1, a fusion protein consisting of human PD-1 and Fc IgG was incubated with tumor cells10 treated with TBM-1. Both flow cytometry and immunofluorescence assays showed remarkably decreased binding of PD-1 to the tumor cell surface in TBM-1-treated H157 cells and A375 cells (Fig. 2A and B, Supporting Information Fig. S2A). Moreover, PD-1/PD-L1 blockage assay also demonstrated that transcriptional-mediated bioluminescent signal was significantly induced in both H157 and A375 cells treated with TBM-1 in a concentration-dependent manner (Fig. 2C). These results suggest that TBM-1 disrupted PD-1/PD-L1 interaction thus suppressed PD-L1's checkpoint activity. Consistently, TBM-1 rendered tumor cells more sensitive to T cell killing, as demonstrated by the cytotoxicity of activated human peripheral blood mononuclear cells (PBMCs) or Jurkat T cells toward co-cultured cancer cells determined by cell impedance assay (Fig. 2D and Fig. S2B). To further evaluate the antitumor effect of TBM-1, H157 or A375 cells were co-cultured with Jurkat T cells and the surviving tumor cells were detected by crystal violet staining. Compared with control, TBM-1 remarkably reduced the survival of H157 or A375 cells (Fig. 2E and Fig. S2C). In summary, the above data demonstrate that TBM-1 alters PD-1 binding and increases the cytotoxicity of T cells toward cancer cells through regulating PD-L1 expression.
Figure 2.

TBM-1 attenuates the ability of tumor cell binding PD-1 and enhances the cytotoxicity of T cells. (A) PD-1 binding to H157 cells treatment with indicated doses of TBM-1 for 24 h was measured by flow cytometry. The y axis represents the MFI of PD-1. (B) Immunostaining of recombinant Fc-PD-1 on H157 cells treated with TBM-1 (2 and 5 μmol/L, 24 h). The nuclei were stained with Hoechst 33,342. (Scale bar, 50 μm). The intensity of green fluorescence indicates bound PD-1. (C) The PD-1/PD-L1 interaction between Jurkat NFAT-luciferase reporter cells and H157 or A375 cells treated with TBM-1 (2 or 5 μmol/L) for 16 h was detected by PD-1/PD-L1blockade assay. Data are presented as fold induction over untreated control. (D) Human PBMC cells toward cancer cells killing in H157 cells treated with TBM-1 (2 or 5 μmol/L) for 24 h were analyzed by cell impedance assay. (E) Activated Jurkat T cell and H157 cells were co-cultured in 12-well plates for 2 days in the presence of TBM-1. Crystal violet staining was used to monitor the surviving cancer cells. Relative surviving cell intensity is shown at the right. Data are presented as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
3.3. TBM-1 inhibits the growth of tumor xenografts in vivo by activating tumor-infiltrating T cells
To determine the antitumor potential of TBM-1 in vivo, we subcutaneously inoculated LLC and B16 melanoma cells into C57BL/6 mice and monitored the tumor growth (Supporting Information Fig. S3A). We found that tumor grew rapidly in PBS-treated mice, but the tumor growth was effectively suppressed in TBM-1 (2 and 4 mg/kg)-treated LLC tumor xenografts mice (Fig. 3A). In addition, TBM-1 treatment significantly decreased the tumor size (Fig. 3B) and tumor weights (Fig. 3C). Similarly, TBM-1 effectively suppressed tumor growth in B16 melanoma-bearing mice (Fig. 3D–F). Consistent with the antitumor efficacy of TBM-1, treatment of TBM-1 did not cause a significant change in body weight (Fig. S3B) or systemic toxicity (Fig. S3C and S3D). Our data thus suggest that TBM-1 possesses inherent antitumor effect. Consistently, in nude mice with defective T cell function, TBM-1failed to suppress the tumor growth of LLC lung carcinoma and B16 melanoma (Fig. 3G and Fig. S4A). T-cell-dependent antitumor effect of TBM-1 was further evidenced via T-cell depletion by CD3+ antibody in C57BL/6 mice (Fig. 3H and Fig. S4B). Tumor-infiltrating lymphocyte (TILs) profile analysis was carried out to investigate TBM-1-mediated antitumor T-cell immunity. An elevation in the population of CD8+ T cell and the ratio of CD8+ to CD4+ T cell were clearly observed from the tumor of TBM-1-treated mice (Fig. S4C), demonstrating that TBM-1 enhances T cell infiltration in tumor tissues. In addition, the population of activated CD69+ CD137+ CD8+ T cell within tumor microenvironment from mice treated with TBM-1 was substantially upregulated compared with control (Fig. 3I and Fig. S4D), implying that TBM-1 enhances the activation of effective T cells in vivo. Moreover, a significant decrease in the levels of PD-L1 and Ki67 (a marker of proliferation) and an increase in cleaved caspase 3 level were found in LLC tumor xenografts after TBM-1 treatment (Fig. 3J), indicating an obvious apoptosis of tumor cells in the TBM-1-treated mice.
Figure 3.

TBM-1 mediated a T-cell dependent antitumor effect. (A)–(C) C57BL/6 mice with subcutaneous LLC tumor were i.p. injected with PBS or TBM-1 (a: 1 mg/kg, b: 2 mg/kg, c: 4 mg/kg), the tumors were ex vivo observed from the PBS or TBM-1 treated mice (A), the tumor volume (B) and tumor weight (C) were monitored for 14 days. Data are presented as mean ± SD, n = 7. (D)–(F) C57BL/6 mice with subcutaneous B16 melanoma were i.p. treated with PBS or TBM-1 (a: 1 mg/kg, b: 2 mg/kg, c: 4 mg/kg), the tumors were ex vivo observed from the PBS or TBM-1 treated mice (D), the tumor volume (E) and tumor weight (F) were monitored for 14 days. (G) BALB/c nude mice bearing LLC tumor were daily i.p. treatment with PBS or TBM-1 (2 mg/kg) for 14 days. Tumor weight was recorded. (H) C57BL/6 mice bearing Leiws tumor were received with anti-CD3 neutralizing antibody and/or TBM-1 for 14 days. Tumor growth was measured. (I) C57BL/6 mice bearing LLC tumor were i.p. treated with PBS or TBM-1 (2 mg/kg). Flow cytometry analyzing the population of CD69+ CD137+ in CD3+CD8+ TILs. Data are presented as mean ± SD, n = 6. (J) Representative IHC staining results for PD-L1, Ki67 and cleaved caspase 3 in PBS or TBM-1 treated mice, Scale bar, 200 μm. Quantification of IHC staining is shown. HPF, high power field. Data are presented as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ns, no significant.
3.4. TBM-1 promotes PD-L1 degradation in a proteasome and autophagosome-independent lysosomal pathway
Next, we investigated how TBM-1 reduced the abundance of PD-L1. It was demonstrated by the real-time PCR that TBM-1 had no effect on the mRNA level of PD-L1 (Supporting Information Fig. S5A). However, half-life analysis in the presence of protein translation inhibitor CHX indicated that PD-L1 degraded faster in cells treated with TBM-1 than in untreated cells (Fig. 4A). In addition, the exogenously overexpressed PD-L1 was also reduced by TBM-1 treatment (Fig. 4B). These data suggest TBM-1-induced PD-L1 degradation primarily occurred at the post-translational level. Previous studies have shown PD-L1 undergoes both proteasome- and autophagic-lysosomal-dependent degradation pathways6,9,28. However, TBM-1-induced PD-L1 degradation was not reversed by the proteasomal degradation inhibitors MG132 or PS-341 (Fig. S5B). Meanwhile, TBM-1 had no effect on ubiquitination of PD-L1 (Fig. S5C). The above data reveal that TBM-1 induces PD-L1 degradation independent of the ubiquitin–proteasome pathway.
Figure 4.

TBM-1 induces lysosome-dependent degradation of PD-L1. (A) Immunoblotting detecting the PD-L1 abundance in H157 cells treatment with DMSO or TBM-1 (5 μmol/L) for the indicated time periods in the presence of CHX (25 μg/mL). The PD-L1 abundance was normalized to GAPDH and quantification of PD-L1 intensity is shown on the right. (B) Myc-PD-L1 was transfected to H157 cells for 24 h, followed by TBM-1 treatment for 16 h. The level of Myc-PD-L1 was measured by immunoblotting. Quantification of Myc-PD-L1 to GAPDH was shown at the right. (C) Immunoblotting detecting the PD-L1 level in H157 and A375 cells pre-treated with indicated dose of 3-MA (10 mmol/L), followed by 5 μmol/L TBM-1 treatment for 24 h. Quantification of PD-L1 to GAPDH was shown at the right. (D) H157 cells were transfected with indicated siRNAs for 48 h, followed by TBM-1 (5 μmol/L) treatment for 24 h. The PD-L1 level was measured by immunoblotting. Quantification of PD-L1 to GAPDH was shown at the right. (E) and (F) Immunoblotting determining the PD-L1 level in H157 and A375 cells pre-treated with indicated dose of Baf (25 nmol/L) (E), or CQ (20 μmol/L) (F) followed by 5 μmol/L TBM-1 treatment for 24 h. Quantifications of PD-L1 to GAPDH were shown on the bottom. (G) Images and quantification of LysoTracker Red in H157 and A375 cells treatment with TBM-1 (2 or 5 μmol/L) for 12 h (scale bar, 50 μm). Quantification of LysoTracker Red staining is shown. (H) H157 and A375 cells were treated with indicated doses of TBM-1 for 6 h and lysosomal NAG activities were measured. (I) Immunofluorescence analyzing the co-localization between PD-L1 and LAMP-1 in H157 cells treatment with TBM-1 (5 μmol/L) for 12 h. The intensity profiles of PD-L1 and LAMP1 are shown in the right panel, with the co-localizing sites marked by green arrows. Data are presented as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Next, we explored whether TBM-1 promoted PD-L1 degradation through autophagic-lysosomal pathway. Inhibiting autophagosomes formation by 3-MA did not reverse the TBM-1-induced PD-L1 degradation (Fig. 4C). Furthermore, when ATG5 and ATG7, two key molecules in autophagy, were knocked down in H157 cells (Fig. S5D), the TBM-1-induced loss of PD-L1 was not reversed (Fig. 4D). Interestingly, TBM-1-mediated PD-L1 degradation was significantly prevented by co-incubation with specific lysosomal acidification and lysosome enzyme inhibitors, such as bafilomycin A1 (Baf) and choloroquin (CQ) (Fig. 4E and F). These results reveal that TBM-1 regulates PD-L1 expression in an autophagosome-independent lysosomal pathway. In support of this conclusion, Lysotracker Red staining was significantly induced by TBM-1 (Fig. 4G). Meanwhile, lysosomal protease activities were obviously increased in H157 and A375 cells treated with TBM-1, as determined by β-N-acetylglucosaminidase (NAG) assays (Fig. 4H), reinforcing the conclusion that TBM-1 triggers biogenesis of lysosomes and PD-L1 degradation.
As previous study revealed that a tyrosine based motif YWHL signal peptide in PD-L1 enables its intracellular locating in the lysosomal compartment29, we next investigated whether TBM-1 may affect PD-L1's distribution to lysosome. A significant increase was found in co-localization of PD-L1 with both LAMP1 in H157 cells (Fig. 4I) and Lysotracker Red in A375 cells (Fig. S5E). These data suggest that TBM-1 triggered PD-L1 translocation to lysosome. Collectively, these findings consistently demonstrate that TBM-1 induces biosynthesis of functional lysosomes and facilitates the translocation of PD-L1 to lysosomes for degradation.
3.5. TBM-1 promotes TFEB nuclear translocation in cancer cells
It has been established that TFEB, TFE3 and MITF trigger lysosome biogenesis and protein degradation30,31. We investigated whether TBM-1-mediated lysosome biogenesis and PD-L1 degradation acts through TFEB, TFE3 or MITF. TBM-1-induced lysosome increase (Fig. 5A) and PD-L1 degradation were significantly suppressed by small interfering RNA (siRNA) silencing of TFEB, but not TFE3 or MITF (Supporting Information Fig. S6A and S6B). These findings reveal that TBM-1 triggers lysosome biogenesis through TFEB. Consistent with this, TBM-1 promoted the nuclear translocation of EGFP-tagged TFEB instead of EGFP-MITF or TFE3 (Fig. 5B and Fig. S6C). Furthermore, cell fractionation analysis revealed that treatment with TBM-1 increased TFEB in the nuclear fraction of H157 and A375 cells (Fig. 5C). Phosphorylation regulates the subcellular localization and activity of TFEB11. Treatment with TBM-1 accelerated the electrophoretic mobility of TFEB (Fig. S6D), and reduced the phosphorylation level of TFEB Ser211 (Fig. 5D), which binds to 14-3-3 protein and retains TFEB in the cytoplasm32,33. Consistently, TBM-1 treatment greatly reduced the interaction between TFEB and 14-3-3 (Fig. 5E and Fig. S6E). These data demonstrate that TBM-1 triggers the nuclear translocation of TFEB by inhibiting TFEB phosphorylation. TFEB can directly bind to CLEAR element in the promoters of lysosomal genes34. Not surprisingly, a remarkable increase in the CLEAR luciferase activity was induced by TBM-1 in a concentration-dependent manner (Fig. S6F).
Figure 5.

TBM-1 triggers TFEB nuclear translocation. (A) H157 cells were transfected with TFEB siRNA, TFE3 siRNA, MITF siRNA, followed by TBM-1 (5 μmol/L) treatment for 12 h. Images and quantification of lysosomes (indicated as LysoTracker staining) were determined (scale bar, 50 μm). (B) Images and quantification of the subcellular locations of EGFP-TFEB, EGFP-MITF in H157 cells treatment with DMSO or TBM-1 (5 μmol/L) for 12 h. (scale bar, 20 μm). (C) Immunoblotting detecting the cytosolic and nuclear distribution of TFEB in H157and A375 cells treatment with DMSO or TBM-1 (5 μmol/L, 12 h). Quantifications of nuclear TFEB to H3 were shown at the right. (D) Immunoblotting determining the levels of TFEB Ser211 phosphorylation in H157 and A375 cells treated with indicated TBM-1 for 12 h. Quantifications of p-TFEB to TFEB are shown at the right. (E) H157 and A375 cells were treated with 5 μmol/L TBM-1 for 12 h. 14-3-3 levels were measured by immunoblotting after immunoprecipitation of TFEB from cell lysates. Quantifications of 14-3-3 to TFEB were shown. (F) and (G) Control siRNA or TFEB siRNA were transfected to H157 cells for 2 days, followed by DMSO or TBM-1 (5 μmol/L) treatment for 12 h. The levels of LAMP1, PD-L1 (F) and relative lysosomal NAG activity (G) were determined. Data are presented as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001, ns, no significant.
In addition, TFEB siRNA was used to further examine the relevance of TFEB on TBM-1 action. Silence of TFEB significantly reversed TBM-1-induced LAMP 1 expression and PD-L1 degradation (Fig. 5F). Furthermore, TFEB knockdown also abolished the lysosome NAG activity induced by TBM-1 (Fig. 5G). In conclusion, TBM-1 promotes TFEB nuclear translocation and induces PD-L1 degradation in cancer cells.
3.6. TBM-1 directly binds to and inactivates mTOR
Protein kinase C (PKC)35, glycogen synthase kinase-3β (GSK-3β), mammalian target of rapamycin complex 1(mTORC1)33, extracellular signal-regulated kinase 2 (ERK2), and lysosomal Ca2+36 have been suggested to be related to regulation of the transcriptional activity of TFEB. To dissect the mechanism by which TBM-1 induces TFEB nuclear translocation, we first tested whether PKC, GSK-3β, ERK1/2 or Ca2+ is required for the action of TBM-1. Co-incubation with the specific inhibitors of PKC (GO6983), GSK-3β (AR-A), ERK1/2 (LY3214996) or Ca2+ signaling (CGP37157) has no effect on TBM-1-induced TFEB nuclear translocation (Fig. S7A). In addition, TBM-1 did not affect the phosphorylation levels of PKC, GSK-3β and ERK1/2 (Fig. S7B). As TBM-1 reduced the phosphorylation of the Ser211 in TFEB (Fig. 5D), we thus investigated whether TBM-1 may inhibit mTOR activity. TBM-1 greatly suppressed the phosphorylation of p70 S6 kinase (p70S6K) and eIF4E-binding protein 1 (4EBP1), the known mTORC1 downstream substrate (Fig. 6A). However, TBM-1 did not affect the phosphorylation of AKT, an mTORC2 substrate (Fig. 6A). These findings suggest that TBM-1 works through a molecular mechanism that involved in inactivation of mTORC1, but not mTOCR2.
Figure 6.

TBM-1 binds to mTOR and inactivates its activity. (A) Immunoblotting determining the phosphorylation of p70S6K, 4EBP1 and AKT in H157 and A375 cells treatment with indicated doses of TBM-1 for 12 h. Quantifications of p-P70S6 K and p-4EBP1 are shown on the bottom. (B) Recombinant human mTOR (rhmTOR) or H157 cell lysate was incubated with Epoxy-activated sepharose beads (Epoxy) or TBM-1-immobilized Epoxy-activated Sepharose beads (Epoxy-TBM-1) and then immunoblotted with anti-mTOR antibody. Quantification of mTOR to GAPDH was shown on the bottom. (C) Molecular docking model revealed that TBM-1 binds to the FRB domain of mTOR. (D) SPR analysis of the TBM-1 and mTOR binding. The activated CM5 sensor chip was used to immobilize the recombinant human mTOR protein and flowed across TBM-1. (E) CETSA indicating mTOR target engagement by TBM-1 in H157 cells and A375 cells. Quantification of the thermal stability of mTOR was indicated on the bottom. (F) Parental and mTORKO H157 cell lysates were incubated with TBM-1-immobilized epoxy-activated sepharose beads and then immunoblotted with anti-mTOR antibody. (G) Parental and mTORKO H157 cells were treated with 5 μmol/L TBM-1 for 12 h. The levels of TFEB ser211 phosphorylation and PD-L1 were measured by immunoblotting. Quantification of PD-L1 to GAPDH was shown on the bottom. Data are presented as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01.
To determine whether TBM-1 binds to mTOR directly, we conjugated TBM-1 to Epoxy-activated Sepharose beads (Fig. S7C) and then incubated with recombinant human mTOR protein or cell lysate. We found that recombinant mTOR and cell lysate-mTOR could be affinity-coupled by Epoxy-TBM-1 (Fig. 6B). In addition, a molecular docking model of TBM-1 with X-ray crystal structure of mTOR (PDB Id: 4JT5) showed that TBM-1 was docked into the FRB domain of mTOR (Fig. 6C). Surface plasmon resonance (SPR) assay was used to further verify the direct interplay between TBM-1 and mTOR. The RU values, a parameter to evaluate the TBM-1 binding to the mTOR protein, displayed a dose-dependent manner and the determined equilibrium dissociation constant (KD) was about 11.34 μmol/L (Fig. 6D). In living cells, thermal stabilization of a cellular protein will be enhanced when binding with a chemical agent37. Cellular thermal shift assay (CETSA)37,38 demonstrated that mTOR was physically engaged and stabilized against thermal changes in H157 and A375 cells treated with TBM-1 (Fig. 6E). Consistent with these results, Epoxy-immobilized TBM-1-captured mTOR was dramatically reduced in CRISPR-Cas9-engineered mTOR knock out H157-cell and A375-cell in contrast to H157 and A375 parental cell (Fig. 6F). In addition, TBM-1-induced loss of TFEB Ser211 phosphorylation and PD-L1 degradation were abolished in CRISPR-Cas9-engineered mTOR knockout H157 cells (Fig. 6G). Collectively, these findings consistently reveal that mTOR is a specific target for the action of TBM-1.
3.7. The combination of TBM-1and anti-CTLA-4 effectively suppresses tumor growth
Combinatorial therapy of anti-PD-1 and anti-CTLA-4 has achieved great success in improving the response and survival rates among cancer patients39, we next investigated whether combination treatments of TBM-1 and anti-CTLA-4 have synergistic antitumor effects. To this end, mice bearing LLC were treated with PBS, TBM-1, anti CTLA-4 antibody, or the combination (Supporting Information Fig. S8A). We found that while TBM-1 or anti-CTLA-4 alone both reduced mouse tumor burden from control, combination treatments of TBM-1 and CTLA-4 antibody further decreased xenograft tumor volume (Fig. 7A and B, and Fig. S8B). In addition, combined treatment with TBM-1 and anti-CTLA-4 led to greater increase in the number of LLC tumor-infiltrating CD8+ T cells in TILs than each treatment alone (Fig. 7C and Fig. S8C). IFN-γ plays a key role in T-cell-mediated antitumor immunity40. TBM-1 and anti-CTLA-4 combination treatment led to a remarkable increase in IFN-γ production by tumor-infiltrating CD8+ T cells compared to that in each agent alone (Fig. 7D), demonstrating that TBM-1 activates cytotoxic T cell in TILs. The analysis of the GzmB level, which indicates cytotoxic T cell activity, reinforced our point that the cytotoxic T cell activity was increased after TBM-1 and anti-CTLA-4combination treatment (Fig. 7E).
Figure 7.

The combination of TBM-1and CTLA-4 blockage suppresses tumor growth. C57BL/6 mice bearing LLC were treated with PBS, anti-CTLA-4, TBM-1 or the combination. (A) and (B) The LLC tumors were ex vivo observed (A) and tumor growth (B) was measured for 14 days. (C) Flow cytometry analyzing the populations of CD8+ T cells in CD3+ CD45+ TILs. (D) and (E) Flow cytometry detecting the IFN-γ (D) and GzmB level (E) in CD3+CD8+ TILs from PBS, anti-CTLA-4, TBM-1 or the combination-treated LLC. (F) and (G) Representative flow cytometric plots and quantification of MDSCs (F) and Tregs (G) populations in PBS, anti-CTLA-4, TBM-1 or the combination-treated LLC. Data are presented as mean ± SD, n = 5. (H) Tumor growth of mTORKO LLC with PBS, anti-CTLA-4, TBM-1 or the combination treated C57BL/6 mice (n = 6). Data are presented as mean ± SD, n = 6; ∗P < 0.05; ∗∗P < 0.01, ∗∗∗P < 0.001, ns, no significant.
MDSCs and Tregs are thought to be two major immunosuppressive populations within tumor microenvironment, promoting tumor immune escape by suppressing T-lymphocyte immunity41,42. Remarkably, mTOR signaling in tumor cells attracts MDSC accumulation16 and induces differentiation of CD4+ T cells into Tregs43, resulting in an immune suppressive environment. As revealed by flow cytometric analysis, a remarkable decrease was found in the accumulation of CD11b+Gr1+ MDSCs and CD4+CD25+FOXP3+ Tregs cells within the tumor tissue in TBM-1 and anti-CTLA-4 combination-treated group compared to those each agent alone (Fig. 7F and G), demonstrating that TBM-1 shifts the immune microenvironment from a suppressed state to an activated state.
To further determine whether mTOR is required for TBM-1-mediated antitumor effect, CRISPR-Cas9-based genome editing was used to knock out mTOR in LLC cells. TBM-1 failed to suppress the tumor growth of mTOR knockout LLC cells in mice (Fig. 7H), indicating that the tumor growth inhibition mediated by TBM-1 is mainly due to mTOR-dependent regulation. Overall, these findings suggest that combining TBM-1 with anti-CTLA-4 could provide additional therapeutic advantages in treating cancers with PD-L1 high expression.
4. Discussion
Targeting PD-L1/PD-1 axis by small-molecule drug is an attractive approach to enhance antitumor immunity. Our flow cytometry-based screening identified TBM-1 as a lead candidate for reducing the abundance of PD-L1, indicating that the natural compound TBM-1 may be repurposed for cancer treatment. TBM-1 possesses intrinsic antitumor activities through inhibiting cell growth, inducting autophagy and cycle arrest, and other mechanisms21. Here, we are the first to show that lower dose of TBM-1 remarkably suppressed the growth of LLC and melanoma by regulating the tumor microenvironment. We provide clear evidences that TBM-1 elicits a T-cell immune response to promote tumor eradication.
The most of previous studies suggest that TBM-1 suppresses the tumor growth via its direct toxicity to tumor cells21. Here, we found TBM-1 showed little or no direct toxicity towards cancer cells at concentration lower than 5 μmol/L (Fig. 1B, Fig. S1B and S1C). In addition, lower dose of TBM-1 attenuated the PD-L1 abundance in cancer cells thus blocked the direct interaction between PD-L1 and PD-1 and activated the immune microenvironment in tumor. Therefore, lower dose of TBM-1 exerts its antitumor effect through its immunomodulatory function. However, given the poor oral bioavailability of TBM-124, certain dosage forms like liposome, nanoparticle and micro emulsion may improve its bioavailability and facilitate the clinical use of TBM-1.
Studies more recently have reported new strategies involving designed proteolysis targeting chimeras (PROTACs)44 and lysosome-degradation system9 to deplete PD-L1. In line with this, our study clarified a role of TBM-1 in modulating TFEB-dependent lysosomal degradation of PD-L1 in cancer cells. We demonstrated that lower dose of TBM-1 treatment led to efficient nuclear translocation of TFEB as well as lysosome biogenesis, thus promoting lysosomal catabolic activity to clear PD-L1 (Figure 4, Figure 5). However, other study showed that high concentration of TBM-1 (10 μmol/L or above) inhibiting lysosomal function45. We thus utilized a highly self-quenched BODIPY-conjugated bovine serum albumin (DQ-BSA) which shows bright red fluorescence only when triggered by proteolytic degradation to test the effect of TBM-1 on lysosome. Indeed, 5 μmol/L TBM-1 increased the DQ-BSA fluorescent signal induced by proteolytic degradation, while high concentration of TBM-1 (10–20 μmol/L) decreased the DQ-BSA fluorescent signal (Fig. S6G). This result indicates that the regulation of lysosome by TBM-1 is highly dose dependent. In addition, TFEB knockdown can reverse the PD-L1 degradation induced by TBM-1 (Fig. 5F). Our findings thus related TFEB to tumor immunotherapeutic modality, which is in line with the latest report that activation of TFEB toggles the ability of TAMs to mediate antitumor immunity46. Interestingly, TBM-1 specifically activates TFEB instead of MITF or TFE3 (Fig. 5A and Fig. S6B), indicating that these three very similar transcription factors can be modulated by different mechanisms.
One of the interesting findings in our study is that TBM-1 selectively binds to and inactivates mTOR activity. Using affinity purification approach, SPR and CETSA assays, we demonstrate that TBM-1 directly associated with mTOR (Fig. 6B–F). Our findings suggest that TBM-1-mediated PD-L1 controlling mechanisms are via mTOR signaling. It appears that the effect of mTOR inhibition on PD-L1 varies among different studies47. In contrast to mTOR negatively regulated PD-L1 expression via reducing the levels of β-TrCP48, TBM-1-mediated mTOR inhibition resulted in decreased PD-L1 abundance in NSCLC and melanoma cells in our hands, which is consistent with previous studies that mTOR positively regulates PD-L1 expression13,49. Therefore, mTOR regulation of the abundance of PD-L1 is context-dependent. Since monotherapies mTOR inhibitors showed limited clinical success of blocking cancer progression50, various mTOR and immune checkpoint inhibitors combinations are in ongoing clinical trials for cancer treatment.
The immunosuppressive microenvironment is a major obstacle for successful tumor immunotherapy51. Reactivation of T cells in tumor microenvironment provides a promising and favorable therapeutic measure for boosting antitumor immunity. We provided evidence that TBM-1 increased T-cell activation in tumors. Besides reducing the PD-L1 level in tumor cells and increasing the CD8+ T-cell infiltration to promote tumor eradication (Figure 2, Figure 3), our data also suggested TBM-1 activated T cell indirectly through inhibiting MDSCs and Treg-mediated immune suppression (Fig. 7F and G). It has been proved that MDSCs and Tregs repress tumor-specific T-cell immune response by producing anti-inflammatory cytokines and growth factors such as IL10 and TGFβ52. Consistent with previous reports that mTORC1 in tumor cells recruits MDSCs through upregulating G-CSF16 and induces differentiation of naïve CD4+ T cell into Tregs by increasing PD-L1 expression13, our findings demonstrated that TBM-1 alleviated MDSCs and Tregs accumulation in tumor microenvironment by inhibition of mTOR-dependent PD-L1/PD-1 interaction. These findings suggest suppressing mTOR signaling in cancer cells regulates the tumor microenvironment. Further studies are warranted to validate whether TBM-1 may also modulate other immune cells in the tumor microenvironment.
It has been reported that ICB by anti-CTLA-4, anti- PD-1, or anti-PD-L1 amplify the antitumor T-cell response53. Because TBM-1 blocks the PD-L1/PD-1 pathway (Fig. 2), the combinatorial therapy of TBM-1 and anti-CTLA-4 may enhance antitumor effect. Our animal model showed that TBM-1 or anti-CTLA-4 alone failed to promote CD8+ T cell infiltration, while the combination treatment increased CD8+ T cell infiltration and induced markedly regression of the established tumors (Fig. 7). Consistent with our findings, CTLA-4 blockage in combination with mTOR inhibitor has synergistic efficacies in treatment of prostate cancer54. Additionally, compared with the immune-related adverse events (irAEs) accompanied by antibody-based ICB, lower dose of TBM-1 did not induce detectable toxicity (Fig. S3C and S3D). Therefore, our findings indicate that the combinational therapy of TBM-1 and CTLA-4 blockade enhances antitumor effect and suggest that TBM-1 has great potential in combination with immunotherapy.
5. Conclusions
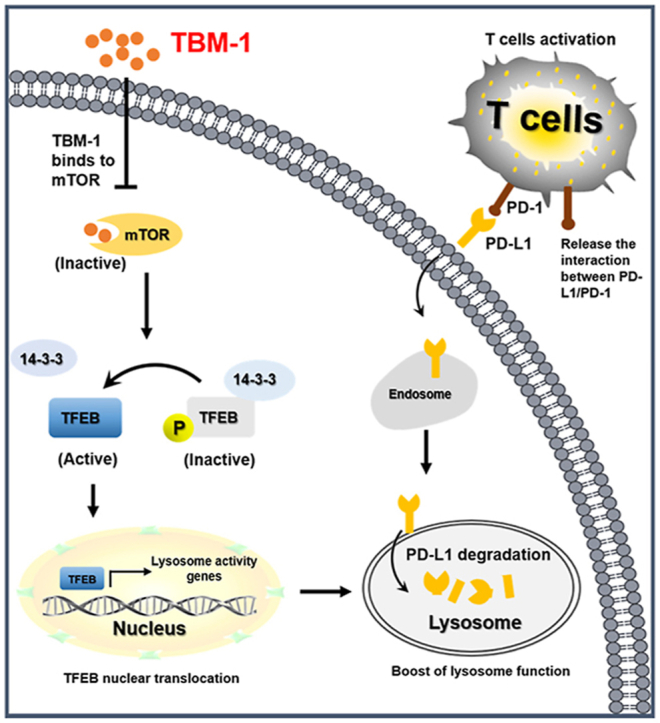
Our findings indicate that TBM-1, by virtue of its ability to bind to and subsequently inactivate mTOR, induces TFEB-dependent lysosomal degradation of PD-L1 in cancer cells and thus ameliorates the immunosuppressive tumor microenvironment and improves antitumor T-cell immunity (Fig. 8). Our study provides the theoretical basis for the potential application of TBM-1 as small molecule immune-modulator in improving the efficacy of current ICB therapy and advance cancer immunotherapy.
Figure 8.

Proposed model of TBM-1-mediated PD-L1 degradation by controlling mTOR-TFEB cascade to activate lysosome function. TBM-1 specific binds to and inactivates mTOR, which results in TFEB dephosphorylation and nuclear translocation, thus boosting lysosome biogenesis. Meanwhile, TBM-1 promotes PD-L1 translocation to lysosome, induces PD-L1 degradation and activation of tumor-infiltrating T cells.
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (81973366, 81773782, 81903695 and 82003792), CAMS Innovation Fund for Medical Sciences (2016-I2M-1-011, China), National Mega-project for Innovative Drugs (2019ZX09721-001, China) and Chinese Pharmaceutical Association-Yiling Pharmaceutical Innovation Fund for Biomedicine (GL-1-B04-20180366, China).
Author contributions
Hongbin Deng overall designed, supervised and coordinated the study. Xiaojia Liu and Mingxiao Yin designed and performed most of the experiments. Jingwen Dong, Zean Kuang and Na Zhang participated in the molecular and cellular biological experiments. Wenjian Min and Peng Yang did the molecular docking. Genxiang Mao provided natural product chemical monomers. Xiaojia Liu and Mingxiao Yin performed the animal studies and data analysis. Hongbin Deng and Xiaojia Liu wrote and revised the manuscript. All authors read and approved the manuscript.
Conflicts of interest
The authors declare no potential conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.03.039.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cha J.H., Chan L.C., Li C.W., Hsu J.L., Hung M.C. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359–370. doi: 10.1016/j.molcel.2019.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X., Pan X., Zhang W., Guo H., Cheng S., He Q. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723–733. doi: 10.1016/j.apsb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schildberg F.A., Klein S.R., Freeman G.J., Sharpe A.H. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. 2016;44:955–972. doi: 10.1016/j.immuni.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maj T., Wang W., Crespo J., Zhang H., Wang W., Wei S. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017;18:1332–1341. doi: 10.1038/ni.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burr M.L., Sparbier C.E., Chan Y.C., Williamson J.C., Woods K., Beavis P.A. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cha J.H., Yang W.H., Xia W., Wei Y., Chan L.C., Lim S.O. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–620. doi: 10.1016/j.molcel.2018.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Settembre C., Fraldi A., Medina D.L., Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H., Yao H., Li C., Shi H., Lan J., Li Z. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat Chem Biol. 2019;15:42–50. doi: 10.1038/s41589-018-0161-x. [DOI] [PubMed] [Google Scholar]
- 10.Zhang N., Dou Y., Liu L., Zhang X., Liu X., Zeng Q. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1. EBioMedicine. 2019;40:151–162. doi: 10.1016/j.ebiom.2019.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Napolitano G., Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–2481. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lastwika K.J., Wilson W., 3rd, Li Q.K., Norris J., Xu H., Ghazarian S.R. Control of PD-L1 expression by cncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76:227–238. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 14.Peng W., Chen J.Q., Liu C., Malu S., Creasy C., Tetzlaff M.T. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Covarrubias A.J., Aksoylar H.I., Yu J., Snyder N.W., Worth A.J., Iyer S.S. Akt–mTORC1 signaling regulates acly to integrate metabolic input to control of macrophage activation. Elife. 2016;5 doi: 10.7554/eLife.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welte T., Kim I.S., Tian L., Gao X., Wang H., Li J. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632–644. doi: 10.1038/ncb3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao H., Wang H., Li C., Fang J.Y., Xu J. Cancer cell-intrinsic PD-1 and implications in combinatorial immunotherapy. Front Immunol. 2018;9:1774. doi: 10.3389/fimmu.2018.01774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Donnell J.S., Massi D., Teng M.W.L., Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91–103. doi: 10.1016/j.semcancer.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Rodrigues T., Reker D., Schneider P., Schneider G. Counting on natural products for drug design. Nat Chem. 2016;8:531–541. doi: 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- 20.Yu T.X., Ma R.D., Yu L.J. Structure-activity relationship of tubeimosides in anti-inflammatory, antitumor, and antitumor-promoting effects. Acta Pharmacol Sin. 2001;22:463–468. [PubMed] [Google Scholar]
- 21.Islam M.S., Wang C., Zheng J., Paudyal N., Zhu Y., Sun H. The potential role of tubeimosides in cancer prevention and treatment. Eur J Med Chem. 2019;162:109–121. doi: 10.1016/j.ejmech.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y., Liu X., Zhang N., Yin M., Dong J., Zeng Q. Berberine diminishes cancer cell PD-L1 expression and facilitates antitumor immunity via inhibiting the deubiquitination activity of CSN5. Acta Pharm Sin B. 2020;10:2299–2312. doi: 10.1016/j.apsb.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X., Zhang N., Liu Y., Liu L., Zeng Q., Yin M. MPB, a novel berberine derivative, enhances lysosomal and bactericidal properties via TGF-beta-activated kinase 1-dependent activation of the transcription factor EB. FASEB J. 2019;33:1468–1481. doi: 10.1096/fj.201801198R. [DOI] [PubMed] [Google Scholar]
- 24.Chen L., Weng Q., Li F., Liu J., Zhang X., Zhou Y. Pharmacokinetics and bioavailability study of tubeimoside I in ICR mice by UPLC–MS/MS. J Anal Methods Chem. 2018;2018:9074893. doi: 10.1155/2018/9074893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E.A., Dan C. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 26.Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 28.Lim S.O., Li C.W., Xia W., Cha J.H., Chan L.C., Wu Y. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durand-Panteix S., Farhat M., Youlyouz-Marfak I., Rouaud P., Ouk-Martin C., David A. B7-H1, which represses EBV-immortalized B cell killing by autologous T and NK cells, is oppositely regulated by c-Myc and EBV latency III program at both mRNA and secretory lysosome levels. J Immunol. 2012;189:181–190. doi: 10.4049/jimmunol.1102277. [DOI] [PubMed] [Google Scholar]
- 30.Perera R.M., Zoncu R. The lysosome as a regulatory hub. Annu Rev Cell Dev Biol. 2016;32:223–253. doi: 10.1146/annurev-cellbio-111315-125125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 32.Roczniak-Ferguson A., Petit C.S., Froehlich F., Qian S., Ky J., Angarola B. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martina J.A., Chen Y., Gucek M., Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 35.Ferron M., Settembre C., Shimazu J., Lacombe J., Kato S., Rawlings D.J. A RANKL-PKCbeta-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013;27:955–969. doi: 10.1101/gad.213827.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundback T., Nordlund P. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 38.Reinhard F.B., Eberhard D., Werner T., Franken H., Childs D., Doce C. Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat Methods. 2015;12:1129–1131. doi: 10.1038/nmeth.3652. [DOI] [PubMed] [Google Scholar]
- 39.Callahan M.K., Postow M.A., Wolchok J.D. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol. 2014;4:385. doi: 10.3389/fonc.2014.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivashkiv L.B. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18:545–558. doi: 10.1038/s41577-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishikawa H., Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. doi: 10.1016/j.coi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Kumar V., Patel S., Tcyganov E., Gabrilovich D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delgoffe G.M., Kole T.P., Zheng Y., Zarek P.E., Matthews K.L., Xiao B. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng B., Ren Y., Cao H., Chen J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem. 2020;199:112377. doi: 10.1016/j.ejmech.2020.112377. [DOI] [PubMed] [Google Scholar]
- 45.Feng X., Zhou J., Li J., Hou X., Li L., Chen Y. Tubeimoside I induces accumulation of impaired autophagolysosome against cervical cancer cells by both initiating autophagy and inhibiting lysosomal function. Cell Death Dis. 2018;9:1117. doi: 10.1038/s41419-018-1151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen D., Xie J., Fiskesund R., Dong W., Liang X., Lv J. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. 2018;9:873. doi: 10.1038/s41467-018-03225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun S.Y. Searching for the real function of mTOR signaling in the regulation of PD-L1 expression. Transl Oncol. 2020;13:100847. doi: 10.1016/j.tranon.2020.100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng L., Qian G., Zhang S., Zheng H., Fan S., Lesinski G.B. Inhibition of mTOR complex 1/p70 S6 kinase signaling elevates PD-L1 levels in human cancer cells through enhancing protein stabilization accompanied with enhanced beta-TrCP degradation. Oncogene. 2019;38:6270–6282. doi: 10.1038/s41388-019-0877-4. [DOI] [PubMed] [Google Scholar]
- 49.Sun C., Mezzadra R., Schumacher T.N. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun S.Y. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 2013;340:1–8. doi: 10.1016/j.canlet.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabrilovich D.I., Ostrand-Rosenberg S., Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guri Y., Nordmann T.M., Roszik J. mTOR at the transmitting and receiving ends in tumor immunity. Front Immunol. 2018;9:578. doi: 10.3389/fimmu.2018.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson K.G., Stromnes I.M., Greenberg P.D. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell. 2017;31:311–325. doi: 10.1016/j.ccell.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu X., Horner J.W., Paul E., Shang X., Troncoso P., Deng P. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543:728–732. doi: 10.1038/nature21676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.